")
Back to Journals » Biologics: Targets and Therapy » Volume 11
Octreotide long-acting repeatable in the treatment of neuroendocrine tumors: patient selection and perspectives
Authors Yau H , Kinaan M, Quinn SL , Moraitis AG
Received 4 July 2017
Accepted for publication 9 November 2017
Published 6 December 2017 Volume 2017:11 Pages 115—122
DOI https://doi.org/10.2147/BTT.S108818
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Doris Benbrook
Hanford Yau,1 Mustafa Kinaan,2 Suzanne L Quinn,3 Andreas G Moraitis3
1Division of Endocrinology, Diabetes, and Metabolism, University of California, San Francisco (Fresno Division), Fresno, CA, USA; 2Division of Internal Medicine, University of Central Florida College of Medicine, Orlando, FL, USA; 3Division of Endocrinology, Diabetes, and Metabolism, Orlando VA Medical Center, Orlando, FL, USA
Abstract: Over the past three decades, the incidence and prevalence of neuroendocrine tumors have gradually increased. Due to the slow-growing nature of these tumors, most cases are diagnosed at advanced stages. Prognosis and survival are associated with location of primary lesion, biochemical functional status, differentiation, initial staging, and response to therapy. Octreotide, the first synthetic somatostatin analog, was initially used for the management of gastrointestinal symptoms associated with functional carcinoid tumors. Its commercial development over time led to long-acting repeatable octreotide acetate, a long-acting version that provided greater administration convenience. Recent research demonstrates that octreotide’s efficacy has evolved beyond symptomatic management to targeted therapy with antitumoral effects. This review examines the history and development of octreotide, provides a synopsis on the classification, grading, and staging of neuroendocrine tumors, and reviews the evidence of long-acting repeatable octreotide acetate as monotherapy and in combination with other treatment modalities in the management of non-pituitary neuroendocrine tumors with special attention to recent high-quality Phase III trials.
Keywords: carcinoid, everolimus, neuroendocrine tumor, octreotide LAR, somatostatin analog, ITMO, NETTER-1, PROMID, RADIANT-2
Introduction
Neuroendocrine tumors (NETs) are a heterogeneous group of neoplasms originating from neuroendocrine cells. Although NETs are slow-growing tumors, they have malignant potential. Diagnosis is challenging and often delayed until distant metastases are present. In addition to their malignant potential, NETs can be either functional or nonfunctional. The majority of NETs are nonfunctional in nature, hormonally inactive, and do not have a distinct clinical presentation. Functional tumors present with clinical signs and symptoms caused by hormone release and are named relative to the hormone that they hypersecrete (Table 1). The gastrointestinal (GI) tract, pancreas, and bronchus are the most common primary sites in patients with NETs. Due to the nonspecific nature of some of the symptoms such as abdominal pain, or unintentional weight loss, diagnosis is sometimes delayed and patients often present with metastatic disease to regional lymph nodes, liver, and bone, conferring a poorer prognosis at the time of diagnosis.
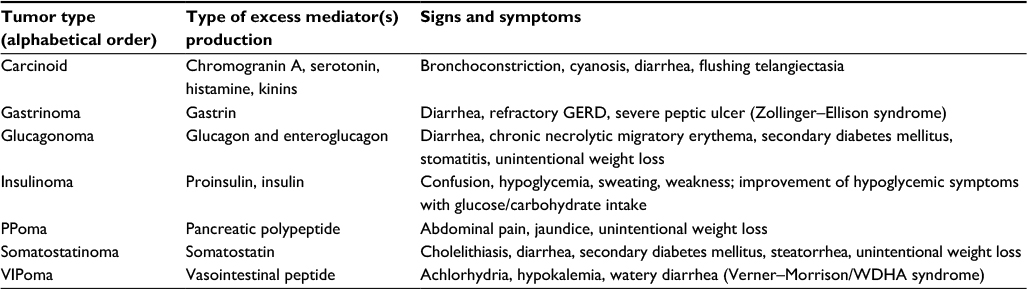 | Table 1 Clinical features of functional neuroendocrine tumors Abbreviations: GERD, gastroesophageal reflux disease; PPoma, pancreatic polypeptidoma; VIPoma, vasoactive intestinal peptide-producing tumor; WDHA, watery diarrhea, hypokalemia, and achlorhydria. |
Endogenous somatostatin is a decatetra-aminopeptide, which inhibits the effects and release of GI hormones (gastrin, motilin, pancreatic polypeptide, secretin, serotonin, and vasoactive intestinal peptide). The inhibitory effects are facilitated through binding as an agonist to the five different G-protein–coupled, somatostatin receptor subtypes (SSTR-1 to -5), leading to decreased activity of adenylate cyclase, stimulation of phosphotyrosine phosphatases, and modulation of mitogen-activated protein kinases. Each receptor subtype triggers several intracellular pathways; the antisecretory effects of somatostatin on various hormones are predominately regulated by SSTR-2 and SSTR-5, which modulate K+ and Ca+2 fluxes in the cell. Approximately 80% of all NETs of the GI tract express these two receptor subtypes. Direct antitumor effects of octreotide occur through inhibition of synthesis of autocrine and paracrine growth factors and induction of apoptosis. SHP-1, a phosphotyrosine phosphatase, is activated through the stimulation of certain somatostatin receptor subtypes such as SSTR-2. This process leads to the arrest of cellular growth and proliferation in human tumors, especially pituitary adenomas. SHP-2, another phosphotyrosine phosphatase, mediates a similar antitumor mechanism by inactivating specific tyrosine kinase receptors, thus depriving the tumor of epidermal growth factor, insulin-like growth factor, and growth hormone. Inhibition of growth hormone occurs through SSTR-2 and SST-5 in the pituitary and insulin-like growth factor-1 in the liver. Indirect tumor effects occur as a result of inhibition of angiogenesis. Several human tumors (neuroendocrine, prostate, and breast) have been reported to overexpress somatostatin receptors with high affinity for SSTR-2- and SSTR-5-binding analogs such as octreotide, especially in the vasculature surrounding these tumors. This pattern has been demonstrated to inhibit proliferating endothelial cells via the downregulation of nitric oxide and vascular endothelial growth factor angiogenetic effects.
History of octreotide and long-acting repeatable octreotide acetate
The first report of the effectiveness of somatostatin to control diarrheal symptoms by inhibition of serotonin and other tachykinins in a carcinoid patient was published in 1978.1,2 Despite its effectiveness, somatostatin is clinically limited due to its short half-life (t½=2–3 minutes). In 1982, Bauer et al3 synthesized octreotide, an octapeptide with three unnatural amino acids, conferring resistance to metabolic degradation and high affinity to SSTR-2 and SSTR-5. The first synthetic commercially available somatostatin analog (SSA) was subcutaneous/intravenous octreotide acetate, a cyclic octapeptide, which was approved by the US Food and Drug Administration (FDA) in October 1988 for on-label treatment of acromegaly, treatment of diarrhea and flushing in patients with metastatic carcinoid tumors, and treatment of watery diarrhea associated with vasoactive intestinal peptide secreting tumors. Octreotide acetate is advantageous over endogenous somatostatin due to its longer half-life (t½=1.7–1.9 hours), and its duration of action can extend up to 12 hours depending upon the tumor type. Treatment with octreotide acetate typically ranged from 50 to 1,500 μg in two to four divided doses per day. By November 1998, further development led to FDA approval of intramuscular long-acting repeatable octreotide acetate (octreotide LAR), a once-monthly (every 4 weeks) injection. The long-acting depot formulation reflects the release of medication from the gradual degradation of the poly-dl-lactide-co-glucolide-glucose microsphere polymer matrix in the muscle postinjection. Once distributed to the systemic circulation, its pharmacokinetics and elimination are identical to octreotide. The concentration plateau of octreotide LAR is reached ~2–3 weeks postinjection and maintained for an additional 2–3 weeks thereafter. After three doses of octreotide LAR are administered monthly, long-term steady-state concentrations are achieved. Pooled data from several trials that were conducted since it was first approved demonstrated that over 70% of the subjects who were treated with octreotide LAR had significant improvement or resolution of symptomatic diarrhea and flushing.4,5 Dose optimization with octreotide LAR is effective with tolerable side effects being mild to moderate. Additionally, dosage escalation did not cause significant changes in adverse events or tolerability while providing beneficial improvement in efficacy. A recent retrospective study by Strosberg et al6 of more than 230 NET patients treated with above-standard doses of octreotide LAR of 30 mg every 4 weeks resulted in improved symptoms of carcinoid syndrome without adverse events, which was consistent with other studies by Chadha et al7 and Ferolla et al.8 In patients with uncontrolled symptoms or increasing tumor burden, measurement of plasma or serum octreotide level may help physicians consider increasing the dosage further beyond the recommended dosage. Studies have demonstrated that plasma concentrations can decrease over time even using the long-acting formulation.9–11 There is also an inverse relationship between patients’ weights and plasma octreotide levels; thus, heavier patients may require higher dosages for its effects.10 In patients who have been on therapy for more than 6 months, loss of response (“escape from response” phenomenon) can occur due to downregulation of SSTR-2 expression or upregulation of other somatostatin receptor subtypes.12,13 Current practice guidelines support the benefit of high-dose somatostatin analog regimen, given its well-established benefit and safety profile.
Classification, grading, and TNM staging of NETs
Care and management of patients with NETs often involves multiple subspecialists. As such, consistent, clear, and effective communication among the health team members is imperative to allow accurate decision making. Well-established grading and staging systems including those by the European Neuroendocrine Tumor Society, World Health Organization (WHO), and the American Joint Committee on Cancer have focused on reporting guidelines and recommendations that contain consistent, uniform, and reproducible data for clinical management and research purposes. Since the clinical and pathologic characteristics of NETs are often reflective of the primary tumor’s origin, research has historically focused on their anatomic origin.
Grading of NETs is a reflection of the biologic aggressiveness of the tumor; it can be expressed by the Ki-67 index which is the percentage of tumor cells demonstrating positive immunolabeling for the proliferation marker Ki-67. Alternatively, counting mitotic figures and expressing them as the number of mitoses per 10 high-power microscopic fields or per 2 mm2 can be used. Thus, Ki-67 index has the advantage of being able to be used in cases of small biopsies, as mitotic rate counting requires a greater amount of tissue sampling due to limited number of high-power microscopic fields in small biopsies.9,10
Differentiation of NETs is based on the histologic resemblance of the tumor cells to their non-neoplastic equivalents. Grade 1 and 2 NETs are classified as well differentiated and histologically uniform, characterized by “organoid” architecture with gyriform, nest-like, or trabecular patterns. They generally express high intensities of neuroendocrine markers, such as chromogranin A (CgA) and synaptophysin, allowing immunohistochemical profiling to be a valuable tool. Grade 3 NETs are poorly differentiated with more of a diffuse architecture, atypical nuclei, and less cytoplasmic granularity. There is less intense and limited expression of neuroendocrine markers compared to differentiated NETs.9,10
Clinical validation of the Ki-67 index criteria in NETs of the GI tract used in the 2010 WHO classification by Yamaguchi et al11 demonstrated and confirmed by a receiver operative characteristics curve analysis that 2.8% was the best Ki-67 index cutoff value for predicting metastasis or recurrence with a sensitivity of ~43% and specificity of about 87%. This confirms that classifying NETs into G1 and G2 based on Ki-67 index of 3% in the 2010 WHO grading system is appropriate for predicting metastases or recurrences. Faggiano et al12 demonstrated in an observational Italian multicenter study in 71 patients treated with octreotide LAR and 35 patients treated with lanreotide autogel with histologically confirmed GI-pancreatic or thoracic NET or NET without a known primary that treatment according to Ki-67 index was efficacious. While objective response and tumor stability (response in 11%, stability in 58%, and progression in 31%) based on Response Evaluation Criteria In Solid Tumors (RECIST) criteria did not show significant difference between G1 and G2 NETs, median progression-free survival (PFS) was longer but not significantly different in G1 than G2 NETs (89 versus 43 months, P=0.15). However, median PFS was significantly longer with G1, G2 NETs with Ki-67 <5% when compared to G2, and G3 NETs with Ki-67 ≥5% (89 versus 35 months, P=0.005). Long-acting somatostatin analog therapy demonstrated significant antiproliferative effects in well-differentiated low/intermediate-proliferating NETs in both G1 and G2 types by using Ki-67 index of 5% or less, suggesting the selection of these patients as best candidates for long-acting somatostatin analog therapy.
Octreotide LAR as monotherapy
Rubin et al13 first demonstrated in a prospective randomized trial comparing double-blinded octreotide LAR at 10, 20, and 30 mg every 4 weeks versus open-label subcutaneous immediate-release octreotide every 8 hours that octreotide LAR was comparably efficacious in symptomatic control of malignant carcinoid syndrome in 79 patients. A 3-year retrospective study by Garland et al14 in which 27 patients with metastatic carcinoid tumors were followed for a median of 23 months demonstrated that octreotide LAR was well tolerated, and the patients expressed improved satisfaction with management of their disease. Anthony and Vinik15 showed in a 6-year retrospective study of about 390 subjects (72% with metastatic disease, n=284) with NETs receiving octreotide LAR Depot for no less than 4 months that symptom resolution or improvement can occur as early as 3 months after initial therapy. Nearly 60% of patients with diarrhea and flushing, 57% of those with bronchoconstriction, and 40% of subjects with carcinoid cardiac disease had resolution or improvement of symptoms. Furthermore, the best tumor response rate using RECIST criteria was >50% for all patients involved, with stable disease rates being 57% at 20 or 30 mg, 55% at 40 mg, and 50% at 60 mg.
The PROMID study (Placebo controlled, Double-Blind, Prospective, Randomized study of the Effect of Octreotide LAR in the Control of Tumor Growth in Patients with Metastatic Neuroendocrine MIDgut Tumors) was the first randomized trial to investigate the antitumor effect of octreotide LAR Depot.16,17 In this Phase III trial, 85 treatment-naïve patients with locally inoperable or metastatic NET were randomized to receive 30 mg octreotide LAR Depot or placebo. The primary endpoint of this study was time to tumor progression or tumor-related death. The median time to progression was 14.3 months in the treatment group versus 6 months in the placebo group (P=0.000072, hazard ratio [HR]=0.34 [95% CI: 0.20–0.59]).16 While overall survival (OS) in the whole cohort did not demonstrate any significant difference with a median time of 84.7 months in the treatment group versus 83.71 months in the placebo group (P=0.511, HR=0.83, [95% CI: 0.47–1.46]), there were significant differences in patients with hepatic tumor burden <10% compared to those with >10% at the time of randomization, with a median time of 107.6 versus 57.5 months, respectively.17 The lack of significance in OS may be confounded by the crossover of patients in the placebo to the treatment arm after tumor progression.17 Results from the PROMID trial were consistent with historical population-based data from the Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute, where median survival time in patients with metastatic jejunal and ileal NETs was only 56 months in the SEER registry.18 PROMID demonstrated for the first time long-term survival data in patients with metastatic midgut NETs, especially in those with low tumor burden. Based on the PROMID study conclusions, multiple medical societies including the European Society for Medical Oncology, National Comprehensive Cancer Network, and the North American Neuroendocrine Tumor Society are recommending octreotide LAR for managing patients with recurrent, inoperable, or metastatic GI NETs.
Combination therapy
Everolimus with octreotide LAR
The findings in the PROMID study altered clinical practice and with emerging evidence suggest that a combination of somatostatin analog, mammalian target of rapamycin inhibitors, and other forms of chemotherapy may improve PFS in patients with NET who fail to respond to somatostatin treatment alone or patients who present with very advanced disease with poor prognosis. The use of octreotide LAR with everolimus (RAD001), an oral mammalian target of rapamycin inhibitor, has been shown to have a positive effect on tumor cell proliferation. In an open-label, Phase II trial in subjects with refractory pancreatic NET, everolimus and octreotide LAR combination enhanced antitumor effects compared with everolimus alone. The RADIANT-1 (RAD001 in Advanced Neuroendocrine Tumors) trial showed that the median PFS was higher in patients treated with a combination of everolimus and octreotide than with everolimus alone (16.7 and 9.7 months, respectively). More studies are necessary to better understand the synergism of octreotide LAR combination with other therapies and its role in achieving better tumor control. In the Phase III RADIANT-2 double-blinded trial, patients with advanced NETs received everolimus plus octreotide LAR or placebo plus octreotide LAR to assess CgA response defined by at least a 50% decrease or normalization of CgA levels. A higher incidence of CgA response was seen in the everolimus group than in the placebo group (45.7% versus 28.8%). Furthermore, on central radiographic review, median PFS increased from 11.3 months on the octreotide LAR plus placebo arm to 16.4 months on the octreotide LAR plus everolimus arm (HR=0.77; P=0.026).19 The changes in CgA response of RADIANT-2 were comparable to the Phase II trial conducted by Yao et al20 in patients with metastatic pancreatic NET who previously failed cytotoxic chemotherapy and were subsequently treated with everolimus monotherapy versus everolimus plus octreotide LAR, where 50.7% of patients (38/75 patients) in the everolimus monotherapy and 60.0% of patients (15/25 patients) in the combined therapy group had a CgA response. Despite changes and improvement based on CgA response, there was no significant difference in OS in everolimus plus octreotide LAR versus placebo plus octreotide LAR arms in the RADIANT-2 study.21 Unlike previous studies which were conducted on patients with advanced disease who had failed other therapies, the Italian Trials in Medical Oncology (ITMO) study group conducted a Phase II, multicenter trial using combination therapy with everolimus and octreotide LAR as the first-line therapy for treatment of NETs of gastroenteropancreatic and pulmonary origin with and without carcinoid syndrome.22 An objective response rate (ORR) of 18% was achieved in 50 patients, with 74% of patients achieving stable disease, 16% achieving partial response, and 2% of patients showing complete response. Subgroup analysis demonstrated no significant ORR difference by primary tumor site and between patients with or without carcinoid syndrome. Tippeswamy et al23 conducted a 3-year study on 16 patients in India who were diagnosed with NETs (low-, intermediate-, or high-grade histologically) and who may have received two or less lines of chemotherapeutic agents. These patients were subsequently given oral everolimus (10 mg/day) plus intramuscular LAR (30 mg once in every 28 days). The study demonstrated clinical benefit in 69% of patients based on RECIST criteria (63% with stable disease, 6% with partial response). The combination of everolimus and octreotide LAR has demonstrated a long history of retrospective and prospective experiences regarding its safety and efficacy, suggesting that it can potentially be used as a first-line therapy in treating patients with advanced NETs as well as for those who failed other treatment modalities.
Radiotherapy with radiolabeled somatostatin analogs
Peptide receptor radionuclide therapy or radiolabeled somatostatin analog therapy has been used for over two decades for the treatment of advanced NETs. This form of systemic radiotherapy delivers radionuclides directly targeting tumor cells with high concentrations of somatostatin receptors. 90Y-DOTA0-Tyr3-octreotide (90Y-DOTATOC), a high energy β emitter, and 177Lu-DOTA0-Tyr3-octreotate (177Lu-DOTATATE), a strong β/weak γ emitter, are the two more commonly used radiopeptides for treating advanced NETs.24 Less commonly used radionuclides include α emitters, Bismuth-213 (213Bi) and Actinium-225 (225Ac), and Auger/γ emitter, Indium-111 (111In).24 Krenning et al were one of the first investigators to publish successful treatment with radiolabeled somatostatin analog therapy.25 In one of the largest studies published, Kwekkeboom et al demonstrated that 177Lu-DOTA0-Tyr3-octreotate treatment in over 500 patients with metastatic gastroenteropancreatic NETs was well tolerated with few adverse effects. Of 310 patients, complete and partial tumor remissions occurred in 2% and 28%, respectively. An additional 16% of patients had tumor response with decrease in size between 25% and 50%. Median OS was 46 months from the start of treatment.26
Despite the published successes of radiolabeled somatostatin analog therapy historically, data from high-quality prospective Phase III trials were limited until the recently conducted landmark international, multicenter open-label Neuroendocrine Tumors Therapy (NETTER-1) trial. Two hundred twenty-nine patients with well-differentiated, metastatic midgut NETs were randomly assigned in a 1:1 ratio to treatment with either four intravenous infusions of 200 mCi (7.4 GBq) 177Lu-DOTATATE every 8 weeks with supportive care including octreotide LAR 30 mg after each treatment followed by monthly injections thereafter or octreotide LAR 60 mg every 4 weeks alone which served as a control group. Interim analysis demonstrated that the estimated rate of PFS after 20 months was 65.2% in the 177Lu-DOTATATE group (95% CI: 50.0–76.8) versus 10.8% in the high-dose octreotide LAR only group (95% CI: 3.5–23.0). Median PFS had not yet been reached in the 177Lu-DOTATATE group at the time of interim analysis and was 8.4 months in the control group (95% CI: 5.8–9.1). The HR for disease progression or death in 177Lu-DOTATATE group versus control was 0.21 (95% CI: 5.8–9.1), which reflected a 79% lower risk of disease progression or death in the treatment group versus control. OS at the time of interim analysis demonstrated 14 deaths in the 177Lu-DOTATATE group versus 26 deaths in the octreotide LAR alone group. The estimated risk of death was 60% lower in the 177Lu-DOTATATE group versus control (HR for death with 177Lu-DOTATATE versus control, 0.4; P=0.004). Tumor response rate to therapy based on RECIST criteria was significantly higher (P<0.001) in the 177Lu-DOTATATE group (18%) versus control (3%).27
The recent interim results of NETTER-1 landmark trial further validate and support previous results of nonrandomized trials of 177Lu-DOTATATE and other radiolabeled somatostatin analog treatments as a relatively well-tolerated and effective targeted treatment modality that has consistently demonstrated efficacious response and improved duration of median PFS.26,28–30 Ongoing support and research is much needed to investigate the role of peptide receptor radionuclide therapy in the treatment of NETs.
Other combination therapies with octreotide LAR
Deutsch et al31 evaluated the long-term survival of 49 patients from a single institution with primary GI (small bowel [44.9%] or pancreas [28.6%]) metastatic neuroendocrine carcinoma treated with aggressive cytoreductive surgery combined with octreotide LAR. With a median follow-up of 112 months, rates of 1-, 5-, 10-, and 15-year disease-specific survival (DSS) were 94%, 78%, 64%, and 31%, respectively, in the group that received aggressive cytoreductive surgery plus octreotide LAR. Compared to a similar cohort from the SEER-Medicare database from 2003 to 2009, the DSS for the patients who received combination therapy was 68.3% at 5 years and 60.6% at 10 years, as compared with 54.7% and 51.8%, respectively, for the 202 patients receiving surgery alone and 50.0% and 36.0%, respectively, for patients receiving long-acting somatostatin analog alone (P<0.0001). The remaining group receiving neither treatment (n=1,093) had 5- and 10-year DSS of 34.3% and 26.3%, respectively.
Another Phase III, prospective, randomized, comparison trial conducted again by Yao et al32 compared octreotide LAR plus bevacizumab or pegylated interferon alfa-2b among patients with metastatic or unresectable carcinoid tumors. The bevacizumab arm produced a superior ORR when compared to the interferon arm (12% versus 4%). The median time to treatment failure (95% CIs) in the bevacizumab arm was 9.9 months (7.3–11.1 months) and was significantly longer when compared to 5.6 months (4.3–6.4 months) in the interferon arm, but no significant differences were noted in PFS.
Toumpanakis et al33 demonstrated in an 8-year study that 59 of 108 patients with progressive metastatic NETs, based on worsening clinical symptoms and/or radiographic assessment using RECIST criteria, already treated with octreotide LAR, received combination therapy with other treatment modalities (23 with Y-90–labeled somatostatin peptides, 15 with I-131 MIBG, 16 had transarterial hepatic embolization with particles, and five patients had interferon) and about a quarter of these patients demonstrated objective response while the remaining patients had disease stabilization. More importantly, combination therapy with these various modalities was safe and tolerable. The PFS (clinical and radiologic) for all patients was 85%±3% in 2 years, 72%±4% in 3 years, 57%±5% in 5 years, and 36%±5% in 7 years. with a statistically significant difference between those on octreotide LAR alone versus those requiring additional therapy (P=0.023). Promising new combination therapies with octreotide LAR suggest the potential for antineoplastic effect and prolonged PFS.
Perspective and future development
SSA was first introduced for symptomatic management of the hormonally induced symptoms caused by NETs. Early studies demonstrated its effectiveness in symptomatic management, but lacked objective tumor response data. In recent years, the landmark findings of the PROMID trial along with other clinical studies have demonstrated the role of octreotide LAR as an antiproliferative agent, both as a single agent and in combination with chemotherapy and cytoreductive surgery, capable of stabilizing and, in some cases, even regressing tumor burden in metastatic carcinoid and pancreatic NETs. When managing well-differentiated NETs, delaying treatment with long-acting somatostatin analog until tumor progression does not adversely affect survival over early aggressive treatment. Individualized treatment decisions taking into account risk factors for tumor progression, such as mitotic count, Ki-67 index, and tumor load as well as the patient’s desire for treatment, are warranted.
Aside from their therapeutic value, radiolabeled somatostatin analogs also play a burgeoning role in diagnostic imaging of NETs. Somatostatin receptor directed positron emission tomography/computed tomography (SSTR PET/CT) imaging is gaining wider acceptance and usage since the approval of 68Ga-DOTA0-DPhe1-Tyr3-octreotate (68Ga-DOTATATE) by the FDA in 2016. Systematic review of nine retrospective and five prospective studies involving a combined 1,561 patients demonstrated that SSTR PET/CT imaging using gallium-based radioconjugates (68Ga-DOTATATE, 68Ga-DOTATOC, and 68Ga-DOTANOC) impacted clinical management changes in 44% of patients (95% CI: 36%–51%).34 While historical studies have demonstrated the superiority of gallium-based radioconjugates imaging modality compared to traditional 111In-DTPA-octreotide (Octreoscan),35–38 a recent systematic analysis by Barrio et al34 highlights how improved imaging not only plays a significant role from a diagnostic imaging perspective, but can also potentially alter clinical management. While there are potential concerns that patients that treatment with synthetic somatostatin analogs prior to SSTR PET/CT imaging may present as a potential source of error in imaging by interfering with tumor detection, Ayati et al39 have demonstrated that while octreotide LAR treatment could significantly decrease the maximum and the mean standardized uptake values (SUVmax and SUVmean) in healthy target organs on 68Ga-DOTATATE PET/CT scan (P<0.05), tracer uptake in primary or metastatic lesions in the liver, bone, lung, or lymph nodes post-octreotide LAR treatment was not affected (P>0.05). Further studies evaluating how clinical management decision changes guided by SSTR PET/CT imaging affect patient outcomes and survival are much needed.
Octreotide LAR has been available for over two decades with a well-established safety profile based on clinical experience. Somatostatin analogs will continue to be key therapeutic agents for symptomatic relief of NET-induced syndromes. Furthermore, they have a survival benefit as shown by comparison of the US-based SEER database from 1988 to 2004 compared with 1973–1987, coinciding with the FDA approval and clinical availability of octreotide as a therapeutic agent.40 Retrospective studies by Shen et al41–43 also demonstrated potential survival benefits of octreotide LAR among elderly patients with distant stage NET with or without carcinoid syndrome. While there are limited data about the duration and predictors of response to octreotide LAR, a recent study by Laskaratos et al44 of treatment-naïve patients with advanced NETs demonstrated that male gender, carcinoid heart disease, and initiation of treatment in the presence of stable disease were predictors of better response to therapy, while advanced disease with extremely elevated (>10 times the upper limit of normal) baseline CgA was associated with unfavorable outcome.
Aside from expressing multiple somatostatin receptor subtypes, bronchial (foregut), GI, and thymic NETs can lead to chronic hypercortisolism resulting from ectopic adrenocorticotropin hormone secretion.45–47 Glucocorticoids have been shown to directly downregulate SSTR-2 expression in human NET cells. The downregulation of SSTR-2 in human neuroendocrine cell lines was reversed with the addition of mifepristone, a glucocorticoid receptor antagonist.48 Mifepristone may also directly influence tumoral SSTR-2 expression levels in human NETs.49 Various published case reports have reported the synergistic effects of glucocorticoid receptor antagonism with somatostatin analogs in ameliorating the metabolic complications associated with hypercortisolism as well as enhancing the efficacy of somatostatin analogs in treatment, diagnostic imaging, and tumor localization.49–51
Based on current and historical data, octreotide LAR remains the cornerstone of first-line treatment for most patients with NETs and for those with advanced NETs as part of combination therapy. Ongoing research efforts to evaluate the use of octreotide LAR as well as other current and future somatostatin analogs with greater affinity and specificity for specific somatostatin receptor subtypes and their downstream molecular effects as monotherapy or in combination with other chemotherapies or treatment modalities are much needed.
Disclosure
Dr Moraitis serves as a Medical Director at Corcept Therapeutics, Menlo Park, CA, USA. Dr Yau is a consultant for Corcept Therapeutics, Menlo Park, CA, USA. The authors report no other conflicts of interest in this work.
References
Thulin L, Samnegard H, Tyden G, Long DH, Efendic S. Efficacy of somatostatin in a patient with carcinoid syndrome. Lancet. 1978; 312(8079):43. | ||
Frolich JC, Bloomgarden ZT, Oates JA, McGuigan JE, Rabinowitz D. The carcinoid flush. Provocation by pentagastrin and inhibition by somatostatin. N Engl J Med. 1978;299(19):1055–1057. | ||
Bauer W, Briner U, Doepfner W, et al. SMS 201–995: a very potent and selective octapeptide analogue of somatostatin with prolonged action. Life Sci. 1982;31(11):1133–1140. | ||
Modlin IM, Latich I, Kidd M, Zikusoka M, Eick G. Therapeutic options for gastrointestinal carcinoids. Clin Gastroenterol Hepatol. 2006;4(5):526–547. | ||
Ludlam WH, Anthony L. Safety review: dose optimization of somatostatin analogs in patients with acromegaly and neuroendocrine tumors. Adv Ther. 2011;28(10):825–841. | ||
Strosberg JR, Benson AB, Huynh L, et al. Clinical benefits of above-standard dose of octreotide LAR in patients with neuroendocrine tumors for control of carcinoid syndrome symptoms: a multicenter retrospective chart review study. Oncologist. 2014;19(9):930–936. | ||
Chadha MK, Lombardo J, Mashtare T, et al. High-dose octreotide acetate for management of gastroenteropancreatic neuroendocrine tumors. Anticancer Res. 2009;29(10):4127–4130. | ||
Ferolla P, Faggiano A, Grimaldi F, et al. Shortened interval of long-acting octreotide administration is effective in patients with well-differentiated neuroendocrine carcinomas in progression on standard doses. J Endocrinol Invest. 2012;35(3):326–331. | ||
Klimstra DS. Pathology reporting of neuroendocrine tumors: essential elements for accurate diagnosis, classification, and staging. Semin Oncol. 2013;40(1):23–36. | ||
Klimstra DS, Beltran H, Lilenbaum R, Bergsland E. The spectrum of neuroendocrine tumors: histologic classification, unique features and areas of overlap. Am Soc Clin Oncol Educ Book. 2015:92–103. | ||
Yamaguchi T, Fujimori T, Tomita S, et al. Clinical validation of the gastrointestinal NET grading system: Ki-67 index criteria of the WHO 2010 classification is appropriate to predict metastasis or recurrence. Diagn Pathol. 2013;8:65. | ||
Faggiano A, Carratu AC, Guadagno E, et al. Somatostatin analogues according to Ki67 index in neuroendocrine tumours: an observational retrospective-prospective analysis from real life. Oncotarget. 2016;7(5):5538–5547. | ||
Rubin J, Ajani J, Schirmer W, et al. Octreotide acetate long-acting formulation versus open-label subcutaneous octreotide acetate in malignant carcinoid syndrome. J Clin Oncol. 1999;17(2):600–606. | ||
Garland J, Buscombe JR, Bouvier C, et al. Sandostatin LAR (long-acting octreotide acetate) for malignant carcinoid syndrome: a 3-year experience. Aliment Pharmacol Ther. 2003;17(3):437–444. | ||
Anthony L, Vinik AI. Evaluating the characteristics and the management of patients with neuroendocrine tumors receiving octreotide LAR during a 6-year period. Pancreas. 2011;40(7):987–994. | ||
Rinke A, Muller HH, Schade-Brittinger C, et al; PROMID Study Group. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–4663. | ||
Rinke A, Wittenberg M, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in Patients with Metastatic Neuroendocrine Midgut Tumors (PROMID): Results of Long-Term Survival. Neuroendocrinology. 2017;104(1):26–32. | ||
Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–3072. | ||
Pavel ME, Hainsworth JD, Baudin E, et al; RADIANT-2 Study Group. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet. 2011;378(9808):2005–2012. | ||
Yao JC, Lombard-Bohas C, Baudin E, et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol. 2010;28(1):69–76. | ||
Pavel ME, Baudin E, Oberg KE, et al. Efficacy of everolimus plus octreotide LAR in patients with advanced neuroendocrine tumor and carcinoid syndrome: final overall survival from the randomized, placebo-controlled phase 3 RADIANT-2 study. Ann Oncol. 2017;28(7):1569–1575. | ||
Bajetta E, Catena L, Fazio N, et al. Everolimus in combination with octreotide long-acting repeatable in a first-line setting for patients with neuroendocrine tumors: an ITMO group study. Cancer 2014;120(16):2457–2463. | ||
Tippeswamy R, Patil S, Sateesh CT, et al. Everolimus plus octreotide long-acting repeatable in advanced neuroendocrine tumors in the routine tertiary cancer care setting: an Indian experience. Indian J Cancer. 2015;52(3):359–362. | ||
Cives M, Strosberg J. Radionuclide therapy for neuroendocrine tumors. Curr Oncol Rep. 2017;19(2):9. | ||
Krenning EP, Kooij PP, Bakker WH, et al. Radiotherapy with a radiolabeled somatostatin analogue, [111In-DTPA-D-Phe1]-octreotide. A case history. Ann N Y Acad Sci. 1994;733:496–506. | ||
Kwekkeboom DJ, de Herder WW, Kam BL, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26(13):2124–2130. | ||
Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125–135. | ||
Bodei L, Cremonesi M, Grana CM, et al. Peptide receptor radionuclide therapy with (1)(7)(7)Lu-DOTATATE: the IEO phase I-II study. Eur J Nucl Med Mol Imaging. 2011;38(12):2125–2135. | ||
Sabet A, Ezziddin K, Pape UF, et al. Long-term hematotoxicity after peptide receptor radionuclide therapy with 177Lu-octreotate. J Nucl Med. 2013;54(11):1857–1861. | ||
Bodei L, Kwekkeboom DJ, Kidd M, Modlin IM, Krenning EP. Radiolabeled somatostatin analogue therapy of gastroenteropancreatic cancer. Semin Nucl Med. 2016;46(3):225–238. | ||
Deutsch GB, Lee JH, Bilchik AJ. Long-term survival with long-acting somatostatin analogues plus aggressive cytoreductive surgery in patients with metastatic neuroendocrine carcinoma. J Am Coll Surg. 2015;221(1):26–36. | ||
Yao JC, Phan A, Hoff PM, et al. Targeting vascular endothelial growth factor in advanced carcinoid tumor: a random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. J Clin Oncol. 2008;26(8):1316–1323. | ||
Toumpanakis C, Garland J, Marelli L, et al. Long-term results of patients with malignant carcinoid syndrome receiving octreotide LAR. Aliment Pharmacol Ther. 2009;30(7):733–740. | ||
Barrio M, Czernin J, Fanti S, et al. The impact of somatostatin receptor-directed PET/CT on the management of patients with neuroendocrine tumor: a systematic review and meta-analysis. J Nucl Med. 2017;58(5):756–761. | ||
Deppen SA, Liu E, Blume JD, et al. Safety and efficacy of 68Ga-DOTATATE PET/CT for diagnosis, staging, and treatment management of neuroendocrine tumors. J Nucl Med. 2016;57(5):708–714. | ||
Buchmann I, Henze M, Engelbrecht S, et al. Comparison of 68Ga-DOTATOC PET and 111In-DTPAOC (Octreoscan) SPECT in patients with neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2007;34(10):1617–1626. | ||
Deppen SA, Blume J, Bobbey AJ, et al. 68Ga-DOTATATE compared with 111In-DTPA-octreotide and conventional imaging for pulmonary and gastroenteropancreatic neuroendocrine tumors: a systematic review and meta-analysis. J Nucl Med. 2016;57(6):872–878. | ||
Krausz Y, Freedman N, Rubinstein R, et al. 68Ga-DOTA-NOC PET/CT imaging of neuroendocrine tumors: comparison with 111In-DTPA-octreotide (OctreoScan®). Mol Imaging Biol. 2011;13(3):583–593. | ||
Ayati N, Lee ST, Zakavi SR, et al. Sandostatin LAR therapy differentially alters 68Ga-DOTATATE uptake in normal tissues compared to primary tumors and metastatic lesions. J Nucl Med. Epub 2017 Jul 20. | ||
Sideris L, Dube P, Rinke A. Antitumor effects of somatostatin analogs in neuroendocrine tumors. Oncologist. 2012;17(6):747–755. | ||
Shen C, Shih YC, Xu Y, Yao JC. Octreotide long-acting repeatable use among elderly patients with carcinoid syndrome and survival outcomes: a population-based analysis. Cancer. 2014;120(13):2039–2049. | ||
Shen C, Shih YC, Xu Y, Yao JC. Octreotide long-acting repeatable among elderly patients with neuroendocrine tumors: a survival analysis of SEER-Medicare data. Cancer Epidemiol Biomarkers Prev. 2015;24(11):1656–1665. | ||
Shen C, Xu Y, Dasari A, Shih YC, Yao JC. Octreotide LAR dosage and survival among elderly patients with distant-stage neuroendocrine tumors. Oncologist. 2016;21(3):308–313. | ||
Laskaratos FM, Walker M, Naik K, et al. Predictive factors of antiproliferative activity of octreotide LAR as first-line therapy for advanced neuroendocrine tumours. Br J Cancer. 2016;115(11):1321–1327. | ||
Ejaz S, Vassilopoulou-Sellin R, Busaidy NL, et al. Cushing syndrome secondary to ectopic adrenocorticotropic hormone secretion: the University of Texas MD Anderson Cancer Center Experience. Cancer. 2011;117(19):4381–4389. | ||
Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab. 2005;90(8):4955–4962. | ||
Isidori AM, Kaltsas GA, Pozza C, et al. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metabo. 2006;91(2):371–377. | ||
de Bruin C, Feelders RA, Waaijers AM, et al. Differential regulation of human dopamine D2 and somatostatin receptor subtype expression by glucocorticoids in vitro. J Mol Endocrinol. 2009;42(1):47–56. | ||
de Bruin C, Hofland LJ, Nieman LK, et al. Mifepristone effects on tumor somatostatin receptor expression in two patients with Cushing’s syndrome due to ectopic adrenocorticotropin secretion. J Clin Endocrinol Metab. 2012;97(2):455–462. | ||
Banerjee RR, Marina N, Katznelson L, Feldman BJ. Mifepristone treatment of cushing’s syndrome in a pediatric patient. Pediatrics. 2015;136(5):e1377–e1381. | ||
Moraitis AG, Auchus RJ. Mifepristone improves octreotide efficacy in resistant ectopic cushing’s syndrome. Case Rep Endocrinol. 2016;2016:8453801. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.