Back to Journals » Biologics: Targets and Therapy » Volume 14
Nucleic Acid Therapy for β-Thalassemia
Authors d'Arqom A ![]()
Received 2 June 2020
Accepted for publication 20 August 2020
Published 15 September 2020 Volume 2020:14 Pages 95—105
DOI https://doi.org/10.2147/BTT.S265767
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Shein-Chung Chow
Annette d’Arqom1,2
1Graduate Program in Molecular Medicine, Faculty of Science, Mahidol University, Bangkok, Thailand; 2Department of Pharmacology and Therapy, Faculty of Medicine, Universitas Airlangga, Surabaya, Indonesia
Correspondence: Annette d’Arqom Department of Pharmacology and Therapy, Faculty of Medicine
Universitas Airlangga, Jl. Mayjen Prof. Dr. Moestopo 47, Surabaya 60131, East Java, Indonesia
Email [email protected]
Abstract: β-thalassemia is caused by mutations in the β-globin gene which diminishes or abolishes β-globin chain production. This reduction causes an imbalance of the α/β-globin chain ratio and contributes to the pathogenesis of the disease. Several approaches to reduce the imbalance of the α/β ratio using several nucleic acid-based technologies such as RNAi, lentiviral mediated gene therapy, splice switching oligonucleotides (SSOs) and gene editing technology have been investigated extensively. These approaches aim to reduce excess free α-globin, either by reducing the α-globin chain, restoring β-globin expression and reactivating γ-globin expression, leading a reduced disease severity, treatment necessity, treatment interval, and disease complications, thus, increasing the life quality of the patients and alleviating economic burden. Therefore, nucleic acid-based therapy might become a potential targeted therapy for β-thalassemia.
Keywords: RNAi, splice switching oligonucleotides, gene editing, targeted therapy, good health and well-being
Introduction
Genetic disease or DNA mutation is one of the major factors for developing diseases. β-thalassemia is one of the most common monogenic disorders found in the Mediterranean, China, Africa, East Asia and Southeast Asia, including Thailand and Indonesia. Approximately 5 to 10% of the world population comprise patients or carriers of thalassemia.1 This disease is caused by mutations in the β-globin gene and causes impaired β-globin chain production.2 Reduced or absent β-globin causes the excess free α-globin chain that can precipitate on the cell membrane, leading to the death of both erythroblast and erythrocytes resulting in anemia.3 As a response to chronic anemic, the body produces erythropoietin (Epo) and induces intramedullary and extramedullary erythropoiesis.4 Furthermore, blood transfusion and increasing iron absorption from the gastrointestinal tract contribute to iron deposition in many organs leading to organ dysfunction causing shorter life expectancy, lower quality of life, and health-economic burdens.5,6
In β-thalassemia, the underlying mechanism is a single point mutation or small deletion in the β-globin gene, so possibilities to target specific mutations might bring benefit for thalassemia therapy.7 The major aim of the therapy is to achieve a balanced α/β ratio, thereby reducing the excess free α-chain. This review elaborates several nucleic acid based-strategies that might bring benefit as precision medicine for β-thalassemia, including reduced α-globin expression; restoration of β-globin expression by replacing the defective gene, modulating β-globin pre-mRNA splicing and correcting mutations using gene editing technology; and reactivating γ-globin expression (Figure 1).
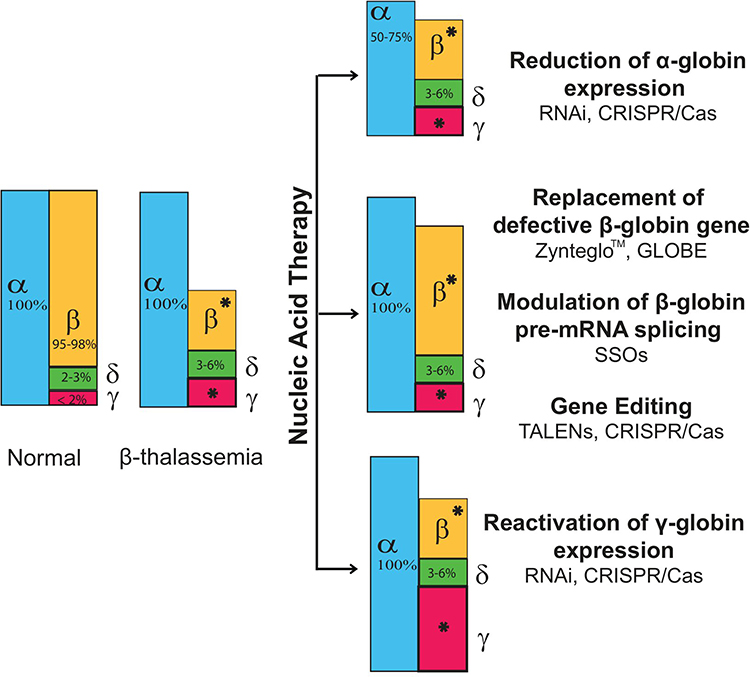 |
Figure 1 Nucleic acid-based targeted therapy for β-thalassemia Pathophysiology of β-thalassemia involves reducing or abolishing the β-globin chain, causing an excess of free α-globin. Therefore, three strategies can be employed to reduce the imbalance of α/β globin ratio: reducing α-globin expression, restoring β-globin expression and reactivating γ-globin. Several technologies can be used to achieve the aim, such as RNA interference (RNAi) to degrade the α-globin mRNA or to inhibit BCL11A to reactivate γ-globin production. Delivery of a normal β-globin gene to replace a defective gene is possible using the lentiviral vector. Moreover, modulating β-globin pre-mRNA splicing by splice switching oligonucleotides (SSOs) might reduce the possibilities of β-globin overexpression. Gene editing technologies such as Zinc Finger Nucleases (ZFNs), Transcription activator-like effector nucleases (TALENs) and Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated protein (CRISPR/Cas) has the ability to restore β-globin expression by correcting the mutation, to reduce α-globin expression by deleting specific α-globin enhancers, such as MCS-R2 and to induce HbF production by disrupting BCL11A, MYB or KLF1 genes. *Level of β-globin and γ-globin varies with the type of mutation.Notes: Adapted from Fucharoen.8 |
Methods
An electronic search of medical literature regarding β-thalassemia, nucleic acid therapy, RNA interference, splice switching oligonucleotides, lentiviral mediated gene therapy, gene editing, ZNF, TALENs and CRISPR/Cas was made on PubMed and Embase. The studies were reviewed and summarized.
Nucleic Acid Therapy for β-Thalassemia
Reduction of α-Globin Chain Expression
The pathology of β-thalassemia is caused by an excess α-globin chain which precipitates in the membrane of erythrocytes and erythroid precursors. In the clinical setting, co-inheritance of α-thalassemia ameliorates the β-thalassemia phenotype by reducing the free α-globin chain.9 Similar finding also found in the murine model of thalassemia.10 To achieve a balanced α/β-globin ratio, 25 to 50% of α-globin mRNA expression should be reduced, without changes in β-globin mRNA expression.11 Currently, two nucleic acid-based approaches have been explored to reduce the α-globin gene expression including post-transcriptional silencing through RNA interference (RNAi)12–15 and genome editing to reduce the α-globin gene expression.16
The first technology is RNAi which regulates gene expression by mRNA degradation and inhibits its translation to protein.17 The mechanism of this system has been reviewed extensively. Briefly, a small double-strand RNA (dsRNA) such as small interfering RNA (siRNA) or short single-stranded RNA such as microRNA (miRNA) makes a complex with RNA-induced silencing complex (RISC) and further binds with the complementary mRNA. The mRNA will be cleaved with the help of RNAse to suppress the gene expression.11,18 Use of this technique to reduce α-globin mRNA expression has been investigated comprehensively during the last decades. One in vitro study in murine erythroleukemic (MEL) cells and murine erythroid progenitor cells demonstrated that siRNA targeting α-globin mRNA reduced α-globin mRNA expression and protein production 50 to 65%. Moreover, the reduced α-globin chain led to declining reactive oxygen species (ROS) levels in thalassemic heterozygous β-KO erythroid progenitor cells.12 A similar result was found in human erythroid progenitor cells, resulting in reduced α-globin mRNA ranging from 30 to 50% with no effect on β-globin chain mRNA expression. The siRNA also reduced the number of hemoglobinized cells in erythroid colony-forming cells (ECFCs).13 Moreover, an in vivo experiment in Hbbth−4 mice, in which the murine adult β globin gene was replaced by the human βIVS2−654-globin gene, showed that intravenous injection of siRNA targeting α-globin sequence and antisense oligonucleotides targeting β-globin sequence improved red cell pathology, such as decreasing the nucleated cells in the bone marrow, increasing reticulocytes number15 and increasing the survival rate of homozygous Hbbth−4/Hbbth−4 and heterozygous Hbb/Hbbth−4.14
Another approach to reduce α-globin expression is using gene editing technologies that are able to insert, delete, disrupt or replace target DNA sequences.19 A study used CRISPR/Cas9 to delete the MCS-R2 α-globin enhancer, which is one of the regulatory elements controlling the expression of α-globin RNA. With 70% deletion efficiency, the deletion of MCS-R2 demonstrated reduced α-globin mRNA expression and restored α/β-globin chain balance in patient-derived hematopoietic stem cells (HSCs) in vitro. Xenograft assay confirmed the editing of long term hematopoietic stem cells (LT-HSCs) because the deletion of MCS-R2 can be found in the primary and secondary transplantation.16
Thus, downregulating α-globin gene expression might bring benefits for β-thalassemia treatment. However, this approach has limitations such as the stability and the ability of siRNA to inhibit α-globin expression continuously. The editing efficiency and off-target of the CRISPR/Cas system are also crucial issues in the gene editing field. Moreover, the number of silenced α-globin expressions and no effect on β-globin expression are essential because the balance of α/β-globin ratio is the goal of this strategy. Even though this approach has not been implemented in the clinical setting, reduced α-globin chain expression using nucleic acid-based technology is a potential strategy as an alternative therapy for β-thalassemia.
Restoration of β-Globin Gene Expression
Another approach to balancing the α/β-globin ratio is by restoring β-globin gene expression. Different from the last approach, reducing α-globin, this strategy works by increasing β-globin gene expression. Several methods are available to restore β-globin gene expression, such as replacing defective genes, modulating β-globin pre-mRNA splicing, and correcting mutations using gene editing technology which will be elaborated in this section.
Replacing the Defective β-Globin Gene
The gene therapy, using own patient hematopoietic stem cells carrying the correct β-globin gene, has been introduced to replace the defective gene. Starting with one patient with HbE/β-thalassemia, allogeneic stem cells were taken from the patient and transduced with the lentivirus carrying the correct β-globin gene and transplanted back to the patient. Several vectors have been used to deliver the correct β-globin gene (Table 1). The first was the βA(T87Q) vector, later known as HPV569 carrying U3 promoter/enhancer for lentivirus expression, with the whole β-globin gene with a mutation at codon 87 for its anti-sickling properties. This vector also carried β-globin promoter; HS2, HS3, and HS4 of LCR; and two copies of the chicken HS4 insulator core.20 A patient with transfusion-dependent β-thalassemia with HbE/β0-thalassemia genotype became transfusion independent one year after gene therapy and total Hb maintained between 9 to 10 g/dL, with 3.7 g/dL of the therapeutic HbAT87Q. A partial clonal dominance upon vector integration within the high mobility group AT hook 2 (HMGA2) gene was observed and declined at 12 years after gene therapy.20 However, due to the specific clonal expansion during the first study, some improvements were made to this vector to increase safety in clinical application. Its U3 promoter/enhancer was changed to CMV promoter/enhancer, and chicken HS4 insulator core was removed, creating the new vector, BB305 or Zynteglo™.21 Moreover, the first phase of the clinical trials among 22 patients was performed to investigate the safety and the efficiency of this vector. Twelve of 13 patients with non β0/β0-thalassemia became transfusion-independent, while only three patients with β0/β0-thalassemia became transfusion-independent and another six still received blood transfusion with decreasing annual transfusion volumes. Importantly, no clonal dominance was observed among all patients.22 The latest vector has been approved by the European Medicines Agency (EMA) in 2020.23
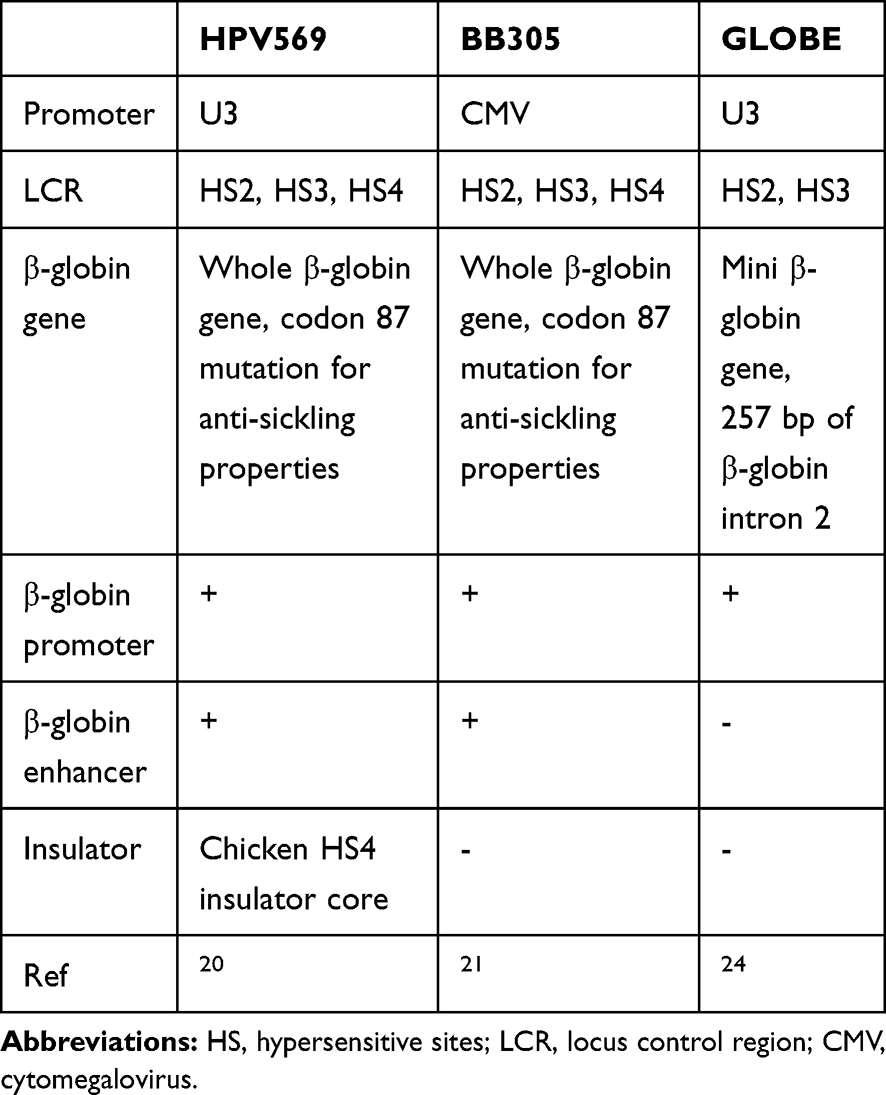 |
Table 1 Lentiviral Vectors Used in Clinical Trials for β-Thalassemia |
The second vector which has been used in a clinical trial was the GLOBE vector. This vector carried minimal LCR, namely, 2.7 kb fragment of HS2 and HS3 with no effect on β-globin expression, and the mini β-globin gene, including the β-globin promoter and absence of β-globin enhancer. Primary and secondary transplantation of GLOBE vector transduced cells resulted in increased hemoglobin level and improved clinical features in thalassemic mice.24 Furthermore, clinical trials among three adults and six pediatric patients with homozygous β-thalassemia showed improved hematological parameters and disease pathology. The reduced transfusion interval and transfusion-dependence was achieved after treatment. Most pediatric patients with β0/β0-thalassemia have achieved transfusion-independence, while adult patients have reduced transfusion requirements.25,26
This breakthrough therapy gives hope to patients with thalassemia for a higher quality of life, even though the cost-benefit study for this approach still needs to be investigated. A clinical trial with a broad range of patients is needed before being clinically implemented and finding better alternatives and exploring safer approaches are always options. The limitation of this strategy is the overexpression of the β-globin gene which might lead to excess free β-globin chain.
Modulating β-Globin Pre-mRNA Splicing
One of the important molecular mechanisms of the defect in β-globin gene expression is mutation-induced aberrant splicing, which activates aberrant splice sites and alters the splicing pathways, even though the correct splice sites might function normally, for example, IVS1-5, IVS1-6, and IVS1-110 mutations in intron 1, IVS2-654, IVS2-705 and IVS2-745 mutations in the intron 2 of the β-globin gene.9 In addition, abnormal hemoglobin such as HbE also has resulted from aberrant splicing.27 Due to the activation of aberrant splice sites, the RNA is incorrectly spliced and some intron fragment is retained leading to nonfunctional β-globin chain production. Thus, this mutation reduces the production of correctly spliced β-globin protein and causes β-thalassemia.
Modulating β-globin pre-mRNA splicing is one potential approach to increase β-globin expression. Short synthetic oligonucleotides (15 to 25 nucleotides) to a specific pre-mRNA sequence might alter the recognition of splice sites by the spliceosome, leading to an alteration of splicing of the targeted transcript and interfering with the ratio of normal protein: RNA or RNA: RNA; therefore, called splice switching oligonucleotides (SSOs) (Figure 2).28,29 However, a chemical modification of SSOs is necessary to prevent degradation of the pre-mRNA-SSOs complex by RNase H.30 Modification of the phosphate backbone with phosphorothioate (PS) backbone modifications or the ribose component of the oligonucleotide might improve the stability of the SSOs in vivo and increase the binding affinity of SSOs to the specific sequence and proteins. This interaction further affects the pharmacokinetic and pharmacodynamic factors of SSOs, such as plasma and tissue distribution and cellular uptake, which further increases efficiency and reduces toxicity and immunostimulatory activity.31 Chemical modification and its effects on SSOs have been reviewed comprehensively.29,32
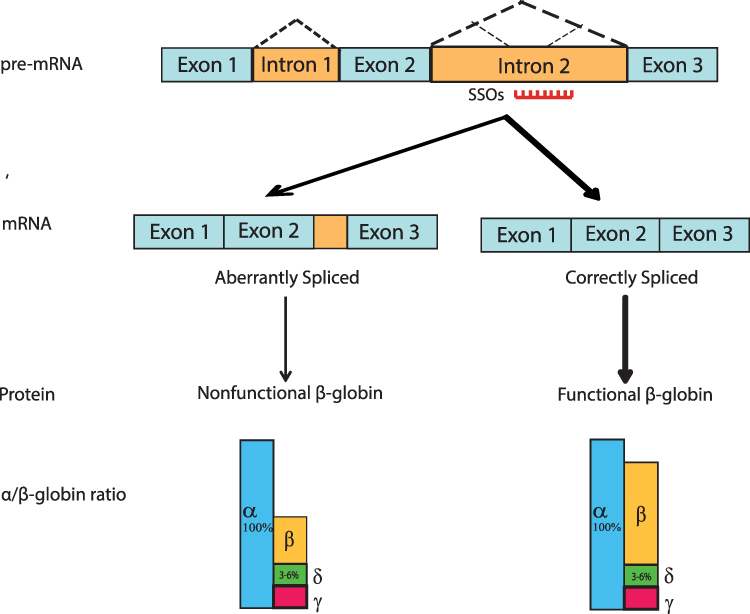 |
Figure 2 Mechanism of splice switching oligonucleotides (SSOs) Mutation in the intron region may activate aberrant splice sites leading to retaining the intron fragment in the mature mRNA and translated to a nonfunctional β-globin chain. SSOs targeted to the aberrant splicing elements block spliceosome to recognize the pseudo-exon and restore correct splicing which is translated to fully functional β-globin protein. Even though 100% of correct splicing cannot be achieved, the correctly spliced β-globin balances the α/β globin ratio, reduces the excess α-globin and increases the HbA level, thus alleviating the disease pathology. Notes: Adapted from Svasti et al.33 Copyright (2009) National Academy of Sciences. |
SSOs modulate RNA splicing by redirecting alternative splicing that can be categorized in four mechanisms: exon skipping, exon retention, restoration of correct splicing and displacement of splicing factors from triplets repeats which have been reviewed extensively.28 The SSOs have been used to correct aberrant splicing in β-thalassemia alleles including the mutations IVS1-110,34,35 IVS2-654,36–38 IVS2-705,39 IVS2-745,40 and βE.36 SSOs successfully induced correct splicing and increased normal β-globin production in cell-free extracts, stable cell lines with a mutated β-globin gene, erythroid mononuclear cells from peripheral blood of patients, thalassemic mouse erythroid progenitors, human iPSCs and β-thalassemic mouse model.36–38,40,41 Among patients with βIVS2−654 thalassemia, erythroid cells, free uptake and syringe load of SSOs into cells increased correct β-globin mRNA up to 77% and HbA production up to 54%.36 Moreover, in the βIVS2−654-thalassemia mouse model, intravenous injection of SSOs restored 12% of correct β-globin mRNA in mice peripheral blood or increased 6-fold compared with untreated controls. Surprisingly, chimeric mouse and human β-globin were found in 1 to 5% of peripheral blood samples.33
However, to maintain the correct splicing, chemical SSOs require life-long and periodic administrations. For that reason, researchers have endeavored to explore vectors that can constantly express the oligonucleotides. Intracellular expression of the antisense sequence is preferable for chronic diseases which can be achieved by the embedding antisense sequence in vectors, such as U7, U1 or U2 small nuclear RNAs (snRNAs).42 In 1998, a study by Gorman proposed using U7 small nuclear RNA (snRNA) which serves as a mediator of histone pre-mRNA processing as a vector to carry antisense sequence for splicing modulation. Because of its location inside the nucleus, this vector demonstrated stability, expressed at a relatively high level and was quite effective to deliver SSOs.43 Using this approach, correctly restoring spliced β-globin pre-mRNA in HeLa cells expressing several mutations in the intron 2 β-globin gene could be observed for at least six months.43–45 However, the correction level depended on the target sequence.45 Double-target engineered U7 snRNAs produced higher correction in HeLa IVS2-654 cells compared with a single targeted sequence (40 and 3%, respectively). Moreover, correctly spliced β-globin mRNA could be translated to hemoglobin A synthesis, improving the thalassemic erythroid cell pathology of HbE/βIVS2−654-thalassemia erythroid progenitor cells.37 The latest construct could restore approximately 80% of correctly spliced βIVS2−654- thalassemic human iPSCs.38 Not only in β-thalassemia, but the modified U7 snRNAs has also been extensively explored in several RNA mis-splicing diseases including DMD and SMA.46–52
Moreover, tagging SSOs in snRNAs improved the nucleus delivery and prevented SSOs from degradation because snRNAs are naturally located and work in the nucleus. Hence, this modification enhances the efficacy of the modulation of the splicing process. Thus, modulating β-globin pre-mRNA splicing might be beneficial in β-thalassemia caused by mutations that create aberrant splicing. Unfortunately, several studies showed a modification of U7 snRNA resulting in two isoforms, full length and truncated, of U7 snRNA that might interfere with the antisenses function to block the splicing machinery in certain animal models.46,53 A similar finding also found in the erythroid progenitor cells of thalassemic mice (unpublished data). Therefore finding the best model to test the SSOs would prove very important before its further application.
Mutation Correction Using Gene Editing Technologies
Gene editing is technology to modify DNA, including deletion, insertion or correction, of living cells, including human, animal and plant.54 This technology combines DNA-binding domains and DNA cleavage nucleases, resulting in creating a double-strand break (DSB). Three well-known techniques in this field are ZFNs, TALENs and CRISPR/CRISPR/Cas9).55
ZFNs are artificial restriction enzymes consisting of a zinc finger DNA binding domain and FokI DNA cleavage domain. The DNA binding domain or so-called finger recognizes a specific DNA triplet (3 bp), and each ZFN usually contains 3 fingers, and is thus able to recognize 9 base pairs.56 Since FokI needs dimerization to functionally cleave the DNA, two individual ZFNs are constructed to recognize DNA on the left and right arms of the target sequence with 5–6 bp spacer between two FokI endonucleases. After dimerization of FokI endonuclease, DSB and DNA repair mechanisms occur.57 Using ZFN technology, reactivation of γ-globin is achieved by inducing indel mutation of the SOX6 binding domain, and γ-globin expression increased up to 6 fold.58
Similar to ZFNs, transcription activator-like effector nucleases (TALENs) also constitute an artificial restriction enzyme composed of a TAL effector DNA-binding domain combined with a FokI endonuclease DNA cleavage domain. The TAL effectors contain 33–35 amino acids and at 12–13 amino acids, known as Repeat Variable Diresidue (RVD), can be modified to recognize specific target sequences. Each TALEN usually contains 13–20 TALE repeats, and similar to ZFNs, to work functionally, FokI endonuclease need dimerization. Therefore, two individual TALENs must bind on opposite sides of the target sequence with a spacer of 14–20 nucleotides. With this characterization, TALENs are very specific to recognize as a specific target sequence. Unfortunately, the size of TALENs might limit delivery to the cells.55–57 In thalassemia, TALENs have been shown to correct βIVS2−654-thalassemia in vitro in human iPSCs and in vivo in double heterozygous TALENs+/Hbbth−4 mice. In these studies, the correction efficiency ranged from 32 to 50%, and the expression of β-globin was comparable to normal controls. Interestingly, no off-target mutation was detected in both studies.59,60
Furthermore, the latest gene editing technology is CRISPR/Cas9 which originally found in bacteria as a defensive system from phage.61,62 Nowadays, with advanced knowledge about CRISPR/Cas9, genome editing becomes a rising star in the field. Editing the diseases caused mutation and introducing the correct donor template have become much more simple and affordable. There are several components for genome editing utilizes CRISPR/Cas9, which are gRNA followed by PAM sequence, Cas9 enzymes, and with or without donor template to promote DNA repair.63 CRISPR/Cas9 recognizes a specific sequence and creates a double-strand break (DSB) on the DNA becoming effective tools for gene editing.
In general, the two types of donor templates are donor plasmid (dsDNA) and single-stranded oligonucleotides (ssODNs).64 The donor template carries the correct sequence with downstream and upstream homologous arms. Donor plasmid can carry several base pairs of homologous arms, including selection markers such as drug resistance genes or the EGFP gene. Moreover, it can carry site-specific genetic manipulation tools such as the Cre-lox system and piggyBac transposase which can be removed after homologous recombination occurs.65–67 In the Cre-lox system, Cre recombinase recognizes 34 bp of loxP resulting in removal sequence between loxP including selection markers. Unfortunately, using this system leaves 34 bp of loxP recognition site on the gene of interest. This remaining sequence is an advantage for the silencing gene of interest but raises a concern for mutation correction.68 This limitation is overcome by piggyBac, a donor plasmid providing flexibility to remove any sequence between ITR of piggyBac, including the selection gene, using transposase and providing a perfect corrected gene. For example, the piggyBac donor, carrying the puromycin resistant gene and ∆TK gene for selection has been used to correct a mutation in CD41/42 β-thalassemia iPSCs.69 With this system, the selection gene can be removed completely and creates seamless correction. Unfortunately, due to its content, the donor plasmid is large and might interfere in its delivery into the cells.
Using ssODNs, which is a single strand donor carry 40–90 bp of upstream and downstream of the mutation site, the delivery rate into the cells might be more effective. However, ssODNs are unable to carry the selection marker; therefore, the selection of the corrected clone might present a challenge. For example, ssODN has been used to correct the mutation in βE and IVS1-110.70,71 In β-thalassemia, CRISPR/Cas9 has been investigated in several mutations such as 4 bp deletion or CD41/42, −28, IVS2-654, IVS1-110 and βE59,65,69-73 using patient-derived iPSCs or CD34+ cells. In CD41/42 and −28 mutation, using a piggyBac system to carry homologous arms, the correction efficiency was 23%.69 In HbE/β-thalassemic patient-derived iPSCs transfected with double expressed plasmid carrying gRNA and Cas9 nuclease and 180 bp of ssODN, HDR efficiency was 2.9%.71 Moreover, in the IVS1-110 mutation, using CD34+ hematopoietic stem cells, 8% of correction can be achieved.70 Surprisingly, in the absence of a donor template, CRISPR/Cas9 successfully induced indel mutation in human IVS1-110 mutation. DNA repair occurred via the nonhomologous end joining (NHEJ) pathway resulting in indel mutation (66.2%) and HbA production increase up to 75.6%. A similar result is achieved when using Cas12 and its suitable gRNA in βIVS2−654-thalassemic erythroid progenitor cells. Without the presence of a donor template, DNA repair occurred via the NHEJ pathway resulting in indel mutation (76.6%) which was sufficient to restore correctly spliced β-globin mRNA (70.1%) and HbA production (9.9–59.1%).73
Unfortunately, low correction efficiency becomes a concern in CRISPR/Cas technology; however, using the appropriate form of Cas nucleases such as DNA plasmid, lentiCRISPR, Cas RNA or Cas protein, and selecting the most suitable delivery methods might overcome this problem.65,74-76 Moreover, small molecules such as β-3 adrenergic receptor agonist or NHEJ inhibitor (RAD51, SCR7) were also found to increase the HDR event.77 In CD41/42 β-thalassemic patient-specific iPSCs, 54% correction efficiency was achieved when combining CRISPR/Cas, ssODN, and β-3 adrenergic receptor agonist.72 Taken together, current advances in the gene editing technology for β-thalassemia are remarkable and show a possible future application to correct mutation causing β-thalassemia, hence restoring the normal β-globin chain.
Reactivating γ-Globin Expression
The clinical manifestation of β-thalassemia usually starts in the first year of life, when fetal hemoglobin (HbF) is replaced by adult hemoglobin (HbA) and declining HbF levels occur. HbF is a tetramer consisting of 2 γ-globin chains incorporating 2 α-globin chains.78 It has a higher oxygen affinity providing advantages for the fetus during pregnancy.79 In clinical settings, a high level of HbF ameliorates the clinical severity of β-thalassemia such as co-inheritance with the hereditary persistence of fetal hemoglobin (HPFH) or Xmn1-HBG2 polymorphism.9 The increasing γ-globin chain binds with the excess α-globin chains, improving the balance of globin chains, and lessening the disease pathology.
Recent findings showed three transcription factors; BCL11A, KLF1, and MYB, regulate the γ-globin gene expression by repressing its expression after birth. Several studies have investigated the possibility to downregulate their expression as a treatment for β-thalassemia.9,80,81 Two powerful tools, RNAi and gene editing technology, are employed to disrupt the BCL11A gene. In vitro study in CD34+ derived erythroid cells demonstrated ability of siRNA to inhibit BCL11A production and successfully increased the HbF levels.82 Studies using a lineage-specific and miRNA-embedded expression of BCL11A-targeting shRNAs have reported that the shRNAs successfully reduced BCL11A mRNA expression up to 90%, induced 60 to 70% γ-globin mRNA expression, and further increased HbF production in cell cultures83 and animal models.84 Unfortunately, one study using RNAi targeting KLF1 gene did not show a reduction of KLF1 mRNA expression.85 Interestingly, study in murine erythroleukemic (MEL) cell line containing an intact 183-kb human β-globin locus showed that MYB-targeting shRNAs successfully reduced 40–70% of BCL11A and KLF1 mRNA expression, however, the MYB knockdown failed to increase γ-globin mRNA expression.86
In addition to correcting mutation causing diseases, gene editing technology, including CRISPR/Cas, ZFNs, and TALENs also can be used to knockdown BCL11A,87–89 KLF1,90 and HBG1/2 genomic regions or promoter91,95 to derepress γ-globin expression, thereby increase HbF production. With the increasing of HbF, the ratio of α-globin and β-like globin can be restored, thus reducing the excess of free α-globin. In 2019, the first clinical trial of CRISPR/Cas9 to disrupt the BCL11A gene was started to treat β-thalassemia and sickle cell disease.96 Thus, modulating the expression of γ-globin using nucleic acid-based therapy might offer an alternative therapy for β-thalassemia in the future.
Conclusion and Future Perspectives
As technology continues to advance, disease-caused mutations might become a target for disease treatment, as specific treatments might provide different benefits for different mutations. In β-thalassemia, the aim of the therapy is to balance the α/β-globin ratio which can be achieved by modulating α-, β-, and γ-globin expression. Several approaches have been investigated to achieve this goal using nucleic acid-based therapy, such as RNA interference, virus-mediated gene therapy, and gene editing. However, limitations such as the stability, delivery methods, suitable vectors, and optimum doses of nucleic acid should be carefully considered. By understanding this nucleic acid-based therapy, personalized medicine or targeted therapy for β-thalassemia can be developed and applied in the clinical setting. Nevertheless, the risk of health inequities and its potential misuse raise concerns among health practitioners. Thus its development and application should be tightly regulated and monitored.
Acknowledgments
I thank Associate Professor Saovaros Svasti and the members of Thalassemia Research Center, Mahidol University for their insightful comments and discussion to enrich the manuscript.
Disclosure
The author reports no conflicts of interest related to this work.
References
1. Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61–76. doi:10.1097/GIM.0b013e3181cd68ed
2. Cao A, Kan YW. The prevention of Thalassemia. Cold Spring Harb Perspect Med. 2013;3(2):a011775. doi:10.1101/cshperspect.a011775
3. Rivella S. Ineffective erythropoiesis and Thalassemias. Curr Opin Hematol. 2009;16(3):187–194. doi:10.1097/MOH.0b013e32832990a4
4. Nienhuis AW, Nathan DG. Pathophysiology and clinical manifestations of the β-thalassemias. Cold Spring Harb Perspect Med. 2012;2:12. doi:10.1101/cshperspect.a011726
5. Riewpaiboon A, Nuchprayoon I, Torcharus K, et al. Economic burden of Beta-thalassemia/Hb E and Beta-thalassemia major in Thai children. BMC Res Notes. 2010;3(1):29. doi:10.1186/1756-0500-3-29
6. Ansari S, Baghersalimi A, Azarkeivan A, et al. Quality of life in patients with Thalassemia major. Iran J Ped Hematol Oncol. 2014;4(2):57–63.
7. Kleanthous M, Phylactides M. Thalassemia and its relevance to personalized medicine. Per Med. 2008;5(2):141–153. doi:10.2217/17410541.5.2.141
8. Fucharoen S. Overview of genotypes and Phenotypes of Thalassemia in Asia. Availabe from: https://www.slideshare.net/Thalassaemia_Intl_Fed/overview-of-genotypes-and-phenotypes-of-thalassemia-inasia. Accessed September 5, 2020.
9. Thein SL. Molecular basis of β-thalassemia and potential therapeutic targets. Blood Cells Mol Dis. 2017.
10. Voon HPJ, Wardan H, Vadolas J. Co-inheritance of α- and β-thalassaemia in mice ameliorates thalassaemic phenotype. Blood Cells Mol Dis. 2007;39(2):184–188. doi:10.1016/j.bcmd.2007.01.006
11. Mettananda S, Gibbons RJ, Higgs DR. α-globin as a molecular target in treatment of β-thalassemia. Blood. 2015;125:3694–3701. doi:10.1182/blood-2015-03-633594
12. Voon HPJ, Wardan H, Vadolas J. siRNA-mediated reduction of α-globin results in phenotypic improvements in β-thalassemic cells. Haematologica. 2008;93(8):1238–1242. doi:10.3324/haematol.12555
13. Sarakul O, Vattanaviboon P, Wilairat P, et al. Inhibition of α-globin gene expression by RNAi. Biochem Biophys Res Commun. 2008;369(3):935–938. doi:10.1016/j.bbrc.2008.02.124
14. Xie S-Y, Ren Z-R, Zhang J-Z, et al. Restoration of the balanced α/β-globin gene expression in β 654 -thalassemia mice using combined RNAi and antisense RNA approach. Hum Mol Genet. 2007;16(21):2616–2625. doi:10.1093/hmg/ddm218
15. Xie SY, Li W, Ren ZR, et al. Correction of β654-thalassaemia mice using direct intravenous injection of siRNA and antisense RNA vectors. Int J Hematol. 2011;93(3):301–310. doi:10.1007/s12185-010-0727-1
16. Mettananda S, Fisher CA, Hay D, et al. Editing an α-globin enhancer in primary human hematopoietic stem cells as a treatment for β-thalassemia. Nat Commun. 2017;8(1):424. doi:10.1038/s41467-017-00479-7
17. Valencia-Sanchez MA, Liu J, Hannon GJ, et al. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20(5):515–524. doi:10.1101/gad.1399806
18. Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10(2):94–108.
19. Li H, Yang Y, Hong W, et al. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Tar. 2020;5(1):1.
20. Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human thalassaemia. Nature. 2010;467(7313):318–322. doi:10.1038/nature09328
21. Negre O, Eggimann A-V, Beuzard Y, et al. Gene therapy of the β-hemoglobinopathies by lentiviral transfer of the β(A(T87Q))-globin gene. Hum Gene Ther. 2016;27(2):148–165. doi:10.1089/hum.2016.007
22. Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378(16):1479–1493. doi:10.1056/NEJMoa1705342
23. Schuessler-Lenz M, Enzmann H, Vamvakas S. Regulators’ advice can make a difference: european medicines agency approval of zynteglo for beta thalassemia. Clin Pharmacol Ther. 2020;107(3):492–494. doi:10.1002/cpt.1639
24. Miccio A, Cesari R, Lotti F, et al. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of β-thalassemia. Proc Natl Acad Sci USA. 2008;105(30):10547–10552. doi:10.1073/pnas.0711666105
25. Marktel S, Cicalese MP, Giglio F, et al. Gene therapy for Beta thalassemia: preliminary results from the PHASE I/II TIGET-BTHAL trial of autologous hematopoietic stem cells genetically modified with globe lentiviral vector. Blood. 2017;130(Suppl 1):355.
26. Marktel S, Scaramuzza S, Cicalese MP, et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat Med. 2019;25(2):234–241. doi:10.1038/s41591-018-0301-6
27. Fucharoen S, Weatherall DJ. The hemoglobin E thalassemias. Cold Spring Harb Perspect Med. 2012;2:8. doi:10.1101/cshperspect.a011734
28. Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125. doi:10.1038/nrd3625
29. Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016;44(14):6549–6563. doi:10.1093/nar/gkw533
30. Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta. 1999;1489(1):141–158. doi:10.1016/S0167-4781(99)00150-5
31. Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018;14(1):9–21.
32. Crooke ST, Vickers TA, Liang X-H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020;48(10):5235–5253. doi:10.1093/nar/gkaa299
33. Svasti S, Suwanmanee T, Fucharoen S, et al. RNA repair restores hemoglobin expression in IVS2–654 thalassemic mice. Proc Natl Acad Sci USA. 2009;106(4):1205–1210. doi:10.1073/pnas.0812436106
34. El-Beshlawy A, Mostafa A, Youssry I, et al. Correction of aberrant pre-mRNA splicing by antisense oligonucleotides in β-thalassemia Egyptian patients with IVSI-110 mutation. J Pediatr Hematol Oncol. 2008;30(4):281–284. doi:10.1097/MPH.0b013e3181639afe
35. Derakhshan SM, Khaniani MS. Restoration of correct splicing in IVSI-110 mutation of β-globin gene with antisense oligonucleotides: implications and applications in functional assay development. Iran J Basic Med Sci. 2017;20(6):700–707. doi:10.22038/IJBMS.2017.8840
36. Suwanmanee T, Sierakowska H, Lacerra G, et al. Restoration of human β-globin gene expression in murine and human IVS2–654 thalassemic erythroid cells by free uptake of antisense oligonucleotides. Mol Pharmacol. 2002;62(3):545–553. doi:10.1124/mol.62.3.545
37. Nualkaew T, Jearawiriyapaisarn N, Hongeng S, et al. Restoration of correct βIVS2−654-globin mRNA splicing and HbA production by engineered U7 snRNA in β-thalassaemia/HbE erythroid cells. Sci Rep. 2019;9(1):7672. doi:10.1038/s41598-019-43964-3
38. Phanthong P, Borwornpinyo S, Kitiyanant N, et al. Enhancement of β‐globin gene expression in thalassemic IVS2‐654 induced pluripotent stem cell‐derived erythroid cells by modified U7 snRNA. Stem Cells Transl Med. 2017;6(4):1059–1069. doi:10.1002/sctm.16-0121
39. Schmajuk G, Sierakowska H, Kole R. Antisense oligonucleotides with different backbones: modification of splicing pathways and efficacy of uptake. J Biol Chem. 1999;274(31):21783–21789. doi:10.1074/jbc.274.31.21783
40. Lacerra G, Sierakowska H, Carestia C, et al. Restoration of Hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients. Proc Natl Acad Sci USA. 2000;97(17):9591–9596. doi:10.1073/pnas.97.17.9591
41. Preedagasamzin S, Nualkaew T, Pongrujikorn T, et al. Engineered U7 snRNA mediates sustained splicing correction in erythroid cells from β-thalassemia/HbE patients. Biochem Biophys Res Commun. 2018;499(1):86–92. doi:10.1016/j.bbrc.2018.03.102
42. Hammond SM, Wood MJA. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011;27(5):196–205. doi:10.1016/j.tig.2011.02.004
43. Gorman L, Suter D, Emerick V, et al. Stable alteration of pre-mRNA splicing patterns by modified U7 small nuclear RNAs. Proc Natl Acad Sci U S A. 1998;95:4929–4934. doi:10.1073/pnas.95.9.4929
44. Suter D, Tomasini R, Reber U, et al. Double-target antisense U7 snRNAs promote efficient skipping of an aberrant exon in three human β-thalassemic mutations. Hum Mol Genet. 1999;8(13):2415–2423. doi:10.1093/hmg/8.13.2415
45. Vacek MM, Ma H, Gemignani F, et al. High-level expression of hemoglobin A in human thalassemic erythroid progenitor cells following lentiviral vector delivery of an antisense snRNA. Blood. 2003;101(1):104–111. doi:10.1182/blood-2002-06-1869
46. Eckenfelder A, Tordo J, Babbs A, et al. The cellular processing capacity limits the amounts of chimeric U7 snRNA available for antisense delivery. Mol Ther Nucleic Acids. 2012;1:e31. doi:10.1038/mtna.2012.24
47. Goyenvalle A, Babbs A, van Ommen G-JB, et al. Enhanced exon-skipping induced by U7 snRNA carrying a splicing silencer sequence: promising tool for DMD therapy. Mol Ther. 2009;17(7):1234–1240. doi:10.1038/mt.2009.113
48. Goyenvalle A, Wright J, Babbs A, et al. Engineering multiple U7snRNA constructs to induce single and multiexon-skipping for Duchenne muscular dystrophy. Mol Ther. 2012;20(6):1212–1221. doi:10.1038/mt.2012.26
49. Imbert M, Dias-Florencio G, Goyenvalle A. Viral vector-mediated antisense therapy for genetic diseases. Genes. 2017;8(2):51. doi:10.3390/genes8020051
50. Madocsai C, Lim SR, Geib T, et al. Correction of SMN2 pre-mRNA splicing by antisense U7 small nuclear RNAs. Mol Ther. 2005;12(6):1013–1022. doi:10.1016/j.ymthe.2005.08.022
51. Meyer K, Marquis J, Trüb J, et al. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum Mol Genet. 2008;18(3):546–555. doi:10.1093/hmg/ddn382
52. Odermatt P, Trüb J, Furrer L, et al. Somatic therapy of a mouse SMA model with a U7 snRNA gene correcting SMN2 splicing. Mol Ther. 2016;24(10):1797–1805. doi:10.1038/mt.2016.152
53. De Angelis FG, Sthandier O, Berarducci B, et al. Chimeric snRNA molecules carrying antisense sequences against the splice junctions of exon 51 of the dystrophin pre-mRNA induce exon skipping and restoration of a dystrophin synthesis in Δ48-50 DMD cells. Proc Natl Acad Sci USA. 2002;99(14):9456–9461. doi:10.1073/pnas.142302299
54. Fang Y, Chen X, Godbey WT. Chapter 42 - Gene editing in regenerative medicine. In: Atala A, Lanza R, Mikos AG, Nerem R, editors. Principles of Regenerative Medicine.
55. Gupta RM, Musunuru K. Expanding the genetic editing tool kit: zFNs, TALENs, and CRISPR-Cas9. J Clin Invest. 2014;124(10):4154–4161. doi:10.1172/JCI72992
56. Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188(4):773–782. doi:10.1534/genetics.111.131433
57. Maeder ML, Gersbach CA. Genome-editing technologies for gene and cell therapy. Mol Ther. 2016;24(3):430–446. doi:10.1038/mt.2016.10
58. Modares Sadeghi M, Shariati L, Hejazi Z, et al. Inducing indel mutation in the SOX6 gene by zinc finger nuclease for gamma reactivation: an approach towards gene therapy of Beta-thalassemia. J Cell Biochem. 2018;119(3):2512–2519. doi:10.1002/jcb.26412
59. Xu P, Tong Y, Liu X-Z, et al. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2–654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci Rep. 2015;5:12065. doi:10.1038/srep12065
60. Fang Y, Cheng Y, Lu D, et al. Treatment of β654-thalassaemia by TALENs in a mouse model. Cell Prolif. 2018;51(6):e12491. doi:10.1111/cpr.12491
61. Bolotin A, Quinquis B, Sorokin A, et al. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151(8):2551–2561. doi:10.1099/mic.0.28048-0
62. Brouns SJJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960–964. doi:10.1126/science.1159689
63. Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protocols. 2013;8(11):2281–2308. doi:10.1038/nprot.2013.143
64. Guan Y, Ma Y, Li Q, et al. CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates Hemophilia in mouse. EMBO Mol Med. 2016;8(5):477–488. doi:10.15252/emmm.201506039
65. Liang X, Potter J, Kumar S, et al. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J Biotechnol. 2017;241:136–146. doi:10.1016/j.jbiotec.2016.11.011
66. Xie N, Zhou Y, Sun Q, et al. Novel epigenetic techniques provided by the CRISPR/Cas9 system. Stem Cells Int. 2018;2018:7834175. doi:10.1155/2018/7834175
67. Yang F, Liu C, Chen D, et al. CRISPR/Cas9-loxP-mediated gene editing as a novel site-specific genetic manipulation tool. Mol Ther Nucleic Acids. 2017;7:378–386. doi:10.1016/j.omtn.2017.04.018
68. Miano JM, Zhu QM, Lowenstein CJ. A CRISPR path to engineering new genetic mouse models for cardiovascular research. Arterioscler Thromb Vasc Biol. 2016;36(6):1058–1075. doi:10.1161/ATVBAHA.116.304790
69. Xie F, Ye L, Chang JC, et al. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014;24:1526–1533. doi:10.1101/gr.173427.114
70. Antony JS, Latifi N, Haque AKMA, et al. Gene correction of HBB mutations in CD34+ hematopoietic stem cells using Cas9 mRNA and ssODN donors. Mol Cell Pediatr. 2018;5(1):9. doi:10.1186/s40348-018-0086-1
71. Wattanapanitch M, Damkham N, Potirat P, et al. One-step genetic correction of hemoglobin E/Beta-thalassemia patient-derived iPSCs by the CRISPR/Cas9 system. Stem Cell Res Ther. 2018;9(1):46. doi:10.1186/s13287-018-0779-3
72. Liu Y, Yang Y, Kang X, et al. One-Step biallelic and scarless correction of a β-Thalassemia mutation in patient-specific iPSCs without drug selection. Mol Ther Nucleic Acids. 2017;6:57–67. doi:10.1016/j.omtn.2016.11.010
73. Xu S, Luk K, Yao Q, et al. Editing aberrant splice sites efficiently restores β-globin expression in β-thalassemia. Blood. 2019;133(21):2255–2262. doi:10.1182/blood-2019-01-895094
74. Kouranova E, Forbes K, Zhao G, et al. CRISPRs for optimal targeting: delivery of CRISPR components as DNA, RNA, and protein into cultured cells and single-cell embryos. Hum Gene Ther. 2016;27(6):464–475. doi:10.1089/hum.2016.009
75. Lino CA, Harper JC, Carney JP, et al. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018;25(1):1234–1257. doi:10.1080/10717544.2018.1474964
76. Yu X, Liang X, Xie H, et al. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol Lett. 2016;38(6):919–929. doi:10.1007/s10529-016-2064-9
77. Li G, Zhang X, Zhong C, et al. Small molecules enhance CRISPR/Cas9-mediated homology-directed genome editing in primary cells. Sci Rep. 2017;7(1):8943. doi:10.1038/s41598-017-09306-x
78. Thein SL. The molecular basis of β-thalassemia. Cold Spring Harb Perspect Med. 2013;3:5. doi:10.1101/cshperspect.a011700
79. Kaufman DP, Khattar J, Lappin SL. Physiology, Fetal Hemoglobin. Treasure Island (FL). StatPearls Publishing; 2020.
80. Lohani N, Bhargava N, Munshi A, et al. Pharmacological and molecular approaches for the treatment of β-hemoglobin disorders. J Cell Physiol. 2018;233(6):4563–4577. doi:10.1002/jcp.26292
81. Bianchi N, Zuccato C, Lampronti I, et al. Fetal hemoglobin inducers from the natural world: A novel approach for identification of drugs for the treatment of β-thalassemia and sickle-cell anemia. Evidence-Based Complementary and Alternative Med. 2009;6(2):141–151. doi:10.1093/ecam/nem139
82. Taghavi SA, Hosseini KM, Tamaddon G, et al. Inhibition of γ/β globin gene switching in CD 34+ derived erythroid cells by BCL11A RNA silencing. Indian J Hematol Blo. 2019;35(4):758–764. doi:10.1007/s12288-019-01131-8
83. Guda S, Brendel C, Renella R, et al. miRNA-embedded shRNAs for lineage-specific BCL11A knockdown and hemoglobin F induction. Mol Ther. 2015;23(9):1465–1474. doi:10.1038/mt.2015.113
84. Brendel C, Guda S, Renella R, et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J Clin Inves. 2016;126(10):3868–3878. doi:10.1172/JCI87885
85. Hu JH, Navas P, Cao H, et al. Systematic RNAi studies on the role of Sp/KLF factors in globin gene expression and erythroid differentiation. J Mol Biol. 2007;366(4):1064–1073. doi:10.1016/j.jmb.2006.12.047
86. Roosjen M, McColl B, Kao B, et al. Transcriptional regulators Myb and BCL11A interplay with DNA methyltransferase 1 in developmental silencing of embryonic and fetal β-like globin genes. FASEB J. 2014;28(4):1610–1620. doi:10.1096/fj.13-242669
87. Khosravi MA, Abbasalipour M, Concordet JP, et al. Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal hemoglobin reactivation: A promising approach for gene therapy of beta thalassemia disease. Eur J Pharmacol. 2019;854:398–405.
88. Psatha N, Reik A, Phelps S, et al. Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in erythroid cells of patients with β-thalassemia major. Mol Ther- Meth Clin D. 2018;10:313–326. doi:10.1016/j.omtm.2018.08.003
89. Chang K-H, Smith SE, Sullivan T, et al. Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34(+) hematopoietic stem and progenitor cells. Molecular Therapy Methods Clinical Development. 2017;4:137–148. doi:10.1016/j.omtm.2016.12.009
90. Shariati L, Khanahmad H, Salehi M, et al. Genetic disruption of the KLF1 gene to overexpress the γ-globin gene using the CRISPR/Cas9 system. J Gene Med. 2016;18(10):294–301. doi:10.1002/jgm.2928
91. Lamsfus-Calle A, Daniel-Moreno A, Antony JS, et al. Comparative targeting analysis of KLF1, BCL11A, and HBG1/2 in CD34+ HSPCs by CRISPR/Cas9 for the induction of fetal hemoglobin. Sci Rep. 2020;10(1):10133. doi:10.1038/s41598-020-66309-x
92. Liu N, Hargreaves VV, Zhu Q, et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018;173(2):430–42.e17. doi:10.1016/j.cell.2018.03.016
93. Weber L, Frati G, Felix T, et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv. 2020;6(7):eaay9392. doi:10.1126/sciadv.aay9392
94. Zhan J, Irudayam MJ, Nakamura Y, et al. High level of fetal-globin reactivation by designed transcriptional activator-like effector. Blood Adv. 2020;4(4):687–695. doi:10.1182/bloodadvances.2019000482
95. Métais JY, Doerfler PA, Mayuranathan T, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3(21):3379–3392. doi:10.1182/bloodadvances.2019000820
96. ClinicalTrials. A safety and efficacy study evaluating CTX001 in subjects with transfusion-dependent β-thalassemia; 2018. Available from: https://clinicaltrials.gov/ct2/show/NCT03655678.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.