")
Back to Journals » OncoTargets and Therapy » Volume 12
NUT midline carcinoma of the head and neck: current perspectives
Authors Napolitano M, Venturelli M , Molinaro E, Toss A
Received 15 January 2019
Accepted for publication 12 March 2019
Published 30 April 2019 Volume 2019:12 Pages 3235—3244
DOI https://doi.org/10.2147/OTT.S173056
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
M Napolitano, M Venturelli, E Molinaro, A Toss
Department of Oncology and Hematology, University Hospital of Modena, Modena, Italy
Abstract: NUT midline carcinoma (NMC) is a rare and aggressive subtype of squamous carcinoma that typically arises from midline supradiaphragmatic structures, frequently from the head and neck area. NMC is genetically driven by a chromosomal rearrangement involving the NUT gene, which forms oncoproteins considered major pathogenic drivers of cellular transformation. Diagnosis of NMC has been made remarkably easier with the availability of a commercial antibody against NUT, and can be established through positive nuclear immunohistochemical staining. Although NMC remains an underrecognized malignancy, in recent years there has appeared to be increasing awareness of disease and frequency of diagnosis in adults. To date, a standard treatment for head and neck NMC has not been established and a multimodal approach with systemic chemotherapy, surgery and radiation therapy is currently adopted in clinical practice. Recently, BET inhibitors and histone deacetylase inhibitors have emerged as two promising classes of targeted agents, currently investigated in clinical trials for adults with head and neck NMC. At the same time, combination approaches and novel targeted agents, such as next-generation BET inhibitors and CDK9 inhibitors, have shown preclinical activity. The present review explores the clinical pathological characteristics of NMC of the head and neck and presents the current state of the art on diagnosis, prognosis, and treatment of this rare but lethal disease.
Keywords: NUT midline carcinoma, head and neck, BRD4-NUT, BET inhibitors, histone deacetylase inhibitors
Introduction
NUT midline carcinoma (NMC) is a genetically defined epithelial malignant neoplasm hallmarked by chromosomal rearrangement of the NUT gene. The most common rearrangement is a translocation between the NUT and BRD4 gene, forming the BRD4–NUT oncoprotein that is considered a major pathogenic driver of cellular transformation.1 In a third of cases, variant NUT rearrangements involve other genes, such as BRD3 and NSD3.2 Consistent with their oncogenic function of blocking epithelial squamous differentiation, fusion proteins maintain the proliferation of immature neoplastic cells, providing a rationale for targeting these proteins.
Initially described in children and adolescents, it is now clear that NMC can develop in males and females of all ages, although its true incidence remains unclear.3 NMC typically arises from midline supradiaphragmatic structures: the upper aerodigestive tract (50%) and the mediastinum (41%).4 However, rarer cases have been diagnosed below the diaphragm (bladder)5 and outside the midline axis (major salivary glands, iliac bone, adrenal gland, and pancreas).6–8
NMC is considered the most aggressive subtype of squamous carcinoma, with >80% of patients dying within 1 year of diagnosis. Treatment approaches have been heterogeneous over the years, and no standard has yet been established. Novel targeted agents, such as histone deacetylase inhibitors (HDACis) and BET inhibitors (BETis), hold great promise alone or in combination with chemotherapy. Because of the disease's rarity, an international NMC registry has been in development since 2010 to pursue a twofold aim. The first is to raise awareness and disseminate information about NMC. The second is to collect clinical data on the disease and its response to treatment, creating a repository of clinical specimens that will support future research.
The present paper reviews the clinical pathological characteristics of head and neck NMC (HNNMC) and presents the current state of the art on diagnosis, prognosis, and treatment of this rare but lethal disease.
Clinical presentation and outcome: a rare and devastating disease
Approximately 39% of NMCs arise from the head and neck region, while the sinonasal area is the most common tumor site, followed by the nasopharynx, oropharynx, hypopharynx, larynx, and unknown primary site.9 The true incidence of HNNMC remains unknown, but increased awareness of the disease and the availability of new easily applicable diagnostic tests could explain both the apparent increase in the frequency of diagnosis, particularly since 2012, and the greater number of adult cases.3,10 In the largest HNNMC cohort reported in the literature to date, the median age at diagnosis was 21.9 years, with the majority of tumors occurring in females.10 The presumed rarity of HNNMC, coupled with its occurrence in young age with minimal smoking history and lack of pathognomonic histopathological features, suggests that the disease remains underrecognized. Only a minority of cases are diagnosed with NMC at the beginning, while the most common incorrect original diagnoses are “poorly differentiated carcinoma” or “poorly differentiated squamous carcinoma”.10 Due to the small number of patients, no epidemiological studies of etiologic factors have been conducted. Nevertheless, there is no evidence that smoking or other environmental factors are associated with HNNMC. In particular, no cases diagnosed to date have been associated with Epstein–Barr virus or human papilloma virus (HPV) infection.
HNNMC runs a devastating clinical course. It usually presents with rapidly enlarging masses, characterized at advanced stages by early metastatic spread to either locoregional lymph nodes or less common distant sites.3 Consequently, most patients present with mass-related symptoms (such as rhinorrhea, epistaxis, nasal obstruction, proptosis, diminished vision, dysphagia, or pain), while aspecific symptoms, such as fever and weight loss, have been seen only occasionally.11
Genetic background: a molecularly defined cancer
The genetic hallmark of NMC is the chromosomal rearrangement of the NUT gene (also known as NUTM1 or Chr15orf55), first described in 2003 by French et al (Figure 1).12 In two-thirds of NMCs, a reciprocal chromosomal translocation involves the NUT gene on chromosome 15q14 and BRD4 on chromosome 19p13.1 (Figure 1). The BRD4 protein, encoded by the BRD4 gene, is the most extensively studied member of the BET protein family, first identified by Jiang et al in 1988.13,14 The classical translocation t(15;19)(q14;p13.1) fuses exon 3 of the NUT gene to exon 11 of the BRD4 gene. This results in an in-frame fusion gene of 6.4 kb that encodes a BRD4–NUT oncoprotein involved in carcinogenesis and driven by the BRD4 promoter.12,3–5,5,6–18 Although three isoforms of the BRD4 protein have been described (called A, B, and C), only isoform C, ubiquitously expressed, is involved in the BRD4–NUT fusion protein.19 The BRD4–NUT fusion gene contains the whole coding region for NUT, which is entirely included in the fusion process, while BRD4 loses its C-terminal domain, including all of its functional domains (Figure 2).
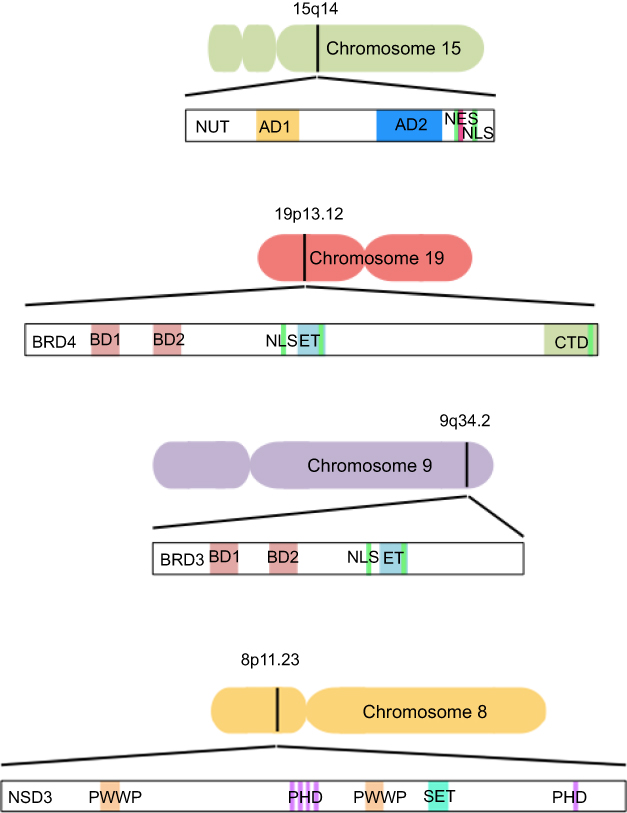 | Figure 1 Schematic representation of native component genes and domain structures of BET proteins (BRD3, BRD4), NSD3, and NUT.Note: Black bars on chromosomes indicate location of translocation-associated break points.Abbreviations: AD, acidic domain; NES, nuclear export signal; NLS, nuclear localization sequence; ET, extraterminal; CTD, C-terminal domain; PWWP, proline–tryptophan–tryptophan–proline; PHD, plant homeodomain. |
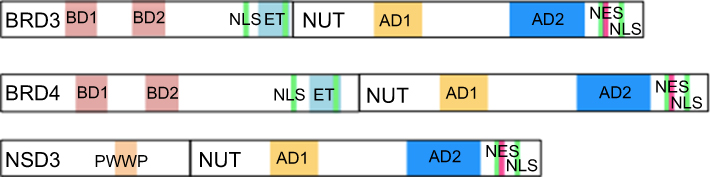 | Figure 2 Schematic representation of fusion oncoproteins involved in NMC.Note: In every fusion oncoprotein, the entire NUT structure is included.Abbreviations: AD, acidic domain; NES, nuclear export signal; NLS, nuclear localization sequence; ET, extraterminal; PWWP, proline–tryptophan–tryptophan–proline. |
On the other hand, in a third of NMCs, the NUT partner genes are BRD3 (Figure 1), NSD3 (Figure 1) or other uncharacterized genes.1,15 In these rearrangements, the entire NUT structure is maintained in the fusion oncogene, along with the bromodomains, the extraterminal domain and the bipartite nuclear localization sequence of BRD3 (Figure 2). Instead, NSD3 is an enzymatic protein involved in the methylation of histone lysine marks, regulating chromatin integrity and gene expression. Only the N-terminus of NSD3 is included in the genetic fusion process, whereas its methyltransferase domain is absent (Figure 2).20
Rare cases of NMC in which NUT is fused to a zinc finger–containing protein, such as ZNF532 and ZNF592, have recently been described in the literature.21,22 ZNF532 is involved in feed-forward regulatory loops that drive propagation of the oncogenic chromatin complex in BRD4–NUT cells. ZNF532, as well as BRD3 and NSD3, interacts with BRD4, and its fusion with NUT results in a powerful oncogenic complex. All these rare fusion partners of NUT are functionally related to BRD4, indicating that the recruitment of NUT to chromatin through the BET family proteins is necessary in NMC pathogenesis. For these reasons, NMC is considered the prototype BET-driven cancer, offering an opportunity to study both oncogenic BRD4 pathways and the effects of its potential inhibition.
Interestingly, several studies have shown that mutation type did not affect prognosis in NMC patients, although a recent work reported a better outcome for NSD3- or BRD3–NUT-positive HNNMC than for those with BRD4–NUT.3,18 However, since most patients carry the BRD4–NUT translocation, leading to a lack of statistical power, the prognostic impact of the translocation type remains unclear.
Several in vitro studies with patient-derived tumor cells using knockdown of BRD3/4–NUT and NSD3–NUT genes have provided evidence of terminal and irreversible squamous differentiation and growth arrest.1,2 This observation indicates that NUT fusion proteins act to maintain growth and block squamous-cell differentiation, by suppressing transcription and decreasing histone acetylation23 in a mechanism dependent on the targeting of MYC and TP63 genes by BRD.18,21
From diagnosis to prognostication
Imaging
Although the radiological features of NMC are not specific, their appearance may be indicative of aggressive malignancy, with a propensity to invade neighboring structures.24 Computed tomography is generally characterized by heterogeneous enhancement of an infiltrative or destructive-appearing primary mass and low attenuation of pathological lymph nodes, related to evidence of necrosis and hemorrhage in the surgery samples. Magnetic resonance imaging represents the gold standard for diagnosis and correct staging of HNNMC, providing superior soft-tissue delineation compared to computed tomography for evaluation of masses involving the head and neck, as well as the musculoskeletal system. Additionally, magnetic resonance imaging plays an important role intreatment planning, providing critical information, such as the presence of vascular invasion, perineural involvement, and skull-base invasion. Fludeoxyglucose positron-emission tomography should be used after confirmed diagnoses of HNNMC to assess the presence of distant metastases, although it may underestimate disease burden, because of low-level fludeoxyglucose uptake in necrotic areas.25
Cytogenetics and histopathology
A distinctive feature of HNNMC is a remarkably simple karyotype, often with a single translocation as its sole cytogenetic aberration, classically a reciprocal t(15;19)(q14;p13.1).5 This finding distinguishes HNNMC from more common head and neck carcinomas, including squamous-cell carcinomas of adulthood, which exhibit complex aneuploid karyotypes and high mutational burden.26
The histopathological features of HNNMC are characteristic, though not diagnostic. The most common appearance is that of a poorly differentiated carcinoma with focal or extensive abrupt keratinization.27 Tumors are usually composed of sheets of undifferentiated medium-size and oval cells with scant amphiphilic or eosinophilic cytoplasm (called “fried egg”-like cells). Characteristically, the cells are monomorphic in appearance, in contrast to other poorly differentiated carcinomas, which consist of striking pleomorphic cells. They have typically vesicular chromatin and prominent nucleoli. Foci of necrosis can be present and mitotic figures are common, reflecting rapid tumor growth. Although infiltrating lymphocytes can occasionally be found, the presence of neutrophilic infiltrates is more common and can be prominent. The unusual pathological pattern occurring in the salivary glands exhibits prominent mesenchymal differentiation, including the presence of myxoid matrix or cartilage.7
When to perform the NUT IHC test
The diagnosis of HNNMC has been made remarkably easier with the availability of a commercial antibody against NUT (clone C52B1) from Cell Signaling Technologies (Danvers, MA, USA). This monoclonal rabbit antibody, developed on the basis of knowledge that NUT expression should not normally be seen outside the testes, can be used routinely in the community to detect NUT expression by immunohistochemistry (IHC).15 While germ-cell tumors and rare poorly differentiated carcinomas can be stained only focally (<10%), NMC diagnosis by IHC with the NUT antibody reveals diffuse nuclear staining, often with a speckled pattern. Haack et al verified the accuracy of the C52B1 antibody to detect NUT rearrangement confirmed by fluorescent in situ hybridization. They demonstrated 87% sensitivity and 100% specificity; therefore, strongly positive staining is virtually diagnostic of NMC.28 In particular, following the World Health Organization, IHC staining >50% of tumor nuclei is considered diagnostic of NMC.29,30 Although not required to confirm diagnosis, various molecular analyses able to demonstrate NUT rearrangement can be used if the monoclonal antibody is not available. These methods include fluorescence in situ hybridization (FISH), reverse-transcription PCR, cytogenetics and next-generation sequencing-based approach. Fluorescence in situ hybridization is the preferred assay, because it detects all NMCs,5 including atypical fusion break points between BRD4/BRD3 and NUTM1, as well as rare fusion partners to NUTM1, whereas reverse-transcription PCR can currently only detect BRD3– or BRD4–NUT tumors and is probably the least sensitive approach.31,32 Fusion-partner characterization may have a clinical impact in view of recent data that indicate a better prognosis for NSD3- or BRD3–NUT-positive HNNMC patients than for those with BRD4-NUT.9
Today, the challenge is no longer diagnosing NMC, but determining when to perform the test. With greater awareness of HNNMC and with the availability of this simple diagnostic assessment, the number of diagnosed HNNMC cases is increasing, and it is likely it will continue to increase. NUT IHC testing is recommended in all poorly differentiated noncutaneous carcinomas of the head and neck with or without squamous differentiation that exhibit a monomorphic pattern. Tumors with glandular differentiation (extremely rare in HNNMC) should not be tested, and viral etiology (such as HPV or Epstein–Barr virus) can be used to exclude HNNMC diagnosis. Interestingly, in many HNNMC cases, strong p16 expression for can be present. However, this are not positive for HPV evaluated by PCR, indicating that HPV infection does not play a role in the pathogenesis of HNNMC.33
Risk categories and prognosis
Due to NMC's rarity and under-diagnosis, there are no existing models to classify patients into risk groups based on baseline clinicopathological factors. Recently, Chau et al developed a prognostic risk classification model for NMC survival outcomes based on the largest cohort of NMC patients analyzed to date, identifying three distinct risk groups: patients without lymph-node or organ metastases, patients with lymph-node or organ metastases and nonthoracic origin, and patients with metastases and thoracic origin.9 The authors concluded that the group of metastatic patients and thoracic primary tumors had markedly poorer prognosis than other subgroups. Another factor that shows an impact on outcome is tumor size, with worse prognosis for larger tumors (>5 cm) than smaller tumors.18
Although data regarding HNNMC are mainly restricted to case reports with limited treatment and follow-up information, a retrospective review of all HNNMC cases in the International NUT Midline Carcinoma Registry was performed. HNNMC showed slightly better survival outcomes than thoracic NMC, based on historical comparison, with median survival of months and 2-year overall survival of 30%.3,10 Despite the small number of cases reported to date and the lack of prospective comparisons, Chau et al showed that complete surgical resection with no residuum was associated with significantly improved survival, in particular for tumors <6 cm in size. The initial therapeutic sequencing strategy seemed to be the most critical factor to affect prognosis. Chemotherapy and radiation alone were often inadequate, even though both may be important as adjuvant treatment. Other clinical or pathologic features, including sex, age, and type of chemotherapy regimen, were not associated with survival outcomes, while the role of NUT-translocation type was unclear because the majority of patients had BRD4–NUT translocation.
Therapeutic management: from orphan to target disease
Despite the fact that a standard treatment for HNMMC has not been established yet, a multimodal approach with systemic chemotherapy, surgery, and radiation therapy is adopted in clinical practice. Due to the rarity of the disease and the ununiform treatments administered, it has been challenging to evaluate the efficacy of various therapeutic approaches.3 When feasible, surgery is usually considered the primary option associated with improved outcome. In a report from the NUT Midline Carcinoma Registry, 40 patients with HNMMC were evaluable, and 2-year overall survival was 30%, with the three long-term survivors (35, 72, and 78 months) undergoing primary gross–total resection and adjuvant therapy.10
Despite the small number of cases and the lack of prospective comparison, this report showed that complete surgical resection was associated with significantly improved survival in contrast with initial radiation or chemotherapy, which did not seem to have an impact on survival. Since survival appears to be affected by response to initial therapy, the treatment sequencing strategy becomes critical to the management of HNNMC. However, the disease is often locally advanced and/or distantly metastatic at diagnosis. Because of that, complete resection often cannot be safely performed and most patients receive postoperative radiation or chemotherapy.18 A variety of radiation therapy and chemotherapy modalities have been used in both the adjuvant setting and the exclusive setting. These heterogeneous systemic therapies included intensive chemotherapy regimens commonly applied in the event of other carcinomas, sarcomas, germ cell tumors and other solid neoplasms. In particular, Cisplatin, taxanes and alkylating agents have been used with some success.11,16,34 However, while rapid response is common, tumor progression occurs early in a treatment-refractory manner and the overall outcome for these patients remains poor.35
Preclinical studies have shown that the NUT-BRD4 fusion is associated with globally decreased histone acetylation and transcriptional repression.Studies have also shown that this acetylation can be restored with histone deacetylase inhibitors, resulting in squamous differentiation and arrested growth in vitro and growth inhibition in xenograft models.23,36 BET inhibitors (BETi) and HDACi represent two promising class of agents that are being investigated for adults with HNNMC, either alone or in combination with chemotherapy.
BET inhibitors (BETi): another way to target BRD-NUT
On the basis of the main role of bromodomain-containing NUT fusion proteins in NMC development, inhibitors of bromodomain of BET proteins have been investigated with promising leads. BET inhibitors are acetyl-lysine mimetic compounds that bind the acetyl-lysing binding pocket of both bromodomains (BD1 and BD2) of all BET proteins including BRDT, BRD2, BRD3 and BRD4.37 Therefore, the antitumor activity of these small molecules derives from the interruption of interactions between BET proteins and acetylated lysine in histones at promoters and enhancers. Originally identified in small molecules used to treat autoimmune disease, they were later discovered for their BET inhibitory properties.37
JQ1, a first-in-class BETi, causes dissolution of BRD4-NUT nuclear speckles resulting in rapid terminal differentiation, apoptosis and growth arrest of cultured NMC cells, as demonstrated by immunocytochemistry with an antikeratin (AE1/AE3) antibody.20 In vivo, JQ1 (administered at 50 mg/kg daily) induced the suppression of tumor growth and improved survival in NMC xenograft models, provided the proof of principle that the inhibition of BRD4 by BETi can be therapeutically targeted.38 Since then, there has been an explosion of clinical trials using BETi in solid and liquid tumors.39–44
Birabresib (MK-8628/OTX015) is a novel BET inhibitor currently in clinical development. It targets BRD2/3/4 with preclinical activity in NMC and several other cancers, particularly selected hematologic tumors.43,45,46 A recent phase Ib study evaluating Birabresib in patients with selected advanced solid tumors, including NMC, highlighted a clinical activity of particular interest for NMC.47 Of the nine evaluable patients with NMC, six demonstrated partial response (PR) or stable disease (SD), with three patients having a PR and a favorable tolerability profile of reversible and self-limiting thrombocytopenia that required dose modification. Previously, Stathis et al described the outcomes of four NMC patients treated with Birabresib outside a clinical trial, reporting dramatic and rapid response with symptomatic improvement in two patients and stable disease in another.38 Interestingly, though all patients eventually died of the disease, two of them had reached an overall survival of 18 and 19 months, markedly longer than the mOS reported in the largest retrospective HNNMC and NMC cohorts published to date (9.7 and 6.7 months respectively).3,10
Since these drugs inhibit all BET proteins expressed in most tissues, it is not surprising that toxicity limited their efficacy in NMC. The most common types of toxicity are diarrhea, nausea, decreased appetite and thrombocytopenia. These derive from a repression of transcription in erythroid and megakaryocytic genes and represent the most common treatment-related serious event requiring dose modification and also treatment interruption.47,48 As for other target therapies, the initial but only transient response to treatment with BETi suggests the emergence of secondary resistance mechanisms that might differ according to cancer type. These mechanisms could include restoration of MYC expression through the WNT pathway or BRD4 phosphorylation.40,49–51 Recently, Liao et al systematically explored about 900 tumor suppressor genes and oncogenes for their ability to mediate resistance of NMC to BETi. By using high-throughput loss-of-function and gain-of-function screening technologies, they identified six pathways mediating resistance.52 Among these, the authors highlighted multiple ways for NMC cells to bypass the cell cycle arrest induced by BETi, including up-regulation of cyclin D1 or cyclin D3 mutant or RB1 loss. In accordance with this finding, CDK4/6 inhibitors showed synergistic activity with JQ1 on NMC in vitro and in vivo, providing a rationale for combination therapy of BETi and CDK4/6 inhibitors for this malignancy.
Several other trials on BETi enrolling NMC patients are ongoing (Table 1) and multiple additional studies are enrolling patients with hematologic and solid cancers.
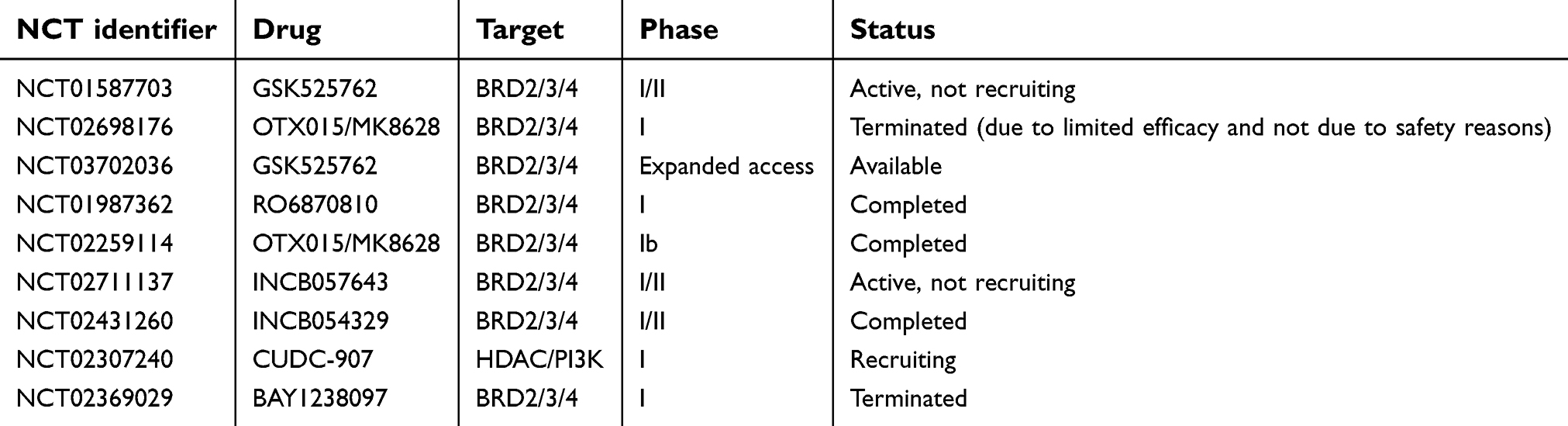 | Table 1 Ongoing or already available clinical trials of targeted therapy for NMC |
Histone deacetylase inhibitors (HDACi)
BRD4/NUT protein is suggested active histone acetyl-transferase p300, thereby sequestering histone acetyltransferase activity to localized regions of BRD4-NUT-acetyl- chromatin binding. This results in an overabundance of HDAC activity outside of these regions, which leads to global hypoacetylation and transcriptional repression of genes required for differentiation.53 It has been hypothesized that these effects can be reversed by hyperacetylating histones artificially with the use of HDACi, restoring chromatin acetylation and increasing the transcription of pro-differentiative genes.
In xenograft models of NMC, HDACi significantly inhibited growth, induced differentiation and improved survival, providing preclinical support for the use of HDACi as targeted therapeutic agents for NMC. Based on these findings, Vorinostat, a histone deacetylase inhibitor, has been used clinically with dramatic objective response, before adverse effects limited its use.23,36 In contrast, Maur et al described a clinical case of ineffective treatment with Romidepsin, another HDACi currently studied in a variety of cancers, possibly due to primary resistance or secondary resistance mechanisms such as parallel pathway activation, drug-induced de novo genetic variations or target gene change.35
Currently, a phase I trial (NCT02307240) evaluating the safety, tolerability and pharmacokinetics of CUDC-907, a dual HDACi/PI3K inhibitor, is open to enrolling NMC patients (Table 1). The antitumor activity of CUDC-907 against NMC cells has been demonstrated in vitro and in animal models and appears to be associated with MYC protein downregulation, a major target of BET proteins, induced by HDAC and PI3K inhibition synergistically.54 HDAC inhibition potently suppresses MYC expression at the transcriptional level, while PI3K inhibition results in enhanced ubiquitin-mediated MYC protein degradation at the post-translational level. Munster et al reported a case of prolonged disease stabilization for over 32 months in an NMC patient treated with CUDC-907 after two prior treatments.55
Future perspectives: novel targeted agents and combination approaches
Despite the initial response, all NMC patients treated with BETi or HDACi develop resistance and relapse during treatment. Therefore, an urgent need to develop effective combination therapies and alternative therapeutic approaches arises. Recently, CDK9 has emerged as new non-oncogenic driver that is potentially and directly druggable. Together with Cyclin-T1, CDK9 forms the positive transcription elongation factor b (P-TEFb) complex, whose recruitment by BRD4 is required for BRD4-dependent transcriptional elongation.56 A study in vitro demonstrated that CDK9 inhibitors lead to robust induction of apoptosis and induction of DNA damage response in NMC cells, suggesting that CDK9 may be an attractive drug target in NMC patients.57 Beesley et al compared the efficacy of the CDK9 inhibitor flavopiridol (FP) with a panel of antitumor agents in NMC cell lines and animal models, finding that FP was one of the most cytotoxic drugs in vitro associated with significant in vivo responses.58 An oral phosphate prodrug of the CDK9 inhibitor (Alvocidib) is being tested in a phase 1 trial enrolling patients with advanced solid tumors, including those with NMC.
Much preclinical evidence of synergism between BETi and different classes of compounds has been reported in different tumor types that have been investigated. In particular, preclinical studies highlighted that BETi shows synergism with immune checkpoint modulators.59,60 JQ1 was seen to regulate expression of the immune checkpoint ligand PD-L1 and to correlate with increased anti–tumor cytotoxic T cells.61 The combination of JQ1 with PD-1 blockade in a KRAS mutant NSCLC xenograft leads to synergistic tumor burden reduction.62 Furthermore, the combined inhibition of histone deacetylases and the proteins of the BET family have recently shown therapeutic efficacy in a number of in vitro and in vivo cancer models including melanoma, pancreatic ductal adenocarcinoma, testicular cancer and lymphoma.63–66 Taken together, these findings highlight the potential of combination therapy for BETi with newer agents.
Recently, Stirweiss and colleagues performed next-generation sequencing on a large panel of NMC cell lines to understand the molecular-genetic landscape of NMC, a critical step towards developing novel therapeutic approaches.67 They identified a recurring high-impact mutation in the RECQL5 gene, which encodes for a DNA-helicase involved in interstrand crosslinking repair, and a network analysis consistent with general failure in DNA-repair.68 These findings provide preliminary evidence of a potential defect in the processes of DNA-repair within the genome of NMC cells. On thesegrounds, it is conceivable that the mutation of RECQL5 promotes the acquisition of additional mutations necessary for the NMC phenotype, leading to genetic instability that could be explored to improve therapeutic strategies. Furthermore, in keeping with a recent publication demonstrating that BETi directly suppresses the aurora kinases genes (AURKA and AURKB) in triple negative breast cancer cells, the authors identified a significant correlation between BETi and AURK inhibitors efficacy.69
It is likely that recent and future investigation on genes that drive cancer development and progression could elucidate the genetic landscape of resistance to BETi and identify combinations that might overcome adaptive resistance mechanisms to NMC therapy. More efforts in research at molecular and clinical levels remain crucial for a rare and lethal cancer such as NMC.
Disclosure
The authors report no conflicts of interest in this work.
References
1. French CA, Ramirez CL, Kolmakova J, et al. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27(15):2237–2242. doi:10.1038/sj.onc.1210852
2. French CA, Rahman S, Walsh EM, et al. NSD3-NUT fusion oncoprotein in NUT midline carcinoma: implications for a novel oncogenic mechanism. Cancer Discov. 2014;4(8):928–941. doi:10.1158/2159-8290.CD-14-0014
3. Bauer DE, Mitchell CM, Strait KM, et al. Clinicopathologic features and long-term outcomes of NUT midline carcinoma. Clin Cancer Res. 2012;18(20):5773–5779. doi:10.1158/1078-0432.CCR-12-1153
4. French CA. Demystified molecular pathology of NUT midline carcinomas. Clin Pathol. 2010;63(6):492–496. doi:10.1136/jcp.2007.052902
5. French CA, Kutok JL, Faquin WC, et al. Midline carcinoma of children and young adults with NUT rearrangement. J Clin Oncol. 2004;22(20):4135–4139. doi:10.1200/JCO.2004.02.107
6. Mertens F, Wiebe T, Adlercreutz C, Mandahl N, French CA. Successful treatment of a child with t(15;19)-positive tumor. Pediatr Blood Cancer. 2007;49(7):1015–1017. doi:10.1002/pbc.20755
7. Den Bakker MA, Beverloo BH, Van Den Heuvel-Eibrink MM, et al. NUT midline carcinoma of the parotid gland with mesenchymal differentiation. Am J Surg Pathol. 2009;33(8):1253–1258. doi:10.1097/PAS.0b013e3181abe120
8. Ziai J, French CA, Zambrano E. NUT gene rearrangement in a poorly-differentiated carcinoma of the submandibular gland. Head Neck Pathol. 2010;4(2):163–168. doi:10.1007/s12105-010-0174-6
9. Chau NG, Ma C, Danga K, et al. A novel prognostic risk classification model for NUT midline carcinoma: a largest cohort analysis from the NMC registry [abstract]. J Clin Onco. 2018;36(suppl):6085. doi:10.1200/JCO.2018.36.15_suppl.6085
10. Chau NG, Hurwitz S, Mitchell CM, et al. Intensive treatment and survival outcomes in NUT midline carcinoma of the head and neck. Cancer. 2016;122(23):3632–3640. doi:10.1002/cncr.30242
11. Kakkar A, Antony VM, Kumar Igrugu DV, Adhikari N, Jain D. NUT midline carcinoma: a series of five cases, including one with unusual clinical course. Head Neck Pathol. 2018;12:230–236. doi:10.1007/s12105-017-0858-2
12. French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 2003;63(2):304–307.
13. Jiang YW, Veschambre P, Erdjument-Bromage H, et al. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc Natl Acad Sci U S A. 1998;95(15):8538–8543.
14. Chaidos A, Caputo V, Karadimitris A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence. Ther Adv Hematol. 2015;6(3):128–141. doi:10.1177/2040620715576662
15. French CA. The importance of diagnosing NUT midline carcinoma. Head Neck Pathol. 2013;7(1):11–16. doi:10.1007/s12105-013-0428-1
16. Edgar M, Caruso AM, Kim E, Foss RD. NUT midline carcinoma of the nasal cavity. Head Neck Pathol. 2017;11(3):389–392. doi:10.1007/s12105-016-0763-0
17. French C. NUT midline carcinoma. Nat Rev Cancer. 2014;14(3):149–150. doi:10.1038/nrc3659
18. Giridhar P, Mallick S, Kashyap L, Rath GK. Patterns of care and impact of prognostic factors in the outcome of NUT midline carcinoma: a systematic review and individual patient data analysis of 119 cases. Eur Arch Otorhinolaryngol. 2018;275(3):815–821. doi:10.1007/s00405-018-4882-y
19. Floyd SR, Pacold ME, Huang Q, et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signaling. Nature. 2013;498(7453):246–250. doi:10.1038/nature12147
20. French CA. Small-molecule targeting of BET proteins in cancer. Adv Cancer Res. 2016;131:21–58. doi:10.1016/bs.acr.2016.04.001
21. Alekseyenko AA, Walsh EM, Zee BM, et al. Ectopic protein interactions within BRD4- chromatin complexes drive oncogenic megadomain formation in NUT midline carcinoma. Proc Natl Acad Sci U S A. 2017;114(21). E4184-E4192. doi:10.1073/pnas.1702086114
22. Shiota H, Elya JE, Alekseyenko AA, et al. “Z4” complex member fusions in NUT carcinoma: implications for a novel oncogenic mechanism. Mol Cancer Res. 2018;16(12):1826–1833. doi:10.1158/1541-7786.MCR-18-0474
23. Schwartz BE, Hofer MD, Lemieux ME, et al. Differentiation of NUT midline carcinoma by epigenomic reprogramming. Cancer Res. 2011;71(7):2686–2696. doi:10.1158/0008-5472.CAN-10-3513
24. Bair RJ, Chick JF, Chauhan NR, French C, Madan R. Demystifying NUT midline carcinoma: radiologic and pathologic correlations of an aggressive malignancy. AJR Am J Roentgenol. 2014;203(4):W391–W99. doi:10.2214/AJR.13.12401
25. Rosenbaum DG, Teruya-Feldstein J, Price AP, Meyers P, Abramson S. Radiologic features of NUT midline carcinoma in an adolescent. Pediatr Radiol. 2012;42(2):249–252. doi:10.1007/s00247-011-2288-8
26. Bielas JH, Loeb KR, Rubin BP, True LD, Loeb LA. Human cancers express a mutator phenotype. Proc Natl Acad Sci U S A. 2006;103(48):18238–18242. doi:10.1073/pnas.0607057103
27. French CA, Carcinoma: NUT. Clinicopathologic features, pathogenesis, and treatment. Pathol Int. 2018;68(11):583–595. doi:10.1111/pin.12727
28. Haack H, Johnson LA, Fry CJ, et al. Diagnosis of NUT midline carcinoma using a NUT- specific monoclonal antibody. Am J Surg Pathol. 2009;33(7):984–991. doi:10.1097/PAS.0b013e318198d666
29. French CA, Den Bakker MA. NUT Carcinoma. In:
30. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization Classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243–1260. doi:10.1097/JTO.0000000000000630
31. Thompson-Wicking K, Rw F, Stirnweiss A, et al. Novel BRD4-NUT fusion isoforms increase the pathogenic complexity in NUT midline carcinoma. Oncogene. 2013;32(39):4664–4674. doi:10.1038/onc.2012.487
32. Stirnweiss A, McCarthy K, Oommen J, et al. A novel BRD4-NUT fusion in an undifferentiated sinonasal tumor highlights alternative splicing as a contributing oncogenic factor in NUT midline carcinoma. Oncogenesis. 2015;4:e174. doi:10.1038/oncsis.2015.33
33. Salles PG, Moura Rde D, Menezes LM, Bacchi CE. Expression of P16 in NUT carcinomas with no association with human papillomavirus (HPV). Appl Immunohistochem Mol Morphol. 2014;22(4):262–265. doi:10.1097/PAI.0b013e3182a4ef2e
34. Seim NB, Philips RHW, Schoenfield L, et al. NUT midline carcinoma of the sublingual gland: clinical presentation and review. Head Neck Pathol. 2017;11(4):460–468. doi:10.1007/s12105-017-0809-y
35. Maur M, Toss A, Dominici M, et al. Impressive response to dose-dense chemotherapy in a patient with NUT Midline Carcinoma. Am J Case Rep. 2015;16:424–429. doi:10.12659/AJCR.893879
36. Maher OM, Christensen AM, Yedururi S, Bell D, Tarek N. Histone deacetylase inhibitor for NUT midline carcinoma. Pediatr Blood Cancer. 2015;62(4):715–717. doi:10.1002/pbc.25350
37. Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi:10.1038/nature09504
38. Stathis A, Zucca E, Bekradda M, et al. Clinical response of carcinomas harboring the BRD4-NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discov. 2016;6(5):492–500. doi:10.1158/2159-8290.CD-15-1335
39. Asangani IA, Dommeti VL, Wang X, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510(7504):278–282. doi:10.1038/nature13229
40. Shu S, Lin CY, He HH, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529(7586):413–417. doi:10.1038/nature16508
41. Fiskus W, Sharma S, Qi J, et al. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Ther. 2014;13(10):2315–2327. doi:10.1158/1535-7163.MCT-14-0258
42. Garcia PL, Miller AL, Kreitzburg KM, et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene. 2016;35(7):833–845. doi:10.1038/onc.2015.126
43. Amorim S, Stathis A, Gleeson M, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3(4):e196–e204. doi:10.1016/S2352-3026(16)00021-1
44. Astorgues-Xerri L, Riveiro ME, Tijeras-Raballand A, et al. OTX008, a selective small- molecule inhibitor of galectin-1, downregulates cancer cell proliferation, invasion and tumour angiogenesis. Eur J Cancer. 2014;50(14):2463–2477. doi:10.1016/j.ejca.2014.06.015
45. Noel JK, Iwata K, Ooike S, Sugahara K, Nakamura H, Daibata M. Development of the BET bromodomain inhibitor OTX015. Mol Cancer Ther. 2013;12(11 Supplement):C244. doi:10.1158/1535-7163.MCT-12-1016-T
46. Berthon C, Raffoux E, Thomas X, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3(4):e186–e195. doi:10.1016/S2352-3026(15)00247-1
47. Lewin J, Soria JC, Stathis A, et al. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins in patients with selected advanced solid tumors. J Clin Oncol. Epub 2018 May 7.
48. Gamsjaeger R, Webb SR, Lamonica JM, Billin A, Blobel GA, Mackay JP. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol Cell Biol. 2011;31(13):2632–2640. doi:10.1128/MCB.05413-11
49. Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol. 2017;28(8):1776–1787. doi:10.1093/annonc/mdx157
50. Fong CY, Gilan O, Lam EY, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525(7570):538–542. doi:10.1038/nature14888
51. Rathert P, Roth M, Neumann T, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibitors. Nature. 2015;525:543–547. doi:10.1038/nature14898
52. Liao S, Maertens O, Cichowski K, Elledge SJ. Genetic modifiers of the BRD4-NUT dependency of NUT midline carcinoma uncovers a synergism between BETis and CDK4/6is. Genes Dev. 2018;32(17–18):1188–1200. doi:10.1101/gad.315648.118
53. Reynoird N, Schwartz BE, Delvecchio M, et al. Oncogenesis by sequestration of CBP/p300 in transcriptionally inactive hyperacetylated chromatin domains. Embo J. 2010;29(17):2943–2952. doi:10.1038/emboj.2010.176
54. Sun K, Atoyan R, Borek MA, et al. Dual HDAC and PI3K inhibitor CUDC-907 downregulates MYC and suppresses growth of MYC-dependent cancers. Mol Cancer Ther. 2017;16(2):285–299. doi:10.1158/1535-7163.MCT-16-0390
55. Munster P, Wu N, McMahon M, Gharavi R, Tuck D. Prolonged disease stabilization and tolerability in a nuclear protein in testis midline carcinoma patient treated with dual histone deacetylase and phosphoinositide 3-kinase inhibitor CUDC-907. Case Reports in Clinical Medicine. 2018;7:451–460. doi:10.4236/crcm.2018.77039
56. Morales F, Giordano A. Overview of CDK9 as a target in cancer research. Cell Cycle. 2016;15(4):519–527. doi:10.1080/15384101.2016.1138186
57. Brägelmann J, Dammert MA, Dietlein F, et al. Systematic kinase inhibitor profiling identifies CDK9 as a synthetic lethal target in NUT midline carcinoma. Cell Rep. 2017;20(12):2833–2845. doi:10.1016/j.celrep.2017.08.082
58. Beesley AH, Stirnweiss A, Ferrari E, et al. Comparative drug screening in NUT midline carcinoma. Br J Cancer. 2014;110(5):1189–1198. doi:10.1038/bjc.2014.54
59. Kagoya Y, Nakatsugawa M, Yamashita Y, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest. 2016;126(9):3479–3494. doi:10.1172/JCI86437
60. Hogg SJ, Vervoort SJ, Deswal S, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017;18(9):2162–2174. doi:10.1016/j.celrep.2017.02.011
61. Zhu H, Bengsch F, Svoronos N, et al. BET bromodomain inhibition promotes anti- tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16(11):2829–2837. doi:10.1016/j.celrep.2016.08.032
62. Shimamura T, Chen Z, Soucheray M, et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin Cancer Res. 2013;19(22):6183–6192. doi:10.1158/1078-0432.CCR-12-3904
63. Mazur PK, Herner A, Mello SS, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21(10):1163–1171. doi:10.1038/nm.3952
64. Bhadury J, Nilsson LM, Muralidharan SV, et al. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc Natl Acad Sci U S A. 2014;111(26):E2721–E2730. doi:10.1073/pnas.1406722111
65. Heinemann A, Cullinane C, De Paoli-Iseppi R, et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget. 2015;6(25):21507–21521. doi:10.18632/oncotarget.4242
66. Jostes S, Nettersheim D, Fellermeyer M, et al. The bromodomain inhibitor JQ1 triggers growth arrest and apoptosis in testicular germ cell tumours in vitro and in vivo. J Cell Mol Med. 2017;21(7):1300–1314. doi:10.1111/jcmm.13059
67. Stirnweiss A, Oommen J, Kotecha RS, Kees UR, Beesley AH. Molecular-genetic profiling and high-throughput in vitro drug screening in NUT midline carcinoma-an aggressive and fatal disease. Oncotarget. 2017;8(68):112313–112329. doi:10.18632/oncotarget.22862
68. Saponaro M, Kantidakis T, Mitter R, et al. RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell. 2014;157(5):1037–1049. doi:10.1016/j.cell.2014.03.048
69. Sahni JM, Gayle SS, Bonk KL, et al. Bromodomain and extraterminal protein inhibition blocks growth of triple-negative breast cancers through the suppression of aurora kinases. J Biol Chem. 2016;291(45):23756–23768. doi:10.1074/jbc.M116.738666
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.