")
Back to Journals » OncoTargets and Therapy » Volume 7
Omacetaxine mepesuccinate in the treatment of intractable chronic myeloid leukemia
Received 21 August 2013
Accepted for publication 16 December 2013
Published 31 January 2014 Volume 2014:7 Pages 177—186
DOI https://doi.org/10.2147/OTT.S41786
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Yaoyu Chen,1 Shaoguang Li2
1Department of Oncology, Novartis Institutes for Biomedical Research, Cambridge, 2Division of Hematology/Oncology, Department of Medicine, University of Massachusetts Medical School, Worcester, MA, USA
Abstract: In a significant proportion of patients with chronic myeloid leukemia, resistance to BCR-ABL tyrosine kinase inhibitors develops due to acquisition of BCR-ABL kinase domain mutations and insensitivity of leukemia stem cells to tyrosine kinase inhibitors. Omacetaxine mepesuccinate (formerly called homoharringtonine) is a natural alkaloid that inhibits protein synthesis and induces cell death. Omacetaxine mepesuccinate has been recently approved by the US Food and Drug Administration to treat patients with chronic myeloid leukemia who failed to respond to multiple tyrosine kinase inhibitors and/or acquired the BCR-ABL-T315I mutation. In this review, we discuss the use and effectiveness of omacetaxine mepesuccinate in the treatment of chronic myeloid leukemia, with coverage of its pharmacology, mode of action, and pharmacokinetics. We believe that omacetaxine mepesuccinate will be beneficial to many patients with chronic myeloid leukemia who do not respond well to tyrosine kinase inhibitors.
Keywords: BCR-ABL, leukemic stem cells, chronic myeloid leukemia, biomarker, hematopoietic stem cells, cancer stem cells
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disease induced by the BCR-ABL oncogene. Pathologically, CML patients develop granulocytosis and splenomegaly. CML often begins with a chronic phase, and without proper treatment, the disease progresses to an accelerated phase and ultimately develops into a terminal phase called blast crisis. A hallmark of CML is acquisition of Philadelphia chromosome (Ph), resulting in formation of BCR-ABL.1 The Ph chromosome is cytogenetically diagnostic of CML. In 1960, Nowell and Hungerford made a landmark discovery of the Ph chromosome and its association with CML.1 This discovery was the first demonstration of a chromosomal rearrangement that is consistently linked to a specific cancer, and sparked searches for associations of additional chromosomal aberrations with specific forms of cancer. BCR-ABL, the product of the Ph chromosome, has deregulated tyrosine kinase activity, and different forms of BCR-ABL protein with different molecular weights can be generated in patients, depending on the precise breakpoints in the translocation or RNA splicing.2 The BCR-ABL transcript was shown to contain the first 13 to 14 BCR exons and exons 1 or 2 through 11 of ABL, generating a large mRNA product that, after splicing, encoded an 8.5 kB BCR-ABL chimeric transcript.3 The fusion of BCR to ABL during translocation increases the tyrosine kinase activity of ABL, and brings new regulatory domains/motifs to ABL, such as the growth factor receptor-bound protein 2 SH2-binding sites.4–6
CML in accelerated phase and blast crisis is much more diverse and aggressive than chronic phase CML. Over 80% of patients in blast phase have definable genetic aberrations in addition to the Ph chromosome.7 Those genetic aberrations including trisomy 8, i(17q),7,8 loss of p53 function,9,10 MYC amplification,8 RB deletion/rearrangement,11 and p16INK4A (CDKN2) rearrangement/deletion12 have been reported to be associated with blast crisis.
Treatment of CML
Before BCR-ABL tyrosine kinase inhibitors (TKIs) were available, allogeneic bone marrow transplantation was the recommended treatment for patients newly diagnosed with CML. The probability of survival and leukemia-free survival of bone marrow transplant recipients at 8 years was 50%–60%, with a low chance of relapse.13,14 After receiving bone marrow transplantation, the majority of long-term survivors could be regarded as operationally “cured”, even if some patients still harbored quiescent leukemia cells.15 In 2001, the first BCR-ABL kinase inhibitor, imatinib mesylate (Gleevec®/Glivec®, formerly STI571; Novartis, Basel, Switzerland), was approved by the US Food and Drug Administration as a first-line standard treatment for CML.16–18 The rate of complete cytogenetic response among patients receiving imatinib was 87% after 5 years of treatment.19 Although it effectively inhibits BCR-ABL kinase activity and improves survival in CML patients, imatinib does not appear to lead to a cure of the disease due to development of point mutations in the ATP binding region of BCR-ABL and the insensitivity of quiescent leukemic stem cells to imatinib. Acquisition of point mutations in the ATP binding region of BCR-ABL has been a major mechanism for development of resistance to imatinib and other BCR-ABL kinase inhibitors. BCR-ABL gene amplification also leads to increased expression of BCR-ABL tyrosine kinase and is associated with drug resistance.20 BCR-ABL-independent resistance mechanisms include effects on drug efflux, import, and binding.21 Further, variations in compound intracellular uptake and retention affect efficacy differences in patients.22 For example, three BCR-ABL mutations (T315I, Y253H, and F317L) have a predicted role in abrogating binding of imatinib to BCR-ABL in resistant patients.23 The second-generation BCR-ABL kinase inhibitor, dasatinib, binds to BCR-ABL with less stringent conformational requirements and was shown to be effective in inhibition of imatinib-resistant mutants.24 Nilotinib is another second-generation BCR-ABL inhibitor and is significantly more potent than imatinib, and also has activity against a number of imatinib-resistant BCR-ABL mutants.25 Compared with imatinib, nilotinib is associated with a reduced incidence of BCR-ABL mutations in patients with newly diagnosed CML in chronic phase.26 Based on a 3-year study, mutations were detected in approximately twice as many patients on imatinib (400 mg once daily) as on nilotinib (300 mg twice daily or 400 mg twice daily).26 Recently, ponatinib, a third-generation TKI, was approved and demonstrated to have remarkable antileukemia activity, particularly in patients with BCR-ABL-T315I mutations that are resistant to other TKIs.27 Ponatinib is a powerful pan-BCR-ABL TKI and is promising for patients with CML or Ph+ acute lymphoblastic leukemia who fail imatinib, dasatinib, and nilotinib.20 It is also active against T315I and other imatinib-resistant mutants.20 However, not all CML patients who are refractory or intolerant to dasatinib or nilotinib are responsive to ponatinib.20 Besides the development of TKI resistance, it has been shown that survival of primitive CML stem cells is not dependent on BCR-ABL kinase activity, and as a result, therapies that aim to inhibit BCR-ABL kinase activity could not eliminate leukemia stem cells in CML.28–31 Thus, a significant number of CML patients will take TKIs for the rest of their lives.32 We have shown that the resistance of leukemia stem cells to imatinib does not appear to involve BCR-ABL kinase domain mutations,33 suggesting that BCR-ABL activates some signaling pathways in a kinase-independent manner in leukemia stem cells.15 Some unique molecular pathways dependent on or independent of BCR-ABL kinase activity were identified to play important roles in the development and survival of leukemia stem cells in CML, including Wnt/β-catenin, Hedgehog, Alox5, PTEN, Src family kinases, and FoxO pathways.33–38 These newly identified genes may serve as potential targets for developing a curative therapy of CML.
Omacetaxine mepesuccinate was recently approved by the US Food and Drug Administration for treating CML patients with resistance or intolerance to two or more TKIs. It is a first-in-class cephalotaxine that has demonstrated efficacy in CML and was shown to produce clinically meaningful responses with acceptable tolerability in patients with chronic phase CML previously treated with two or more TKIs.39 In this paper, we discuss the pharmacology, mode of action, and pharmacokinetics of omacetaxine mepesuccinate, efficacy studies in both patients and animal models of CML, and the safety and tolerability of omacetaxine mepesuccinate as well as its future perspectives.
Pharmacology, mode of action, and pharmacokinetics
Pharmacology
Omacetaxine mepesuccinate is also known as homoharringtonine (HHT) [cephalotaxine, 4-methyl (2R)-2-hydroxy-2-(4-hydroxy-4-methylpentyl) butanedioate (ester)]. It is a natural cephalotaxine alkaloid (Figure 1), and is originally derived from the bark of several cephalotaxus species that are indigenous to Asia (mostly mainland China, and also Japan, Korea, and India).40 The molecular weight of omacetaxine mepesuccinate is 545.62 (g/mol).40 Historically, extracts from the bark of cephalotaxus were used in cancer patients by practitioners of traditional Chinese medicine in the Fujian Province of China.41 However, further development of HHT was hampered by a limited source supply and difficult production for high purity. The successful semisynthesis of HHT by direct esterification of the abundant biosynthetic precursor cephalotaxine was reported in 1999. The development of semisynthetic HHT was paralleled by development of the more convenient subcutaneous route of administration, with pharmacokinetic properties, clinical efficacy, and safety comparable with that of continuous infusion. Semisynthetic HHT is now produced as omacetaxine mepesuccinate (Synribo®, Teva Pharmaceutical Industries Ltd, Petah Tikva, Israel). It is worth mentioning that omacetaxine mepesuccinate is the only effective natural therapeutic agent used in patients with CML.
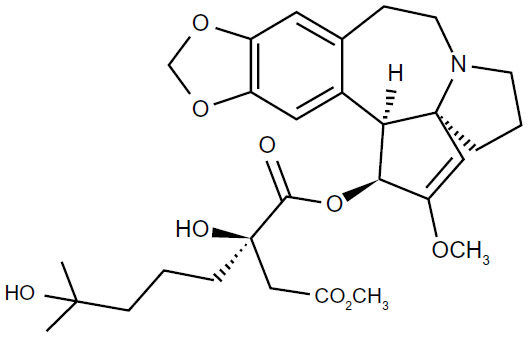 | Figure 1 The structure of omacetaxine mepesuccinate. |
Mode of action
Omacetaxine mepesuccinate functions as an inhibitor of protein synthesis, mechanistically preventing aminoacyl-tRNA binding to the ribosomal acceptor site and peptide bond formation at the early stage of protein elongation.41,42 Omacetaxine mepesuccinate may inhibit the elongation phase of translation by preventing substrate binding to the acceptor site on the 60-S ribosome subunit, leading to blockade of aminoacyl-tRNA binding and peptide bond formation.42 Further, omacetaxine mepesuccinate was shown to affect diphenylalanine synthesis and puromycin reaction instead of eEF-1-dependent Phe-tRNAPhe binding, and to exert a significant influence on polypeptide chain elongation.43,44 A recent study showed that omacetaxine mepesuccinate blocks protein synthesis by competing with the amino acid side chains of incoming aminoacyl-tRNAs for binding at the A-site cleft in the peptidyl transferase center.45
The mechanism for the antileukemic effect of omacetaxine mepesuccinate is mainly related to induction of apoptosis in leukemia cells (Figure 2).46 In imatinib-resistant K562 cells, omacetaxine mepesuccinate caused degradation of BCR-ABL proteins by inhibiting heat shock protein 90 in a dose-dependent manner,47 whereas heat shock protein 70, which mediates the resistance of BCR-ABL-expressing cells to apoptosis, was not changed by treatment with the drug. Omacetaxine mepesuccinate also induced intrinsically apoptotic pathways by activating caspase-9, caspase-8, and caspase-3 and downregulating myeloid cell leukemia-1 (MCL-1) in chronic lymphocytic leukemia48 and in myeloid leukemia cells.49 Although downregulation of MCL-1 was associated with a substantial decrease in viability of K562 cells, loss of MCL-1 did not significantly enhance the efficacy of omacetaxine mepesuccinate in primary B-cells or neutrophils.48 In BCR-ABL-induced B-cell acute lymphoblastic leukemia, treatment with omacetaxine mepesuccinate slightly reduced the level of nonmutated BCR-ABL proteins, but the level of BCR-ABL-T315I was markedly decreased, indicating that the mutant BCR-ABL is more sensitive to inhibition by omacetaxine mepesuccinate.47 Interestingly, compared with myeloid leukemia cells, the level of heat shock protein 90 in B-cell acute lymphoblastic leukemia cells was not reduced by treatment with omacetaxine mepesuccinate. The stability of the BCR-ABL-T315I mutant may be more sensitive to treatment with omacetaxine mepesuccinate and depends on alternative oncoprotein chaperones, such as heat shock protein 70 or Hsc70.
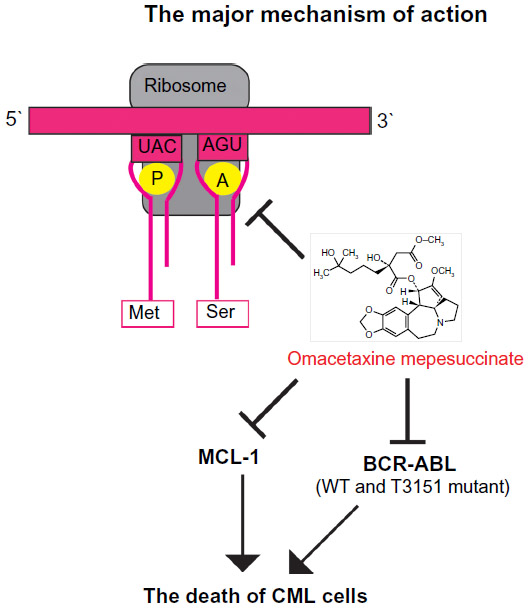 | Figure 2 The major mechanism of action of omacetaxine mepesuccinate. |
Omacetaxine mepesuccinate also had a significant inhibitory effect on leukemia stem cells. In a mouse model of CML, omacetaxine mepesuccinate was shown to greatly reduce the numbers of both leukemia stem cells and total leukemia cells, and treatment of CML mice using both imatinib and omacetaxine mepesuccinate did not further reduce the number of leukemia stem cells or total leukemia cells compared with mice treated using omacetaxine mepesuccinate alone.47 Omacetaxine mepesuccinate also had a similar inhibitory effect on BCR-ABL T315I-expressing leukemia stem cells compared with nonmutant BCR-ABL-expressing leukemia stem cells. On the other hand, when samples from patients with primary CML were tested, omacetaxine mepesuccinate effectively induced apoptosis of primary human CML stem cells (CD34+38low) by downregulation of MCL-1 proteins.50 In contrast with findings for TKIs, omacetaxine mepesuccinate did not cause accumulation of undivided cells in vitro. Further, the functions of surviving stem cells following treatment with omacetaxine mepesuccinate was significantly reduced in a dose-dependent manner, as determined by a colony-forming assay and the more stringent long-term culture-initiating cell colony assay.50
The cytotoxicity of omacetaxine mepesuccinate was thought to be caused by inhibition of cells in the G1 and G2 phases42 or differentiated noncycling cells,51 and through reduction in levels of pro-oncogenic or prosurvival proteins through shortening their half-lives, including those of MCL-1, cyclin D1, and c-MYC.52 Although more mechanistic studies are needed to understand fully the molecular mechanisms by which omacetaxine mepesuccinate suppresses CML cells, the various studies described above have consistently shown that MCL-1 is involved in inhibition of CML cells by omacetaxine mepesuccinate. Identification of other signaling pathways that interact with MCL-1 in CML cells is a logical next step to take in better understanding the mode of action of omacetaxine mepesuccinate.
Pharmacokinetics
Omacetaxine mepesuccinate is administered subcutaneously, and maximum concentration in blood is achieved within one hour of injection.53 Omacetaxine mepesuccinate is primarily hydrolyzed to 4′-DMHHT through plasma esterases with hepatic microsomal oxidative and/or esterase-mediated metabolism.53 On average, less than 15% of omacetaxine mepesuccinate is excreted unchanged in the urine, and its mean half-life following subcutaneous administration is around 6 hours.53 To understand its pharmacokinetics, omacetaxine mepesuccinate (1.25 mg/m2) was administered subcutaneously twice a day on days 1–14 every 28 days for two cycles, until disease progression or unacceptable toxicity occurred. Blood and urine were collected to measure concentrations of omacetaxine mepesuccinate and its inactive metabolites. Pharmacokinetic parameters were estimated from 21 patients with a diagnosis of relapsed or refractory CML, acute promyelocytic leukemia, acute myeloid leukemia, or myelodysplastic syndrome, or advanced solid tumors who had exhausted or become intolerant to all available therapies.53 Omacetaxine mepesuccinate is rapidly absorbed and widely distributed, as evidenced by an apparent volume of distribution of 126.8 L/m.53 Plasma concentration versus time data demonstrated biexponential decay and a mean steady-state terminal half-life of 7 hours. Concentrations of inactive metabolites, 4′-DMHHT and cephalotaxine, were approximately 10% those of omacetaxine mepesuccinate and undetectable in most patients.53
The clinical pharmacokinetics of omacetaxine mepesuccinate was similarly studied in eight patients who received uniformly labeled omacetaxine mepesuccinate at 3–4 mg/m2 by continuous 6-hour infusion.54 Unchanged omacetaxine mepesuccinate declined biphasically in plasma, with an α-half-life of 0.5±0.1 hours and a β-half-life of 9.3±1.4 hours. The total clearance of omacetaxine mepesuccinate was 177.4±27.7 mL/hour × kg, and the apparent volume of distribution, estimated from the area under the drug concentration versus time curve, was 2.4±0.4 L/kg.54
Omacetaxine mepesuccinate is mainly metabolized in the liver, but does not cause clinical liver toxicity.55 A major metabolite of omacetaxine mepesuccinate was identified as omacetaxine mepesuccinate acid, which was 700 times less toxic than omacetaxine mepesuccinate. Urinary excretion of unchanged omacetaxine mepesuccinate accounted for 15% of the dose.55 The major drug-related adverse effects included thrombocytopenia (48%) and neutropenia (33%).55 Although omacetaxine mepesuccinate appears to have more side effects than TKIs, it has a unique activity against TKI-insensitive CML stem cells (see below), making it a useful therapeutic agent in some patients who have failed to respond to TKIs.
Efficacy studies
Efficacy of omacetaxine mepesuccinate in treating CML
Omacetaxine mepesuccinate has been studied using BCR-ABL-expressing myeloid and lymphoid cell lines and mouse models of CML and B-cell acute lymphoblastic leukemia induced by nonmutant BCR-ABL or mutant BCR-ABL-T315I.47 It is exciting to see that more than 90% of leukemia stem cells are killed after treatment with omacetaxine mepesuccinate in vitro. Omacetaxine mepesuccinate effectively caused reduction of leukemia cells in CML and mice with B-cell acute lymphoblastic leukemia.47 Omacetaxine mepesuccinate also inhibited BCR-ABL T315I-expressing leukemia stem cells.47
Omacetaxine mepesuccinate was initially used in the early 1980s to treat CML patients in the People’s Republic of China,56 and two clinical studies later showed that omacetaxine mepesuccinate was an effective treatment for CML, as shown by induction of hematologic remission for longer than 12 months in the majority of patients in chronic phase.57,58 In a Phase I/II study of subcutaneous administration of omacetaxine mepesuccinate in CML patients who had failed prior therapy, efficacy and good tolerance were observed for omacetaxine mepesuccinate, and could be achieved when the same doses were administered by intravenous injection.59 The maximal tolerated dose was 1.25 mg/m2 subcutaneously twice daily. Six patients (median age 53 years) who had failed imatinib were treated with omacetaxine mepesuccinate, and five turned out to be evaluable.59 Among the five patients, a complete hematologic response (CHR) was achieved in all patients and three patients had a cytogenetic response (one complete and two minor).59 In two patients who were found to have BCR-ABL kinase domain mutations at the start of treatment with omacetaxine mepesuccinate, a cytogenetic response were achieved and accompanied by no detectable BCR-ABL mutations.60 The efficacy of omacetaxine mepesuccinate was assessed further in a Phase II study of CML patients with the BCR-ABL-T315I mutation.61 In total, 62 patients received a median of seven cycles of omacetaxine mepesuccinate. A CHR was achieved in 48 of these patients and their median response duration was 9.1 months. Fourteen patients achieved a major cytogenetic response, including a complete cytogenetic response in ten patients (16%). Median progression free-survival in this trial was 7.7 months.
So far, a couple of Phase II studies have been conducted to test HHT/omacetaxine mepesuccinate either alone or in combination with other antitumor agents in 828 CML patients with different disease stages (Tables 1 and 2). Four single-agent Phase II studies tested the effect of HHT/omacetaxine mepesuccinate in early or late chronic phase CML patients.57,58,60,62 In total, 212 CML patients were treated with HHT (2.5 mg/m2 ×14 days) or omacetaxine mepesuccinate (1.25 mg/m2 twice/day ×14 days). The average CHR was 80%, and 42% of patients achieved a cytogenetic response. Four single-agent Phase II studies demonstrated the effectiveness of omacetaxine mepesuccinate in treating CML patients resistant or intolerant to two or more TKIs.63–66 A total of 252 CML patients were treated with omacetaxine mepesuccinate (1.25 mg/m2 twice daily, 28 days cycle for induction, ≤7 days/cycle as maintenance), and the average cytogenetic response rate was 20.5%.
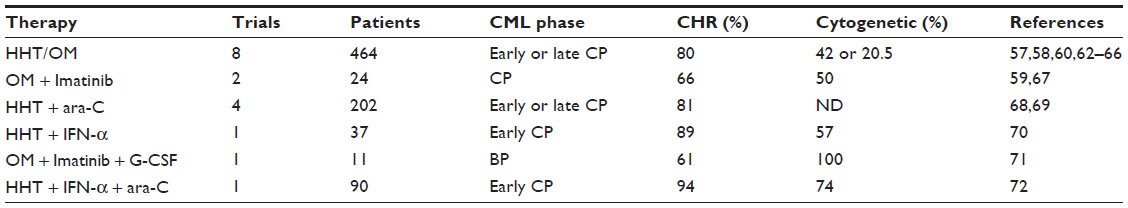 | Table 1 Summary of OM related clinic trials |
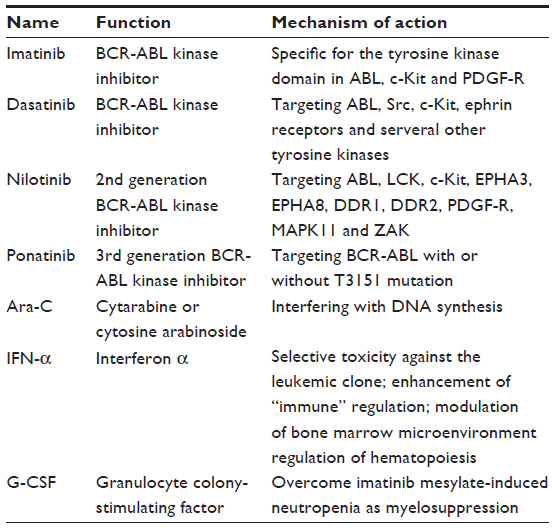 | Table 2 Summary of current CML treatment |
Omacetaxine mepesuccinate were also tested in combination with other active agents. Two Phase II trials tested the efficacy of omacetaxine mepesuccinate (1.25 mg/m2 twice daily ×14 days) combined with imatinib in 24 patients with chronic phase CML.59,67 The average CHR of drug-treated patients was 66%. Fifty percent of these patients achieved a cytogenetic response. Four Phase II trials tested the effect of a combination of HHT 2.5 mg/m2 and ara-C in 202 patients with chronic phase CML.68,69 The average CHR was 81%. One Phase II trial tested the effect of HHT 2.5 mg/m2 combined with interferon-α in 37 patients with early chronic phase CML.70 The average CHR was 89%, with 57% of patients achieving a cytogenetic response. One Phase II trial tested the effect of omacetaxine mepesuccinate combined with imatinib and granulocyte colony-stimulating factor in eleven patients with blast phase CML.71 The average CHR was 61%, and 100% of patients achieved a cytogenetic response. Another Phase II trial tested the effect of a combination of HHT, interferon-α, and ara-C in 90 patients with early chronic phase CML.72 The average CHR was 94%, and 74% of patients achieved a cytogenetic response.
Efficacy of omacetaxine mepesuccinate in other blood malignancies
In addition to being used to treat CML, omacetaxine mepesuccinate has also been used to treat other types of blood malignancy, such as myelodysplastic syndrome, acute myeloid leukemia, and multiple myeloma. Although CML is the focus of this review, we believe that it would be beneficial if we also discuss, at least briefly, other diseases in which omacetaxine mepesuccinate has a therapeutic effect.
In a study using HHT at a dose of 5 mg/m2 by 24-hour continuous infusion for 9 days in 28 patients (16 with myelodysplastic syndrome, 12 with myelodysplastic syndrome/acute myeloid leukemia), a 28% overall response rate (8/28) was achieved, with complete remission in seven patients and partial remission in one patient.73 In another Phase II pilot study of HHT in myelodysplastic syndrome, HHT was given at a dose of 2.5 mg/m2 via continuous infusion for 7 days and maintenance every 4 weeks. One patient (11%) responded with a CHR and cytogenetic remission after one course and eight patients did not respond.74 Similarly, the effect of omacetaxine mepesuccinate was tested in patients with acute myeloid leukemia. HHT was evaluated at a dose of 5 mg/m2 by continuous infusion daily for 9 days in 66 patients with relapsed/refractory acute myeloid leukemia or blastic phase CML. Seven of 43 patients with relapsed acute myeloid leukemia achieved a complete remission (16%). Two of three patients primarily resistant to low-dose cytarabine also achieved complete remission, while eleven patients with acute myeloid leukemia primarily resistant to an anthracycline-cytarabine combination did not respond.75 The effect of omacetaxine mepesuccinate was also shown to have a stronger effect on acute myeloid leukemia when combined with other therapeutic agents such as cytarabine, aclarubicin, or granulocyte-colony stimulating factor.76–78
The efficacy of omacetaxine mepesuccinate was also studied in multiple myeloma. Omacetaxine mepesuccinate significantly inhibited proliferation of human multiple myeloma cell lines and tumor cells from patients with relapsed refractory multiple myeloma in a dose-dependent manner.79 Omacetaxine mepesuccinate further reduced the levels of cellular FLICE-like inhibitory protein, activated caspase-8, and induced active truncated-Bid in multiple myeloma cells, including RPMI8226 and U266.79 When combined with other antitumor agents, omacetaxine mepesuccinate enhanced the efficacy of melphalan, bortezomib, and ABT-737.80 The cytotoxicity of omacetaxine mepesuccinate was related to downregulation of AKT phosphorylation/activation and various substrates of AKT, including nuclear factor kappa B, XIAP, cIAP, and cyclin D1.81
Safety and tolerability
Myelosuppression and nonhematologic toxicity
Myelosuppression is a major dose-related side effect of omacetaxine mepesuccinate. In CML patients treated with omacetaxine mepesuccinate alone or in combination with TKIs or other agents, intravenous administration of omacetaxine mepesuccinate caused granulocytopenia (<0.5×109/L) in 27%–39% of patients, and thrombocytopenia (30×109/L) in 13%–25% of patients.57,58 Myelosuppression is also the principal side effect with subcutaneous administration of omacetaxine mepesuccinate.39
The most common nonhematologic toxicities in CML patients treated with omacetaxine mepesuccinate include diarrhea, fatigue, pyrexia, nausea, asthenia, headache, anorexia, hyperglycemia, injection site erythema, and tachycardia/chest pain.59,60,82–84 Recently, a pooled safety analysis in patients with TKI-resistant CML treated using subcutaneous omacetaxine mepesuccinate (1.25 mg/m2) showed an acceptable safety profile in all phases of CML.85 Adverse events were primarily hematologic, and grade 3/4 nonhematologic adverse events were uncommon.85 However, a concern is the potential adverse effects in patients after long-term use of omacetaxine mepesuccinate. It is likely that some patients may not tolerate these adverse effects, and need to switch to other therapies. On the other hand, some patients may benefit from this treatment, simply because they have run out of therapeutic options and the drug is effective. Further, in contrast with TKIs, omacetaxine mepesuccinate has an inhibitory activity against leukemia stem cells, making it an attractive therapy for being close to curing the disease.
Potential use of omacetaxine mepesuccinate
Imatinib at 400 mg daily is the standard of care for patients with CML in chronic phase, and dasatinib and nilotinib represent alternatives that have replaced imatinib as first-line therapy in patients with imatinib resistance or intolerance.19,86–88 Clearly, omacetaxine mepesuccinate will not substitute for imatinib and other TKIs. However, omacetaxine mepesuccinate has a unique role in certain patients with CML. First, because omacetaxine mepesuccinate primarily targets signaling molecules downstream of BCR-ABL and has a unique mode of action, it could be a choice for patients carrying BCR-ABL mutation and resistant to TKIs, because some patients including BCR-ABL-T315I carriers do not respond to available TKIs. For these patients, omacetaxine mepesuccinate could be used as a single agent for treating CML. Second, TKIs do not eradicate the leukemia stem cells that initiate CML. Finally, omacetaxine mepesuccinate has been shown to have an inhibitory effect on leukemia stem cells in preclinical models.47,50 Therefore, omacetaxine mepesuccinate should be used in combination with a TKI. A recent study showed that omacetaxine mepesuccinate in combination with nilotinib led to undetectable residual disease at a molecular level in an imatinib-resistant CML patient harboring the BCR-ABL-T315I mutation.89 As the third-generation TKI ponatinib came to the market and responses were observed in highly refractory patients with either no mutations or other mutations resistant to first- or second-generation FDA approved TKIs.20 Thus, a combinatorial therapeutic strategy using omacetaxine mepesuccinate and a new-generation TKI would be an attractive approach in helping to cure CML. In fact, omacetaxine mepesuccinate has been shown to be effective in treating CML as a single agent57,58,60,62 and in combination with imatinib,58,90 interferon-α,70 cytarabine,68,69 and both interferon-α and cytarabine.72
Conclusion
After the US Food and Drug Administration granted accelerated approval for omacetaxine mepesuccinate (Synribo) in the treatment of adult patients in CML chronic phase or accelerated phase with resistance and/or intolerance to two or more TKIs, its effectiveness in treating these patients has been observed. In addition, omacetaxine mepesuccinate has been shown to have inhibitory activity in TKI-insensitive CML stem cells, suggesting that this agent in combination with TKIs provides a curative therapeutic strategy for CML patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Nowell PC, Hungerford DA. Chromosome studies in human leukemia. IV. Myeloproliferative syndrome and other atypical myeloid disorders. J Natl Cancer Inst. 1962;29:911–931. | |
Li S, Ilaria RL Jr, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med. 1999;189:1399–1412. | |
Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88:2375–2384. | |
Pendergast AM, Quilliam LA, Cripe LD, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75:175–185. | |
Gross AW, Zhang X, Ren R. Bcr-Abl with an SH3 deletion retains the ability to induce a myeloproliferative disease in mice, yet c-Abl activated by an SH3 deletion induces only lymphoid malignancy. Mol Cell Biol. 1999;19:6918–6928. | |
Ghaffari S, Daley GQ, Lodish HF. Growth factor independence and BCR/ABL transformation: promise and pitfalls of murine model systems and assays. Leukemia. 1999;13:1200–1206. | |
Mitelman F. The cytogenetic scenario of chronic myeloid leukemia. Leuk Lymphoma. 1993;11 Suppl 1:11–15. | |
Jennings BA, Mills KI. c-MYC locus amplification and the acquisition of trisomy 8 in the evolution of chronic myeloid leukaemia. Leuk Res. 1998;22:899–903. | |
Prokocimer M, Rotter V. Structure and function of p53 in normal cells and their aberrations in cancer cells: projection on the hematologic cell lineages. Blood. 1994;84:2391–2411. | |
Feinstein E, Cimino G, Gale RP, et al. p53 in chronic myelogenous leukemia in acute phase. Proc Natl Acad Sci U S A. 1991;88:6293–6297. | |
Ahuja HG, Jat PS, Foti A, Bar-Eli M, Cline MJ. Abnormalities of the retinoblastoma gene in the pathogenesis of acute leukemia. Blood. 1991;78:3259–3268. | |
Sill H, Goldman JM, Cross NC. Homozygous deletions of the p16 tumor-suppressor gene are associated with lymphoid transformation of chronic myeloid leukemia. Blood. 1995;85:2013–2016. | |
Mughal TI, Yong A, Szydlo RM, et al. Molecular studies in patients with chronic myeloid leukaemia in remission 5 years after allogeneic stem cell transplant define the risk of subsequent relapse. Br J Haematol. 2001;115:569–574. | |
van Rhee F, Szydlo RM, Hermans J, et al. Long-term results after allogeneic bone marrow transplantation for chronic myelogenous leukemia in chronic phase: a report from the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 1997;20:553–560. | |
Chen Y, Peng C, Sullivan C, Li D, Li S. Critical molecular pathways in cancer stem cells of chronic myeloid leukemia. Leukemia. 2010;24:1545–1554. | |
Druker BJ. Chronic myeloid leukemia. Sceptical scientists. Lancet. 2001;358 Suppl:S11. | |
Mauro MJ, Druker BJ. STI571: a gene product-targeted therapy for leukemia. Curr Oncol Rep. 2001;3:223–227. | |
Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. | |
Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. | |
Ohanian M, Cortes J, Kantarjian H, Jabbour E. Tyrosine kinase inhibitors in acute and chronic leukemias. Expert Opin Pharmacother. 2012;13:927–938. | |
Bixby D, Talpaz M. Mechanisms of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia and recent therapeutic strategies to overcome resistance. Hematology Am Soc Hematol Educ Program. 2009:461–476. | |
White DL, Saunders VA, Dang P, et al. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006;108:697–704. | |
Branford S, Rudzki Z, Walsh S, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–3475. | |
Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004; 305:399–401. | |
Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–141. | |
Hochhaus A, Saglio G, Larson RA, et al. Nilotinib is associated with a reduced incidence of BCR-ABL mutations vs imatinib in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Blood. 2013;121:3703–3708. | |
Frankfurt O, Licht JD. Ponatinib – a step forward in overcoming resistance in chronic myeloid leukemia. Clin Cancer Res. 2013;19:5828–5834. | |
Hamilton A, Helgason GV, Schemionek M, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119:1501–1510. | |
Holyoake T, Jiang X, Eaves C, Eaves A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood. 1999;94:2056–2064. | |
Graham SM, Jorgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. | |
Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121:396–409. | |
Rousselot P, Huguet F, Rea D, et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood. 2007;109:58–60. | |
Hu Y, Chen Y, Douglas L, Li S. Beta-catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia. 2009;23:109–116. | |
Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12:528–541. | |
Naka K, Hoshii T, Muraguchi T, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463:676–680. | |
Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. | |
Peng C, Chen Y, Yang Z, et al. PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood. 2010;115:626–635. | |
Chen Y, Hu Y, Zhang H, Peng C, Li S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41:783–792. | |
Cortes JE, Nicolini FE, Wetzler M, et al. Subcutaneous omacetaxine mepesuccinate in patients with chronic-phase chronic myeloid leukemia previously treated with 2 or more tyrosine kinase inhibitors including imatinib. Clin Lymphoma Myeloma Leuk. 2013;13:584–591. | |
Kim TD, Frick M, le Coutre P. Omacetaxine mepesuccinate for the treatment of leukemia. Expert Opin Pharmacother. 2011;12:2381–2392. | |
Huang MT. Harringtonine, an inhibitor of initiation of protein biosynthesis. Mol Pharmacol. 1975;11:511–519. | |
Fresno M, Jimenez A, Vazquez D. Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem. 1977;72:323–330. | |
Tujebajeva RM, Graifer DM, Karpova GG, Ajtkhozhina NA. Alkaloid homoharringtonine inhibits polypeptide chain elongation on human ribosomes on the step of peptide bond formation. FEBS Lett. 1989;257:254–256. | |
Tujebajeva RM, Graifer DM, Matasova NB, et al. Selective inhibition of the polypeptide chain elongation in eukaryotic cells. Biochim Biophys Acta. 1992;1129:177–182. | |
Gurel G, Blaha G, Moore PB, Steitz TA. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J Mol Biol. 2009;389:146–156. | |
Wetzler M, Segal D. Omacetaxine as an anticancer therapeutic: what is old is new again. Curr Pharm Des. 2011;17:59–64. | |
Chen Y, Hu Y, Michaels S, Segal D, Brown D, Li S. Inhibitory effects of omacetaxine on leukemic stem cells and BCR-ABL-induced chronic myeloid leukemia and acute lymphoblastic leukemia in mice. Leukemia. 2009;23:1446–1454. | |
Chen R, Guo L, Chen Y, Jiang Y, Wierda WG, Plunkett W. Homoharringtonine reduced Mcl-1 expression and induced apoptosis in chronic lymphocytic leukemia. Blood. 2011;117:156–164. | |
Tang R, Faussat AM, Majdak P, et al. Semisynthetic homoharringtonine induces apoptosis via inhibition of protein synthesis and triggers rapid myeloid cell leukemia-1 down-regulation in myeloid leukemia cells. Mol Cancer Ther. 2006;5:723–731. | |
Allan EK, Holyoake TL, Craig AR, Jorgensen HG. Omacetaxine may have a role in chronic myeloid leukaemia eradication through downregulation of Mcl-1 and induction of apoptosis in stem/progenitor cells. Leukemia. 2011;25:985–994. | |
Lindqvist LM, Vikstrom I, Chambers JM, et al. Translation inhibitors induce cell death by multiple mechanisms and Mcl-1 reduction is only a minor contributor. Cell Death Dis. 2012;3:e409. | |
Robert F, Carrier M, Rawe S, Chen S, Lowe S, Pelletier J. Altering chemosensitivity by modulating translation elongation. PLoS One. 2009;4:e5428. | |
Nemunaitis J, Mita A, Stephenson J, et al. Pharmacokinetic study of omacetaxine mepesuccinate administered subcutaneously to patients with advanced solid and hematologic tumors. Cancer Chemother Pharmacol. 2013;71:35–41. | |
Savaraj N, Lu K, Dimery I, et al. Clinical pharmacology of homoharringtonine. Cancer Treat Rep. 1986;70:1403–1407. | |
Ni D, Ho DH, Vijjeswarapu M, Felix E, Rhea PR, Newman RA. Metabolism of homoharringtonine, a cytotoxic component of the evergreen plant Cephalotaxus harringtonia. J Exp Ther Oncol. 2003;3:47–52. | |
Zhang ZY, Hou CH, Zhu YF. [A preliminary therapeutic analysis of 82 cases of chronic granulocytic leukemia treated with harringtonine]. Zhonghua Nei Ke Za Zhi. 1986;25:156–157, 190. Chinese. | |
O’Brien S, Kantarjian H, Keating M, et al. Homoharringtonine therapy induces responses in patients with chronic myelogenous leukemia in late chronic phase. Blood. 1995;86:3322–3326. | |
O’Brien S, Kantarjian H, Koller C, et al. Sequential homoharringtonine and interferon-alpha in the treatment of early chronic phase chronic myelogenous leukemia. Blood. 1999;93:4149–4153. | |
Marin D, Kaeda JS, Andreasson C, et al. Phase I/II trial of adding semisynthetic homoharringtonine in chronic myeloid leukemia patients who have achieved partial or complete cytogenetic response on imatinib. Cancer. 2005;103:1850–1855. | |
Quintas-Cardama A, Kantarjian H, Garcia-Manero G, et al. Phase I/II study of subcutaneous homoharringtonine in patients with chronic myeloid leukemia who have failed prior therapy. Cancer. 2007;109:248–255. | |
Cortes J, Lipton JH, Rea D, et al. Phase 2 study of subcutaneous omacetaxine mepesuccinate after TKI failure in patients with chronic-phase CML with T315I mutation. Blood. 2012;120:2573–2580. | |
Cortes J, Digumarti R, Parikh PM, et al. Phase 2 study of subcutaneous omacetaxine mepesuccinate for chronic-phase chronic myeloid leukemia patients resistant to or intolerant of tyrosine kinase inhibitors. Am J Hematol. 2013;88:350–354. | |
Nicolini FE LJ, Kantarjian H, et al. Subcutaneous omacetaxine mepesuccinate in patients with chronic phase (CP) or accelerated phase (AP) chron myeloid leukemia (CML) resistant/intolerant to two or three approved tyrosine-kinase inhibitors (TKIs). ASCO Annual Meeting. 2012;30:6513. | |
Nicolini FE, Khoury HJ, Akard L, et al. Omacetaxine mepesuccinate for patients with accelerated phase chronic myeloid leukemia with resistance or intolerance to two or more tyrosine kinase inhibitors. Haematologica. 2013;98(7):e78–e79. | |
Nicolini FE, Chomel JC, Roy L, et al. The durable clearance of the T315I BCR-ABL mutated clone in chronic phase chronic myelogenous leukemia patients on omacetaxine allows tyrosine kinase inhibitor rechallenge. Clin Lymphoma Myeloma Leuk. 2010;10:394–399. | |
Akard LP KH, Nicolini FE, et al. Omacetaxine mepesuccinate in chronic-phase chronic myeloid leukemia (CML) in patients resistant, intolerant, or both to two or more tyrosine-kinase inhibitors (TKIs). ASCO Annual Meeting. 2012;30:6596. | |
Li YF, Liu X, Liu DS, Din BH, Zhu JB. The effect of homoharringtonine in patients with chronic myeloid leukemia who have failed or responded suboptimally to imatinib therapy. Leuk Lymphoma. 2009;50:1889–1891. | |
Kantarjian HM, Talpaz M, Smith TL, et al. Homoharringtonine and low-dose cytarabine in the management of late chronic-phase chronic myelogenous leukemia. J Clin Oncol. 2000;18:3513–3521. | |
Stone RM, Donohue KA, Stock W, et al. A phase II study of continuous infusion homoharringtonine and cytarabine in newly diagnosed patients with chronic myeloid leukemia: CALGB study 19804. Cancer Chemother Pharmacol. 2009;63:859–864. | |
O’Brien S, Talpaz M, Cortes J, et al. Simultaneous homoharringtonine and interferon-alpha in the treatment of patients with chronic-phase chronic myelogenous leukemia. Cancer. 2002;94:2024–2032. | |
Fang B, Li N, Song Y, Han Q, Zhao RC. Standard-dose imatinib plus low-dose homoharringtonine and granulocyte colony-stimulating factor is an effective induction therapy for patients with chronic myeloid leukemia in myeloid blast crisis who have failed prior single-agent therapy with imatinib. Ann Hematol. 2010;89:1099–1105. | |
O’Brien S, Giles F, Talpaz M, et al. Results of triple therapy with interferon-alpha, cytarabine, and homoharringtonine, and the impact of adding imatinib to the treatment sequence in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in early chronic phase. Cancer. 2003;98:888–893. | |
Feldman EJ, Seiter KP, Ahmed T, Baskind P, Arlin ZA. Homoharringtonine in patients with myelodysplastic syndrome (MDS) and MDS evolving to acute myeloid leukemia. Leukemia. 1996;10:40–42. | |
Daver N, Vega-Ruiz A, Kantarjian HM, et al. A phase II open-label study of the intravenous administration of homoharringtonine in the treatment of myelodysplastic syndrome. Eur J Cancer Care (Engl). 2013;22:605–611. | |
Feldman E, Arlin Z, Ahmed T, et al. Homoharringtonine in combination with cytarabine for patients with acute myelogenous leukemia. Leukemia. 1992;6:1189–1191. | |
Zhang WG, Wang FX, Chen YX, et al. Combination chemotherapy with low-dose cytarabine, homoharringtonine, and granulocyte colony-stimulating factor priming in patients with relapsed or refractory acute myeloid leukemia. Am J Hematol. 2008;83:185–188. | |
Wu L, Li X, Su J, et al. Effect of low-dose cytarabine, homoharringtonine and granulocyte colony-stimulating factor priming regimen on patients with advanced myelodysplastic syndrome or acute myeloid leukemia transformed from myelodysplastic syndrome. Leuk Lymphoma. 2009;50:1461–1467. | |
Yu W, Mao L, Qian J, et al. Homoharringtonine in combination with cytarabine and aclarubicin in the treatment of refractory/relapsed acute myeloid leukemia: a single-center experience. Ann Hematol. 2013;92:1091–1100. | |
Lou YJ, Qian WB, Jin J. Homoharringtonine induces apoptosis and growth arrest in human myeloma cells. Leuk Lymphoma. 2007;48:1400–1406. | |
Kuroda J, Kamitsuji Y, Kimura S, et al. Anti-myeloma effect of homoharringtonine with concomitant targeting of the myeloma-promoting molecules, Mcl-1, XIAP, and beta-catenin. Int J Hematol. 2008;87:507–515. | |
Meng H, Yang C, Jin J, Zhou Y, Qian W. Homoharringtonine inhibits the AKT pathway and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Leuk Lymphoma. 2008;49:1954–1962. | |
Levy V, Zohar S, Bardin C, et al. A phase I dose-finding and pharmacokinetic study of subcutaneous semisynthetic homoharringtonine (ssHHT) in patients with advanced acute myeloid leukaemia. Br J Cancer. 2006;95:253–259. | |
Kantarjian HM, Keating MJ, Walters RS, Koller CA, McCredie KB, Freireich EJ. Phase II study of low-dose continuous infusion homoharringtonine in refractory acute myelogenous leukemia. Cancer. 1989;63:813–817. | |
Sylvester RK, Lobell M, Ogden W, Stewart JA. Homoharringtonine-induced hyperglycemia. J Clin Oncol. 1989;7:392–395. | |
Wetzler M, Kantarjian H, Nicolini FE, et al. Pooled safety analysis of omacetaxine mepesuccinate in patients with chronic myeloid leukemia (CML) resistant to tyrosine-kinase inhibitors (TKIs). J Clin Oncol. 2012;30 Suppl:Abstr 6604. | |
Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–2270. | |
Nicolini FE, Turkina A, Shen ZX, et al. Expanding Nilotinib Access in Clinical Trials (ENACT): an open-label, multicenter study of oral nilotinib in adult patients with imatinib-resistant or imatinib-intolerant Philadelphia chromosome-positive chronic myeloid leukemia in the chronic phase. Cancer. 2012;118:118–126. | |
Koren-Michowitz M, le Coutre P, Duyster J, et al. Activity and tolerability of nilotinib: a retrospective multicenter analysis of chronic myeloid leukemia patients who are imatinib resistant or intolerant. Cancer. 2010;116:4564–4572. | |
Coude MM, Luycx O, Cariou ME, et al. Undetectable molecular residual disease after omacetaxine and nilotinib combination therapy in an imatinib-resistant chronic myeloid leukaemia patient harbouring the BCR-ABL1 T315I gatekeeper mutation. Br J Haematol. 2012;157:407–410. | |
Li YF, Deng ZK, Xuan HB, et al. Prolonged chronic phase in chronic myelogenous leukemia after homoharringtonine therapy. Chin Med J (Engl). 2009;122:1413–1417. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.