Back to Journals » Journal of Inflammation Research » Volume 14
Nucleic Acid-Sensing Pathways During SARS-CoV-2 Infection: Expectations versus Reality
Authors Mdkhana B ![]() , Saheb-Sharif Askari N
, Saheb-Sharif Askari N ![]() , Ramakrishnan RK
, Ramakrishnan RK ![]() , Goel S, Hamid Q, Halwani R
, Goel S, Hamid Q, Halwani R ![]()
Received 19 August 2020
Accepted for publication 16 October 2020
Published 26 January 2021 Volume 2021:14 Pages 199—216
DOI https://doi.org/10.2147/JIR.S277716
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Bushra Mdkhana,1 Narjes Saheb Sharif-Askari,1 Rakhee K Ramakrishnan,1 Swati Goel,1 Qutayba Hamid,1– 3 Rabih Halwani1,2
1Sharjah Institute of Medical Research, College of Medicine, University of Sharjah, Sharjah, United Arab Emirates; 2Department of Clinical Sciences, College of Medicine, University of Sharjah, Sharjah, United Arab Emirates; 3Meakins-Christie Laboratories, Research Institute of the McGill University Health Center, Montreal, Quebec, Canada
Correspondence: Rabih Halwani
College of Medicine, University of Sharjah, Sharjah, United Arab Emirates
Email [email protected]
Abstract: The coronavirus disease 2019 (COVID-19) pandemic has affected millions of people and crippled economies worldwide. The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) responsible for this pandemic has triggered avid research on its pathobiology to better understand the pathophysiology of COVID-19. In the absence of approved antiviral therapeutic strategies or vaccine platforms capable of effectively targeting this global threat, the hunt for effective therapeutics has led to many candidates being actively evaluated for their efficacy in controlling or preventing COVID-19. In this review, we gathered current evidence on the innate nucleic acid-sensing pathways expected to be elicited by SARS-CoV-2 and the immune evasion mechanisms they have developed to promote viral replication and infection. Within the nucleic acid-sensing pathways, SARS-CoV-2 infection and evasion mechanisms trigger the activation of NOD-signaling and NLRP3 pathways leading to the production of inflammatory cytokines, IL-1β and IL-6, while muting or blocking cGAS-STING and interferon type I and III pathways, resulting in decreased production of antiviral interferons and delayed innate response. Therefore, blocking the inflammatory arm and boosting the interferon production arm of nucleic acid-sensing pathways could facilitate early control of viral replication and dissemination, prevent disease progression, and cytokine storm development. We also discuss the rationale behind therapeutic modalities targeting these sensing pathways and their implications in the treatment of COVID-19.
Keywords: COVID-19, SARS-CoV-2, innate immune system, nucleic acid sensing, immune evasion, NLRP3, cGAS-STING, coronavirus
Introduction
Belonging to the Coronaviridae family, coronaviruses (CoVs) are enveloped, positive-sense single-stranded RNA viruses with a 5′‐cap structure and a 3′‐poly‐A tail. They are further characterized by one of the largest RNA viral genomes of up to 26–32 kilobases in length.1 Seven members of this family have been identified to infect humans till date2 including, highly pathogenic severe acute respiratory syndrome coronavirus (SARS-CoV) and middle east respiratory syndrome coronavirus (MERS-CoV). These coronaviruses were responsible for the outbreaks of SARS in 20023 and MERS in 2012,4 respectively.
In December 2019, the outbreak of pneumonia of unknown origin in Wuhan, China5 accelerated interest in a novel strain of coronavirus, which was later named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). World Health Organization (WHO) has declared SARS-Cov2 a pandemic on March 11, 2020 due to its rampant dissemination. Since its discovery, the virus has also exerted a strong global crisis crippling both our healthcare systems and economies. SARS-CoV-2 is a member of the Coronaviridae family, Coronavirinae subfamily, Betacoronavirus genus and subgenus Sarbecovirus. The latest coronavirus disease 2019 (COVID-19) pandemic caused by the SARS-CoV-2 virus has affected over sixteen million people and caused over half a million death worldwide as of 31st July, 2020.6
The coronavirus genome encodes nonstructural (nsp), structural and accessory viral proteins. The open reading frames (ORF) 1a and 1b at the 5′-terminal of the CoV genome constitute the viral replicase gene that encodes 16 nonstructural enzymes. The CoV virion is composed of primarily four structural proteins – spike (S), envelope (E), membrane (M) and nucleocapsid (N). The nucleocapsid representing the viral genome is enclosed within the viral envelope containing the M and E proteins. The S glycoprotein that protrudes from the viral envelope is a class 1 viral fusion protein that is processed by host proteases into two functional domains, S1 receptor binding unit and S2 membrane fusion unit.7 The accessory proteins 3a, 3b, 6, 7a, 7b, and 8 (or 8a and 8b in some isolates) have been identified to be involved in immune evasion. This aids viral pathogenesis through inhibition of type I interferons (3b and 6), modulation of cellular DNA synthesis (6, 8b), induction of apoptosis (3a, 3b, 8a), stimulation of chemokine synthesis (3a), induction of arms of unfolded protein response,8 and inflammation.7–10 SARS-CoV-2 appears to have evolved from the recent bat CoV isolate RaTG13 and shares 80% homology to SARS-CoV (retrospectively named SARS-CoV-1), and 50% to MERS-CoV.11
The S protein on the surface of these viruses engages with the respective receptor proteins on the host cell surface enabling viral attachment and subsequent fusion with the host cells resulting in viral infection. While the SARS-CoV uses the angiotensin-converting enzyme 2 (ACE2) receptor to gain entry into ciliated bronchial epithelial cells and alveolar type II cells,12,13 the dipeptidyl peptidase 4 (DPP4; also known as CD26) serves as the functional receptor for MERS-CoV entry into unciliated bronchial epithelial cells and alveolar type II cells.14,15 SARS-CoV-2 also shows strong binding affinity to ACE2 receptor.16 Post viral entry, translation of the two 5′-terminal ORFs 1a and 1b leads to the synthesis of multiple copies of new RNA genomes which encapsulate with the N protein forming a helical nucleocapsid. The M protein then plays a key role in assembling the virion particles which bud off from the endoplasmic reticulum (ER) and the Golgi bodies, and new viruses are released by exocytosis.17,18
The pathogenic human coronaviruses are known to cause acute pulmonary disease characterized by atypical pneumonia in humans. Respiratory failure is leading cause of mortality during SARS-Cov2 infection and is associated with immune dysregulation and cytokine storm. While the human immune system is equipped to control and eliminate such infections, dysregulated immune response can pave the way to immunopathology and subsequent impairment in pulmonary gas exchange. Despite the global efforts from different sectors to develop a vaccine and to control the rapid spread of infection, to this date there is no specific medication for the prevention or treatment of COVID-19 infection. Understanding the immune antiviral sensing mechanisms, the immunological pathways elicited, and the immune evasion mechanisms regulated by SARS-CoV-2 are needed to guide the proper design and development of a vaccine against it.
In this review, we first summarize the current state of knowledge on the known viral nucleic acid-sensing mechanisms and speculate their evasion by the novel SARS-CoV-2 based on the genomic similarity between SARS-CoV-2 and other human Betacoronaviruses (SARS-CoV-1, and MERS-CoV). The immune recognition and evasion mechanisms of SARS-CoV-2 being a relatively nascent field, studies related to other CoVs such as SARS-CoV-1 and MERS-CoV provide references to gain insights on the possible mechanisms involved. We further address the therapeutic modulation of the innate sensing mechanisms as prospective therapy. We have critically reviewed the preprint literature together with published peer-reviewed articles in order to gather updated information on the rapidly evolving field of COVID-19 pathogenesis.
Innate Immune Recognition of SARS-CoV-2
The observation of non-specific inflammation in pulmonary lesions and pulmonary tissue damage in SARS cases highlights the importance of immune recognition and subsequent inflammatory response throughout the course of coronavirus infection.19 Although the pathogenesis of SARS-CoV-2 is not yet completely understood, it is evident that both viral and host components play a crucial role in the severity of COVID-19 infection. In general, viral infection of a host cell triggers an immune reaction against the virus post viral recognition by the innate immune system. However, pathogenesis of these viral infections is usually associated with an immune response that gets uncurbed, thereby resulting in tissue damage and functional impairment in lungs, as well as reduced lung capacity. Here, we review the activity of nucleic acid-sensing pathways during COVID-19 infection. The RNA-sensing arm is activated by RNA viruses such as SARS-CoV-2, while DNA-sensing arm is triggered by host damage-associated molecular patterns (DAMPs) released as byproducts of viral reproduction and tissue injury.
The innate immune system recognizes the pathogen-associated molecular patterns (PAMPs) comprising viral nucleic acids, protein, lipids, structural components and other viral intermediates including single-stranded RNA (ssRNA) or double-stranded RNA (dsRNA), via distinct pattern recognition receptors (PRRs). These receptors include RIG‐I‐like receptor (RLR), toll‐like receptor (TLR), NOD‐like receptor (NLR), C‐type lectin‐like receptors (CLmin) as well as certain cytoplasmic free‐molecule receptors, such as stimulator of interferon genes (STING), cyclic GMP-AMP synthase (cGAS), gamma-interferon-inducible protein 16 (IFI16) and DNA-dependent activator of interferon-regulatory factors (DAI), to name a few.20–22 Viral RNA recognition occurs primarily in the cytoplasmic or endosomal compartments within an infected cell (Figure 1). CoVs which are typically ssRNA viruses and form dsRNA during their replicative stage. While ssRNA can be detected by TLR7 or TLR8 and potentially RIG-I and PKR, dsRNA engages TLR3 in the endosome and RIG-I, MDA5, and PKR in the cytosol. These PRRs initiate a signaling cascade culminating in primarily type I interferon (IFN-α/β) and inflammatory cytokine production. Type I IFNs are key in the establishment of an antiviral state through their induction of interferon stimulated genes (ISGs) and their gene products that inhibit viral infection robustly by impacting both the early and late stages of the virus life cycle.23 Type I IFNs bind to their cognate IFNAR, thereby activating the JAK-STAT pathway leading to the transcription of ISGs. Although type I (IFN-α, IFN-β, IFN-ε, IFN-κ, IFN-ω in humans) and also type III (IFN-γ) IFNs are key to antiviral defense, other pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), and interleukin-1 beta (IL-1β), IL-6, and IL-18, also play a supporting role. This helps in the establishment of an antiviral state in the infected cell and its immediate surroundings as well as potentiate an adaptive immune response.
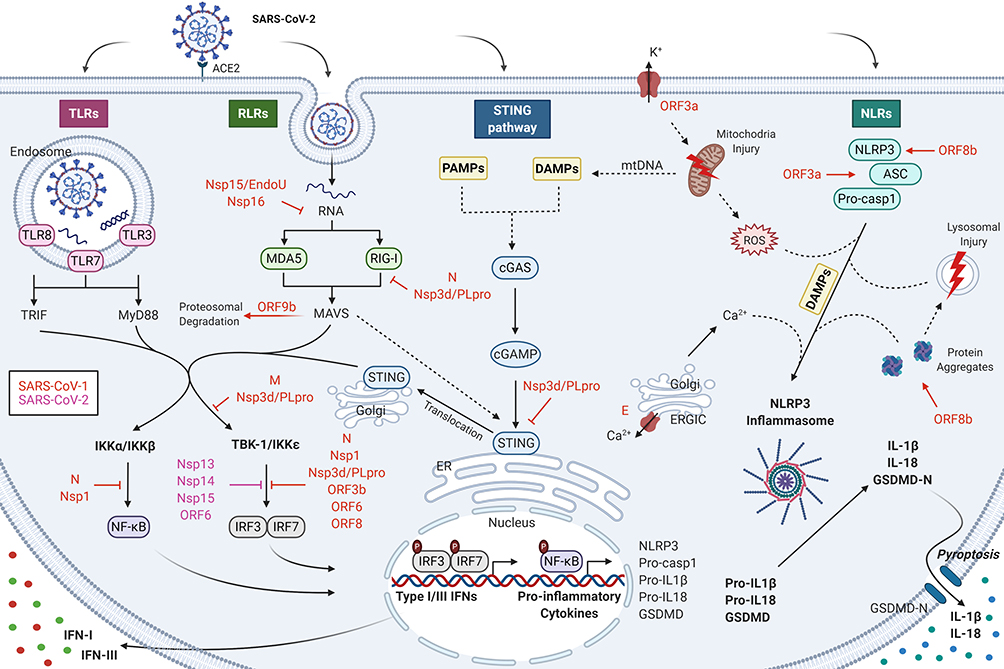 |
Figure 1 Nucleic acid-sensing pathways and their evasion by SARS-CoV-2 during COVID-19 infection. The nucleic acid-sensing pathways within the innate immune system recognizes the pathogen-associated molecular patterns (PAMPs) comprising of viral nucleic acids and other viral intermediates including single-stranded RNA (ssRNA) or double-stranded RNA (dsRNA), via distinct pattern recognition receptors (PRRs). The RNA-sensing arm is activated by RNA viruses such as SARS-CoV-2, while DNA-sensing is triggered by host genome released as byproducts of viral reproduction and tissue injury. These receptors include toll‐like receptor (TLR), RIG‐I‐like receptor (RLR), C‐type lectin‐like receptors (CLmin) as well as certain cytoplasmic free‐molecule receptors, such as stimulator of interferon genes (STING), cyclic GMP-AMP synthase (cGAS), and NOD‐like receptor (NLR). Coronaviruses (CoVs) which are typically ssRNA viruses, form dsRNA during their replicative stage. While ssRNA can be detected by TLR7 or TLR8 and potentially RIG-I and PKR, dsRNA engages TLR3 in the endosome and RIG-I, MDA5, and PKR in the cytosol. The cGAS-STING pathway is incapable of directly sensing RNA of CoVs; however, they are likely to get activated by danger-associated molecular patterns (DAMP) signals, such as mitochondrial DNA (mtDNA), released as a result of CoV infection. These PRRs initiate a signaling cascade culminating in primarily type I interferon (IFN-α/β) and inflammatory cytokine production (IL-6 and IL-1β). The presence of low innate antiviral defenses to SARS-CoV-2 suggests the presence of effective evasion mechanisms by SARS-CoV-2 to escape immune surveillance. SARS-CoV-2 is suggested to suppress IFN-I response by means of similar mechanisms as used by SARS-CoV-1, namely Nsp1, Nsp3d/PLpro, Nsp7, Nsp15/EndoU, Nsp16, ORF3, ORF6, ORF8, ORF9b, M, and N. |
The role of the various PRRs in CoV infection is derived from genetic studies where increased susceptibility to infection was noted in the absence of a specific PRR and its associated signaling. Despite the limited literature on innate immune response specific to SARS-CoV-2, their shared sequence homology with other CoVs and the conserved innate immune signaling mechanisms enable us to recapitulate the findings from related CoVs.
RIG-I-Like Receptors (RLRs)
Retinoic acid-inducible gene (RIG)-I-like receptors (RLRs) include cytoplasmic viral sensors such as RIG-I, melanoma differentiation-associated gene 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2), that primarily detect viral RNA.24,25 RLRs thus constitute key PRRs for RNA viruses that can also recognize some DNA viruses.
While RIG-I senses long dsRNA molecules and short dsRNAs bearing a tri- or di-phosphorylated 5′ end, MDA5 is more specific for long dsRNAs as well as higher-order structured RNA.26–28 Recognition of viral PAMP RNA activates RIG‐I and MDA5 that converge upon the adaptor protein, mitochondrial activator of virus signaling (MAVS). Interaction with MAVS then leads to the recruitment and activation of protein kinases such as TANK-binding kinase 1 (TBK1)/IKKɛ and IKKα/IKKβ, which in turn activate interferon regulatory factor (IRF)-3, IRF-7 and NF-κB, respectively. These activated transcriptional factors translocate to the nucleus and induce the expression of various innate immune response genes, such as IFN-I, IFN-III and various other pro-inflammatory cytokines (PIC), including TNF-α, IL-1, IL-6, and IL-18; leading to the establishment of an antiviral state in the infected cell and surrounding tissue.29–31 SARS-CoV-1 infection of bronchial epithelial cells upregulated the transcription of several genes associated with the RLR signaling pathway, including RIG-I, MDA5, IRF-3/7, IFN-β, -λ, STATs and several other ISGs.32 SARS-CoV-2 being similar to SARS-CoV-1 by 80%,11 is likely to be detected by the RLR sensors, RIG-I and MDA5.
Toll‐Like Receptors (TLRs)
Toll‐like receptors (TLRs) are widely expressed in various cellular compartments including plasma membranes, endosomes, lysosomes, cytosol and endocytolysosomes. The various TLRs recognize a variety of PAMP signals, including nucleic acids, lipids, proteins and lipoproteins.33 RNA sensing TLR3, 7, 8 and 9, anchored on the intracellular membranes of endosomes, detect viral nucleic acid patterns associated with viral infections.34 While TLR3 detects dsRNA, TLR7 and 8 detect ssRNA and TLR9 detects unmethylated CpG containing ssDNA. Different TLRs are equipped with distinct adaptor proteins, including myeloid differentiation primary response gene 88 (MyD88), toll-interleukin 1 receptor (TIR) domain-containing adaptor-inducing interferon-β (TRIF), TIR domain-containing adaptor protein (TIRAP), TRIF-related adaptor molecule (TRAM), and thyroid hormone receptor interactor (TRIP), which induces distinct immune responses. For instance, TLR4, TLR7 and TLR8 use the MyD88 adaptor for downstream signaling, and TLR3 and TLR4 use TRIF. MyD88 activates the transcription factors NF‐κB and mitogen‐activated protein kinases (MAPKs) pathways, while TRIF activates the transcription factors IRF-3 and NF‐κB to induce the expression of type I interferon.35
TLR3 senses dsRNA leading to the activation of NF‐κB and type I IFN production.36 TLR3 and MyD88 were differentially expressed in response to SARS-CoV-1 infection in vivo,37 indicating the activation of at least two distinct TLR signaling pathways in SARS-CoV pathogenesis. TLR3 signaling via TRIF adaptor was also shown to elicit a balanced protective immune response against pathogenic CoV, SARS-CoV-1 and MERS-CoV, infections.37,38 The intracellular RNA in endosomes is known to activate TLR7 leading to the instant production of type I IFNs, which is key to control potentially lethal CoV infections by SARS-CoV-1 and MERS-CoV.39 Rapid clinical whole-exome sequencing of young men with severe COVID-19 led to the identification of a rare putative loss-of-function variants of TLR7 and transcriptional downregulation of type I IFN signaling.40 Furthermore, MyD88, a vital component to transduce signaling downstream of multiple TLRs, was found to be vital in limiting the lethality of mouse-adapted SARS-CoV-1 infection.41 Similarly, TRIF deficient mice were highly susceptible to SARS-CoV-1 infection and displayed an aberrant pro-inflammatory profile similar to those seen in patients infected with SARS-CoV-1 or MERS-CoV, and with poor disease outcomes.37 Furthermore, cell surface TLRs, in particular TLR4, have also been suggested to detect molecular patterns associated with S protein from SARS-CoV-2 to induce inflammatory responses.16
NOD-Like Receptors (NLRs)
NOD-like receptors (NLRs) are a type of PRR that detect a wide range of both cytosolic pathogen- and danger-associated molecular patterns (PAMP and DAMP). NLRs such as NACHT, LRR, and PYD domains-containing protein 1 (NLRP1), NLRP3, NLR family CARD domain-containing protein 4 (NLRC4), and absent in melanoma 2 (AIM2) interact with apoptosis-associated speck-like protein (ASC) and caspase-1 forming a multiprotein complex called inflammasome.42,43 The inflammasome aids in viral eradication by the secretion of pro-inflammatory cytokines, IL-1β and IL-18, and induces a form of inflammatory cell death called pyroptosis.42,43 NLRP3 inflammasome is the most studied inflammasome that is known to mediate inflammation and antiviral responses.43 NLRP3 is prospectively activated in response to SARS-CoV-2.
The activation of NRLP3 inflammasome requires two steps – priming and activation. Initially, the priming step involves the activation of PRRs (RLRs and TLRs) via sensing RNA of SARS-CoV-2. This eventually leads to NF-κB activation and induces the expression of NLRP3, pro-caspase-1, pro-IL-1β and pro-IL-18. Then, the activation step is triggered by sensing intracellular DAMPs induced by SARS-CoV-2.
Similar to SARS-CoV-1 and MERS-CoV, SARS-CoV-2 causes cellular alterations in host cells, including abnormal ion concentrations, and accumulation of protein aggregates, which are sensed as DAMPs in the infected host cells and lead to NLRP3 inflammasome activation.43 The main SARS-CoV-2 proteins that could activate NLRP3 inflammasome and induce IL-1β secretion are envelope protein (E protein), open reading frame-3a (ORF3a), ORF8 and non-structure protein-3c (Nsp3c) (Table 1). SARS-CoV-2 E protein has 94% similarity to SARS-CoV-1,44–46 suggesting that it may induce the activation of NLRP3 inflammasome by similar mechanisms. The ion channel (IC) activity of E protein, through pore formation in ER-Golgi intermediate compartment (ERGIC)/Golgi membranes, results in releasing calcium ions (Ca2+) in the cytosol. Ca2+ is the major key ion that triggers the secretion of IL-1β in an NLRP3 inflammasome-dependent manner and is also a major contributor for acute respiratory distress syndrome (ARDS).47,48 Further, SARS-CoV-1 ORF3a activates NLRP3 through its ion channel feature as well. The ion channel activity of ORF3a mediates the disruption of intracellular potassium ion (K+) concentration when localized to plasma membrane.49 Thus, the alteration in intracellular K+ concentration triggers mitochondrial damage and reactive oxygen species (ROS) release which augments the NLRP3 activation.49,50 Interestingly, ORF3a also stimulates IL-1β secretion independently of NLRP3 activation through ubiquitination of ASC which facilitates ASC specks formation.51,52 SARS-CoV-2 ORF3a shares homology by 72.04% to SARS-CoV-1 ORF3a and is speculated to use similar mechanisms for NLRP3 activation.44–46 Upon SARS-CoV-1 infection, ORF8b mediates formation of intracellular aggregates triggering endoplasmic reticulum (ER) stress and lysosomal damage, thereby resulting in NLRP3 activation.53,54 Also, ORF8b directly interacts with NLRP3 by binding to its LRR domain.54 However, SARS-CoV-2 ORF8 has low similarity to SARS-CoV-1 ORF8 (about 40%),45,46 and hence may not behave similarly between the two viruses. Recently, Chang and his colleagues reported that the SARS unique domain (SUD) of SARS-CoV-1, which is similar to SARS-CoV-2 by 82%,55,56 activates NLRP3 inflammasomes. SUD is the c domain of non-structural protein 3 (Nsp3c) which aids in viral genome replication. SUD induces IL-1β expression, its secretion and mediates the upregulation of CXCL10.57 It also induces macrophage/monocyte cell infiltration to bronchial and lung interstitial spaces leading to lung injury.57 The activation of complement (C5a) by MERS-CoV infection also contributes to pyroptosis via overexpression of caspase-1 and IL-1β.58,59
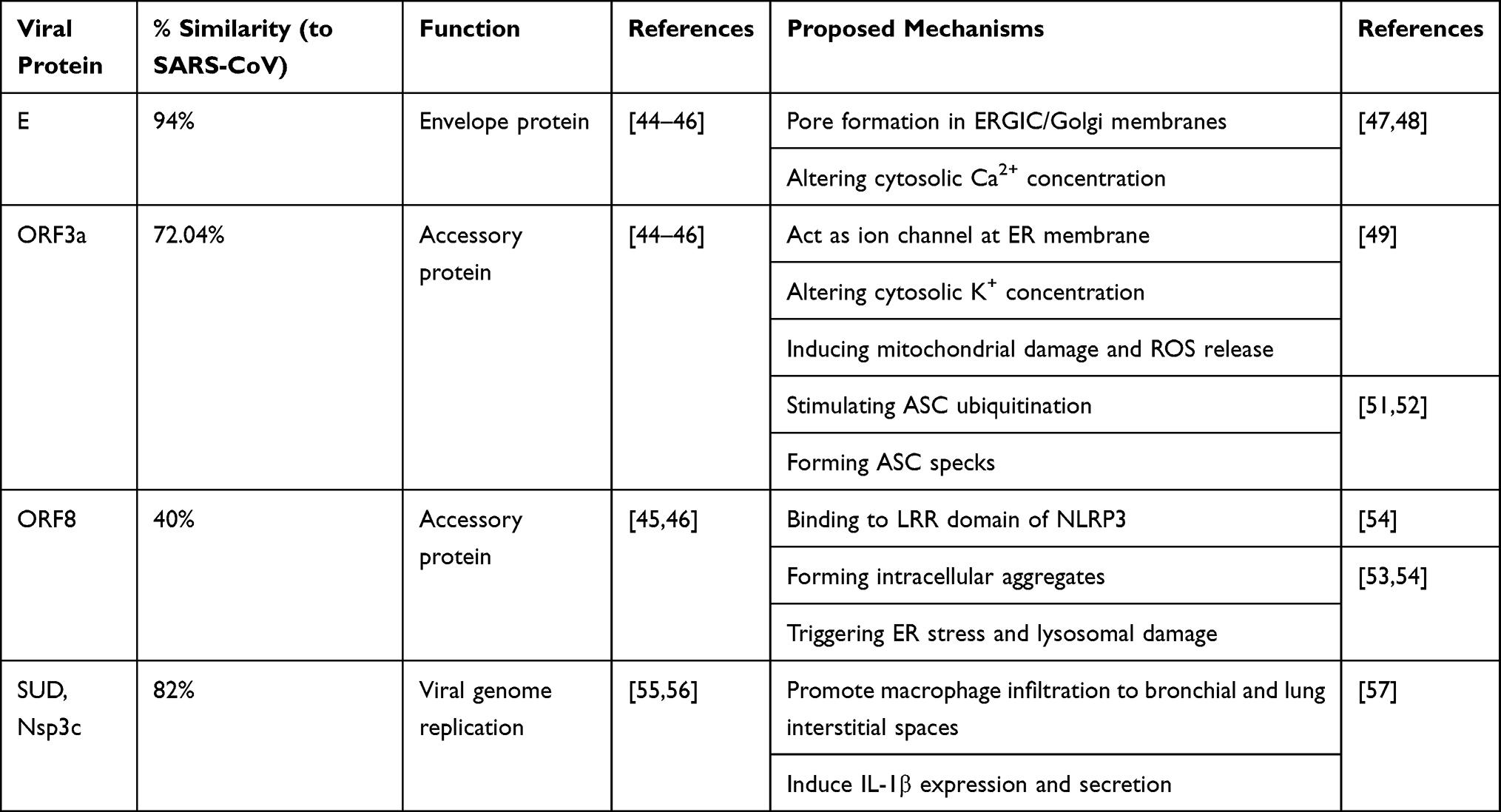 |
Table 1 The Mechanism of Action of SARS-CoV-2 Viral Proteins Activating NLRP3 Inflammasomes |
The SARS-CoV-2 activated NLRP3 is expected to interact with ASC forming ASC specks. The recruitment of pro-caspase-1 by ASC speck induces caspase-1 activation by self-cleavage which subsequently cleaves the pro-IL-1β, pro-IL-18 and inactive gasdermin D (GSDMD) into IL-1β, IL-18 and active GSDMD. Further, activated GSDMD induces pyroptosis by forming pores in cell membrane allowing proinflammatory cytokines, IL-1β and IL-18, to be secreted into the extracellular space.42,43
STING Pathway
In the last decade, stimulator of interferon genes (STING) was identified as an adapter protein involved in innate immune signaling triggering IFN-I expression in response to mislocalized nucleic acids in the cytosol. STING is well documented as a cytosolic DNA sensing pathway consequent to DNA viral infections in a cyclic GMP-AMP synthetase (cGAS) dependent manner. This canonical STING pathway involves cGAS activation upon recognizing viral DNA in the cytosol of infected cells which leads to the production of the second messenger cyclic GMP-AMP (cGAMP). cGAMP then binds to STING on the ER resulting in its translocation to the Golgi apparatus where TBK1 is recruited and activated by autophosphorylation. Activated TBK1 phosphorylates IRF-3 and inhibitor of NF- κB kinase (IκB kinase) releasing NF-κB, which then translocate to the nucleus to induce transcription of inflammatory genes.60,61
The cGAS-STING pathway is incapable of directly sensing RNA of CoVs; however, they are likely to get activated by DAMP signals, such as mitochondrial DNA (mtDNA), released as a result of CoV infection.62 Additionally, STING also plays a crucial role in limiting the infection of RNA viruses through the non-canonical STING pathway, which represents the crosstalk between the RNA and DNA sensing pathways. Upon RNA viral infection, RLRs sense the cytosolic RNA and then activate MAVS, which interact with STING forming a complex at mitochondrial-associated endoplasmic reticulum membranes (MAM). These in turn recruit and activate TBK1 and IKKɛ, that activate the transcriptional factors, IRF-3, IRF-7 and NF-κB.63 The induction of IFN-I production is the main antiviral response mediated by STING activation. Besides the role of STING in the induction of IFN-I expression, STING inhibits the initiation of translation of viral proteins independent of the initiation factor eIF2.64 Thus, the innate immune sensing pathways, RLRs and STING, harmonize to restrict RNA viral infection through several antiviral mechanisms, including IFNs and restriction of viral protein synthesis.
Collectively, SARS-CoV-2, an enveloped RNA virus, is likely to activate innate immune responses by distinct mechanisms, including RLRs, TLRs, NLRP3, cGAS and STING pathways.
Innate Immune Evasion of SARS-CoV-2
Unsurprisingly, CoVs have evolved to encode multiple factors in order to evade the immune sensing pathways, antagonize the IFN pathway, and thus, avoid recognition.65,66 This in turn favors viral immunopathogenesis through a dysregulated immune response. The host response to SARS-CoV-2 is defined by low type-I/III IFN levels and exuberant pro-inflammatory chemokine and cytokine production.67 Low plasma titers of neutralizing antibodies against SARS-CoV-2 were also noted in 30% of recovered COVID-19 patients.68 The presence of low innate antiviral defenses to SARS-CoV-2 suggests the presence of effective evasion mechanisms by SARS-CoV-2 to escape immune surveillance.
CoVs have evolved various strategies to antagonize IFN induction, to the establishment of a cell-intrinsic state of viral resistance, and its consequent signaling. CoVs have developed virulence factors antagonizing each step of the pathway right from PRR sensing to IFN secretion to IFN signal transduction and ISG activation (Figure 1). The virus is known to use physical strategies, such as the formation of double-membrane vesicles, to avoid immune recognition.69 The Nsps are responsible for rearranging membranes originating from host cellular membranes such as the rough ER, into double-membrane vesicles. These may shield viral replication and transcription from detection by the surrounding host innate immune sensors.69–71
SARS-CoV-1 have been shown to effectively suppress innate IFN response in numerous in vitro and in vivo studies via multiple mechanisms.72–75 SARS-CoV-2 is suggested to suppress IFN-I response by means of similar mechanisms as used by SARS-CoV-1, namely Nsp1, Nsp3d/PLpro, Nsp7, Nsp15/EndoU, Nsp16, ORF3, ORF6, ORF8, ORF9b, M, and N (Table 2).
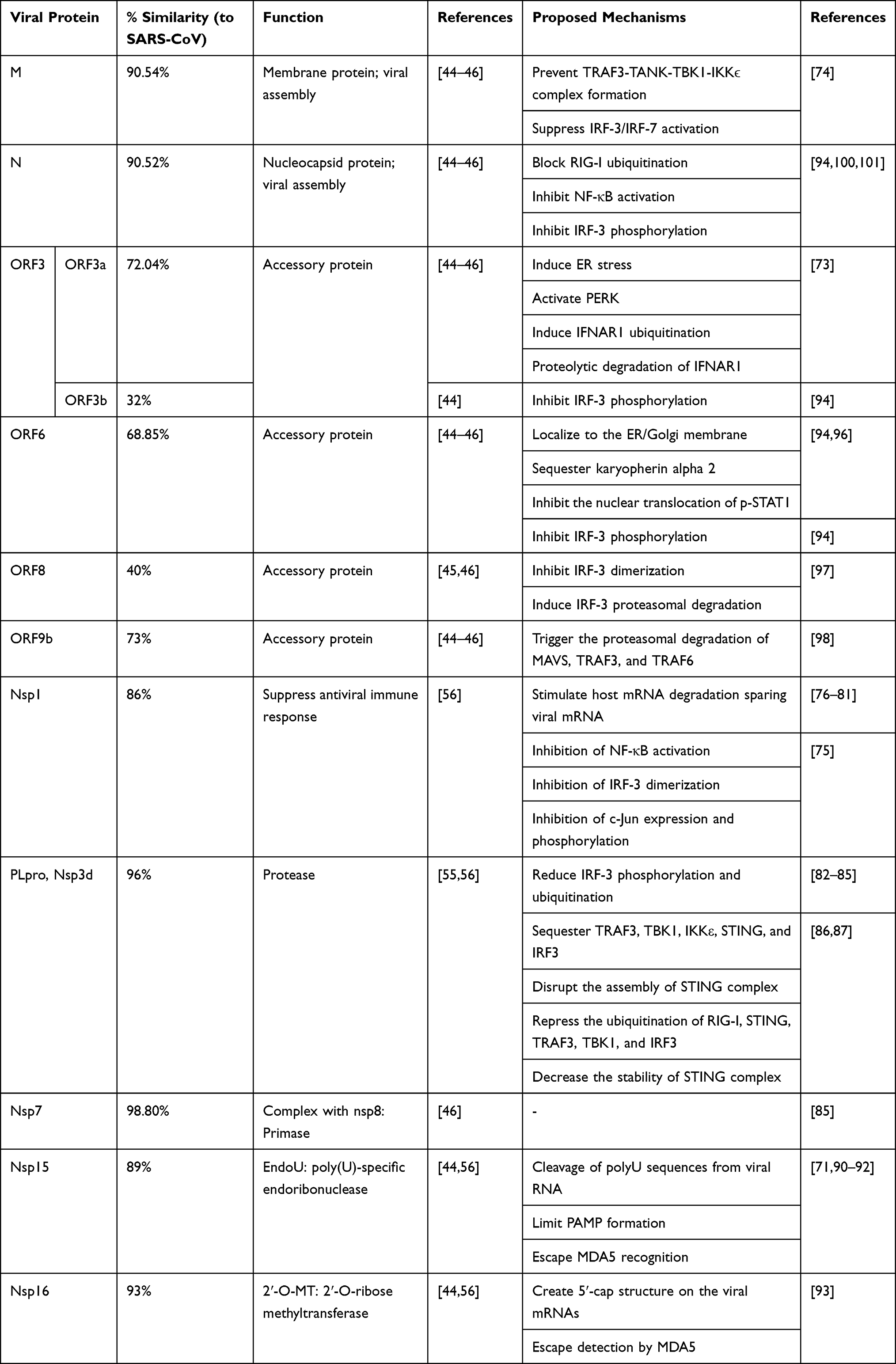 |
Table 2 The Mechanism of Action of SARS-CoV-2 Viral Proteins Evading Interferon Production and Signaling |
Nsp1 antagonizes IFN-I through multiple antiviral mechanisms. Nsp1 was shown to inhibit host protein translation by stimulating host mRNA degradation, including that of type I IFN. It selectively targets host mRNA while sparing viral mRNA.76–81 Further, Nsp1 blocks IFN-β expression upon SARS-CoV-1 infection through inhibition of NF-κB activation, IRF-3 dimerization, and c-Jun expression and phosphorylation.75 However, when exposed to IFN-I treatment, Nsp1 suppresses STAT1 phosphorylation.75 SARS-CoV-2 Nsp1 is 86% similar to SARS-CoV-1 Nsp1, suggesting the induction of similar evasion mechanisms.56
Papain-like protease (PLpro) is the d domain of Nsp3 that cleaves the N-terminal of replicase polyprotein at three different sites releasing essential non-structural proteins (Nsp) for replication.56 SARS-CoV-1 PLpro is a potent IFN antagonist independent of its protease activity. PLpro inhibits nuclear translocation of IRF-3 by inhibiting IRF-3 phosphorylation and ubiquitination.82–85 The membrane anchored PLpro domain (PLpro-TM) sequesters TRAF3, TBK1, IKKε, STING, and IRF-3 preventing STING dimerization and the assembly of STING-TRAF3-TBK1 complex. Also, PLpro-TM reduces the stability of RIG-I, STING, TRAF3, TBK1, and IRF-3 in the STING-TRAF3-TBK1 complex by suppressing their ubiquitination.86,87 Further, Yang and his colleagues showed that MERS-CoV PLpro has the same activity in decreasing IFN-β expression as SARS-CoV-1 PLpro.88 SARS-CoV-2 PLpro sequence, being 96% similar to that of SARS-CoV-1,55,56 would be expected to induce similar effects. However, despite sharing high amino acid sequence similarity with SARS-CoV-1, SARS-CoV-2 PLpro was found to lack both IFN-antagonizing and deubiquitinase properties.89
Nsp7 and Nsp15 were also suggested to impede IFN induction in SARS-CoV-1, but the mechanism is yet to be completely understood.85 Both Nsp7 and Nsp15 aid in viral replication as Nsp7 is a primase that forms a complex with Nsp8, while Nsp15 is poly(U)-specific endoribonuclease (EndoU).46 SARS-CoV-2 Nsp7 is 98.80% similar to SARS-CoV-1 Nsp7,46 while SARS-CoV-2 Nsp15 has 89% similarity to SARS-CoV-1 Nsp15;44,56 suggesting that both Nsp7 and Nsp15 may induce similar mechanisms.
EndoU is a highly conserved enzyme in all CoVs which is encoded by Nsp15.90 EndoU plays a key role in suppressing both the type I and type III IFN response in epithelial cells and macrophages.91 Its activity limits viral RNA sensing by host dsRNA sensors, including MDA5, PKR and OAS/RNaseL. Coronaviruses with mutated Nsp15 were found to have poor replication and induces rapid apoptosis in mouse bone marrow-derived macrophages.71 In this study, Nsp15 mutant virus activated the host immune viral sensors, MDA5, PKR, and the OAS/RNase L system, resulting in an early induction of type I IFN and PKR-mediated apoptosis. During the early stage of infection, majority of the dsRNA was associated with the viral replication complex in the WT virus-infected macrophages, whereas the dsRNA was dispersed in the Nsp15 mutant virus-infected cells. This indicated that coronavirus Nsp15 was critical for viral evasion of host dsRNA sensors in macrophages. Moreover, the CoV EndoU was found to target the polyuridine (polyU) extensions on 5′-polyU-containing, negative-sense (PUN) RNAs, where the PUN RNA constitutes the PAMP recognized by MDA5.92 Therefore, the cleavage of polyU sequences from PUN RNAs by EndoU limits the PAMP formation and their immune recognition, and thereby hinder the ability of MDA5 to activate the innate immune response in response to CoV infection. EndoU is a highly conserved virulence factor and thus, may present a potential target for vaccine and antiviral strategies.
Furthermore, SARS-CoV-1 Nsp16 which is a 2ʹ-O-ribose methyltransferase counteracts IFN induction via preventing MDA5 detection by creating 5′-cap structure on the viral mRNAs.93 SARS-CoV-2 Nsp16 is 93% similar to that of SARS-CoV-1.44,56 This similarity suggests that the evasion strategy of SARS-CoV-1 Nsp16 is likely to be retained in SARS-CoV-2 as well.
ORF3 is an accessory protein that has two domains, ORF3a and ORF3b. As ORF3a localizes to ER-Golgi compartment, it causes ER stress which in turn activates PKR-like ER kinase (PERK). The activated PERK induces increased IFN alpha-receptor subunit 1 (IFNAR1) ubiquitination. This causes the IFNAR1 to translocate to lysosomal compartment for proteolytic degradation. ORF3a induces the IFNAR1 degradation independent of its cognate ligand IFN-I.73 ORF3b of SARS-CoV-1 inhibit IRF-3 phosphorylation thus inhibiting its nuclear translocation.94 Although SARS-CoV-2 ORF3b is similar to SARS-CoV-1 by only 32%,44 ORF3b of SARS-COV-2 is reportedly more potent in antagonizing IFN-I, which may explain the poor IFN response in COVID-19 patients.95
ORF6 inhibits both IFN-I synthesis and signaling. The downregulation of IFN-I expression by SARS-CoV-1 ORF6 is associated with the prevention of IRF-3 phosphorylation.94 The IFN-I signaling is further inhibited by ORF6 via sequestering karyopherin alpha 2 preventing the nuclear translocation of p-STAT1.94,96 We speculate similar activity by SARS-CoV-2 ORF6 due to their amino acid homology with SARS-CoV-1 ORF6 by 68.85%.44–46
SARS-CoV-1 ORF8 hinders IRF-3 dimerization and induces IRF-3 proteasomal degradation,97 while ORF9 triggers the proteasomal degradation of MAVS, TRAF3, and TRAF6.98 SARS-CoV-1 ORF8 is 40%45,46 while ORF9b is 73%44–46 similar to SARS-CoV-2. This low homology could contribute to increased SARS-CoV-2 susceptibility to type I IFN when compared to SARS-CoV-1.99
Both nucleocapsid (N) and membrane (M) proteins of SARS-CoV-2 are similar to SARS-CoV-1 proteins by 90.5%.44–46 N protein counteracts IFN-I at different levels – inhibits RIG-I stabilization by blocking its ubiquitination, and inhibits NF-κB activation and IRF-3 phosphorylation.94,100,101 Further, viral M protein has also been reported to inhibit IFN-1 production by impeding the formation of TRAF3, TANK, TBK1/IKKε complex and subsequently inhibiting TBK1/IKKε-dependent activation of IRF-3/IRF-7 transcription factors.74
A very recent report has also identified the role of SARS-CoV-2 Nsp13, Nsp14, Nsp15 and ORF6 as potent IFN antagonists.89 Amongst the 27 SARS-CoV-2 proteins investigated in this study, ORF6 was revealed to be the strongest suppressor of primary IFN production and IFN signaling.
Severe and critically ill COVID-19 patients presented with a characteristic phenotype of impaired type I IFN production and activity, accompanied by downregulation of ISGs, compared to mild or moderate cases.102 The lack of a robust type I/III IFN response in SARS-CoV-2-infected cells suggests the likelihood of similar evasion mechanism as seen in SARS-CoV-1.67 Collectively, these evasion strategies could be responsible for the low levels of IFN-I/III response in in vitro and in vivo models of SARS-CoV-2 infection as well as in COVID-19 patients.67
Interferon Signaling in the Early and Late COVID-19 Phases
In addition to their role in establishing an effective antiviral defense, chronic type I IFN response can also be deleterious owing to its systemic, pro-inflammatory actions.103 It appears that rather than the complete absence of IFN response, the response may be delayed enabling the creation of sufficient viral titer levels. SARS-CoV-1 infection was found to be characterized by temporal and spatial variation in the activation of NF-κB, activator protein (AP)-1, and IRF-3/7 with time.32 Here, the expression of IFNs was delayed in comparison to that of pro-inflammatory cytokines. In fact, the kinetics of CoV replication and IFN signaling is believed to determine the severity of CoV infection. While an early IFN-I response can aid in controlling CoV infections, delayed signaling paves way for viral replication and disease progression. Therefore, in addition to the timing of IFN-I response, the associated viral titer levels in the tissue also play a dictating role in determining the severity of infection. A delayed IFN-1 response accompanied by robust viral replication orchestrated an inappropriate inflammatory response through the accumulation of pathogenic inflammatory monocyte-macrophages (IMMs) and subsequent lung immunopathology together with reduced survival in a SARS-CoV-1 infection model in mice.104 In this study, an early IFN-I response was seen to reduce viral titers and ameliorate immunopathology. It was also interesting to note that the depletion of IFN-α/β receptor (IFNAR) reduced the IMM response, and morbidity and mortality associated with SARS-CoV-1 infection. Similarly, in MERS-CoV infection, the timing of IFN-I response relative to viral replication appears to dictate the infection outcome in mouse models. While early IFN-I signaling is protective during MERS-CoV infection, late induction of IFN-I failed to inhibit viral replication resulting in fatal pneumonia and inflammatory cytokine storm.105
Early studies on SARS-CoV-2 also suggest an improved sensitivity to early IFN-I signaling. While SARS-CoV-2 exhibited similar viral replication kinetics to SARS-CoV-1 in vitro (Vero cells), it showed increased sensitivity to IFN-I pre-treatment relative to SARS-CoV-1.99,106 This could perhaps be explained by the lack of ORF3b, a key IFN antagonist, in SARS-CoV-2 as opposed to SARS-CoV-1. Type III IFN response was found to better control SARS-CoV-2 replication and spread compared to type I IFN.107 Nevertheless, the specific ISGs activated and those that control viral spread need further elucidation. Lymphocyte antigen 6 complex, locus E (LY6E), was found to impair the cellular fusion of multiple CoVs, including SARS-CoV-2, SARS-CoV-1, and MERS-CoV, by interfering with the S protein-mediated membrane fusion.108 This suggests that IFN-induced transmembrane (IFITM) protein family members, such as LY6E, could potentially restrict the entry of SARS-CoV-2 and their replication, as was previously shown in the case of SARS-CoV-1.109 However, IFITM proteins have also been implicated in promoting infection by other human CoVs, such as HCoV-OC43.110,111 Specific mutations in IFITM1 or IFITM3 were shown to convert them from an entry restriction factor to an entry facilitating factor and thus, aiding MERS-CoV or SARS-CoV S protein-mediated entry.111
At the same time, an early upregulation of IFN and other cytokines was found to promote severe respiratory stress and lethality in older individuals and aged mouse models of SARS.112 The acute phase of SARS-CoV infection is usually associated with an IFN-γ-related cytokine and chemokine storm causing immunopathological damage in SARS patients.113 Thus, although IFN-α and β reportedly inhibit in vitro viral replication and protects type I pneumocytes against SARS-CoV infection in macaques,114,115 IFN- γ appears to be a double-edged sword. This imbalance between IFN and pro-inflammatory response with age may also have important implications in COVID-19 pathogenesis.
The innate recognition of viral factors elicits the release of chemotactic factors that amplify the immune response against viral infection. Therefore, the spectral changes in these chemotactic factors during the course of infection may set in motion an unstable immune response. This immune misdirection may in turn favor viral replication and aggravate tissue damage. It is, therefore, of utmost importance to ascertain the specific molecular mechanisms employed by SARS-CoV-2 to evade immune surveillance to be able to successfully curb the COVID-19 infection.
Implications of Nucleic Acid-Sensing Pathway Modulation in the Treatment of COVID-19
The presence of reduced innate viral resistance with regards to low levels of IFN induction together with a robust pro-inflammatory chemokine and cytokine profile appears to define and drive key features of COVID-19 pathogenesis. After careful evaluation of the innate recognition of SARS-CoV-2 and its subsequent evasion, we rationalize treatment strategies currently under investigation as antivirals for COVID-19 and their clinical efficacy is carefully considered in this section.
Inhibition of NLRP3 Activation
Pyroptotic cell death and high level of IL-1β secretion are derived upon the activation of NLRP3 inflammasomes. Colchicine is a drug used clinically for gout and familial Mediterranean fever. Research has suggested its ability to inhibit NLRP3 inflammation non-selectively.116 Colchicine inhibits NLRP3 through inhibiting the activation of P2X7 receptor and thereby the interaction between ASC and NLRP3; more specifically, the transportation of ASC to NLRP3.117,118 The GRECCO-19 study investigated the use of colchicine to prevent COVID-19 associated complications, such as myocardial necrosis and pneumonia development (NCT04326790).119 This clinical trial recommended the use of colchicine as it significantly improved the time taken for clinical deterioration; however, its results should be interpreted with caution.120
Chloroquine and hydroxychloroquine are antimalarial drugs which also have anti-inflammatory activity. These drugs suppress NLRP3 inflammasomes by targeting the two signals of NLRP3 activation, namely priming followed by activation as discussed previously.121 Chloroquine was reported to inhibit SARS-CoV-1 effectively in in vitro studies.121,122 However, hydroxychloroquine showed more potency than chloroquine in inhibiting SARS-CoV-2.123 Nevertheless, the RECOVERY clinical trial investigating the use of hydroxychloroquine along with other repurposed drugs for the treatment of COVID-19 has halted its hydroxychloroquine intervention arm as no added benefit was observed compared to placebo in the hospitalized patients (NCT04381936).124 Therefore, using these drugs in COVID-19 treatment still stands controversial and further studies need to be conducted.125
Further, NLRP3 could be inhibited by select small molecules such as MCC950 and OLT1177 which also appear promising.126,127 Knocking out NLRP3 or using MCC950 to inhibit NLRP3 in a murine model of SARS-CoV-1 infection resulted in increased survival and decreased pulmonary edema and injury.57 OLT1177 also suppressed systemic inflammation in an LPS mouse model by preventing NLRP3 inflammasome formation and was found to be safe for use in humans.127 However, treating patients infected with SARS-CoV-2 with these drugs need further pre-clinical and clinical studies to ascertain their safety and efficacy.
There are many more investigational drugs targeting NLRP3 activation, such as Inzomelid and IFM-2427, which are currently being considered in the treatment of COVID-19 and these have been extensively reviewed in.128
Anti-IL-1β
Since IL-1β is one of the major cytokines in the hyperinflammatory cytokine storm associated with ARDS in COVID-19 patients, IL-1β-neutralizing antibody or recombinant IL-1 receptor antagonist could potentially be used as a COVID-19 treatment regimen.128 Canakinumab is a monoclonal antibody neutralizing IL-1β that was found to be a promising anti-inflammatory treatment in diseases, including lung cancer (NCT01327846), atherosclerotic disease (NCT01327846), and autoinflammatory recurrent fever syndromes (NCT02059291). Canakinumab when tested in a subgroup of COVID-19 patients with ARDS receiving hydroxychloroquine and lopinavir–ritonavir demonstrated a rapid anti-inflammatory response and improvement in oxygenation in addition to being safe and well tolerated when compared to those not receiving canakinumab.129
The recombinant antagonist of IL-1 receptor, anakinra, is used in the treatment of rheumatoid arthritis. Various clinical trials have indicated a reduction in systemic inflammatory response upon using anakinra in cases of sepsis and macrophage activating syndrome (MAS).130–132 There are currently two registered COVID-19 clinical studies using anakinra but the results are yet to be published (NCT04339712, NCT04330638). Notably, using these anti-inflammatory treatments in COVID-19 may weaken the cytokine storm post SARS-CoV-2 infection.
Type I and Type III Interferons (IFN-I/III)
The downregulation of interferons is a common evasion mechanism used by SARS-CoV-2, SARS-CoV-1 and MERS-CoV. Using recombinant IFNs is the treatment of choice to overcome this obstacle and it was extensively tested previously in SARS-CoV-1 and MERS-CoV. While recombinant IFN-α and IFN-β have been approved for therapy in diseases such as multiple sclerosis and viral hepatitis,133 recombinant IFN-γ is yet to be approved for any indication.
In vitro studies have concluded that IFN-β is the most effective in inhibiting SARS-CoV-1 replication comparing to IFN-α/γ.134,135 Also, combining IFN-α/β with the antiviral drug, ribavirin, is a promising regimen in SARS-CoV-1.136,137 A preliminary clinical study on combining IFN alfacon-1, a synthetic IFN-α, and corticosteroids in SARS patients resulted in rapid restoration of lung abnormalities, oxygen saturation and creatinine kinase levels at an early stage of infection.138 However, the use of this intervention at a later stage of infection was noted to be ineffective; thus, stressing the usage of this combination at an early stage of SARS only.
MERS-CoV replication was also found to be inhibited by IFN-α/β,139 particularly IFN-β was the strongest MERS-CoV replication inhibitor compared to other IFNs.140 It is interesting to note that MERS-CoV are more sensitive to IFN-α than SARS-CoV which may be explained by the absence of ORF6 in MERS-CoV.141 Using recombinant IFN-α/β in combination with antiviral drugs such as lopinavir/ritonavir, ribavirin and cyclosporin A were also shown to be effective against MERS-CoV.141–143 Two clinical studies on combining IFN-α and ribavirin in MERS patients reported the increase of overall survival at an early stage of the disease while the critically ill patients with ARDS who were diagnosed late did not benefit from this combination.144,145 Indeed, these findings stress on the usage of IFN with antiviral drugs at an early stage of MERS infection.
While two in vitro studies have demonstrated improved sensitivity of SARS-CoV-2 to IFN-1 with drastically reduced viral titers upon pre-treatment, in comparison with SARS-CoV-1,99,106 the results from the clinical studies are highly awaited to conclusively ascertain the safety and efficacy of IFN-I as a therapy in COVID-19. Several clinical trials are ongoing across the world investigating the use of IFN-1 either as a single agent or in combination therapy. The DisCoVeRy trial is examining interventions of IFN-β1A in combination with lopinavir-ritonavir, lopinavir-ritonavir alone, remdesivir alone, or hydroxychloroquine alone as a treatment for COVID-19 in hospitalized adults (NCT04315948). Another Phase II trial also studies inhaled IFN-β1A for COVID-19 treatment (NCT04385095).146 Potential experimental therapeutics IFN-α2b, arbidol or a combination of IFN-α2b plus arbidol was repurposed for COVID-19 patients, and IFN-α2b treatment significantly reduced the duration of both detectable viral levels as well as that of elevated blood levels of IL-6 and C-reactive protein (CRP).147 There are also two registered clinical trials using IFN-β in triple combination for the treatment of COVID-19. A Phase II, open-label, randomized trial on treatment of COVID-19 patients with IFN-β combines it with lopinavir/ritonavir and ribavirin (NCT04276688).148,149 This study reported the safety of the triple combination and it also shortened the hospital stay in mild-to-moderate patients. The other randomized controlled trial on moderate-to-severe COVID-19 patients treated them with a triple combination of IFN-β, lopinavir/ritonavir and hydroxychloroquine, but the results are yet to be published (NCT04343768).150 A third phase, open-label, non-randomized clinical trial on the use of IFN-α as prophylaxis in high-risk healthcare staff over 28 days reported a protective role on medical staff without any side effects (NCT04320238).151
Type III IFNs have also been widely studied in SARS and MERS, where it was found to be largely protective.152,153 Although type III IFNs are not yet approved for use in the clinic, IFN-λ provides an attractive intervention strategy in SARS-CoV-2 infection as well. SARS-CoV-2 infection of intestinal epithelial cells was found to be sensitive to IFN-III treatment.107 Another mouse model of SARS-CoV-2 infection also showed similar results and both prophylactic and therapeutic administration of IFN-λ1a effectively inhibited SARS-CoV-2 replication.154 Therefore, clinical studies are under way to investigate further the efficacy of IFN-λ1 in COVID-19 (NCT04343976, NCT04331899).
Type I IFN being more potent but inflammatory and type III IFN with their sustained action, we propose an intervention strategy that brings out the strengths of both IFN-I and III. Hence, treating COVID-19 patients with IFN appears to be a promising treatment option at an early stage of infection. IFN-1 therapy, in particular, is proposed to be effective as a prophylactic agent or an early treatment option for SARS-CoV-2.
Inhibition of EndoU Activity
The structural similarity of RNase A with EndoU presents with the opportunity to use small molecule inhibitors of RNase A to inhibit Nsp15 activity. In a study by Ortiz-Alcantara et al, some previously characterized RNase A inhibitors, such as Benzopurpurin B, C-467,929, C-473,872, and congo red showed efficacy in inhibiting the endoribonuclease activity of SARS-CoV-1 Nsp15.155 In silico analysis of FDA-approved compounds targeting EndoU suggested clinically available drugs, Glisoxepide and Idarubicin, that are currently used for treating diabetes and leukemia, respectively, as strong candidates for repurposed COVID-19 drugs due to their strong affinity to bind EndoU.156
Conclusions
The unprecedented spread of SARS-CoV-2 and the global impact of COVID-19 have urged the scientific community to accelerate their basic science and clinical research on this pandemic. The wealth of scientific data generated have led to remarkable improvements in our understanding of COVID-19 pathogenesis. Being a relatively nascent field, references to related CoVs, particularly SARS-CoV-1 and MERS-CoV, have also greatly helped in this regard.
Although SARS-CoV-2 does share a lot of similarities with SARS-CoV-1 and MERS-CoV, it is far more pathogenic than its predecessors and also, elicit differential host immune responses in certain regards. For instance, the asymptomatic nature of a significant proportion of those infected with SARS-CoV-2 suggests rapid transmission rates and longer incubation period, thereby adding to the challenge of controlling the COVID-19 spread in the community. It is, therefore, of utmost importance to develop a holistic understanding of the immune responses elicited by SARS-CoV-2 to be able to successfully develop therapeutic modalities against COVID-19. Here, we have reviewed the recent literature and made educated guesses where applicable from related CoVs on the mechanisms entailing innate sensing of SARS-CoV-2, the viral escape from innate detection, the role of IFN signaling over the course of infection and lastly, addressed some of the currently being investigated therapeutic strategies modulating these sensing pathways to control COVID-19 infection. COVID-19 still poses a lot of unanswered questions with respect to its differential pathogenesis across patients as well as its differences with other related CoVs, necessitating further studies to be able to timely meet the need for the development of an effective antiviral therapeutic or vaccine to curb this global threat.
Acknowledgments
This research has been financially supported by Tissue Injury and Repair (TIR) group operational grant (Grant code: 150317); COVID-19 research grant (CoV19-0307); Seed grant (Grant code: 2001090275); and by collaborative research grant (Grant code: 2001090278) to RH, University of Sharjah, UAE; and by a Sandooq Al Watan Applied Research & Development grant to RH (SWARD-S20-007); and by Al Jalila Foundation Seed Grant (AJF202019).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1–23.
2. Ciotti M, Angeletti S, Minieri M, et al. COVID-19 outbreak: an overview. Chemotherapy. 2019;64(5–6):215–223. doi:10.1159/000507423
3. Zhong NS, Zheng BJ, Li YM, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet. 2003;362(9393):1353–1358. doi:10.1016/S0140-6736(03)14630-2
4. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–1820. doi:10.1056/NEJMoa1211721
5. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733. doi:10.1056/NEJMoa2001017
6. WHO. WHO coronavirus disease (COVID-19) dashboard. Available from: https://covid19.who.int/?gclid=CjwKCAjwx9_4BRAHEiwApAt0ztGCqDKOG3tvC1Gxgs-X7bgQVwEFSi96HD50ZZQUndU-KIrvQa5HmRoCmYgQAvD_BwE.
7. Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol. 2003;77(16):8801–8811.
8. Luk HKH, Li X, Fung J, Lau SKP, Woo PCY. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infect Genet Evol. 2019;71:21–30. doi:10.1016/j.meegid.2019.03.001
9. de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14(8):523–534. doi:10.1038/nrmicro.2016.81
10. Brian DA, Baric RS. Coronavirus genome structure and replication. Curr Top Microbiol Immunol. 2005;287:1–30. doi:10.1007/3-540-26765-4_1
11. Li C, Yang Y, Ren L. Genetic evolution analysis of 2019 novel coronavirus and coronavirus from other species. Infect Genet Evol. 2020;82:104285. doi:10.1016/j.meegid.2020.104285
12. Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi:10.1038/nature02145
13. Qian Z, Travanty EA, Oko L, et al. Innate immune response of human alveolar type II cells infected with severe acute respiratory syndrome-coronavirus. Am J Respir Cell Mol Biol. 2013;48(6):742–748. doi:10.1165/rcmb.2012-0339OC
14. Raj VS, Mou H, Smits SL, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495(7440):251–254. doi:10.1038/nature12005
15. Lu G, Hu Y, Wang Q, et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature. 2013;500(7461):227–231. doi:10.1038/nature12328
16. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020;92(10):2105–2113. doi:10.1002/jmv.25987
17. Neuman BW, Kiss G, Kunding AH, et al. A structural analysis of M protein in coronavirus assembly and morphology. J Struct Biol. 2011;174(1):11–22. doi:10.1016/j.jsb.2010.11.021
18. Kindler E, Thiel V, Weber F. Interaction of SARS and MERS coronaviruses with the antiviral interferon response. Adv Virus Res. 2016;96:219–243.
19. Li G, Fan Y, Lai Y, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92(4):424–432. doi:10.1002/jmv.25685
20. Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 2012;86(6):2900–2910. doi:10.1128/JVI.05738-11
21. Barber GN. Cytoplasmic DNA innate immune pathways. Immunol Rev. 2011;243(1):99–108. doi:10.1111/j.1600-065X.2011.01051.x
22. Tan X, Sun L, Chen J, Chen ZJ. Detection of microbial infections through innate immune sensing of nucleic acids. Annu Rev Microbiol. 2018;72(1):447–478. doi:10.1146/annurev-micro-102215-095605
23. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32(1):513–545. doi:10.1146/annurev-immunol-032713-120231
24. Kell AM, Gale M
25. Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227(1):54–65. doi:10.1111/j.1600-065X.2008.00727.x
26. Binder M, Eberle F, Seitz S, et al. Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J Biol Chem. 2011;286(31):27278–27287. doi:10.1074/jbc.M111.256974
27. Goubau D, Schlee M, Deddouche S, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5ʹ-diphosphates. Nature. 2014;514(7522):372–375. doi:10.1038/nature13590
28. Pichlmair A, Schulz O, Tan CP, et al. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol. 2009;83(20):10761–10769. doi:10.1128/JVI.00770-09
29. Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20(1):17–22. doi:10.1016/j.coi.2008.01.002
30. Belgnaoui SM, Paz S, Hiscott J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr Opin Immunol. 2011;23(5):564–572. doi:10.1016/j.coi.2011.08.001
31. Liu S, Chen J, Cai X, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife. 2013;2:e00785. doi:10.7554/eLife.00785
32. Yoshikawa T, Hill TE, Yoshikawa N, et al. Dynamic innate immune responses of human bronchial epithelial cells to severe acute respiratory syndrome-associated coronavirus infection. PLoS One. 2010;5(1):e8729. doi:10.1371/journal.pone.0008729
33. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi:10.1016/j.cell.2006.02.015
34. Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. 2020;180(6):1044–1066. doi:10.1016/j.cell.2020.02.041
35. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11(5):373–384. doi:10.1038/ni.1863
36. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature. 2001;413(6857):732–738. doi:10.1038/35099560
37. Totura AL, Whitmore A, Agnihothram S, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. 2015;6(3):e00638–15. doi:10.1128/mBio.00638-15
38. Zhao J, Li K, Wohlford-Lenane C, et al. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc Natl Acad Sci U S A. 2014;111(13):4970–4975. doi:10.1073/pnas.1323279111
39. Cervantes-Barragan L, Zust R, Weber F, et al. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. 2007;109(3):1131–1137. doi:10.1182/blood-2006-05-023770
40. van der Made CI, Simons A, Schuurs-Hoeijmakers J, et al. Presence of genetic variants among young men with severe COVID-19. JAMA. 2020;324(7):663. doi:10.1001/jama.2020.13719
41. Sheahan T, Morrison TE, Funkhouser W, et al. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog. 2008;4(12):e1000240. doi:10.1371/journal.ppat.1000240
42. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi:10.1038/s41577-019-0165-0
43. Zhao C, Zhao W. NLRP3 inflammasome-a key player in antiviral responses. Front Immunol. 2020;11:211. doi:10.3389/fimmu.2020.00211
44. Chan JF, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221–236. doi:10.1128/JVI.77.16.8801-8811.2003
45. Grifoni A, Sidney J, Zhang Y, Scheuermann RH, Peters B, Sette A. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe. 2020;27(4):671–80e2. doi:10.1016/j.chom.2020.03.002
46. Xu J, Zhao S, Teng T, et al. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses. 2020;12(2):244. doi:10.3390/v12020244
47. Nieto-Torres JL, DeDiego ML, Verdia-Baguena C, et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014;10(5):e1004077. doi:10.1371/journal.ppat.1004077
48. Nieto-Torres JL, Verdia-Baguena C, Jimenez-Guardeno JM, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–339. doi:10.1016/j.virol.2015.08.010
49. Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019;10:50. doi:10.3389/fmicb.2019.00050
50. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
51. Siu KL, Yuen KS, Castano-Rodriguez C, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33(8):8865–8877. doi:10.1096/fj.201802418R
52. Yue Y, Nabar NR, Shi CS, et al. SARS-coronavirus open reading frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018;9(9):904. doi:10.1038/s41419-018-0917-y
53. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021. doi:10.1016/j.tibs.2016.09.002
54. Shi CS, Nabar NR, Huang NN, Kehrl JH. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019;5(1):101. doi:10.1038/s41420-019-0181-7
55. Neuman BW, Joseph JS, Saikatendu KS, et al. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J Virol. 2008;82(11):5279–5294. doi:10.1128/JVI.02631-07
56. Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10(5):766–788. doi:10.1016/j.apsb.2020.02.008
57. Chang YS, Ko BH, Ju JC, et al. (SUD) of severe acute respiratory syndrome coronavirus induces NLRP3 inflammasome-dependent CXCL10-mediated pulmonary inflammation. Int J Mol Sci. 2020;21:9.
58. Jiang Y, Li J, Teng Y, et al. Complement receptor C5aR1 inhibition reduces pyroptosis in hDPP4-transgenic mice infected with MERS-CoV. Viruses. 2019;11(1):39. doi:10.3390/v11010039
59. Jiang Y, Zhao G, Song N, et al. Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg Microbes Infect. 2018;7(1):77. doi:10.1038/s41426-018-0063-8
60. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–770. doi:10.1038/nri3921
61. Lee J, Ghonime MG, Wang R, Cassady KA. The antiviral apparatus: STING and oncolytic virus restriction. Mol Ther Oncolytics. 2019;13:7–13. doi:10.1016/j.omto.2019.02.002
62. Sun B, Sundstrom KB, Chew JJ, et al. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci Rep. 2017;7(1):3594. doi:10.1038/s41598-017-03932-1
63. Zevini A, Olagnier D, Hiscott J. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol. 2017;38(3):194–205. doi:10.1016/j.it.2016.12.004
64. Franz KM, Neidermyer WJ, Tan YJ, Whelan SPJ, Kagan JC. STING-dependent translation inhibition restricts RNA virus replication. Proc Natl Acad Sci U S A. 2018;115(9):E2058–E67. doi:10.1073/pnas.1716937115
65. Rose KM, Elliott R, Martinez-Sobrido L, Garcia-Sastre A, Weiss SR. Murine coronavirus delays expression of a subset of interferon-stimulated genes. J Virol. 2010;84(11):5656–5669. doi:10.1128/JVI.00211-10
66. Menachery VD, Eisfeld AJ, Schafer A, et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio. 2014;5(3):e01174–14.
67. Blanco-Melo D, Nilsson-Payant BE, Liu WC, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181(5):1036–45e9.
68. Wu F, Wang A, Liu M, Wang Q, Chen J, Xia S, et al. Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications. medRxiv. 2020. doi:10.1101/2020.03.30.20047365
69. Snijder EJ, van der Meer Y, Zevenhoven-Dobbe J, et al. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J Virol. 2006;80(12):5927–5940.
70. Knoops K, Kikkert M, Worm SH, et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6(9):e226. doi:10.1371/journal.pbio.0060226
71. Deng X, Hackbart M, Mettelman RC, et al. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc Natl Acad Sci U S A. 2017;114(21):E4251–E60. doi:10.1073/pnas.1618310114
72. Cameron MJ, Kelvin AA, Leon AJ, et al. Lack of innate interferon responses during SARS coronavirus infection in a vaccination and reinfection ferret model. PLoS One. 2012;7(9):e45842. doi:10.1371/journal.pone.0045842
73. Minakshi R, Padhan K, Rani M, Khan N, Ahmad F, Jameel S. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One. 2009;4(12):e8342. doi:10.1371/journal.pone.0008342
74. Siu KL, Kok KH, Ng MH, et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem. 2009;284(24):16202–16209. doi:10.1074/jbc.M109.008227
75. Wathelet MG, Orr M, Frieman MB, Baric RS. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol. 2007;81(21):11620–11633. doi:10.1128/JVI.00702-07
76. Huang C, Lokugamage KG, Rozovics JM, Narayanan K, Semler BL, Makino S. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. 2011;7(12):e1002433. doi:10.1371/journal.ppat.1002433
77. Kamitani W, Huang C, Narayanan K, Lokugamage KG, Makino S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat Struct Mol Biol. 2009;16(11):1134–1140. doi:10.1038/nsmb.1680
78. Kamitani W, Narayanan K, Huang C, et al. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc Natl Acad Sci U S A. 2006;103(34):12885–12890. doi:10.1073/pnas.0603144103
79. Lokugamage KG, Narayanan K, Huang C, Makino S. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J Virol. 2012;86(24):13598–13608. doi:10.1128/JVI.01958-12
80. Narayanan K, Huang C, Lokugamage K, et al. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J Virol. 2008;82(9):4471–4479. doi:10.1128/JVI.02472-07
81. Tanaka T, Kamitani W, DeDiego ML, Enjuanes L, Matsuura Y. Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J Virol. 2012;86(20):11128–11137. doi:10.1128/JVI.01700-12
82. Clementz MA, Chen Z, Banach BS, et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J Virol. 2010;84(9):4619–4629. doi:10.1128/JVI.02406-09
83. Devaraj SG, Wang N, Chen Z, et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem. 2007;282(44):32208–32221. doi:10.1074/jbc.M704870200
84. Matthews K, Schafer A, Pham A, Frieman M. The SARS coronavirus papain like protease can inhibit IRF3 at a post activation step that requires deubiquitination activity. Virol J. 2014;11(1):209. doi:10.1186/s12985-014-0209-9
85. Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J Virol. 2009;83(13):6689–6705. doi:10.1128/JVI.02220-08
86. Chen X, Yang X, Zheng Y, Yang Y, Xing Y, Chen Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell. 2014;5(5):369–381. doi:10.1007/s13238-014-0026-3
87. Sun L, Xing Y, Chen X, et al. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One. 2012;7(2):e30802. doi:10.1371/journal.pone.0030802
88. Yang X, Chen X, Bian G, et al. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J Gen Virol. 2014;95(Pt 3):614–626. doi:10.1099/vir.0.059014-0
89. Yuen CK, Lam JY, Wong WM, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect. 2020;9(1):1418–1428. doi:10.1080/22221751.2020.1780953
90. Deng X, Baker SC. An “old” protein with a new story: coronavirus endoribonuclease is important for evading host antiviral defenses. Virology. 2018;517:157–163. doi:10.1016/j.virol.2017.12.024
91. Deng X, van Geelen A, Buckley AC, et al. Coronavirus endoribonuclease activity in porcine epidemic diarrhea virus suppresses type i and type III interferon responses. J Virol. 2019;93(8):8. doi:10.1128/JVI.02000-18
92. Hackbart M, Deng X, Baker SC. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc Natl Acad Sci U S A. 2020;117(14):8094–8103. doi:10.1073/pnas.1921485117
93. Zust R, Cervantes-Barragan L, Habjan M, et al. Ribose 2ʹ-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol. 2011;12(2):137–143. doi:10.1038/ni.1979
94. Kopecky-Bromberg SA, Martinez-Sobrido L, Frieman M, Baric RA, Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81(2):548–557. doi:10.1128/JVI.01782-06
95. Konno Y, Kimura I, Uriu K, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is further increased by a naturally occurring elongation variant. bioRxiv. 2020;
96. Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol. 2007;81(18):9812–9824. doi:10.1128/JVI.01012-07
97. Wong HH, Fung TS, Fang S, Huang M, Le MT, Liu DX. Accessory proteins 8b and 8ab of severe acute respiratory syndrome coronavirus suppress the interferon signaling pathway by mediating ubiquitin-dependent rapid degradation of interferon regulatory factor 3. Virology. 2018;515:165–175. doi:10.1016/j.virol.2017.12.028
98. Shi CS, Qi HY, Boularan C, et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol. 2014;193(6):3080–3089. doi:10.4049/jimmunol.1303196
99. Lokugamage KG, Hage A, Schindewolf C, Rajsbaum R. SARS-CoV-2 is sensitive to type I interferon pretreatment. bioRxiv. 2020.
100. Lu X, Pan J, Tao J, Guo D. SARS-CoV nucleocapsid protein antagonizes IFN-beta response by targeting initial step of IFN-beta induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes. 2011;42(1):37–45. doi:10.1007/s11262-010-0544-x
101. Hu Y, Li W, Gao T, et al. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type i interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J Virol. 2017;91(8):8. doi:10.1128/JVI.02143-16
102. Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and exacerbated inflammatory responses in severe Covid-19 patients. medRxiv. 2020;
103. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49. doi:10.1038/nri3581
104. Channappanavar R, Fehr AR, Vijay R, et al. Dysregulated type i interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19(2):181–193. doi:10.1016/j.chom.2016.01.007
105. Channappanavar R, Fehr AR, Zheng J, et al. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. 2019;129(9):3625–3639. doi:10.1172/JCI126363
106. Mantlo E, Bukreyeva N, Maruyama J, Paessler S, Huang C. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antiviral Res. 2020;179:104811. doi:10.1016/j.antiviral.2020.104811
107. Stanifer ML, Kee C, Cortese M. et al. Critical role of type III interferon in controlling SARS-CoV-2 infection, replication and spread in primary human intestinal epithelial cells. bioRxiv. 2020;
108. Pfaender S, Mar KB, Michailidis E. et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. bioRxiv. 2020;
109. Huang IC, Bailey CC, Weyer JL, et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011;7(1):e1001258. doi:10.1371/journal.ppat.1001258
110. Zhao X, Guo F, Liu F, et al. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc Natl Acad Sci U S A. 2014;111(18):6756–6761. doi:10.1073/pnas.1320856111
111. Zhao X, Sehgal M, Hou Z, et al. Identification of residues controlling restriction versus enhancing activities of IFITM proteins on entry of human coronaviruses. J Virol. 2018;92:6.
112. Rockx B, Baas T, Zornetzer GA, et al. Early upregulation of acute respiratory distress syndrome-associated cytokines promotes lethal disease in an aged-mouse model of severe acute respiratory syndrome coronavirus infection. J Virol. 2009;83(14):7062–7074. doi:10.1128/JVI.00127-09
113. Huang KJ, Su IJ, Theron M, et al. An interferon-gamma-related cytokine storm in SARS patients. J Med Virol. 2005;75(2):185–194. doi:10.1002/jmv.20255
114. Haagmans BL, Kuiken T, Martina BE, et al. Pegylated interferon-alpha protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat Med. 2004;10(3):290–293. doi:10.1038/nm1001
115. Tan EL, Ooi EE, Lin CY, Tan HC, Ling AE, Lim B. Inhibition of SARS coronavirus infection in vitro with clinically approved antiviral drugs. Emerg Infect Dis. 2004;10(4):581–586. doi:10.3201/eid1004.030458
116. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15(4):203–214. doi:10.1038/nrcardio.2017.161
117. Misawa T, Takahama M, Kozaki T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14(5):454–460. doi:10.1038/ni.2550
118. Marques-da-Silva C, Chaves MM, Castro NG, Coutinho-Silva R, Guimaraes MZ. Colchicine inhibits cationic dye uptake induced by ATP in P2X2 and P2X7 receptor-expressing cells: implications for its therapeutic action. Br J Pharmacol. 2011;163(5):912–926. doi:10.1111/j.1476-5381.2011.01254.x
119. Deftereos SG, Siasos G, Giannopoulos G, et al. The Greek study in the effects of colchicine in Covid-19 complications prevention (GRECCO-19 study): rationale and study design. Hellenic J Cardiol. 2020;61(1):42–45. doi:10.1016/j.hjc.2020.03.002
120. Deftereos SG, Giannopoulos G, Vrachatis DA, et al. Effect of colchicine vs standard care on cardiac and inflammatory biomarkers and clinical outcomes in patients hospitalized with coronavirus disease 2019: the GRECCO-19 randomized clinical trial. JAMA Netw Open. 2020;3(6):e2013136. doi:10.1001/jamanetworkopen.2020.13136
121. Chen X, Wang N, Zhu Y, Lu Y, Liu X, Zheng J. The antimalarial chloroquine suppresses LPS-induced NLRP3 inflammasome activation and confers protection against murine endotoxic shock. Mediators Inflamm. 2017;2017:6543237. doi:10.1155/2017/6543237
122. Vincent MJ, Bergeron E, Benjannet S, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. 2005;2(1):69. doi:10.1186/1743-422X-2-69
123. Yao X, Ye F, Zhang M, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis. 2020;71(15):732–739. doi:10.1093/cid/ciaa237
124. Jp K. NIH halts clinical trial of hydroxychloroquine. Available from: https://www.nih.gov/news-events/news-releases/nih-halts-clinical-trial-hydroxychloroquine. 2020.
125. Lucchesi A, Silimbani P, Musuraca G, et al. Clinical and biological data on the use of hydroxychloroquine against SARS-CoV-2 could support the role of the NLRP3 inflammasome in the pathogenesis of respiratory disease. J Med Virol. 2020. doi:10.1002/jmv.26217
126. Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. doi:10.1038/nm.3806
127. Marchetti C, Swartzwelter B, Gamboni F, et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U S A. 2018;115(7):E1530–E9. doi:10.1073/pnas.1716095115
128. Freeman TL, Swartz TH. Targeting the NLRP3 inflammasome in severe COVID-19. Front Immunol. 2020;11:1518. doi:10.3389/fimmu.2020.01518
129. Ucciferri C, Auricchio A, Di Nicola M, et al. Canakinumab in a subgroup of patients with COVID-19. Lancet Rheumatol. 2020;2(8):e457–ee8. doi:10.1016/S2665-9913(20)30167-3
130. Rajasekaran S, Kruse K, Kovey K, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children. Pediatr Crit Care Med. 2014;15(5):401–408. doi:10.1097/PCC.0000000000000078
131. Shakoory B, Carcillo JA, Chatham WW, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med. 2016;44(2):275–281. doi:10.1097/CCM.0000000000001402
132. Lind-Holst M, Hartling UB, Christensen AE. High-dose anakinra as treatment for macrophage activation syndrome caused by refractory Kawasaki disease in an infant. BMJ Case Rep. 2019;12(8):8. doi:10.1136/bcr-2019-229708
133. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity. 2019;50(4):907–923. doi:10.1016/j.immuni.2019.03.025
134. Cinatl J, Morgenstern B, Bauer G, Chandra P, Rabenau H, Doerr HW. Treatment of SARS with human interferons. Lancet. 2003;362(9380):293–294. doi:10.1016/S0140-6736(03)13973-6
135. Spiegel M, Pichlmair A, Muhlberger E, Haller O, Weber F. The antiviral effect of interferon-beta against SARS-coronavirus is not mediated by MxA protein. J Clin Virol. 2004;30(3):211–213. doi:10.1016/j.jcv.2003.11.013
136. Morgenstern B, Michaelis M, Baer PC, Doerr HW, Cinatl J
137. Stroher U, DiCaro A, Li Y, et al. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon-alpha. J Infect Dis. 2004;189(7):1164–1167. doi:10.1086/382597
138. Loutfy MR, Blatt LM, Siminovitch KA, et al. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. JAMA. 2003;290(24):3222–3228. doi:10.1001/jama.290.24.3222
139. Zielecki F, Weber M, Eickmann M, et al. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J Virol. 2013;87(9):5300–5304. doi:10.1128/JVI.03496-12
140. Hart BJ, Dyall J, Postnikova E, et al. Interferon-beta and mycophenolic acid are potent inhibitors of Middle East respiratory syndrome coronavirus in cell-based assays. J Gen Virol. 2014;95(Pt 3):571–577. doi:10.1099/vir.0.061911-0
141. de Wilde AH, Raj VS, Oudshoorn D, et al. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J Gen Virol. 2013;94(Pt 8):1749–1760. doi:10.1099/vir.0.052910-0
142. Chan JF, Yao Y, Yeung ML, et al. Treatment with lopinavir/ritonavir or interferon-beta1b improves outcome of MERS-CoV infection in a nonhuman primate model of common marmoset. J Infect Dis. 2015;212(12):1904–1913. doi:10.1093/infdis/jiv392
143. Falzarano D, de Wit E, Rasmussen AL, et al. Treatment with interferon-alpha2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nat Med. 2013;19(10):1313–1317. doi:10.1038/nm.3362
144. Omrani AS, Saad MM, Baig K, et al. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect Dis. 2014;14(11):1090–1095. doi:10.1016/S1473-3099(14)70920-X
145. Al-Tawfiq JA, Momattin H, Dib J, Memish ZA. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. Int J Infect Dis. 2014;20:42–46. doi:10.1016/j.ijid.2013.12.003
146. Sallard E, Lescure FX, Yazdanpanah Y, Mentre F, Peiffer-Smadja N. Type 1 interferons as a potential treatment against COVID-19. Antiviral Res. 2020;178:104791. doi:10.1016/j.antiviral.2020.104791
147. Zhou Q, Wei X-S, Xiang X, et al. Interferon-a2b treatment for COVID-19. medRxiv. 2020;
148. Hung IF, Lung KC, Tso EY, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, Phase 2 trial. Lancet. 2020;395(10238):1695–1704. doi:10.1016/S0140-6736(20)31042-4
149. Shalhoub S. Interferon beta-1b for COVID-19. Lancet. 2020;395(10238):1670–1671. doi:10.1016/S0140-6736(20)31101-6
150. Irvani SSN, Golmohammadi M, Pourhoseingholi MA, Shokouhi S, Darazam IA. Effectiveness of interferon beta 1a, compared to interferon beta 1b and the usual therapeutic regimen to treat adults with moderate to severe COVID-19: structured summary of a study protocol for a randomized controlled trial. Trials. 2020;21(1):473. doi:10.1186/s13063-020-04382-3
151. Acharya D, Liu G, Gack MU. Dysregulation of type I interferon responses in COVID-19. Nat Rev Immunol. 2020;20(7):397–398. doi:10.1038/s41577-020-0346-x
152. Mordstein M, Neugebauer E, Ditt V, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol. 2010;84(11):5670–5677. doi:10.1128/JVI.00272-10
153. Jeon YJ, Gil CH, Jo A, Won J, Kim S, Kim HJ. The influence of interferon-lambda on restricting Middle East respiratory syndrome coronavirus replication in the respiratory epithelium. Antiviral Res. 2020;180:104860. doi:10.1016/j.antiviral.2020.104860
154. Dinnon KH, Leist SR, Schäfer A, et al. A mouse-adapted SARS-CoV-2 model for the evaluation of COVID-19 medical countermeasures. bioRxiv. 2020;
155. Ortiz-Alcantara JBK, Palaninathan S, Frieman M, Baric R, Kao C. Small molecule inhibitors of the SARS-CoV Nsp15 endoribonuclease. Virus Adapt Treat. 2010;2:125–133.
156. Chandra A, Gurjar V, Qamar I, Singh N. Identification of potential inhibitors of SARS-COV-2 endoribonuclease (EndoU) from FDA approved drugs: a drug repurposing approach to find therapeutics for COVID-19. J Biomol Struct Dyn. 2020;1–11.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.