Back to Journals » International Journal of Nanomedicine » Volume 15
Nucleic Acid Delivery with α-Tocopherol-Polyethyleneimine-Polyethylene Glycol Nanocarrier System
Authors Hossian AKMN ![]() , Jois SD, Jonnalagadda SC
, Jois SD, Jonnalagadda SC ![]() , Mattheolabakis G
, Mattheolabakis G
Received 29 April 2020
Accepted for publication 30 July 2020
Published 11 September 2020 Volume 2020:15 Pages 6689—6703
DOI https://doi.org/10.2147/IJN.S259724
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
A K M Nawshad Hossian,1 Seetharama D Jois,1 Subash C Jonnalagadda,2 George Mattheolabakis1
1School of Basic Pharmaceutical and Toxicological Sciences, College of Pharmacy, University of Louisiana Monroe, Monroe, LA, USA; 2Department of Chemistry and Biochemistry, Rowan University, Glassboro, NJ, USA
Correspondence: George Mattheolabakis
School of Basic Pharmaceutical and Toxicological Sciences, College of Pharmacy, University of Louisiana Monroe, 1800 Bienville Dr, Monroe, LA 71201, USA
Tel +1 318 342-7930
Email [email protected]
Purpose: Nucleic acid-based therapies are a promising therapeutic tool. The major obstacle in their clinical translation is their efficient delivery to the desired tissue. We developed a novel nanosized delivery system composed of conjugates of α-tocopherol, polyethyleneimine, and polyethylene glycol (TPP) to deliver nucleic acids.
Methods: We synthesized a panel of TPP molecules using different molecular weights of PEG and PEI and analyzed with various analytical approaches. The optimized version of TPP (TPP111 - the 1:1:1 molecular ratio) was self-assembled in water to produce nanostructures and then evaluated in diversified in vitro and in vivo studies.
Results: Through a panel of synthesized molecules, TPP111 conjugate components self-assembled in water, forming globular shaped nanostructures of ∼ 90 nm, with high nucleic acid entrapment efficiency. The polymer had low cytotoxicity in vitro and protected nucleic acids from nucleases. Using a luciferase-expressing plasmid, TPP111-plasmid nano-complexes were rapidly up-taken by cancer cells in vitro and induced strong transfection, comparable to PEI. Colocalization of the nano-complexes and endosomes/lysosomes suggested an endosome-mediated uptake. Using a subcutaneous tumor model, intravenously injected nano-complexes preferentially accumulated to the tumor area over 24 h.
Conclusion: These results indicate that we successfully synthesized the TPP111 nanocarrier system, which can deliver nucleic acids in vitro and in vivo and merits further evaluation.
Keywords: nanoparticles, gene delivery, plasmid, tocopherol, polyethyleneimine, transfection
Introduction
Advances in our understanding of nucleic acid constructs and their respective activities have allowed for the identification, development, and utilization of an entire novel group of therapeutic moieties.1 Nucleic acid delivery poses significant challenges. Briefly, nucleic acids are high-molecular-weight molecules, hydrophilic and negatively charged, which are unable to cross the negatively charged, lipophilic-bilayered cell membrane. Furthermore, unprotected nucleic acids are rapidly degraded by nucleases in the circulation in vivo.2
Not surprisingly, intensive work takes place on the development of polymer-based systems for the cellular delivery of nucleic acids, such as plasmids, siRNAs, and miRNAs. Efficient in vitro transfection and favorable in vivo biodistribution to the tumor area remains an elusive goal for nucleic acid delivery. In fact, in recent years, there has been an increasing complexity on the molecular structures of the potential nucleic acid delivery carriers, which may not necessarily translate to improved therapeutic benefits.
α-Tocopherol is the predominant form of the Vitamin E family of molecules, which consists of four tocopherols and four tocotrienols.3 Early on since its discovery, this highly hydrophobic molecule was established as an antioxidant, free radical scavenger, protecting organisms against oxidative damage.3 Due to its biocompatibility and apparent lack of toxicity, α-tocopherol has been widely used successfully in drug formulations. A prominent example is the α-tocopherol-Polyethylene glycol molecule, also referred to as TPGS (D-ɑ-tocopheryl polyethylene glycol succinate). Its amphiphilic structure has been widely used in wetting, solubilizing, and emulsifying hydrophobic molecules, as well as an alternative source of the fat-soluble Vitamin E.4 In addition, this simple Vitamin E derivative can significantly improve the pharmacokinetics, biodistribution, and efficacy of active compounds following intravenous administration, such as paclitaxel,5 curcumin, gemcitabine,6 and other.
PEI has consistently demonstrated a strong capacity to transfect cells in vitro and in vivo. This positive-charged polymer complexes with the negatively charged nucleic acids, forming polyplexes.7 The PEI-nucleic acid complexes enter the cells through endocytosis and release the nucleic acids through the “proton-sponge” mechanism.8
In this study, we developed Vitamin E derivatives conjugated with polyethyleneimine (PEI) and polyethylene glycol (PEG) for the delivery of nucleic acids (Figure 1). We capitalize on the promising simplicity and behavior of the TPGS molecule, to develop a simple Vitamin E derivative capable of harboring both hydrophobic compounds and nucleic acids while protecting against DNA/RNA-se degradation and with improved pharmacokinetics. The entrapment of nucleic acids inside the PEI corona will accommodate their protection from degradation and their endosomal escape through the proton-sponge effect,2 while the hydrophilic neutral PEG corona will facilitate the carrier’s prolonged systemic circulation, and accumulation to the tumor area through the enhanced permeability and retention effect.9
 |
Figure 1 Schematic representation of TPP synthesis process. α-tocopherol succinate was synthesized by reacting succinic anhydride and (±) α-tocopherol. mPEG and PEI were conjugated using a diisocyanate crosslinker. Following purification, α-tocopherol succinate was activated using EDC/NHS and reacted with PEI-PEG, followed by purification through dialysis. |
In this part of our work, we describe the synthesis and characterization of the Vitamin E derivatives, annotated at TPP polymers, aiming for the development of nanocarriers for intravenous administration of nucleic acids with the smallest nano-sized dimensions, and with excellent nucleic acid complexation and protection from enzymatic degradation. In our study, we used the plasmid pGL-3 (Promega, Madison, WI) as a model nucleic acid.
Materials and Methods
Materials
Cell culture reagents were purchased from GibcoTM (Life technologies, Carlsbad, CA) and VWR. Opti-MEM, and Lipofectamine 2000 reagent were purchased from ThermoFisher. Bovine Serum Albumin (BSA) was obtained by Atlanta Biologicals. Luciferin, solvents and other chemicals and kits were all of analytical grade, obtained from Fisher or Sigma.
Cell Cultures
A549 cell line was cultured in DMEM/F12K media and supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin, whereas, H358 cell line was cultured in RPMI media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Both cell lines were maintained at 37 ºC with 5% CO2 supply in humidified conditions. The cell lines were purchased from ATCC.
Plasmid DNA
Plasmid pGL-3 is a luciferase reporter vector that contains the modified coding region for firefly luciferase, which we obtained from Promega (Madison, WI). The plasmids were amplified in competent Escherichia coli K strain (JM109-Promega) and purified using a plasmid extraction kit (QIAGEN-Chatsworth, Calif).
Synthesis of α-Tocopherol-Polyethyleneimine-Polyethylene Glycol (TPP) Polymers
We synthesized (±)-α-tocopherol succinate by reacting succinic anhydride with (±)-α-tocopherol, as previously described.10 Briefly, 4.3 g (10.0 mmol) of (±) α-tocopherol reacted with 1.50 g (15.0 mmol) of succinate anhydride in 20 mL of Toluene. 0.35 mL (2.5 mmol) of Triethylamine was added in the reaction with continuous stirring at 22℃. Then, the reaction was continued for 5 h at 60℃, under reflux. We extracted the reaction mixture with CH2Cl2, and washed with water, 1 N HCl and again with water, finally dried with Na2SO4. We obtained a yellow viscous liquid after concentrating in a rotary evaporator. The reaction mixture was further purified with flash chromatography using 10–30% of EtOAc/Hexane. We obtained ~5 g of a white solid, (±) α-tocopherol succinate, after drying.
We conjugated mPEG and PEI of varying molecular weights with diisocyanate linker, as previously described.11 Briefly, we dissolved 4 mmol of mPEG in 10 mL of dichloromethane (DCM), and added 12 mmol of HMDI, under stirring. The reaction was continued for 8 h at 50 ºC under reflux, followed by dropwise precipitation in ice-cold petroleum ether (3x in 250 mL). We collected the precipitate and dried it under reduced pressure.
We dissolved the HMDI-modified mPEG in a large volume of DCM, and, in a separate flask, we dissolved an equimolar amount of PEI in a large volume of chloroform. The PEG solution was then added dropwise to the PEI solution, and the reaction was continued for 12 h at 50℃, under stirring and reflux. We precipitated the reaction mixture dropwise in ice-cold petroleum ether (3x in 250 mL) and dried under vacuum. Finally, we activated the (±)-α-tocopherol succinate with EDC, NHS, and reacted it with PEG-PEI at different molar ratios. Briefly, we dissolved (±)-α-tocopherol succinate in DMSO and 2x molar ratio of EDC and 1.4x molar ratio NHS, and allowed the mixture to react for 30 min at room temperature. In a separate flask, we dissolved PEG-PEI in DMSO. The (±) α-tocopherol succinate solution was added dropwise, and the solution of the PEG-PEI and the mixtures was allowed to react for 18 h at room temperature. We utilized different molecular ratios between (±)-α-tocopherol succinate and PEG-PEI to achieve the different ratios in the final conjugate products. The reaction mixture was purified by dialysis in water and freeze-dried until further use.
Nuclear Magnetic Resonance Spectroscopy (NMR)
The NMR spectra of the TPP were recorded in JEOL Eclipse ECS-400 after dissolving in deuterated chloroform (CDCl3) (Acros Organics).
Fourier Transformed Infrared Spectroscopy (FTIR)
We performed the FTIR spectroscopy using a Spectrum Two FTIR spectrometer (PerkinElmer, Waltham, MA). Briefly, all samples were prepared without the use of solvents. Individual components of TPP, like, activated PEG, PEI, Toc, and TPP analyzed by directly compressing on the ATR crystal and scanned from 400 to 4000 cm-1. Data were analyzed with PerkinElmer Spectrum Quant software.
Differential Scanning Calorimetry (DSC)
DSC measurements were conducted using a TA instrument-waters LLC (New Castle, DE). Scans took place under a nitrogen atmosphere with temperature ranging from 0 to 150 ºC, with a 2 ºC/minute increment. Individual components of TPP, such as PEG, Toc-succinate, and PEI-PEG, were analyzed along with TPP. We also analyzed a physical mixture of Toc-succinate and PEG-2000-PEI-1800. Approximately 10 mg of the different samples were analyzed, and the melting temperatures (Tm) were derived from the heating curve.
Differential Light Scattering (DLS) and Transmission Electron Microscopy (TEM)
We identified the particle size, size distribution, and zeta potential of TPP micelles using Nanobrook 90plus PALS (Brookhaven, Holtsville, NY) at 25 ºC. We analyzed the nanoparticles using TEM, where 3 μL of the suspension were placed on a 300-mesh carbon filmed TEM grid (EMS #CF300-CU) and dried at room temperature. Then, the grid was inserted in a JEOL JEM-1400, 120kV Transmission electron microscope (Tokyo, Japan), and imaged with gatan digital camera.
Preparation of Nano-Complexes
We prepared nano-complexes of the TPP polymers. Briefly, 5 mg of each TPP polymer was placed in 1 mL of water or 50 mM HEPES buffer (pH 7.2). The samples were subsequently bath sonicated for 1 min at room temperature. Where plasmid was used, we subsequently physically mixed the plasmid in HEPES solution with the polymer, vortexed, and incubated for 20 min at room temperature, before further use.
Critical Micellar Concentration (CMC)
We determined the CMC as a function of the TPP111 concentration in an aqueous solution at room temperature, as previously described.12 Briefly, we prepared serial dilutions of the TPP at different concentrations and plotted the concentration against kCPS value, as detected using DLS.
Buffering Capacity Analysis
We dissolved 10 mg of TPP or PEI in 0.1 M NaCl solution. We decreased the pH to 3, using 1 N HCl, and then we performed pH titration with the gradual addition of a 3 μL 1 N NaOH solution. Following each addition, we measured the pH using a Mettler Toledo pH meter (Columbus, OH). The titration was stopped upon reaching pH 9.
Cell Viability Assay by MTT
We determined the cytotoxicity of the polymer with or without the plasmid, using a standard MTT assay. Briefly, we seeded 10^4 A549 cells in a 96-well tissue culture plate and allowed the cells to attach overnight. TPP or TPP complexes with pGPL-3 plasmid were prepared in HEPES buffer, as described above, then diluted into media and added into their respective wells. The cells were then incubated for 24, 48, or 72 h. For TPP+pGL-3, cells were treated for 6 h, to mimic with transfection conditions, as described in the Methods, and then replaced with fresh DMEM/F12K complete media, and incubated for 24, 48, or 72 h. After the respective incubation period, we added 10 µL of sterile MTT solution (5 mg/mL) into each well and incubated for 3 h at 37 ºC. We determined the cell survival using 10% acidified SDS solution and detected the color at 570/630 nm using a plate reader (Biotek synergyH1 plate reader-Winooski, VT). The cell viability (%) was calculated as a percentile ratio of sample optical density (OD) over control OD using the following equation.

Gel Retardation and DNase I Stability Assay
Electrostatic interaction between the positively charged TPP111 and the negatively charged plasmid was evaluated by the agarose gel electrophoresis method. TPP111/pGL-3 complexes were prepared using different N/P ratios from 0.5 to 60. N/P ratio corresponds to the atomic ratio of nitrogen (originating from the polymer) to phosphates (originating from the nucleic acids). The complexes were prepared and loaded into 1% agarose gel with a loading dye. For the gel retardation assay, 0.1 µg/mL of Ethidium Bromide was added in the gel, as well as in 1X TBE running buffer. The gel electrophoresis was performed for 1 h at 100 V. After the run, the gel was visualized using an imaging system (Chemidoc Touch Imaging system-Biorad, Hercules, CA).
We also evaluated the protecting effect of the TPP111 to the nucleic acids against enzymatic degradation, as previously described.13 We prepared nano-complexes of TPP and pGL-3 plasmid at different N/P ratios. Naked and TPP conjugated plasmids were incubated with 2.2 µL of DNase I (2 U of DNase/600 ng of plasmid) for 30 min at 37 ºC. The reaction was stopped by adding 4.2 μL 50 mM of EDTA. Samples were run in 1% agarose gel, as described above.
Plasmid Transfection with TPP and PEI
We performed transfections of the luciferase-expressing pGL-3 plasmid in two cell lines, A549 and H358, using TPP111 and PEI-1800. Complexation of TPP111 or PEI with pGL-3 was achieved, as described above. We seeded 104 cells in 96-Well Optical-Bottom Plates (Fisher, Hampton, NH) and incubated overnight for attachment. The following day, we washed each well with 1X PBS and treated the cells with 20 µg of pGL-3 plasmid complexed with TPP111 or PEI, at different N/P ratios. Each sample was replicated 5 times, and the plates were subsequently incubated in 37 ºC for 6 h. Following the incubation period, we replaced the media in each well with fresh complete media. After 24 or 48 h, we detected the luciferase activity and cell survival according to the manufacturer’s protocol (ONE-GLO + Tox Luciferase Reporter and cell viability Assay kit-Promega, Madison, WI). Where applicable, we obtained the ratio of luminescence over survival for each well.
Cellular Uptake Study of TPP Nano-Complexes
We conjugated the Cy-5.5-NHS fluorophore (Lumiprobe, Cockeysville, MD) onto TPP111, following the manufacture’s protocol. We investigated the cellular uptake using confocal laser scanning microscopy (CLSM), as previously described.14 Briefly, we prepared TPP111-Cy5.5 (TPPc) nano-complexes with pGL-3 plasmid, as described above. We seeded 5x104 of A549 or H358 cells in chambered cell culture slides (Falcon, Corning, NY). We added the TPPc-pGL-3 complexes at the concentration of 1 mg/mL and incubated the cells. Following the incubation period, all wells were washed with 1x PBS and fixed with 4% formaldehyde for 10 minutes at room temperature. Each well was washed with PBS and incubated with Lysotracker (Red DND-99 Invitrogen, Eugene, OR) to detect lysosomes and Cell Light Early Endosomes GFP (Invitrogen, Eugene, OR) to detect endosomes, respectively, according to the manufacturer’s protocol. After washing with PBS, we used DAPI-containing mountain media to stain the nuclei before analyzing under CLSM.
Biodistribution Analysis of TPP
Biodistribution analysis was performed in anthemic female nude mice (4–6 wks old; Envigo, Indianapolis, IN). The experimental protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Louisiana Monroe, based on Office of Laboratory Animal Welfare (OLAW), National Institute of Health (NIH) guidelines and the Guide for the Care and Use of Laboratory Animals, 8th ed. We conjugated the TPP111 with Cy-5.5 dye, as described above. We injected 4x105 A549 cells in each flank of female nude mice for developing tumors. Two weeks post-injection, when tumor volume was approximately 250 mm3, as determined using caliper measurements, we treated each mouse with 1 mg/kg of pGL-3 plasmid complexed with 1:30 N/P ratio of TPP111. The formulations were injected intravenously in the tail vein of each mouse, at a volume ~50 µL/mouse. At predetermined time points (0 h-no injection, 1 h, 2 h, 4 h, 8 h and 24 h), we sacrificed 3 mice per time point, and obtained full-body fluorescent images using IVIS. Subsequently, tumors, heart, liver, kidneys, lung, spleen, and brain were harvested from each animal and imaged for fluorescence.
Results
Synthesis and Characterization of TPP
We prepared a panel of TPP polymers, by changing the ratio between Toc-succinate and PEG-PEI, as well as the molecular weights of either PEG or PEI. Our objective was to identify a conjugate with the minimum size (<150 nm15) and strong complexation with nucleic acids. First, we modified the hydroxyl group of Toc with succinic anhydride to obtain a carboxyl group available for further derivatization, following established protocols.10 The Toc-succinate was characterized by 1H-NMR (Figure 2A) to confirm appropriate conjugation, with the characteristic peaks of Toc at 1.24 ppm (H-CH-C-H and H-CH-CH-H) and succinate peaks at 2.92 and 2.81 ppm (H-CH-CH-H).
 |
Figure 2 NMR, FTIR and DSC characterization of TPP. (A) Representative NMR spectroscopic analysis of PEG-2000-PEI-1800, tocopherol succinate, and TPP. TPP presents the characteristic peaks of tocopherol at 1.24 ppm (H-CH-C-H and H-CH-CH-H) and succinate peaks at 2.92 and 2.81 ppm (H-CH-CH-H). PEG-PEI peaks are at 3.6 ppm (-CH2CH2O- of PEG) and at 2.5–3.8 ppm (-CH2CH2NH- of PEI). (B) FTIR analysis of TPP and its components. (C) DSC analysis of the melting curves, indicating the exothermic peaks of TPP and its individual components. |
Activation of the PEG polymers and attachment to PEI was performed using the HMDI crosslinker reaction, as previously described.16 During this reaction, we maintained an approximate 1:1 molar ratio for any of the PEG-PEI products. The molecular weights of PEG spanned between 550 and 2000 and for PEI we studied the molecular weights 1800 and 10,000 (Table 1). Subsequently, we reacted the Toc-succinate with the different PEG-PEI molecules, at several ratios (Table 1). Increasing the Toc-succinate ratio indicated a rapid increase in particle size. Although the change in the molecular weight of PEI or PEG maintained the nanosized dimensions of the produced carriers in several of the cases, only in the case of the PEG2000-PEI1800 conjugated with a-tocopherol towards TPP produced self-assembled nanostructures less than half of any other studied polymer and below the established cut-off limit of <150 nm,15 with an average diameter of ~90 nm. Thus, for any further analysis, we chose this polymer, which we annotate as TPP111. Additional considerations towards this selection were that the molar content of PEI per polymer structure in all of the constructs remained constant, and TPP based on PEI 10,000 caused significantly larger particles, while PEI10,000 is known to have higher systemic toxicity vs PEI1,800 but comparable transfection capacities.17,18
 |
Table 1 Size and Zeta Potential of TPP Polymers in HEPES Buffer |
We analyzed the three-step synthesis process of the TPP polymers with various spectroscopic methods. For PEG-PEI, the 1H NMR indicated for PEG a single peak at 3.6 ppm (-CH2CH2O-) and for PEI multiple peaks at 2.2–2.8 ppm (-CH2CH2NH-, Figure 2A). Additionally, we evaluated the final polymeric product using FTIR spectroscopy (Figure 2B). We found (I) Amine stretching of N-H peak at 3298 cm−1, (II) C-H stretching 2888 cm−1, (III) O=C=N isocyanate stretching 2272 cm−1 which is not present in PEG-PEI conjugate, (IV) C=O urethane stretching 1693 cm−1, (V) C=O urea stretching 1566 cm−1, (VI) C-O ether stretching 1111 cm−1 (VII) C=O stretching of -COOH 1746 cm−1 which disappeared in TPP. We further analyzed the products using differential scanning calorimetry (Figure 2C). We identified the PEG-2000 indicated a maximum of heat flow at ~54°C, indicating the melting point for the material, while PEI-1800 did not produce a definite melting point at the scanned temperatures. The PEG-2000-PEI-1800 produced a heat flow maximum at ~40°C. The drop of the melting point of the material compared to PEG2000 alone confirmed the reaction and conjugation of the materials. Toc-succinate analysis indicated a heat flow maximum at ~60°C. The final TPP111 product indicated a heat flow maximum at ~44°C, confirming the reaction between PEG-PEI and Toc-succinate. Finally, we analyzed the physical mixture between PEG2000-PEI1800 with Toc-succinate at an approximate molar ratio between the two molecules at 1:1, and it indicated the peak of PEG2000-PEI1800 only approximately at the same temperature, while the heat flow was sustained above the baseline until the Toc-succinate melting point. These confirm the successful synthesis of the molecules.
Formation and Characterization of TPP/DNA Complexes
The prerequisite of the negatively charged nucleic acid delivery with cationic molecules is to have strong and stable electrostatic interactions.19 We observed the self-assembling capacity of the TPP111 to produce nanoparticles in aqueous solution through DLS and TEM. The TPP111 produced nanoparticles with a diameter of 90 ± 3.5 nm, where the polydispersity index (PDI) was at 0.29 ± 0.02 (Supplementary Figure 1). The Zeta potential of the polymer in water was 40.45 ± 1.3 mV. Transmission electron microscopy (TEM) analysis indicated nano-sized globular-shaped nanostructures of approximately similar dimensions, as detected by the DLS (Figure 3A). The TPP111 complexed with plasmid DNA increased the structures’ size, as detected by TEM analysis (Figure 3B). For comparison, DLS indicated that TPP111-pGL3 complexes had an average size of 160 ± 4.2 nm, with a PDI of 0.210 ± 0.01.
 |
Figure 3 TEM analysis of the self-assembled nanoparticles of TPP111 and its complexes with plasmid in HEPES buffer. (A) The size of TPP111 nanoparticles without the presence of nucleic acid was approximately similar to the DLS measured size. (B) The size of TPP111 complexed with plasmid DNA. (C) Gel retardation was performed to determine the complexation of TPP111 with plasmid DNA, as different N/P ratios. The analysis indicated complete plasmid complexation with the TPP polymer, at N/P ratio as low as 7. We used PAA as a positive control to confirm the complexation, as PAA dissociates the plasmids from the polymer. (D) We evaluated the protective properties of the TPP111 polymer on the complexed plasmids against DNase-mediated degradation. The TPP111 protected the plasmids from degradation, following incubation with DNases for 30 min. |
We analyzed the minimum concentration of TPP111 polymer required to generate nanostructures in water, using a standard critical micellar concentration (CMC) analysis. The kilo counts/second (kCPS) values generated by the light scattering of DLS were recorded from serially diluted samples of TPP111, starting at 1 mg/mL. The log10 of the polymer concentration vs the kCPS was plotted to identify the sharp inflection point. We found that at ~17 µg/mL is the CMC for the TPP111 (Figure 4A).
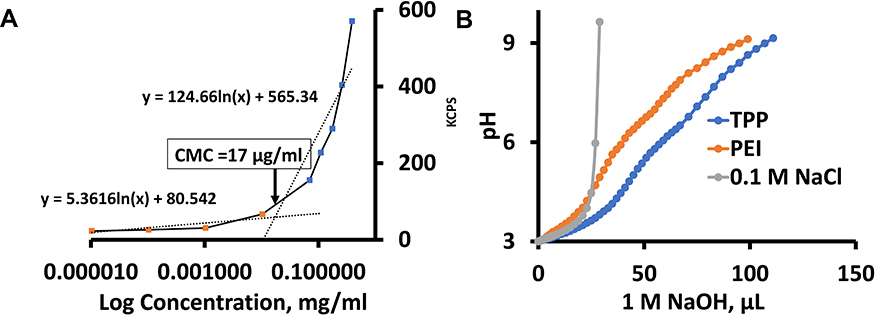 |
Figure 4 CMC and buffering analysis of the TPP111 polymer indicated a CMC at ~17 μg/mL and buffering capacity comparable to its parent compound, PEI. (A) Plot of log10 concentration (µg/mL) of TPP111 in water vs kilo count per second (kCPS), as measured by DLS. (B) Buffering capacity of TPP111, PEI and normal saline (NaCl) was measured using acid-base titration. 1 N HCl was used to reach the starting point at pH 3. Titration was performed by gradually adding 1 N NaOH to reach pH at 9. |
We evaluated the complexation of TPP111 with nucleic acids using gel retardation assay at different N/P (nitrogen/phosphate) ratios. As shown in Figure 3C, at N/P ratio of 7, TPP111 completely prevented the migration of plasmids through the agarose gel, indicating complete complexation between the polymer and the plasmid. This was confirmed by treating complexes with polyacrylic acid (PAA), which is highly anionic in charge and can cause the dissociation of the plasmid from the TPP111. We identified the release of the intact plasmids after the addition of the PAA, which confirms the plasmids’ complexation with TPP111. Finally, we evaluated the DNase-mediated degradation of naked and the protection provided by TPP111 to the plasmids from nucleases. After incubation with DNase for 30 minutes at 37℃, we observed that the TPP111 prevents the degradation of plasmids in the presence of DNases, as shown in Figure 3D. In contrast, we found DNases completely degraded the naked plasmid (lack of any band signal). Indicatively, the presence of plasmid band after PAA addition, followed by DNase treatment, confirms the protection of the plasmids by TPP111.
Buffering Capacity of TPP111
We evaluated the buffering capacity of TPP111. 1 M HCl was used to reduce the pH to 3 of TPP111 or PEI solutions in a 0.1 M NaCl solution. The TPP111 concentration used here was above the CMC. Subsequently, the continuous addition of 1 N of NaOH solution was used to titrate, and we determined the quantity of NaOH required to change the pH of the solutions/suspensions to 9. For our analysis of the TPP111’s and PEI’s buffering capacity, we only used the required volume of NaOH required to adjust the pH from 5 to 7, as previously described.20 For the pH change, 30 μL of 1 N NaOH for TPP111 vs 26 μL of 1 N NaOH for PEI were required, indicating that the TPP111 maintains similar or slightly improved buffering capacity as its parent material, the PEI. As a negative control, we used NaCl saline solution and found no buffering resistance on pH change (Figure 4B).
In vitro Cytotoxicity and Transfection of TPP111
We evaluated the cell viability of TPP111 with (Figure 5A) or without (Figure 5B) complexation with the pGL-3 plasmid. In both cases, TPP111 exhibited modest cytotoxicity for all studied time points, with IC50 >200 μg/mL in A549 cells, in the majority of the studied cases (Table 2). PEI’s cytotoxicity is presented in Supplementary Figure 2.
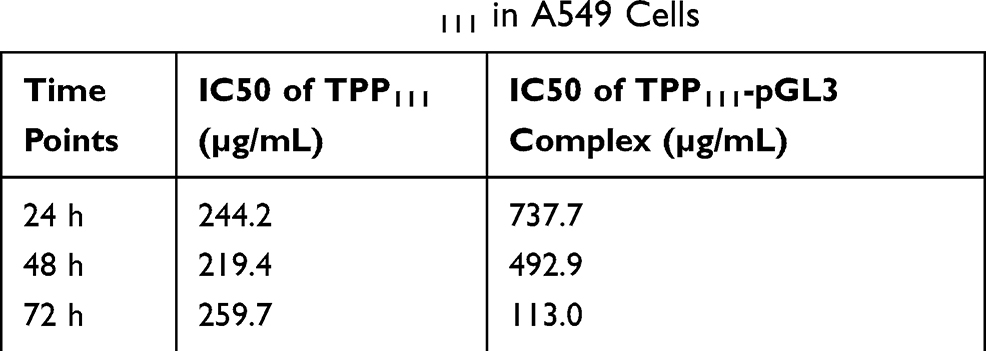 |
Table 2 IC50 Values of TPP111 in A549 Cells |
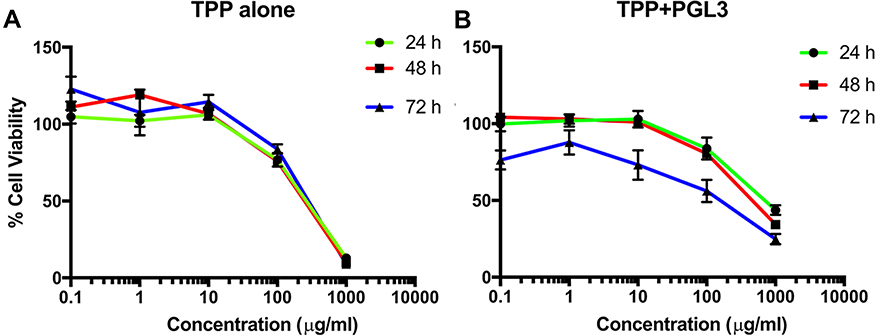 |
Figure 5 Survival study indicated low cytotoxicity of the TPP111 in A549 cells with or without the presence of nucleic acids. (A) % Cell viability TPP111 alone was evaluated with different concertation and plotted as logarithmic form to determine the IC50 at 24, 48 and 72 h time points. (B) IC50 calculation was performed with TPP111 -PGL-3 complex for 24, 48 and 72 h time points. |
The transfection activity of TPP111 was analyzed using different cell lines. We cultured A549 and H358 cells, according to the techniques described above. The transfection activity of the polymer was determined and compared to PEI, using different N/P ratios. We maintained a constant amount of plasmid in all of the studied cases, only changing the polymer content. For N/P ratios below 1:15, we measured the luminescence intensity only. For ratios above this value, we also included the measurement of cell viability, to compensate for any potential cytotoxic effect from either TPP111 or PEI, even though we were below the IC50 value. Thus, the luminescence intensity from the luciferase activity, as detected by a plate reader, was divided by the cell viability (%) for each of the respective 96 wells in each plate, which indicates luciferase activity over survived cells. The result corresponds to the luminescence per number of cells. The TPP111 induced an overall strongest transfection compared to PEI, when compared for the same N/P ratios and time points (p<0.05 is presented in the figure). In Figure 6, we performed statistical analysis between the TPP111 vs PEI for each respective N/P ratio and time point only, since all of the polymer groups were significantly higher than the untreated cells (no plasmid). Similar results were obtained for both cell lines (please see Supplementary Figure 3 for H358 cell line).
 |
Figure 6 Transfection efficiency of TPP111 was evaluated to determine the delivery of pGL-3 luciferase-expressing plasmid in A549 cells for 24, 48 and 72 h time points. (A) Transfection was performed using N/P ratios spanning between 0.1 to 15 for TPP111 or PEI. (B) Transfection was performed using N/P ratios spanning between 15 to 45 of TPP or PEI. “Luminescence/% cells” indicates luminescence intensity over number of live cells. *: p<0.05, ***:p<0.001 for comparison between TPP and PEI respective groups; #: p< 0.05, ##: p<0.01, ###:p<0.001 for comparison to pGL-3 alone group. All statistical analyses are two-tailed t-tests. |
Cellular Uptake of TPP111 in vitro and Biodistribution in vivo
We evaluated the cellular uptake of the TPP111 and PGL-3 plasmid complexes by incubating them with A549 and H358 for different time periods (0, 0.5, 1, 2, 4 and 6 h). We conjugated the Cy-5.5 fluorescent dye with TPP111 to track the micellar complexes inside the cells. We found the TPP111 complexes enter cells within 30 min (Figure 7 and Supplementary Figure 4). We observed a steady increase in the cellular uptake, as indicated by a continuous increase of fluorescence intensity by the Cy-5.5 over time, and reached its maximum at the 4 h sample, which was maintained up to our last time point (6 h). To study the uptake pathway for the nanoparticles, we labeled lysosomes and early endosomes with Red DND-99 and GFP, respectively. We observed a strong overlap in the merged figures for TPP111-Cy5.5 complexes with the GFP-labeled endosomes in all time points, with the strongest colocalization taking place at the 4 and 6 h samples (Figure 7, white arrows indicate the colocalization). Thus, this indicates that the uptake of the TPP111-plasmid complexes takes place through the endosomes. The limited presence of TPP111 fluorescence outside the endosomal structures indicates that endosomal-mediated cellular uptake is the primary mechanism. We also observed TPP111 entrapment in lysosomes at the 4 h time points, which indicates some TPP111 complexes still remain within the endosomal structure post their maturation to lysosomes. The same behavior was also observed in H358 cell line (Supplementary Figure 4).
 |
Figure 7 Strong cellular uptake analysis of Cy-5.5-conjugated TPP111 complexed with pGL-3 plasmid was detected using Confocal Laser Scanning Microscopy (CLSM), potentiated through endosomal uptake. Lysosomes and endosomes were stained with Red DND-99 (sudo color green) and GFP (green), respectively. Nuclei (blue) were stained with DAPI. A549 cells were incubated with TPP111-plasmid complexes for different incubation periods. The scale bar is at 40 µm. Colocalization of TPP complex and endosomes/lysosomes is indicated with white arrows. |
We evaluated the biodistribution of TPP111-pGL-3 complexes in vivo. We injected intravenously Cy5.5-conjugated TPP111 complexed with pGL-3 plasmids in A549 tumor-bearing athymic nude mice. As shown in Figure 8 and Supplementary Figure 5, up to 1 h following the injection of the complexes, the complexes were distributed throughout the body, including the liver, lung, kidneys, and tumors. Subsequently, there was a steady decrease of the TPP111 complexes’ fluorescence in the major organs, such as liver, kidneys, and spleens, while the levels of fluorescence intensity in the tumors were maintained. Specifically, at the 8 and 24 h time points, the TPP111 complexes were barely detected in the liver and kidneys, and complete absence of fluorescence was observed in the other organs, while significant fluorescence was maintained in the tumor area. This represents the prolonged residence of TPP111 complexes in the tumors, most likely due to the EPR effect and the small size of the complexes.9
 |
Figure 8 In vivo biodistribution of Cy5.5 conjugated TPP111 complexed with pGL3 plasmid was performed in female athymic nude mice carrying subcutaneous lung cancer tumors, and visualized using IVIS imaging system. Representative ex vivo fluorescence images of different organs (liver, heart, lung, spleen, kidney, tumor and brain) at 0, 1, 2, 4, 8 and 24 h after injection indicated a favorable accumulation of the TPP111 complexes in the tumor area. |
Discussion
Nucleic acids have emerged as powerful tools for disease treatment and studying molecular mechanisms.21 Their inherent instability in circulation has prompted strong research on their delivery applications, with innovative approaches constantly being developed. Nucleic acids have to transverse in vivo harsh conditions, whether they are administered orally, intraperitoneally, intravenously or by any other route of administration.2,8 The ubiquitous presence of DNA/RNA-ses in our bodies limit their prolonged presence in the circulation in vivo. Furthermore, the large size of the nucleic acid constructs, their hydrophilicity, and negative charge hinders their ability to enter into cells.8,22,23
Nanotechnology approaches present the potential to overcome most of these limitations. We capitalized on the success of an existing FDA-approved, Generally-Regarded-As-Safe (GRAS) Vitamin E derivative, called TPGS, which repeatedly and consistently has proven efficient in protecting, delivering and prolonging the systemic circulation of a plethora of compounds.24,25 In our study, as presented in this paper, we developed, characterized and evaluated a Vitamin E derivative, composed of α-Tocopherol, PEI, and PEG, for delivering nucleic acids in vitro and in vivo. PEGylation of cations-nucleic acid complexes enhances their solubility, decreased plasma protein binding, and can improve their biodistribution profile.26
We prepared a panel of Vitamin E derivatives and selected the most promising to analyze its ability to produce nanostructures, capable of complexing and protecting nucleic acids, while being able to enter cells and release their load. We also analyzed the in vivo biodistribution profile using a subcutaneous lung cancer mouse model.
From the different prepared polymer compositions, we identified that the Tocopherol:PEI:PEG 1:1:1, annotated as TPP111, demonstrated self-assembling properties, developing nanostructures of approximately 90 nm, with spherical size, as determined by DLS and TEM analysis. The other formulations did not produce sizes of similar small dimensions, in many cases with significantly larger diameters and aggregates. The TPP111 polymer synthesis was analyzed by NMR, FTIR, and DSC, confirming the successful synthesis of the product. More importantly, the TPP111 in water maintained its cationic nature, necessary for nucleic acid complexation and endosomal escape. Finally, we determined the CMC of the TPP111 polymer, through serial dilutions of the polymer, while being analyzed by DLS. The TPP111 presented a low CMC, at 17 μg/mL, which is far below any in vitro or in vivo analysis described in this paper.
The ability to induce endosomal escape is one of the most important actions for carriers to release nucleic acids into the cytoplasm.27 PEI is capable of inducing endosomal escape through the proton-sponge effect, where negatively charged ions like Cl- influx into endosomes until their rupture, due to higher presence of the positive charges originating from the cationic polymer inside the endosomes.28 The cationic behavior of PEI stems from the presence of secondary or tertiary amines.29 For these reasons, PEI possesses a strong capacity for endosomal escape in vitro.30 To evaluate whether the TPP111 maintains the cationic and buffering characteristics of the PEI, we dissolved the TPP111 polymer in saline water, while adjusting its pH by the stable addition of NaOH. We measured the total required volume for the transition from pH 5 to 7, which was used as a measure for its buffering capacity. We compared this to the PEI, analyzed under the same conditions, and we detected no significant differences in the buffering capacity for the TPP111 polymer compared to its PEI parent material. The comparable activity of the TPP111 to PEI indicates that the addition of the lipophilic a-tocopherol and the relatively large molecule of PEG did not interfere with the buffering capacity of the parent polymer.
We evaluated the capacity of the TPP111 to complex with and protect nucleic acids from degradation. Nucleic acid stability is instrumental during in vivo application, and strong complexation between the polymer/carrier and the nucleic acids, while shielding the latter from the environment, is important.8 PEI and PEI constructs have been reported to not only complex with nucleic acids but also protect them from nuclease degradation.31 We determined through a standard gel retardation assay that the TPP111 polymer strongly complexed with the pGL-3 plasmid, at N/P ratios as low as 7. For perspective, this ratio corresponds to a weight ratio of 1:2 for plasmid:polymer. Subsequently, using the same analysis, we incubated the TPP111-complexes in the presence of DNase I, to determine the ability of the polymer to protect the nucleic acids from degradation. Following incubation of the TPP111-complexes with DNases, we released the plasmids from the complexes and analyzed them through the gel electrophoresis. TPP111 protected the nucleic acids from DNases, whereas the plasmid alone in the presence of the nucleases wascompletely degraded.
Successful cellular uptake is critical of any drug delivery carrier to deliver their load. We complexed TPP111 polymer labeled with Cy-5.5 with plasmid and incubated cells in the presence of the complexes. Confocal microscopy indicated that the complexes rapidly enter into the cells, as early as 30 min post-incubation initiation. Using the Red DND-99 lysosomal tracker and the GFP-early endosome tracker kits, we identified strong colocalization of the Cy-5.5 fluorescence signal with the green GFP fluorescence signal, primarily at the earlier time points (<4 h). This indicates that the TPP111-plasmid complexes are primarily up-taken through the endosomal pathway. At later time points, colocalization of the Cy-5.5 fluorescence signal and the Red DND-99 takes place (>4 h). We believe this is the result of the natural progression and development of the endosomes to late endosomes and potential fusion with lysosomes. This analysis confirms that the complexes are up-taken by the cells through the endosomes, but does not ensure the endosomal escape.
To this end, we evaluated the transfection capacity of the TPP111 polymer using the luciferase-expressing plasmid. We transfected A549 and H358 cells using the TPP111-plasmid complexes for 6 h, and detected the expression of the firefly luciferase protein through a luciferase assay kit. We used several N/P ratios between TPP111 and the plasmid, and compared our results to the parent PEI. We confirmed that there is endosomal escape, as there are strong transfection and production of the luciferase protein, which can only take place if the plasmid escapes the endosomes to be transcribed. Furthermore, the TPP111 polymer demonstrated comparable and, in some cases, improved transfection compared to PEI (p<0.05), while in some other cases, the opposite took place. For example, in A549 cells, TPP111’s strongest transfection compared to PEI was a 7-fold increase to luciferase activity at 72 h and N/P ratio of 1:15 (p<0.05), while PEI’s strongest transfection compared to TPP111 was a 4-fold increase in luciferase activity compared to TPP111 at 24 h and N/P ratio of 1:0.5 (no significance). In H358, we did not observe significant differences between the two polymers. From our analysis, we identified the optimal N/P ratio for TPP111 to nucleic acids to be 30, which we used for our in vivo biodistribution studies. We need to point out that the TPP111 appeared to have a stronger transfection at the 48 and 72 h compared to PEI, while PEI was frequently giving the strongest signal at 24 and 48 h. This is not an exhaustively consistent behavior, but an overall observation.
PEI is highly cationic, which is detrimental for its in vivo biodistribution, following intravenous injection.32 In fact, it has been reported that the PEI rapidly accumulated to the liver, spleen and kidneys,33,34 presenting challenges for prolonged tumor accumulation. We developed a subcutaneous mouse model of lung cancer, by injecting A549 cells into both flanks of female athymic nude mice. Once the tumors reached an average volume of ~250 mm3, we intravenously injected the Cy-5.5 modified TPP111-plasmid complexes, and at predetermined time points, we sacrificed the animals and harvested their major organs. Through fluorescent imaging, we detected how the fluorescently labeled nano-complexes distributed throughout the animals’ bodies as a function of time. Our analysis indicated a strong accumulation of the nano-complexes into the tumor area. More importantly, this accumulation was maintained throughout the 24 h study, with minimal reduction at the later time points. For comparison, the Cy5.5 alone has previously been shown to have limited accumulation to the tumor area.35–37 We also detected liver and kidney accumulation of the nano-complexes in the early time points, which diminished over time. At the 24 h time point, the fluorescence signal from the liver had greatly diminished compared to the tumors, while the kidneys maintained a strong signal, as potentially the nano-complexes were excreted through this organ.
Our analysis indicates that the TPP111 polymer constitutes a propitious solution for delivery of nucleic acids in vivo, due to its strong tumor-accumulating properties. The lipophilic core at the center of the micelles will allow for the encapsulation of lipophilic compounds, similar to the TPGS molecule. This will find applicability in combinatorial treatments, which include drug and nucleic acid delivery. Compared to PEI, which accumulates in the liver, spleen or kidneys,33,34 our polymer demonstrated strong tumor-accumulating properties. Furthermore, the derivatization of PEI did not impact its transfection capacity, with the TPP111 polymer presenting comparable and frequently improved transfection in vitro. Finally, the moderate cost of the tocopherol, PEI and PEG materials, as well as their use in FDA-approved applications, establishes the proposed nanocarrier as a promising approach in drug and nucleic acid delivery that merits further evaluation.
Conclusion
In this study, we successfully synthesized different TPP conjugates and identified their optimal structure (1:1:1 molar ratio conjugate). This formulation demonstrated low cytotoxicity, strong buffering capacity, and strong protective capability of nucleic acids from enzymatic degradation, which are the key parameters of a safe and efficient nucleic acid delivery carrier. Moreover, enhanced cellular uptake in vitro and favorable accumulation in tumors in vivo make this carrier a promising choice for nucleic acid delivery for cancer therapeutics.
Statistical Analysis
The statistical analysis was performed with a Student two-tailed t-test to determine any significant differences among groups. We compared the mean values ± standard errors and p values < 0.05 were considered statistically significant.
Acknowledgments
This work was supported by the College of Pharmacy, University of Louisiana Monroe start-up funding, and the National Institutes of Health (NIH) through the National Institute of General Medical Science Grants 5 P20 GM103424-15, 3 P20 GM103424-15S1. We would also like to acknowledge and thank for their support at the microscopy core at Louisiana State University.
Disclosure
The authors declare no potential conflicts of interest for this work.
References
1. Sharma VK, Rungta P, Prasad AK. Nucleic acid therapeutics: basic concepts and recent developments. RSC Adv. 2014;4(32):16618–16631. doi:10.1039/c3ra47841f
2. Labatut AE, Mattheolabakis G. Non-viral based miR delivery and recent developments. Eur J Pharm Biopharm. 2018;128:82–90. doi:10.1016/j.ejpb.2018.04.018
3. Wolf G. How an increased intake of alpha-tocopherol can suppress the bioavailability of gamma-tocopherol. Nutr Rev. 2006;64(6):295–299. doi:10.1111/j.1753-4887.2006.tb00213.x
4. Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of Vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49(2):175–186. doi:10.1016/j.ejps.2013.02.006
5. Liu Y, Huang L, Liu F. Paclitaxel nanocrystals for overcoming multidrug resistance in cancer. Mol Pharm. 2010;7(3):863–869. doi:10.1021/mp100012s
6. Xu Y, Meng H, Du F, et al. Preparation of intravenous injection nanoformulation of VESylated gemcitabine by co-assembly with TPGS and its anti-tumor activity in pancreatic tumor-bearing mice. Int J Pharm. 2015;495(2):792–797. doi:10.1016/j.ijpharm.2015.09.030
7. Dunlap DD, Maggi A, Soria MR, Monaco L. Nanoscopic structure of DNA condensed for gene delivery. Nucleic Acids Res. 1997;25(15):3095–3101. doi:10.1093/nar/25.15.3095
8. Hossian A, Mackenzie GG, Mattheolabakis G. miRNAs in gastrointestinal diseases: can we effectively deliver RNA-based therapeutics orally? Nanomedicine (Lond). 2019;14(21):2873–2889. doi:10.2217/nnm-2019-0180
9. Mattheolabakis G, Rigas B, Constantinides PP. Nanodelivery strategies in cancer chemotherapy: biological rationale and pharmaceutical perspectives. Nanomedicine (Lond). 2012;7(10):1577–1590. doi:10.2217/nnm.12.128
10. Lipshutz BH, Ghorai S, Abela AR, et al. TPGS-750-M: a second-generation amphiphile for metal-catalyzed cross-couplings in water at room temperature. J Org Chem. 2011;76(11):4379–4391. doi:10.1021/jo101974u
11. Brus C, Petersen H, Aigner A, Czubayko F, Kissel T. Physicochemical and biological characterization of polyethylenimine-graft-poly(ethylene glycol) block copolymers as a delivery system for oligonucleotides and ribozymes. Bioconjug Chem. 2004;15(4):677–684. doi:10.1021/bc034160m
12. Yu J, Zhou Y, Chen W, et al. Preparation, characterization and evaluation of alpha-tocopherol succinate-modified dextran micelles as potential drug carriers. Materials (Basel). 2015;8(10):6685–6696. doi:10.3390/ma8105332
13. Zhang X, Pan SR, Hu HM, et al. Poly(ethylene glycol)-block-polyethylenimine copolymers as carriers for gene delivery: effects of PEG molecular weight and PEGylation degree. J Biomed Mater Res A. 2008;84(3):795–804. doi:10.1002/jbm.a.31343
14. Zhang L, Yang X, Lv Y, et al. Cytosolic co-delivery of miRNA-34a and docetaxel with core-shell nanocarriers via caveolae-mediated pathway for the treatment of metastatic breast cancer. Sci Rep. 2017;7:46186. doi:10.1038/srep46186
15. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. doi:10.1038/nbt.3330
16. Iqbal J, Hombach J, Matuszczak B, Bernkop-Schnurch A. Design and in vitro evaluation of a novel polymeric P-glycoprotein (P-gp) inhibitor. J Control Release. 2010;147(1):62–69. doi:10.1016/j.jconrel.2010.06.023
17. Wen Y, Pan S, Luo X, Zhang X, Zhang W, Feng M. A biodegradable low molecular weight polyethylenimine derivative as low toxicity and efficient gene vector. Bioconjug Chem. 2009;20(2):322–332. doi:10.1021/bc800428y
18. Morimoto K, Nishikawa M, Kawakami S, et al. Molecular weight-dependent gene transfection activity of unmodified and galactosylated polyethyleneimine on hepatoma cells and mouse liver. Mol Ther. 2003;7(2):254–261. doi:10.1016/S1525-0016(02)00053-9
19. Sunshine JC, Bishop CJ, Green JJ. Advances in polymeric and inorganic vectors for nonviral nucleic acid delivery. Ther Deliv. 2011;2(4):493–521. doi:10.4155/tde.11.14
20. Singh B, Maharjan S, Park TE, et al. Tuning the buffering capacity of polyethylenimine with glycerol molecules for efficient gene delivery: staying in or out of the endosomes. Macromol Biosci. 2015;15(5):622–635. doi:10.1002/mabi.201400463
21. Sridharan K, Gogtay NJ. Therapeutic nucleic acids: current clinical status. Br J Clin Pharmacol. 2016;82(3):659–672. doi:10.1111/bcp.12987
22. Hossian A, Sajib MS, Tullar PE, Mikelis CM, Mattheolabakis G. Multipronged activity of combinatorial miR-143 and miR-506 inhibits lung cancer cell cycle progression and angiogenesis in vitro. Sci Rep. 2018;8(1):10495. doi:10.1038/s41598-018-28872-2
23. Hossian A, Muthumula CMR, Sajib MS, et al. Analysis of combinatorial miRNA treatments to regulate cell cycle and angiogenesis. J Vis Exp. 2019;145.
24. Yan A, Von Dem Bussche A, Kane AB, Hurt RH. Tocopheryl polyethylene glycol succinate as a safe, antioxidant surfactant for processing carbon nanotubes and fullerenes. Carbon N Y. 2007;45(13):2463–2470. doi:10.1016/j.carbon.2007.08.035
25. Yang C, Wu T, Qi Y, Zhang Z. Recent advances in the application of Vitamin E TPGS for drug delivery. Theranostics. 2018;8(2):464–485. doi:10.7150/thno.22711
26. Tang GP, Zeng JM, Gao SJ, et al. Polyethylene glycol modified polyethylenimine for improved CNS gene transfer: effects of PEGylation extent. Biomaterials. 2003;24(13):2351–2362. doi:10.1016/S0142-9612(03)00029-2
27. Smith SA, Selby LI, Johnston APR, Such GK. The endosomal escape of nanoparticles: toward more efficient cellular delivery. Bioconjug Chem. 2019;30(2):263–272. doi:10.1021/acs.bioconjchem.8b00732
28. Freeman EC, Weiland LM, Meng WS. Modeling the proton sponge hypothesis: examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J Biomater Sci Polym Ed. 2013;24(4):398–416. doi:10.1080/09205063.2012.690282
29. Sunshine JC, Peng DY, Green JJ. Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol Pharm. 2012;9(11):3375–3383. doi:10.1021/mp3004176
30. Kafil V, Omidi Y. Cytotoxic impacts of linear and branched polyethylenimine nanostructures in a431 cells. Bioimpacts. 2011;1(1):23–30. doi:10.5681/bi.2011.004
31. Aldawsari HM, Dhaliwal HK, Aljaeid BM, Alhakamy NA, Banjar ZM, Amiji MM. Optimization of the conditions for plasmid DNA delivery and transfection with self-assembled hyaluronic acid-based nanoparticles. Mol Pharm. 2019;16(1):128–140. doi:10.1021/acs.molpharmaceut.8b00904
32. Di Gioia S, Conese M. Polyethylenimine-mediated gene delivery to the lung and therapeutic applications. Drug Des Devel Ther. 2009;2:163–188. doi:10.2147/dddt.s2708
33. Liu S, Huang W, Jin MJ, et al. High gene delivery efficiency of alkylated low-molecular-weight polyethylenimine through gemini surfactant-like effect. Int J Nanomedicine. 2014;9:3567–3581. doi:10.2147/IJN.S64554
34. Jeong GJ, Byun HM, Kim JM, et al. Biodistribution and tissue expression kinetics of plasmid DNA complexed with polyethylenimines of different molecular weight and structure. J Control Release. 2007;118(1):118–125. doi:10.1016/j.jconrel.2006.12.009
35. Lee BR, Jo E, Yoon HY, et al. Nonimmunogenetic viral capsid carrier with cancer targeting activity. Adv Sci (Weinh). 2018;5(8):1800494. doi:10.1002/advs.201800494
36. Ma Y, Mou Q, Zhu L, et al. Polygemcitabine nanogels with accelerated drug activation for cancer therapy. Chem Commun (Camb). 2019;55(46):6603–6606. doi:10.1039/C9CC01506J
37. Mou Q, Ma Y, Pan G, et al. DNA trojan horses: self-assembled floxuridine-containing DNA polyhedra for cancer therapy. Angew Chem Int Ed Engl. 2017;56(41):12528–12532. doi:10.1002/anie.201706301
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.