Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Nucleotide-Oligomerizing Domain-1 Activation Exaggerates Cigarette Smoke-Induced Chronic Obstructive Pulmonary-Like Disease in Mice
Authors Han G, Li M, Du J, Chen Y, Xu C
Received 14 June 2021
Accepted for publication 16 August 2021
Published 16 September 2021 Volume 2021:16 Pages 2605—2615
DOI https://doi.org/10.2147/COPD.S323616
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Guangchao Han, Min Li, Junfeng Du, Yang Chen, Chen Xu
Department of Respiratory and Critical Care Medicine, Cangzhou Central Hospital, Cangzhou, 061000, Hebei, People’s Republic of China
Correspondence: Guangchao Han
Department of Respiratory and Critical Care Medicine, Cangzhou Central Hospital, No. 16 Xinhua West Road, Cangzhou, 061000, Hebei, People’s Republic of China
Tel +86-15833371555
Email [email protected]
Introduction: Chronic obstructive pulmonary disease (COPD) is a progressive condition related to abnormal inflammatory responses. As an inflammatory driver, nucleotide-binding oligomerizing domain-1 (NOD1) is highly expressed in pulmonary inflammatory cells; however, the roles of NOD1 in COPD are unknown.
Methods: A COPD mouse model was established by lipopolysaccharides tracheal instillation plus cigarette smoke (CS) exposure. NOD1 activation was induced by C12-iE-DAP (iE) treatment in both control and COPD mice. Inflammatory infiltration, pulmonary histological damage and gene expression were measured to evaluate the lung function of treated mice.
Results: The results showed that NOD1 was up-regulated in COPD mice, which significantly exaggerated CS-induced impairment of lung function, demonstrated by increased airway resistance, functional residual capacity and pulmonary damages. Mechanistically, NOD1 activation strongly activated the TLR4/NF-κB signaling pathway and then increased inflammatory responses and promoted the secretion of inflammatory cytokines.
Discussion: This study demonstrates that NOD1 is an important risk factor in the progression of COPD; therefore, targeting NOD1 in lung tissues is a potential strategy for COPD treatment.
Keywords: COPD, pulmonary damage, inflammation, NOD1, TLR4/NF-κB signaling
Introduction
Chronic obstructive pulmonary disease (COPD) is an incurable and debilitating disease characterized by airflow limitation and persistent respiratory symptoms.1 As one of the most prevalent diseases, COPD is the fifth leading cause of death in the world, and it is more common in adults over 40 years old.2 COPD is highly related to the exposure to toxic gases and particles that result in abnormalities in the respiratory tract.3–5 Although multiple therapies have been developed, including β2-agonists, corticosteroids and PDE4 inhibitors, they cannot effectively attenuate the progression of COPD and have some side effects.6,7 Therefore, there is an urgent need for exploring the underlying pathological mechanism and developing new drugs for COPD treatment.
COPD is characterized by progressively decreasing lung function and chronic airflow limitation, which is related to uncontrollable inflammatory responses to harmful particles and/or gases in the lungs and airways.8,9 The harmful ingredients of cigarette smoke (CS) activate the innate immune system and mediate the inflammatory process.10,11 Therefore, inflammation has been thought to be the major determinant for the frequent occurrence of COPD among patients.12 Many studies have shown that inhibiting the inflammatory response can greatly attenuate the development of CS-induced COPD.13–15
Nucleotide-binding oligomerizing domain-1 (NOD1) is a member of nucleotide-binding oligomerization domain-like receptors (NLRs) family, which plays important roles in recognizing abnormal or damaged host cells and microorganism components.16–18 NOD1 is highly expressed in inflammatory cells, which activates the NF-κB signaling and results in the secretion of inflammatory cytokines.19–21 Recent studies have reported that NOD1 activation could cause tissue inflammation in the heart and aggravate cardiac ischemia-reperfusion injury.16,22
NOD1 is widely expressed in lung tissues.23 Cardenas et al demonstrated that NOD1 activation was able to activate the inflammatory response of human alveolar macrophages.19 Recently, Stefano et al reported that the expression level of NOD1 was significantly increased in bronchial mucosa in patients with moderate and severe stable COPD compared to healthy controls.24 In the Japanese population, polymorphisms of the NOD gene were associated with the severity and prevalence of COPD.25 In addition, the rs2075820 variant in NOD1 was closely associated with high risk of childhood asthma, and both the mRNA and protein levels of NOD1 were significantly increased in asthma patients.26 The above studies demonstrated the potential correlation between NOD1 and COPD progression. However, little is known about the role of NOD1 in COPD, or whether it is involved in the occurrence and development of the disease. Here, we established the COPD mouse model by using CS exposure and lipopolysaccharides (LPS) intratracheal instillation, and stimulated NOD1 expression in the lung tissues through the agonist treatment to investigate the regulatory role of NOD1 in CS-induced COPD mice. We found that NOD1 activation exaggerated CS-induced COPD in mice and described the underlying molecular mechanism of NOD1 involvement in COPD inflammatory responses.
Materials and Methods
Reagents
C12-iE-DAP (iE), the agonist of NOD1, was purchased from InvivoGen (San Diego, USA) and dissolved in sterile saline to make the stock solution. LPS was ordered from Millipore Sigma (Burlington, USA) and dissolved in sterile saline for the following animal treatment.
Animal and COPD Models
The 8–10 weeks old male C57BL/6J mice (18–20 g) were obtained from Cyagen Biosciences (Suzhou, China) and housed under 12/12 hours of light/dark cycles with free access to food and water. This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (8th edition, NIH). All animal studies were reviewed and approved by the Ethical Committee of Cangzhou Central Hospital.
The COPD mouse model was established by tracheal instillation of 50 μL LPS (7.5 μg) on day 1 and 14, and exposed to CS circumstance for three months. Mice were put in a closed organic glass chamber (80×60×50 cm) and exposed under the smoke with 9 cigarettes per hour for 4 hours/day (6 days/week) except for the days of LPS tracheal instillation, as described in previous studies.27–30 For the iE dose testing, mice were divided into 4 groups, and treated with vehicle, 50 μg, 150 μg and 200 μg iE intragastrically for 14 days, respectively, following the protocol published by Tukhvatulin et al.31 For the COPD and iE co-treatment, there were four groups of mice, and 10 mice per group: Control group, mice were exposed with normal air plus saline gavage; iE group, mice were exposed with normal air plus 150 μg iE gavage; COPD group, mice were exposed with CS plus saline gavage; COPD+iE group, mice were exposed with CS plus 150 μg iE gavage. After 2 months of CS exposure, we started the saline or iE gavage, which was performed 30 min before CS exposure and lasted for two weeks (day 61–74).
Lung Function Measurement
After three months of COPD or COPD+iE treatment, mice were anesthetized by using pentobarbital sodium (50 mg/kg). Then, mice were placed in a body chamber and tracheotomized below the larynx, and then intubated with a trachea cannula. Forced Pulmonary Maneuver System (Buxco R.S., Wilmington, USA) was used to examine the lung function parameters (total lung capacity, FEV20/FVC, airway resistance and functional residual capacity) following the protocol described by Gebel et al.32 Breathing frequency of the tested mice was maintained at 150 breaths per min during the analysis. The average alveolar intercept represented the degree of emphysema, which was quantified by the Mean Linear Intercept function of Image Pro Plus 6.0 software, following the protocol described in previous studies.28,33
Bronchoalveolar Lavage Fluid (BALF) Cell Counting
Bronchoalveolar lavage (BAL) was performed to examine the cellular and acellular content in the lung lumen, following the protocol established by Hoecke et al.34 At the end of experiment, mice were anesthetized, and BAL fluid was isolated by inserting a catheter in the trachea, through which 600 μL saline solution was instilled into the bronchioles. The instilled fluid was gently retracted to maximize BALF retrieval. BALF cells were collected and counted with a hemocytometer (Weber Scientific, Hamilton, USA). H&E staining was used to determine the cell classification.
qRT-PCR
Total RNA of lung tissues from the above treated mice was extracted via Invitrogen Trizol (Carlsbad, USA) following the protocol of manufacturer. 1 μg RNA was reversely transcribed into cDNA by using the M-MLV reverse transcriptase (Promega, Madison, USA). The Bio-Rad qPCR Master Mix (Hercules, USA) was used to conduct qRT-PCR on the 7500 Real-Time PCR system. GAPDH were used to normalize the expression level of NOD1 and TLR4. The relative expression levels of NOD1 and TLR4 were calculated by using the 2−ΔΔCT method. Primers were listed below: NOD1: Forward: 5’-AGGAGGCCAACAGACGCC-3’, Reverse: 5’-CTGACCTAGAGGGTATCG-3’; TLR4: Forward: 5’-AGCTCCTGACCTTGGTCTTG-3’, Reverse: 5’- CGCAGGGGAACTCAATGAGG-3’. GAPDH: Forward: 5’-CATCACTGCCACCCAG AAG ACTG-3’, Reverse: 5’-ATGCCAGTGAGCTTCCCGTTCAG-3’.
Western Blot
Lung tissues were harvested and homogenized by using LabGEN 125 Homogenizer (Cole-Parmer, Vernon Hills, USA), and then lysed by the Beyotime RIPA buffer supplemented with fresh protease inhibitor cocktails (Promega, Madison, USA). Total protein concentration was determined by Pierce™ BCA Protein Assay Kit. Target proteins were detected by Western blot as described previously.35 Primary antibodies against NOD1 (ab217798, 1:1500 dilution), TLR4 (ab22048, 1:1000 dilution) and total p65 (ab32536, 1:1500 dilution) were ordered from Abcam (Cambridge, UK); p-p65 (#3033, 1:1000 dilution) and β-actin (#4970, 1:2000 dilution) were obtained from Cell Signaling Technology, Inc. (Danvers, USA).
H&E Staining
The lung pathology of the treated mice was determined by H&E staining. Paraffin-embedded lung tissues were cut into 12 μm sections using the Leica RM2145 microtome (Leica Biosystems Division of Leica, Buffalo Grove, USA). H&E staining and pathological analysis were performed as previously described.27 The scores of inflammatory infiltrations were classified as follows: score 0 is absent; score 1 is minimal, with a single-layer clustering of inflammatory cells; score 2 is moderate, with localized clustering of inflammatory cells; and score 3 is abundant, with large clusters of inflammatory cells.
Immunohistochemistry
In order to detect the expression of NOD1 in the COPD mouse model, immunohistochemistry was performed. Primary antibody against of NOD1 (ab217798, 1:200 dilution) was purchased from Abcam (Cambridge, UK). Paraffin section was performed as previously described.36 Imaging was conducted by using a Zeiss LSM 710 Confocal microscope (Carl Zeiss, Jena, Germany) with appropriate filter sets.
Enzyme-Linked Immunosorbent Assay (ELISA)
The concentrations of pulmonary inflammatory factors, Ccl-2, Cxcl-10, IL-1β, and IFN-γ, were detected by using the commercial ELISA kits following the protocols provided by the manufacturers. Ccl-2 and IL-1β were measured by using the Mouse MCP-1/CCL2 ELISA Kit and Mouse IL-1 β ELISA Kit (Millipore Sigma, Burlington, USA), respectively. Cxcl-10 and IFN-γ were detected by using the Mouse IP-10 ELISA Kit (CXCL10) (ab260067) and Mouse Interferon gamma ELISA Kit (IFNG) (ab100689, Abcam, Cambridge, UK).
Statistical Analysis
GraphPad Prism 8.0 was used for all the statistical analyses in this study. One-way analysis of variance (ANOVA) followed by Dunn’s multiple comparisons test (for multiple groups) and Student’s t-test methods (for two groups) were used to analyze the differences between groups. All the data were represented as mean ± standard deviation (SD).
Results
NOD1 is Induced in the Lungs of Mice Exposed to CS
To investigate the regulatory role of NOD1 in COPD, we established the COPD mouse model through a combination of CS exposure and LPS tracheal instillation. Gene expression analysis showed that NOD1 mRNA level was increased by nearly 3-fold in COPD mice compared to control mice (Figure 1A). We also detected the protein level of NOD1 by performing Western blot, and the result showed that NOD1 protein was also significantly increased in COPD mice (Figure 1B and C). Moreover, immunohistochemical data showed that there was more NOD1 signal in the bronchial mucosa of COPD mice than control mice (Figure S1). These data indicated that NOD1 was up-regulated in the lung tissues of COPD mice, which might be involved in the occurrence and development of COPD.
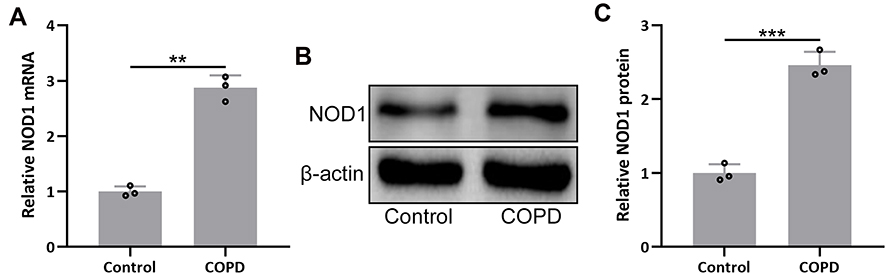 |
Figure 1 NOD1 was up-regulated in the lungs of mice exposed to cigarette smoke (CS). RT-qPCR and Western blotting were used to analyze the mRNA (A) and proteins expressions (B and C) of NOD1 in the lung tissues between the controls and CS exposed mice. Data shown are mean ± SD. n = 3 for each group. **p < 0.01 and ***p < 0.001 between the indicated groups. Unpaired t-test. |
iE Stimulates NOD1 in the Lung Tissues
iE is an agonist of NOD1, which has been reported to increase the expression level of NOD1 in vitro and in vivo.31 Here, we investigated the NOD1 stimulation and dose-dependent effects of iE (50, 150 and 200 μg per day) in the lung tissues. As shown in Figures 2A, 2 weeks of iE treatment could significantly stimulate the expression of NOD1 in the lung tissues. Moreover, the NOD1 stimulation effect of iE displayed a dose-dependent pattern (Figure 2A). Similarly, iE treatment increased the protein expression level of NOD1 in a dose-dependent manner (Figure 2B). In addition, upon iE treatment, the NOD1 signal was significantly increased in the bronchial mucosa compared to that in control and COPD mice (Figure S1). There was no difference in NOD1 stimulation at the doses of 150 and 200 μg, and mouse body weight was comparable in control and iE treated groups (data not shown), indicating that the doses of iE used in this study did not induce any systemic impairment for the study period. Therefore, 150 μg per day was chosen in the following treatment.
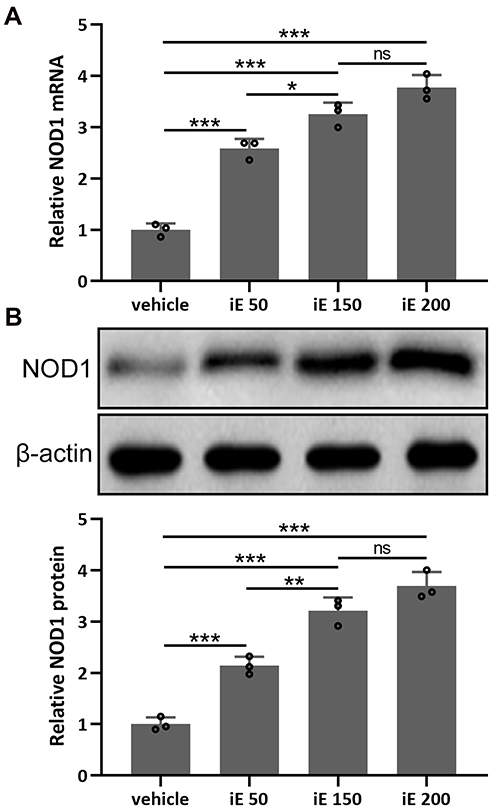 |
Figure 2 iE stimulated NOD1 in the lung tissues. Animals received i.p. 50, 150, 200 μg of iE/day, or vehicle. After 2 weeks of treatment, RT-qPCR and Western blotting were used to analyze the mRNA (A) and proteins expressions (B) of NOD1 in mouse lung tissues. Data shown are mean ± SD. n = 3 from 6 mixed tissues for each group. *p < 0.05, **p < 0.01, ***p < 0.001 and ns means no significance between the indicated groups. One-way ANOVA followed Dunn’s multiple comparisons test. |
NOD1 Activation Exaggerates CS-Induced Lung Function Impairment and Pulmonary Histological Damage
Since iE treatment can increase the expression of NOD1, which was also up-regulated in COPD mice, we asked whether iE treatment could exaggerate the CS-induced COPD in mouse model. We performed two weeks of iE treatment on control and COPD mice, and then examined the lung functions using the Forced Pulmonary Maneuver system. COPD mice showed higher total lung capacity than control mice. Interestingly, COPD+iE co-treatment could further increase the total lung capacity over that of the COPD group (Figure 3A). Similar promotional effects of iE on COPD were observed in terms of airway resistance and functional residual capacity (Figure 3C and D). In contrast, COPD decreased the FEV20/FVC in mice, and COPD+iE co-treatment further reduced this ratio (Figure 3B). It is worth noting that NOD1 activation exhibited a certain effect on the lung function of normal mice, but there was no significant difference (Control vs iE).
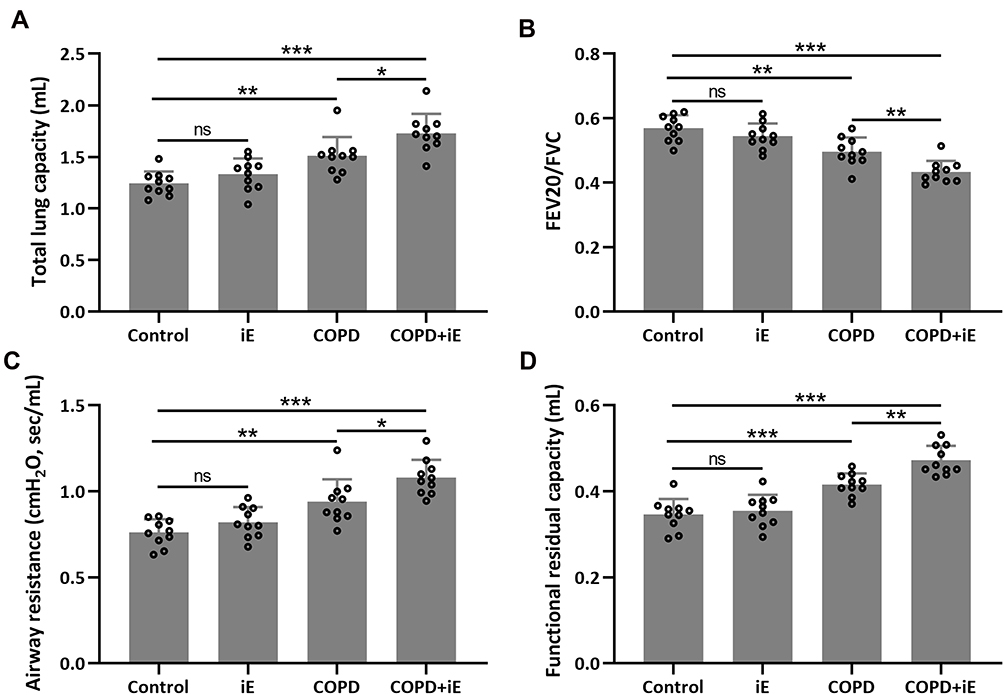 |
Figure 3 NOD1 activation exaggerated CS-induced lung function impairment. Lung function parameters including total lung capacity (A), FEV20/FVC (B), airway resistance (C) and functional residual capacity (D) among different groups were calculated three months after CS inhalation. Data shown are mean ± SD. n = 10 for each group. *p < 0.05, **p < 0.01, ***p < 0.001 and ns means no significance between the indicated groups. One-way ANOVA followed Dunn’s multiple comparisons test. |
Next, we evaluated the pulmonary damage of COPD and COPD+iE treated mice by examining the inflammatory infiltration, average alveolar intercept and airway wall thickness through H&E staining. Inflammatory score of COPD mice was significantly higher than those of control mice, and COPD+iE co-treatment could further increase the inflammatory score in COPD mice over those of control mice (Figure 4A and B). Similar promotional effects of iE treatment on COPD-induced pulmonary damage were observed in the average alveolar intercept and airway wall thickness of treated mice (Figure 4A, C, and D). All these results indicated that CS exposure caused significant damage of lung function, furthermore, NOD1 activation by iE could further aggravate lung function impairment and pulmonary damage in COPD mice.
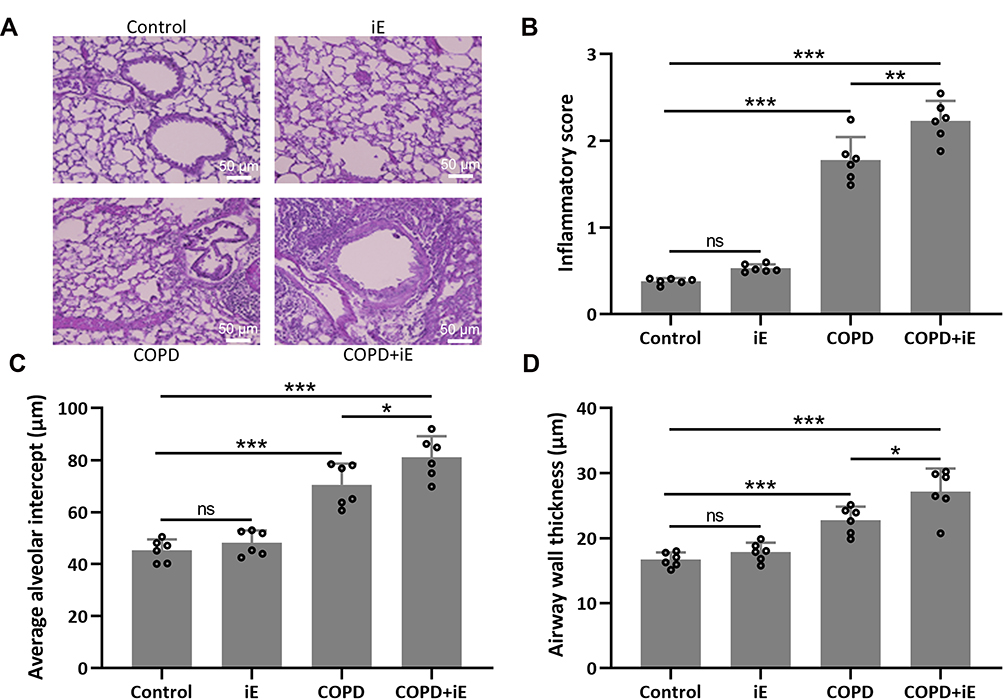 |
Figure 4 NOD1 activation exaggerated CS-induced pulmonary histological damage. Three months after CS inhalation, lung pathology was determined by H&E staining. The representative images are shown in (A). Comparisons of inflammatory score (B), average alveolar intercept (C) and airway wall thickness (D). Data shown are mean ± SD. n = 6 for each group. *p < 0.05, **p < 0.01, ***p < 0.001 and ns means no significance between the indicated groups. One-way ANOVA followed Dunn’s multiple comparisons test. |
NOD1 Activation Facilitates CS-Induced Inflammation in BALF and Pulmonary Inflammatory Responses
We detected the effects of iE on COPD-induced inflammatory responses by counting the inflammation cells in BALF and performing ELISA assays of inflammatory factors in the lung tissues. As shown in Figure 5A, both iE treatment and COPD could significantly increase the amount of inflammatory cells in BALF, and COPD+iE co-treatment showed stronger promotion than COPD alone. We further investigated different types of inflammatory cells, including macrophages (Figure 5B), neutrophils (Figure 5C) and lymphocytes (Figure 5D), and found that the numbers of all these inflammatory cells were significantly increased in the BALF of COPD and iE treated mice, with COPD+iE co-treatment showing even more pronounced enhancement in these inflammatory cells (Figures 5B–D). ELISA assays showed that the iE-induced NOD1 activation and COPD could promote the inflammatory response in the lung tissues significantly (Figure 6). Moreover, the COPD and iE combined treatment was able to further enhance the inflammatory response in the lung tissues of the treated mice, as evidenced by the induction of four inflammatory factors Ccl-2 (Figure 6A), Cxcl-10 (Figure 6B), IL-1β (Figure 6C) and IFN-γ (Figure 6D). These results indicated that NOD1 activation led to significantly increased inflammatory cells in BALF and inflammatory responses in normal mice, which promoted the inflammatory responses in COPD mice.
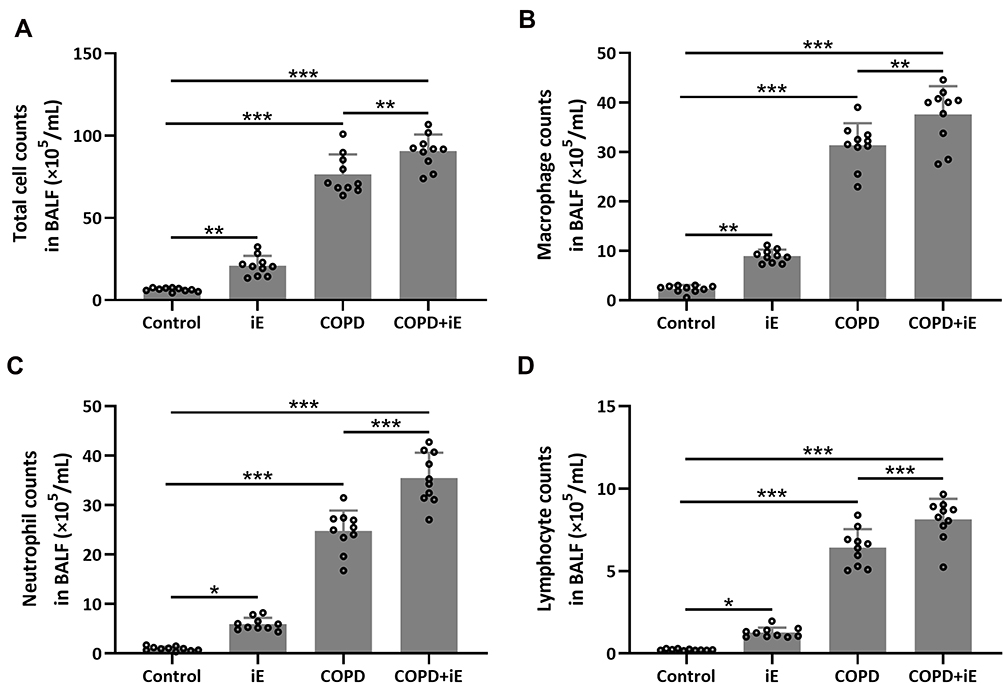 |
Figure 5 NOD1 activation exaggerated CS-induced inflammation cells in BALF. Three months after CS inhalation. Counts of total cells (A), macrophages (B), neutrophils (C) and lymphocytes (D) in BALF were compared. Data shown are mean ± SD. n = 10 for each group. *p < 0.05, **p < 0.01, ***p < 0.001. One-way ANOVA followed Dunn’s multiple comparisons test. |
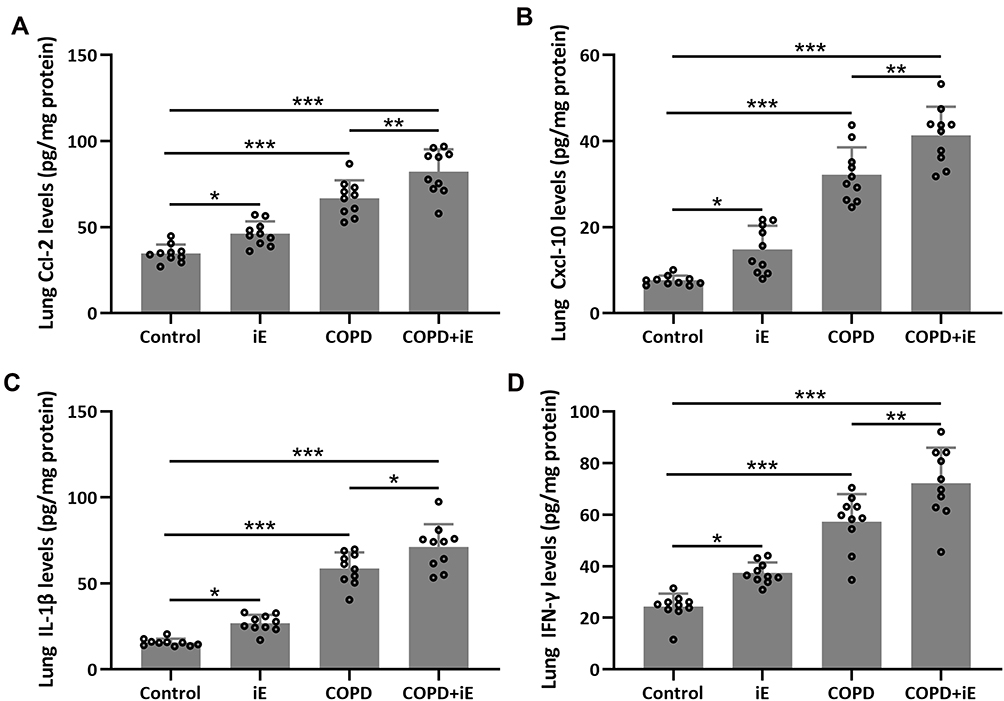 |
Figure 6 NOD1 activation exaggerated CS-induced pulmonary inflammatory responses. Three months after CS inhalation, pulmonary Ccl-2 (A), Cxcl-10 (B), IL-1β (C) and IFN-γ (D) were measured by ELISA. Data shown are mean ± SD. n = 10 for each group. *p < 0.05, **p < 0.01, ***p < 0.001. One-way ANOVA followed Dunn’s multiple comparisons test. |
NOD1 Activation Promotes the CS-Induced Pulmonary TLR4/NF-κB Signaling
NOD1 has been reported to be involved in the regulation of TLR4/NF-κB signaling.37 We therefore detected the expression of TLR4, which leads to the activation of the NF-κB pathway and the production of inflammatory cytokines, in lung tissues by using qRT-PCR. The result showed that both iE-induced NOD1 activation and COPD could significantly up-regulate the expression of TLR4, and COPD+iE co-treatment resulted in further induction of TLR4 (Figure 7A). Next, we investigated the activation of TLR4/NF-κB signaling through Western blot. In line with the qRT-PCR data, the protein levels of TLR4 and p-p65 were significantly elevated within the lung tissues of iE and COPD mice, while the strongest activation of TLR4/NF-κB signaling was observed in the mice with COPD+iE co-treatment (Figure 7B–D). The above data suggested that both NOD1 activation and COPD could activate the TLR4/NF-κB signaling in the lungs of normal mice, furthermore, NOD1 activation could aggravate the activation of TLR4/NF-κB signaling in COPD mice.
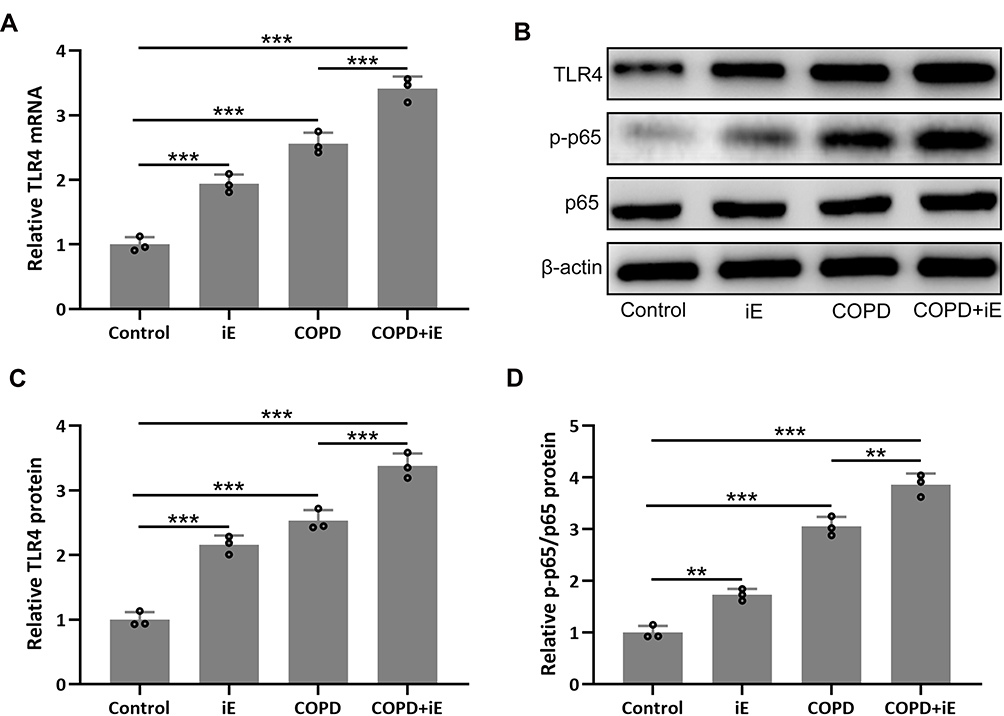 |
Figure 7 NOD1 exaggerated CS-induced pulmonary TLR4/NF-κB signaling activation. Three months after CS inhalation, pulmonary mRNA expression of TLR4 (A) were measured by RT-qPCR. Pulmonary protein expressions of TLR4, p-p65 and p65 (B) were measured by Western blotting. The expressions were normalized to control (C and D). Data shown are mean ± SD. n = 3 for each group. **p < 0.01, ***p < 0.001. One-way ANOVA followed Dunn’s multiple comparisons test. |
Discussion
COPD is a progressive and lethal chronic lung disease, which is characterized by declined pulmonary function and chronic airflow limitation. Inflammation has been thought to be the major determinant of multi-morbidities in COPD patients.12 NOD1 is highly expressed in inflammatory cells, which can active multiple inflammation associated with signaling pathways and result in inflammatory cytokine secretion.19,20 In this study, we investigated the regulatory role of NOD1 in COPD, and found that the expression level of NOD1 was significantly increased in the lung tissues of COPD mice. Similar to COPD treatment, NOD1 activation could facilitate inflammatory responses and increase the number of macrophages, neutrophils and lymphocytes in BALF of mice. However, NOD1 activation alone could not induce pulmonary histological damage or lung function impairment. Interestingly, NOD1 activation in COPD mice was able to elevate the average alveolar intercept, increase airway wall thickness, worsen inflammatory score and finally result in serious lung function impairment. Mechanistically, NOD1 activation can strongly active the TLR4/NF-κB signaling pathway, which then increased inflammatory responses and promoted the secretion of inflammatory cytokines. Our findings suggest that NOD1 is an important risk factor in the progression of COPD; hence, targeting NOD1 and inhibiting its expression in the lung tissues is a potential strategy for COPD treatment.
The NLRs trigger the activation of numerous signaling pathways and result in inflammation and apoptosis.17,38 NOD1 is one of the important members of NLR family, which can be stimulated by the meso-diaminopimelic acid (DAP) containing peptidoglycan fragments.39 Activation of NOD1 initiates multiple signaling pathways and induces the secretion of pro-inflammatory chemokines and cytokines.40,41 In the mouse heart, NOD1 activation could induce cardiomyocyte apoptosis and cardiac fibrosis, leading to cardiac dysfunction.16 Yang et al reported that DAP-induced NOD1 activation contributed to the development of myocardial ischemia/reperfusion damage through activation of NF-κB, MAPK and JNK signaling pathways.22 In this study, we found that NOD1 activation promoted the secretion of pulmonary inflammatory chemokines and cytokines, including Ccl-2, Cxcl-10, IL-1β and IFN-γ. Moreover, NOD1 was up-regulated in the lungs of COPD mice, the activation of which could further exaggerate CS-induced pulmonary inflammatory responses and lung function impairment. Similarly, Juárez et al demonstrated that the activation of NOD1 receptor was able to promote pro-inflammatory response and autophagy in human alveolar macrophages.42 On the other hand, the activation of NOD1 by agonist can induce inflammatory responses and autophagy, which might also be involved in the progression of COPD.43,44 Roussel et al reported that pretreatment of airway epithelium cells with IL-17 and NOD1 agonist resulted in a 10-fold increase of IL-8 production, which indicated the importance of NOD1 as a major contributor of abnormal inflammatory responses in the cystic fibrosis and CODP.45 NOD1 agonist, iE-DAP, can up-regulate the expression of IL-6, IFN-γ and TNF-α in the mucosal epithelial cells.46 These findings suggest that NOD1 is a risk factor for stimulating inflammatory responses, which might contribute to the pathogenic mechanism of NOD1 in COPD progression.
Serious pulmonary histological damage could be induced by CS inhalation. In the current study, 3 months of CS exposure could significantly increase the inflammatory cell infiltration in BALF, promote the secretion of pro-inflammatory chemokines and cytokines, and result in obvious airway resistance, which was consistent with previous studies.29,30 Although NOD1 activation could worsen most of the COPD-related phenotypes in COPD mice, there was very weak effect on pulmonary damage and lung function impairment. This observation indicates that, as a risk factor, NOD1 might be involved in the development rather than the initiation of COPD, which could be addressed by using NOD1 agonist in animal models to see whether it could further accelerate the progression of COPD-like lung disease in future studies. It would also be interesting to address the regulatory mechanism of NOD1 up-regulation in the lung tissues of COPD mice.
Conclusion
NOD1 is up-regulated in the lung tissues of COPD mouse model, which in turn exaggerates CS-induced chronic obstructive pulmonary-like disease in COPD mice.
Funding
This study was supported by Key Research and Development Program of Cang Zhou City (#172302134).
Disclosure
The authors declare that they have no competing interests.
References
1. MacNee W. Pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2(4):258–266. doi:10.1513/pats.200504-045SR
2. Wang C, Xu J, Yang L, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): a national cross-sectional study. Lancet. 2018;391(10131):1706–1717. doi:10.1016/S0140-6736(18)30841-9
3. Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379(9823):1341–1351. doi:10.1016/S0140-6736(11)60968-9
4. McCarthy B, Casey D, Devane D, Murphy K, Murphy E, Lacasse Y. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2015;2.
5. Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest. 2012;122(8):2749–2755. doi:10.1172/JCI60324
6. MacNee W. Accelerated lung aging: a novel pathogenic mechanism of chronic obstructive pulmonary disease (COPD). Biochem Soc Trans. 2009;37(Pt 4):819–823. doi:10.1042/BST0370819
7. Price D, Yawn B, Brusselle G, Rossi A. Risk-to-benefit ratio of inhaled corticosteroids in patients with COPD. Prim Care Respir J. 2013;22(1):92–100. doi:10.4104/pcrj.2012.00092
8. Rodriguez E, Ferrer J, Zock JP, et al. Lifetime occupational exposure to dusts, gases and fumes is associated with bronchitis symptoms and higher diffusion capacity in COPD patients. PLoS One. 2014;9(2):e88426. doi:10.1371/journal.pone.0088426
9. Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. doi:10.1164/rccm.201204-0596PP
10. Tashkin DP. Smoking cessation in chronic obstructive pulmonary disease. Semin Respir Crit Care Med. 2015;36(4):491–507. doi:10.1055/s-0035-1555610
11. Zuo L, He F, Sergakis GG, et al. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am J Physiol Lung Cell Mol Physiol. 2014;307(3):L205–L218. doi:10.1152/ajplung.00330.2013
12. Liu Y, Pleasants RA, Croft JB, et al. Smoking duration, respiratory symptoms, and COPD in adults aged >/=45 years with a smoking history. Int J Chron Obstruct Pulmon Dis. 2015;10:1409–1416. doi:10.2147/COPD.S82259
13. Amatngalim GD, Broekman W, Daniel NM, et al. Cigarette smoke modulates repair and innate immunity following injury to airway epithelial cells. PLoS One. 2016;11(11):e0166255. doi:10.1371/journal.pone.0166255
14. Sethi S, Mahler DA, Marcus P, Owen CA, Yawn B, Rennard S. Inflammation in COPD: implications for management. Am J Med. 2012;125(12):1162–1170. doi:10.1016/j.amjmed.2012.06.024
15. Tamimi A, Serdarevic D, Hanania NA. The effects of cigarette smoke on airway inflammation in asthma and COPD: therapeutic implications. Respir Med. 2012;106(3):319–328. doi:10.1016/j.rmed.2011.11.003
16. Fernandez-Velasco M, Prieto P, Terron V, et al. NOD1 activation induces cardiac dysfunction and modulates cardiac fibrosis and cardiomyocyte apoptosis. PLoS One. 2012;7(9):e45260. doi:10.1371/journal.pone.0045260
17. Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227(1):106–128. doi:10.1111/j.1600-065X.2008.00734.x
18. Shigeoka AA, Kambo A, Mathison JC, et al. Nod1 and nod2 are expressed in human and murine renal tubular epithelial cells and participate in renal ischemia reperfusion injury. J Immunol. 2010;184(5):2297–2304. doi:10.4049/jimmunol.0903065
19. Cardenas I, Mulla MJ, Myrtolli K, et al. Nod1 activation by bacterial iE-DAP induces maternal-fetal inflammation and preterm labor. J Immunol. 2011;187(2):980–986. doi:10.4049/jimmunol.1100578
20. Tukhvatulin AI, Logunov DY, Gitlin II, et al. A in vitro and in vivo study of the ability of NOD1 ligands to activate the transcriptional factor NF-kB. Acta Naturae. 2011;3(1):77–84. doi:10.32607/20758251-2011-3-1-77-84
21. Yu JS, Jin J, Li YY. The physiological functions of IKK-selective substrate identification and their critical roles in diseases. STEMedicine. 2020;1(4):e49. doi:10.37175/stemedicine.v1i4.49
22. Yang H, Li N, Song LN, et al. Activation of NOD1 by DAP contributes to myocardial ischemia/reperfusion injury via multiple signaling pathways. Apoptosis. 2015;20(4):512–522. doi:10.1007/s10495-015-1089-1
23. Moreira LO, Zamboni DS. NOD1 and NOD2 signaling in infection and inflammation. Front Immunol. 2012;3:328. doi:10.3389/fimmu.2012.00328
24. Di Stefano A, Ricciardolo FLM, Caramori G, et al. Bronchial inflammation and bacterial load in stable COPD is associated with TLR4 overexpression. Eur Respir J. 2017;49(5):1602006. doi:10.1183/13993003.02006-2016
25. Kinose D, Ogawa E, Hirota T, et al. A NOD2 gene polymorphism is associated with the prevalence and severity of chronic obstructive pulmonary disease in a Japanese population. Respirology. 2012;17(1):164–171. doi:10.1111/j.1440-1843.2011.02069.x
26. Belhaj R, Kaabachi W, Khalfallah I, Hamdi B, Hamzaoui K, Hamzaoui A. Gene variants, mRNA and NOD1/2 protein levels in tunisian childhood asthma. Lung. 2019;197(3):377–385. doi:10.1007/s00408-019-00209-4
27. Liu Y, Huang ZZ, Min L, Li ZF, Chen K. The BRD4 inhibitor JQ1 protects against chronic obstructive pulmonary disease in mice by suppressing NF-kappaB activation. Histol Histopathol. 2021;36(1):101–112.
28. Liang X, Wang J, Guan R, et al. Limax extract ameliorates cigarette smoke-induced chronic obstructive pulmonary disease in mice. Int Immunopharmacol. 2018;54:210–220. doi:10.1016/j.intimp.2017.11.004
29. Nie YC, Wu H, Li PB, et al. Characteristic comparison of three rat models induced by cigarette smoke or combined with LPS: to establish a suitable model for study of airway mucus hypersecretion in chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2012;25(5):349–356. doi:10.1016/j.pupt.2012.06.004
30. Zhang WG, He L, Shi XM, et al. Regulation of transplanted mesenchymal stem cells by the lung progenitor niche in rats with chronic obstructive pulmonary disease. Respir Res. 2014;15:33. doi:10.1186/1465-9921-15-33
31. Tukhvatulin AI, Gitlin II, Shcheblyakov DV, et al. Combined stimulation of Toll-like receptor 5 and NOD1 strongly potentiates activity of NF-kappaB, resulting in enhanced innate immune reactions and resistance to Salmonella enterica serovar Typhimurium infection. Infect Immun. 2013;81(10):3855–3864. doi:10.1128/IAI.00525-13
32. Gebel S, Diehl S, Pype J, et al. The transcriptome of Nrf2-/- mice provides evidence for impaired cell cycle progression in the development of cigarette smoke-induced emphysematous changes. Toxicol Sci. 2010;115(1):238–252. doi:10.1093/toxsci/kfq039
33. Knudsen L, Weibel ER, Gundersen HJ, Weinstein FV, Ochs M. Assessment of air space size characteristics by intercept (chord) measurement: an accurate and efficient stereological approach. J Appl Physiol. 2010;108(2):412–421. doi:10.1152/japplphysiol.01100.2009
34. Van Hoecke L, Job ER, Saelens X, Roose K. Bronchoalveolar lavage of murine lungs to analyze inflammatory cell infiltration. J Vis Exp. 2017;123:e55398.
35. Wang G, Mohammadtursun N, Lv Y, Zhang H, Sun J, Dong J. Baicalin exerts anti-airway inflammation and anti-remodelling effects in severe stage rat model of chronic obstructive pulmonary disease. Evid Based Complement Alternat Med. 2018;2018:7591348. doi:10.1155/2018/7591348
36. Zhou X, Moore BB. Lung section staining and microscopy. Bio Protoc. 2017;7(10).
37. Caruso R, Warner N, Inohara N, Nunez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. 2014;41(6):898–908. doi:10.1016/j.immuni.2014.12.010
38. Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27(4):549–559. doi:10.1016/j.immuni.2007.10.002
39. Wietzerbin J, Das BC, Petit JF, Lederer E, Leyh-Bouille M, Ghuysen JM. Occurrence of D-alanyl-(D)-meso-diaminopimelic acid and meso-diaminopimelyl-meso-diaminopimelic acid interpeptide linkages in the peptidoglycan of Mycobacteria. Biochemistry. 1974;13(17):3471–3476. doi:10.1021/bi00714a008
40. Sorbara MT, Ellison LK, Ramjeet M, et al. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity. 2013;39(5):858–873. doi:10.1016/j.immuni.2013.10.013
41. Grubman A, Kaparakis M, Viala J, et al. The innate immune molecule, NOD1, regulates direct killing of Helicobacter pylori by antimicrobial peptides. Cell Microbiol. 2010;12(5):626–639. doi:10.1111/j.1462-5822.2009.01421.x
42. Juarez E, Carranza C, Hernandez-Sanchez F, et al. Nucleotide-oligomerizing domain-1 (NOD1) receptor activation induces pro-inflammatory responses and autophagy in human alveolar macrophages. BMC Pulm Med. 2014;14:152. doi:10.1186/1471-2466-14-152
43. Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. 2008;27(2):373–383. doi:10.1038/sj.emboj.7601962
44. Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11(1):55–62. doi:10.1038/ni.1823
45. Roussel L, Rousseau S. IL-17 primes airway epithelial cells lacking functional cystic fibrosis transmembrane conductance regulator (CFTR) to increase NOD1 responses. Biochem Biophys Res Commun. 2010;391(1):505–509. doi:10.1016/j.bbrc.2009.11.088
46. Gao Y, Jiang W, Qian Y, et al. NOD1 agonist iE-DAP reverses effects of cigarette smoke extract on NOD1 signal pathway in human oral mucosal epithelial cells. Int J Clin Exp Med. 2015;8(8):12519–12528.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.