")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Nuclear Pore Glycoprotein 62 Genetic Variant rs9523 is Associated with Clinical Outcomes of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Lung Adenocarcinoma Patients
Authors Park JE, Hong MJ, Lee SY, Lee JH, Choi JE, Kang HG, Do SK, Jeong JY , Shin KM, Lee WK, Choi SH, Lee YH, Seo HW, Yoo SS, Lee J , Cha SI, Kim CH, Park JY
Received 14 July 2021
Accepted for publication 18 September 2021
Published 1 October 2021 Volume 2021:14 Pages 1291—1302
DOI https://doi.org/10.2147/PGPM.S329055
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Ji Eun Park,1,* Mi Jeong Hong,2,3,* Shin Yup Lee,1,4 Jang Hyuck Lee,2,4 Jin Eun Choi,2,3 Hyo-Gyoung Kang,2,3 Sook Kyung Do,2,3 Ji Yun Jeong,5 Kyung Min Shin,6 Won Kee Lee,7 Sun Ha Choi,1 Yong Hoon Lee,1 Hye won Seo,1 Seung Soo Yoo,1 Jaehee Lee,1 Seung Ick Cha,1 Chang Ho Kim,1 Jae Yong Park1,2
1Department of Internal Medicine, School of Medicine, Kyungpook National University, Daegu, Republic of Korea; 2Department of Biochemistry, School of Medicine, Kyungpook National University, Daegu, Republic of Korea; 3Cell and Matrix Research Institute, School of Medicine, Kyungpook National University, Daegu, Republic of Korea; 4BK21 Plus KNU Biomedical Convergence Program, Department of Biomedical Science, Kyungpook National University, Daegu, Republic of Korea; 5Department of Pathology, School of Medicine, Kyungpook National University, Daegu, Republic of Korea; 6Department of Radiology, School of Medicine, Kyungpook National University, Daegu, Republic of Korea; 7Department of Medical Informatics, School of Medicine, Kyungpook National University, Daegu, Republic of Korea
*These authors contributed equally to this work
Correspondence: Shin Yup Lee; Jae Yong Park
Department of Internal Medicine, School of Medicine, Kyungpook National University, Daegu, Republic of Korea
Tel +82-53-200-2632
Fax +82-53-200-2027
Email [email protected]; [email protected]
Introduction: Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have represented the prototype of targeted therapy in NSCLC. Patients with EGFR-mutant lung adenocarcinoma extract an extraordinary clinical benefit from EGFR-TKIs. However, the extent and duration of these responses are heterogeneous, suggesting the existence of genetic modifiers affecting an individual’s response to TKIs. We investigated whether genetic variants in miRNA binding sites are associated with the clinical outcome of EGFR-TKIs in lung adenocarcinoma patients.
Methods: One hundred SNPs at miRNA binding sites in cancer-related genes were selected for the analysis using the crosslinking, ligation and sequencing of hybrids (CLASH) and CancerGenes database. qRT-PCR and luciferase assays were conducted to evaluate the functional relevance of the SNPs.
Results: NUP62 rs9523A>G were significantly associated with worse response to EGFR-TKIs, overall survival (OS), and progression-free survival (PFS). The other three SNPs (DVL2 rs2074216G>A, ARF1 rs11541557G>T, and UHRF1 rs2261988C>A) were significantly associated with worse OS and PFS. The rs9523A>G was significantly associated with decreased NUP62 expression in tumor tissues. In addition, a significantly decreased luciferase activity was noted in NUP62 rs9523 G allele compared to A allele.
Conclusion: Genetic variants in miRNA binding sites, especially NUP62 rs9523A>G, may be useful in predicting the clinical outcomes of EGFR-mutant lung adenocarcinoma patients treated with EGFR-TKIs.
Keywords: lung adenocarcinoma, EGFR-TKI, clinical outcome, miRNA binding site, polymorphism
Introduction
During the last decades, pronounced development regarding cancer genomics and molecular biology has proposed a fundamental change in the paradigm of care in NSCLC, including targeted therapy and immunotherapy. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have represented the prototype of targeted therapy in lung adenocarcinoma. EGFR-TKIs have an extraordinary effect in patients with EGFR-mutant NSCLC and prolong progression-free survival (PFS) significantly compared to conventional platinum-based chemotherapy.1–5 However, those who initially respond to EGFR-TKIs will eventually develop acquired resistance in approximately 12 months.1–5 Intensive researches have focused on the mechanisms of acquired resistance to EGFR-TKI to identify several mechanisms such as T790M gatekeeper mutation6 which explains the resistance in almost half of cases, mesenchymal–epithelial transition (MET) amplification,7 and transformation into small-cell lung cancer,8 among others.
Meanwhile, although most EGFR-mutant tumors exhibit dramatic initial response to EGFR-TKIs, the magnitude and duration of the responses varies considerably, suggesting the existence of genetic factors modifying an individual’s response to EGFR-TKIs. Primary resistance occurs in approximately 20% of patients with EGFR-mutated NSCLC.1–5 Several coexisting genetic variations have been suggested for the mechanism of primary resistance to EGFR-TKIs, including de novo EGFR T790M mutation,9 MET amplification,10 PTEN loss,11 KRAS mutations,12 and germline variation such as BIM deletion polymorphism.13 In addition, even among the patients with EGFR mutation who achieve initial response to EGFR-TKIs, the duration of response varies widely. However, the underlying mechanism has been largely unknown.
MicroRNAs (miRNAs) play important roles in various biological functions, such as cell proliferation and survival, DNA repair, and immune response.14,15 Evidence indicates that miRNAs are critically involved in the development and progression of diverse human cancers.15,16 Studies have suggested that single nucleotide polymorphisms (SNPs) at miRNA target sites are associated with the risk and the prognosis of many types of cancer, including lung cancer.17–20 In contrast to the computational prediction methods for miRNA target recognition, which were developed to predict miRNA-mRNA binding based primarily on the complementarity to seed sequence, crosslinking, ligation, and sequencing of hybrids (CLASH) provided direct experimental observation of transcriptome-wide miRNA-target pairs, revealing that the interactions occurred more frequently in coding sequence than 3ʹ UTR and the majority of miRNA-target bindings were noncanonical.21 Based on the important roles of miRNA network in carcinogenesis, we hypothesized that polymorphisms at miRNA target sites may influence miRNA-mRNA binding and consequently the expression of target genes, thereby influencing the clinical outcomes in EGFR-mutant lung adenocarcinoma patients who are treated EGFR-TKIs. To test this hypothesis, we selected SNPs at miRNA binding sites using CLASH data and evaluated their association with the clinical outcome of EGFR-TKIs in EGFR-mutant lung adenocarcinoma patients.
Materials and Methods
Study Populations
In this study, 217 lung adenocarcinoma patients with available genomic DNA samples, who were treated with EGFR-TKI at Kyungpook National University Hospital (KNUH) in Daegu, Korea, between March 2007 and July 2015, were enrolled. The patients had stage III/IV or recurred disease after surgery. Among 217 patients, 169 had positive EGFR mutation status. Since EGFR mutation analysis was not widely adopted in the early part of this period, 48 patients with unknown EGFR mutation status who had not progressed for longer than 6 months on EGFR-TKIs as a second- or further-line therapy were included in this study.22 Patients received either first-generation (erlotinib, gefitinib) or second-generation (afatinib) TKIs until disease progression, occurrence of major toxicity, or according to the patient’s or physician’s decision. Clinical data, including age at diagnosis, gender, smoking status, clinical staging, performance status, presence of weight loss, EGFR mutation status, were obtained retrospectively by reviewing medical records. Assessment of tumor response was performed by computed tomography, and responses were assessed using Response Evaluation Criteria in Solid Tumors.23 The best overall response was reported and patients with a complete response (CR) or a partial response (PR) were defined as responders, and patients with stable disease (SD) or progressive disease (PD) were defined as nonresponders. Genomic DNA samples from the patients were provided by the National Biobank of Korea, KNUH, which is supported by the Ministry of Health, Welfare and Family Affairs. This study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines and was approved by the institutional review board (KNUCH 2019-04-014). All patients provided written informed consent.
SNP Selection and Genotyping
Potentially functional polymorphisms at miRNA target sites were assessed using PolymiRTS database 3.0 (http://compbio.uthsc.edu/miRSNP),24 and 24,027 SNPs at experimentally validated miRNA target sites were selected by downloading data from CLASH experiment, which has been integrated into PolymiRTS database 3.0. Among these, 1574 SNPs in cancer-related genes were selected using a list of cancer genes from the CancerGenes database (http://cbio.mskcc.org/cancergenes).25 Finally, 100 SNPs with a minor allele frequency ≥ 0.05 in the HapMap JPT were collected after excluding those in linkage disequilibrium (LD, r2 ≥ 0.8). Genotyping was performed using the iPLEX® Assay and MassARRAY® System (Agena Bioscience, San Diego, CA, USA).
RNA Preparation and Quantitative Reverse Transcription-PCR (qRT-PCR)
Nucleoporin 62 (NUP62), disheveled 2 (DVL2), ADP-ribosylation factor 1 (ARF1), and ubiquitin-like with PHD and ring finger domains 1 (UHRF1) mRNA expression levels were measured by quantitative reverse transcription-PCR in tumor and corresponding normal lung tissues of lung adenocarcinoma patients who underwent surgical resection in Kyungpook National University Hospital (n = 82). Total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA, USA). Real time-PCR was performed for each gene and beta-actin with QuantiFast SYBR® Green PCR Master Mix (Qiagen, Hilden, Germany) in a LightCycler 480 (Roche Applied Science, Mannheim, Germany) using the following primers: NUP62 forward, 5′-AGAAATCTTCCCAAGGCTGC-3′; NUP62 reverse, 5′-GTGCCTCCAAAATTAAACCCG-3′; DVL2 forward, 5′-GCGAGTTCTTTGTGGATGTTATG-3′; DVL2 reverse, 5′-ACAATCTCCTGTATGGCAGC-3′; ARF1 forward, 5′-ACAGGAACTGGTACATTCAGG-3′; ARF1 reverse, 5′-CACATGAGAGTAAAGCAGAGGG-3′; UHRF1 forward 5′-GAAACTCACCAACACCAACAG-3′; UHRF1 reverse, 5′-TGCTATTCTTGCCACCCTTG-3′; beta-actin forward, 5′-TTGTTACAGGAAGTCCCT.
TGCC-3′; beta-actin reverse, 5′-ATGCTATCACCTCCCCTGTGT-3′. The relative mRNA expression was normalized with beta-actin expression and then calculated by the 2−∆∆CT method.
Cloning of the Luciferase Reports Gene and Dual Luciferase Assay
Luciferase report assay was performed to investigate whether rs9523A>G modulates the binding of miR-1914 and therefore changes the expression of NUP62. The psiCHECKTM-2 vector (Promega, Madison, WI, USA) was used to construct luciferase reporter plasmids. NUP62 3ʹ-UTR sequence containing rs9523A or rs9523G was synthesized by PCR from human genomic DNA and cloned into the psiCHECKTM-2 vector. The psiCHECKTM-2-NUP62 constructs containing rs9523A>G were generated and co-transfected with miR-1914 into an EGFR mutant (PC9) cell line and an EGFR wild-type (H1299) cell line based on the manufacturer’s instructions. The human lung carcinoma cell lines PC9 and H1299 were purchased from Korean Cell Line Bank (KCLB, Seoul, Korea). After the incubation period, relative Renilla luciferase values were measured using the firefly luciferase activities as a normalization control.
Statistical Analysis
Hardy–Weinberg equilibrium was tested using a goodness-of-fit χ2 test with 1 degree of freedom. The genotypes for each SNP were analyzed as three-group categorical variable, and analyzed under dominant and recessive model. The association between clinical variables or genotypes and chemotherapy response was tested by odds ratio (OR) and 95% confidence intervals (CIs) using unconditional logistic regression analysis. For survival assessment, overall survival (OS) was defined as the interval between the first EGFR-TKI dose and the date of death, and progression-free survival (PFS) was defined as the duration between the initiation of EGFR-TKI and the date of objective disease progression or death. Kaplan–Meier method was used to calculate survival estimates, and the difference in OS and PFS according to different clinical variables or genotypes was compared using Log rank tests. Cox’s proportional hazard regression model was used for the multivariate survival analyses. The hazard ratio (HR) and 95% CI were also estimated. A cut-off P value of 0.05 was adopted for all statistical analyses. Statistical data were obtained using the Statistical Analysis System for Windows, version 9.4 (SAS Institute, Cary, NC, USA).
Results
Clinical characteristics and the associations with clinical outcomes are shown in Table 1. The overall response rate of EGFR-TKIs was 84.8%, and median survival time (MST) was 35.4 months (95% CI = 30.8–39.7 months) for OS and 14.3 months (95% CI = 12.1–16.9 months). Response to EGFR-TKIs was not associated with clinical variables, such as age, gender, smoking status, stage, performance status, or weight loss. Compared with patients with EGFR mutation, response rate was significantly higher in those without the mutation test results, probably because only patients who experienced treatment responses or stable disease for longer than 6 months comprised this subgroup. However, PFS was significantly better in patients with EGFR mutation than those without confirmed mutation status. Patients diagnosed with stage III/IV lung adenocarcinoma had worse OS and PFS compared with those with recurrent disease after surgery, suggesting tumor burden at the beginning of EGFR-TKI treatment may have affected the survival outcome.26 Patients with ECOG 0 performance status had better OS and PFS than those with ECOG 1–2. Patients who experienced weight loss had worse OS than those who did not.
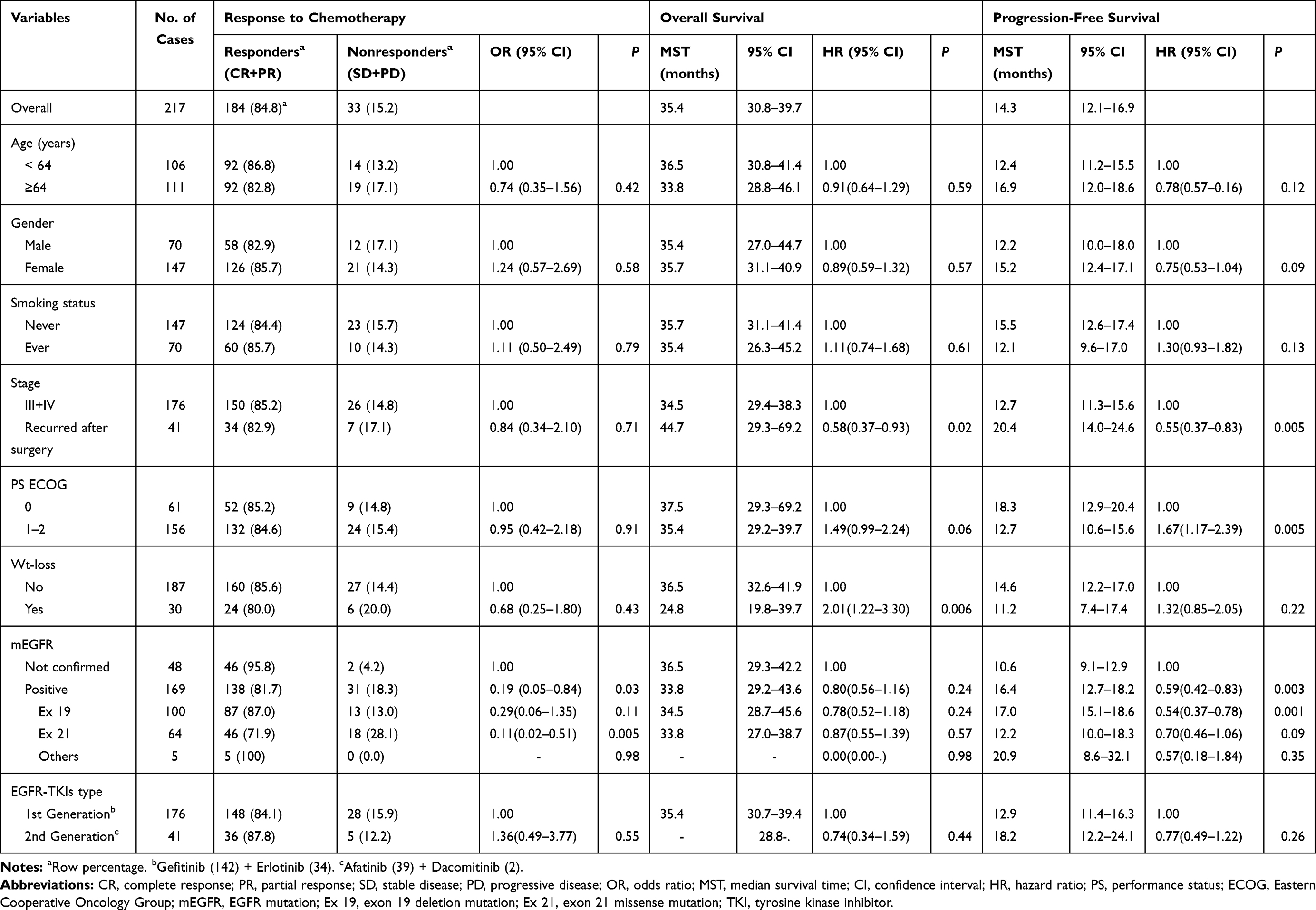 |
Table 1 Univariate Analysis for Response to Chemotherapy, Overall Survival, and Progression-Free Survival by Clinical Variables |
Among the 100 SNPs genotyped, 75 SNPs were further analyzed after excluding 2 SNPs with genotyping failure and 23 SNPs, which were deviated from the Hardy–Weinberg equilibrium (P < 0.05) or low call rates (<95%) (Supplementary Table 1). Of the 75 SNPs analyzed, 6 SNPs were significantly associated with the response to EGFR-TKIs (Table 2), 19 SNPs with OS (Table 3), and 13 SNPs with PFS (Table 4), respectively. Among these SNPs, NUP62 rs9523A>G were significantly associated with worse response to TKIs (adjusted odds ratio [aOR] = 0.26, 95% confidence interval [CI] = 0.11–0.64, P = 0.003), worse OS (adjusted hazard ratio [aHR] = 1.98, 95% confidence interval [CI] = 1.27–3.08, P = 0.003), and worse PFS (aHR = 1.59, 95% CI = 1.05–2.40, P = 0.029). Another three SNPs - DVL2 rs2074216G>A, ARF1 rs11541557G>T, and UHRF1 rs2261988C>A - were significantly associated with worse OS (aHR = 2.19, 95% CI = 1.32–3.66, P = 0.003; aHR = 1.92, 95% CI = 1.24–2.97, P = 0.004; and aHR = 3.8, 95% CI = 1.11–13.04, P = 0.034, respectively) and worse PFS (aHR = 1.72, 95% CI = 1.07–2.76, P = 0.025; aHR = 2.5, 95% CI = 1.65–3.78, P < 0.0001; and aHR = 6.38, 95% CI = 1.85–22.06, P = 0.003, respectively) in multivariate analysis adjusted for age, gender, smoking status, stage, performance status, and weight loss (Table 5 and Figure 1). Next, we performed an exploratory analysis investigating the combined effects of the 4 SNPs. We considered the rs9523 GG, rs2074216 AA, rs11541557 GT+TT, and rs2261988 AA genotypes as bad genotypes and then evaluated their combined effects by grouping the patients based on the number of bad genotypes. Compared with the reference group that had no bad genotypes, OS and PFS decreased in a dose-dependent manner as the number of bad genotypes increased (Ptrend = <0.0001 for both) and those with at least one bad genotype had HR of 2.38 for OS and 1.93 for PFS (Supplementary Table 2).
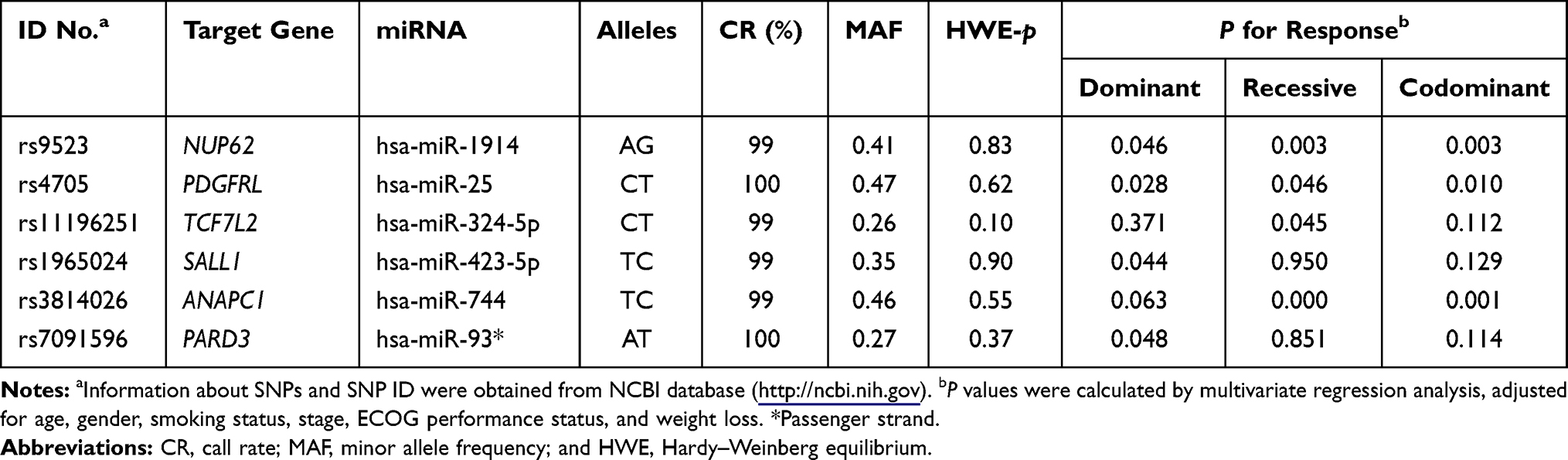 |
Table 2 Summary of 6 SNPs and the Response to EGFR-TKIs |
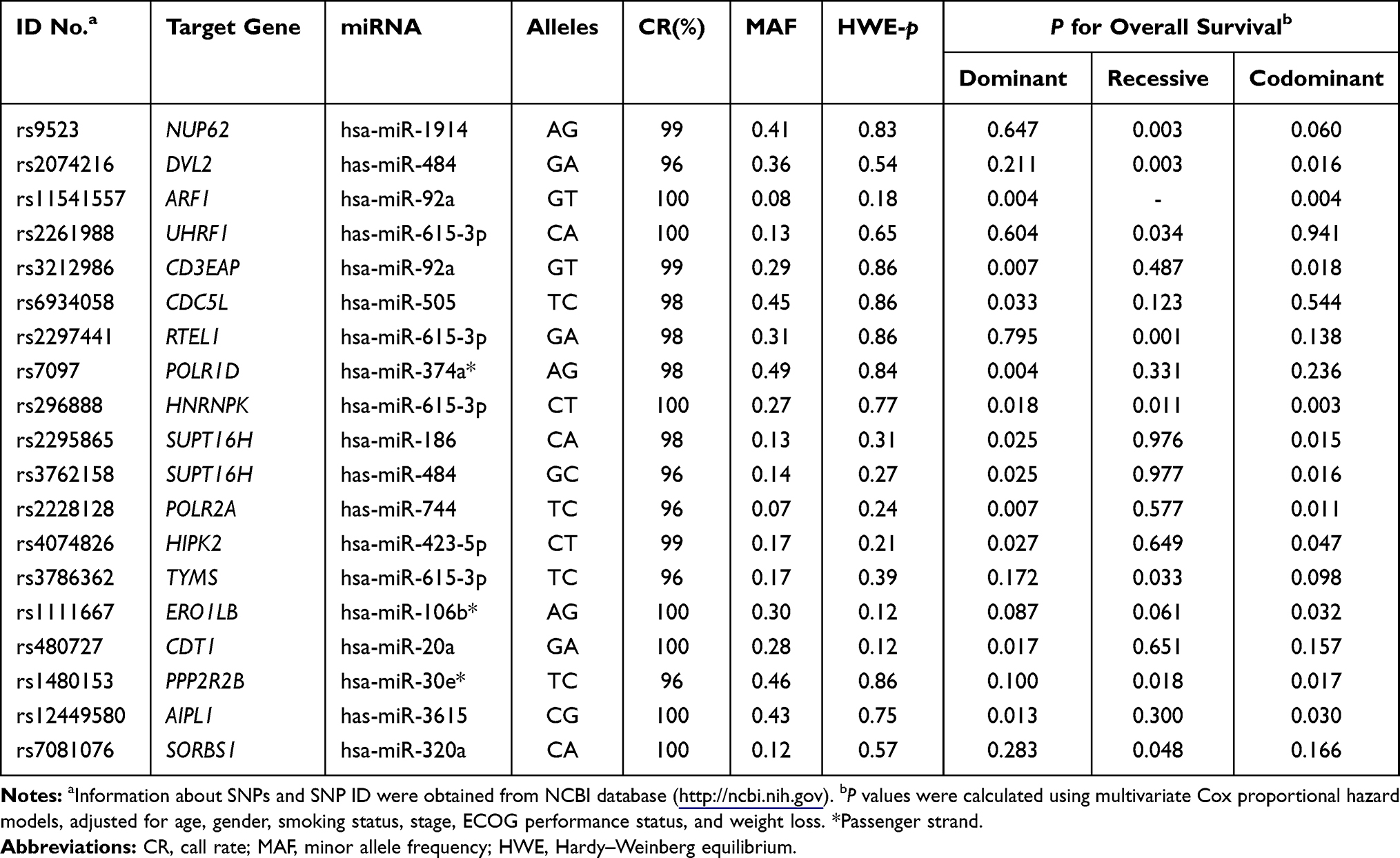 |
Table 3 Summary of 19 SNPs and Overall Survival |
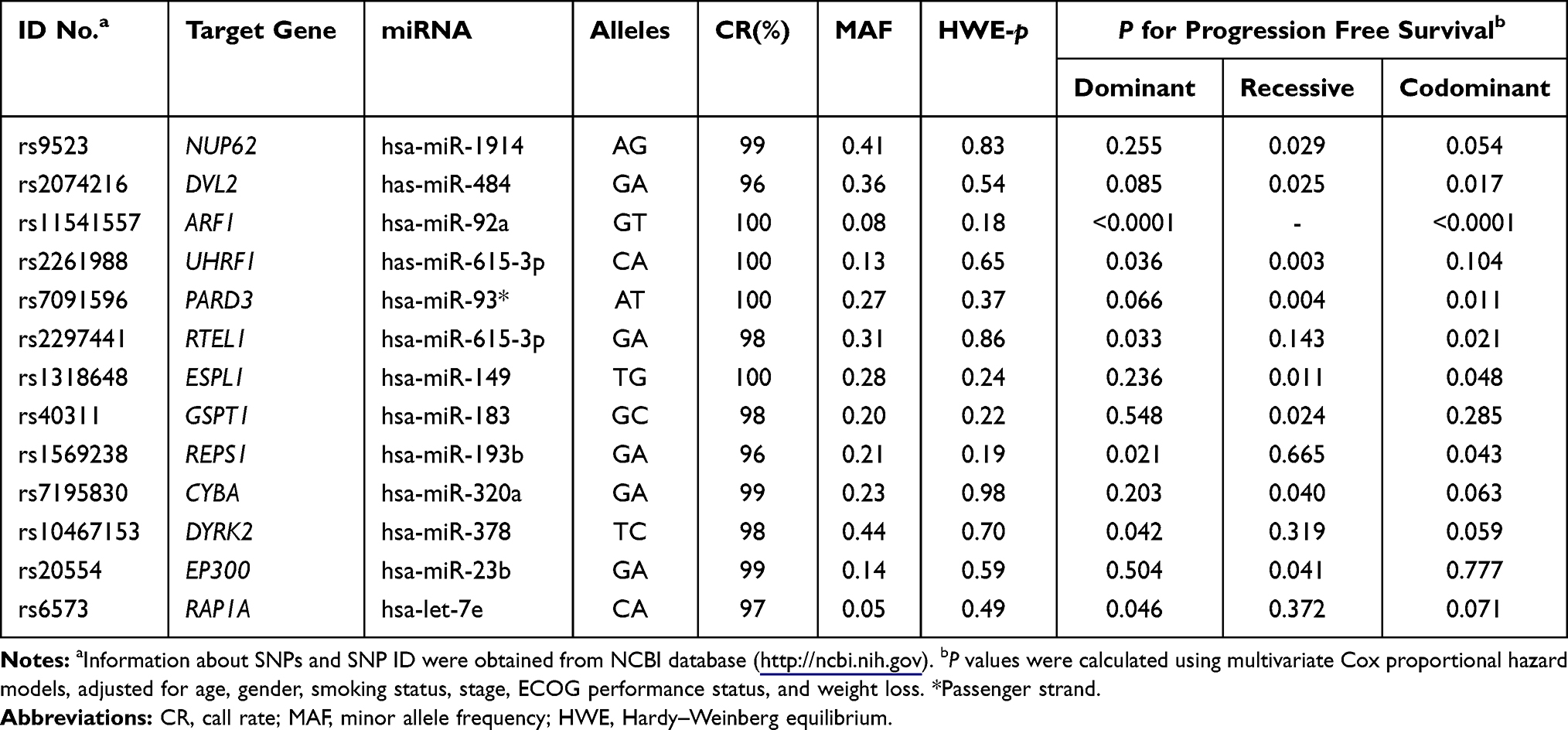 |
Table 4 Summary of 13 SNPs and Progression-Free Survival |
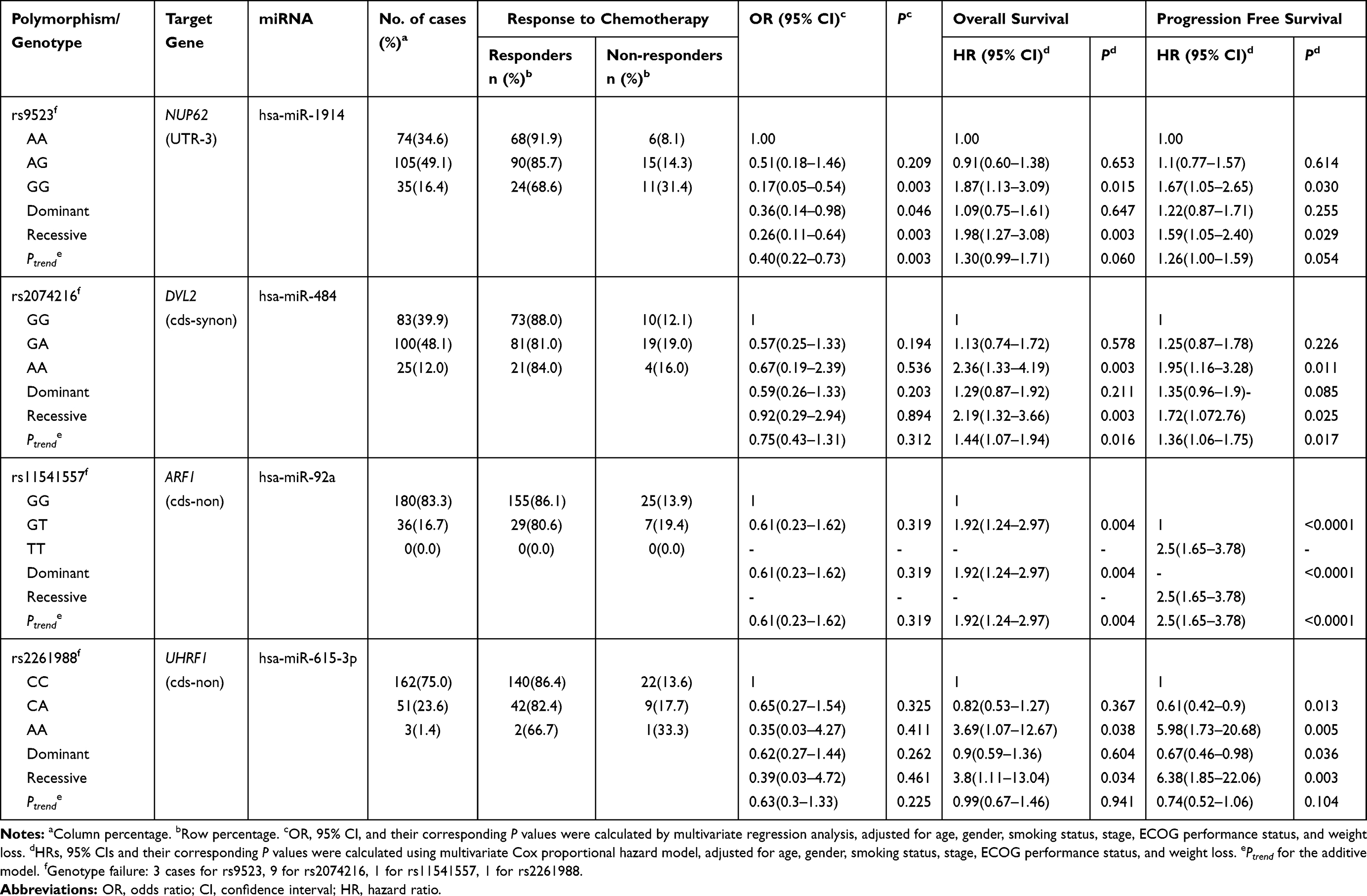 |
Table 5 Genotypes of Polymorphisms and Their Associations with Clinical Outcomes of EGFR-TKIs |
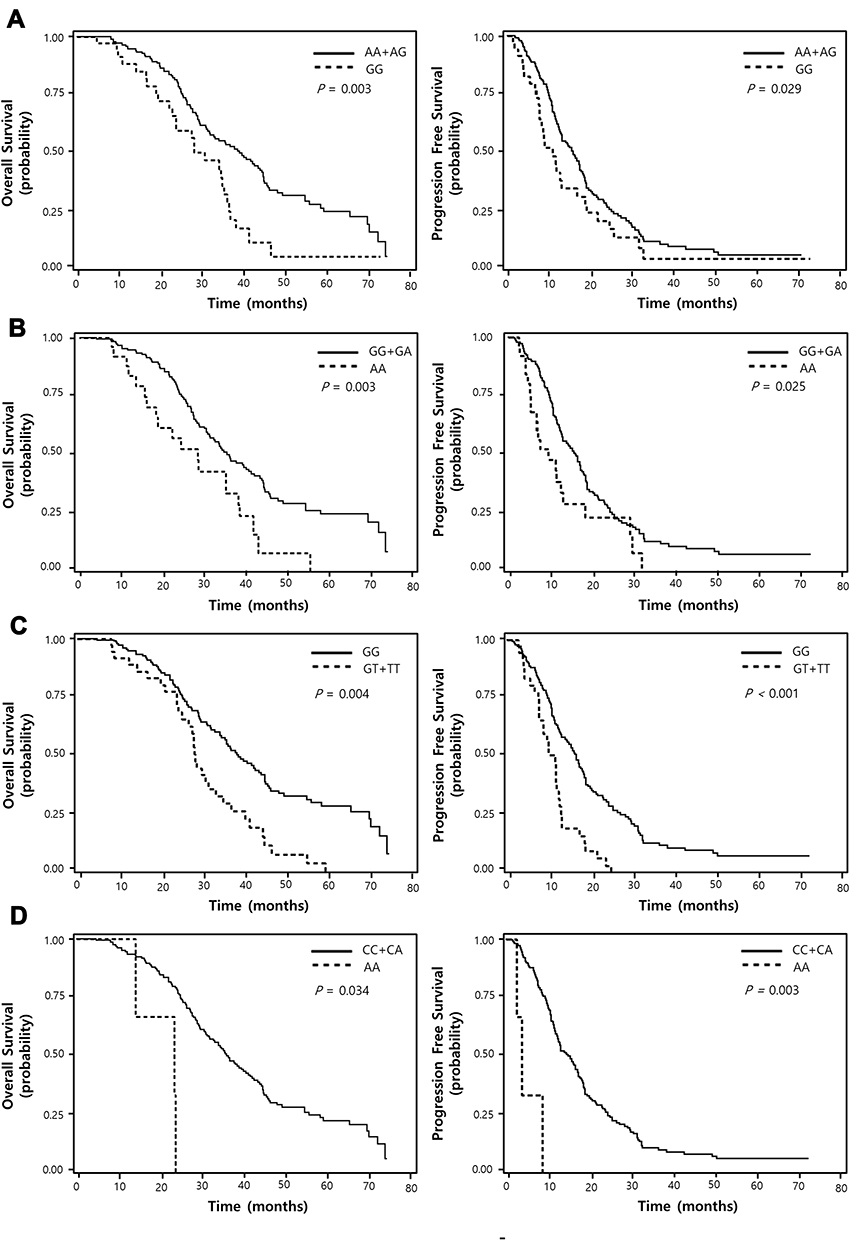 |
Figure 1 Overall survival and progression-free survival curves according to NUP62 rs9523A>G (A), DVL2 rs2074216 (B), ARF1 rs11541557 (C), and UHRF1 rs2261988 (D) genotypes. P values by multivariate Cox proportional hazard models. |
To evaluate the functional relevance of NUP62 rs9523A>G, DVL2 rs2074216G>A, ARF1 rs11541557G>T, and UHRF1 rs2261988C>A, we compared the relative expression level of NUP62, DVL2, ARF1, and UHRF1 mRNA in tumor and paired non-malignant lung tissues. The expression level of NUP62, ARF1, and UHRF1was significantly higher in tumor tissues than in non-malignant lung tissues (P = 0.011, P = 0.044, and P = 2×10−5, respectively), but there was no significant difference in DVL2 expression level between tumor and normal lung tissues (Figure 2A). According to the genotypes, NUP62 rs9523A>G was significantly related with decreased NUP62 expression in tumor tissues (Ptrend = 0.016, and P = 0.043 under recessive model; Figure 2B). When divided into the high and low NUP62 expression groups based on the median expression level in tumor tissues, the survival outcome of the low NUP62 mRNA expression group was worse than that of the high-expression group (P = 0.019, Figure 2C). Next, we evaluated the effect of rs9523A>G on miR-1914 binding and NUP62 gene expression using a dual-luciferase reporter assay. psiCHECKTM-2-NUP62 constructs containing rs9523A>G were generated and co-transfected with miR-1914 into PC9 and H1299 cells. As shown in Figure 3, the Renilla luciferase activity was significantly decreased in NUP62 rs9523 G allele compared to A allele (P = 0.004, and P = 0.04, respectively). This result implicates that rs9523A>G in 3ʹUTR of NUP62 gene modulates the miR-1914 binding and consequently suppresses the expression of NUP62.
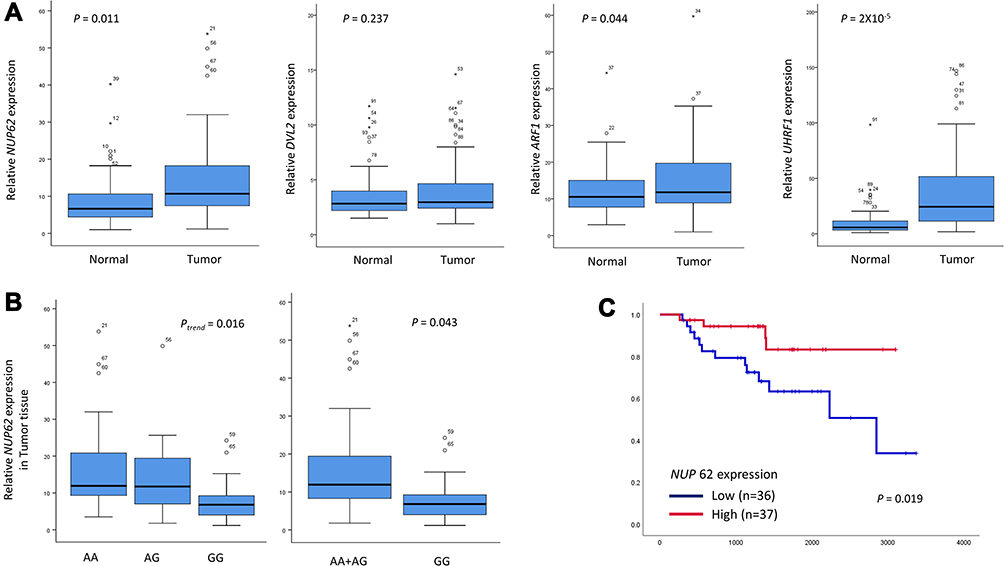 |
Figure 2 The mRNA expression levels of NUP62, DVL2, ARF1, and UHRF1 in tumor and corresponding non-malignant lung tissues (A), NUP62 mRNA expression level according to rs9523A>G genotypes (24AA, 30AG, and 14GG) in tumor tissues (B), and the Kaplan-Meier plot for overall survival according to the expression level of NUP62 (C). The horizontal lines within the boxes represent median values; the upper and lower boundaries of the boxes represent 75th and 25th percentiles, respectively; the upper and lower bars represent the largest and smallest observed values, respectively, except outliers. Circles are the outliers, and asterisks are the extreme outliers. P values by Student’s t-test, trend test, and Log rank test. |
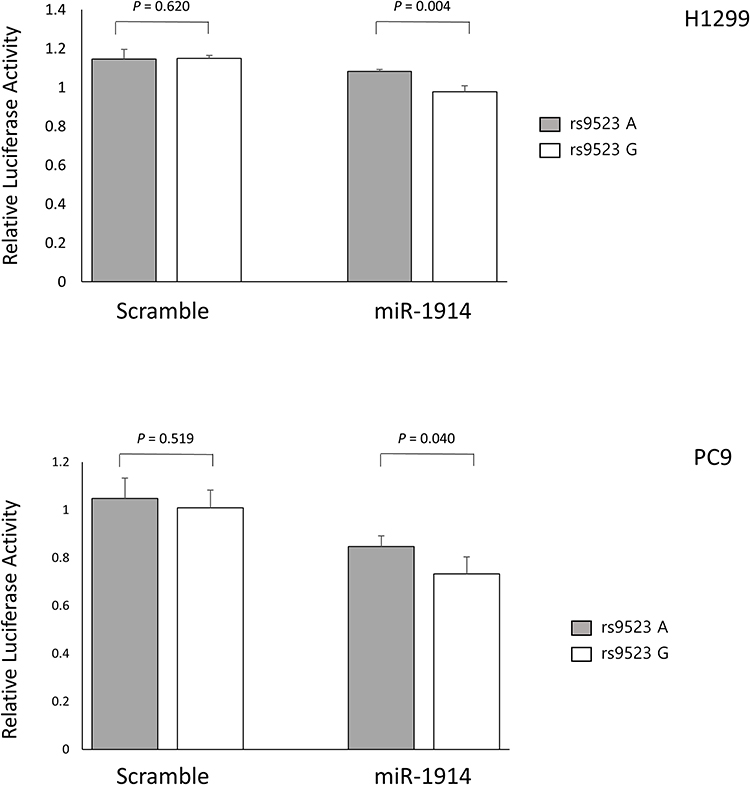 |
Figure 3 Functional analysis of NUP62 rs9523A>G by dual luciferase reporter assay. Renilla luciferase assay for the effect of miRNA binding on rs9523A>G using H1299 and PC9 cells. Renilla luciferase activity was normalized to firefly luciferase activity and data are presented relative to the Mock control. Each bar represents mean ± SE. P values by Student’s t-test. |
Discussion
In the present study, we investigated whether polymorphisms in miRNA binding sites have an impact on clinical outcomes in lung adenocarcinoma patients who were treated with EGFR-TKIs. This study showed that NUP62 rs9523A>G could predict worse response to EGFR-TKIs, PFS, and OS. In addition, DVL2 rs2074216G>A, ARF1 rs11541557G>T, and UHRF1 rs2261988C>A were associated with PFS and OS. Functional analysis using clinical samples and in vitro assays supported the biological relevance of NUP62 rs9523A>G. Those four SNPs, particularly NUP62 rs9523A>G, may be useful in predicting the clinical outcomes in patients treated with EGFR-TKIs.
Nucleoporins are structural components of the nuclear pore complex (NPC), which regulates transport of a wide array of macromolecules including mRNA between nucleoplasm and cytoplasm. NPC also plays a role in transcriptional regulation, chromatin silencing, and DNA damage repair.27 Export of mRNAs from the nucleus to the cytoplasm through NPC is a key regulatory step in protein expression.28 It serves as a surveillance mechanism to sort out aberrant mRNAs, and controls translation and consequently the response to extracellular signals by permitting altered flow of specific mRNAs into the cytoplasm.28 RNA export factors and NPC components regulate the export of selected mRNAs involved in nearly all aspects of malignancy, such as survival, proliferation, metastases, and invasion.28,29 Aberrant mRNA export associated with altered nucleoporin expression or function has been linked to cancers.27,28 Nucleoporin 62 (NUP62), a protein complex that belongs to the class of nucleoporins, was highly expressed in squamous cell carcinomas, including head, neck and cervix, and was a key regulator of cell proliferation and differentiation via controlling the nuclear transport of p63.30 The expression of NUP62 was notably increased in specimens of advanced prostate cancer by immunohistochemistry.31 However, a study showed that NUP62 expression was decreased in ovarian carcinomas and that the partial knockdown of NUP62 confers cisplatin resistance in high-grade ovarian carcinoma cells,32 suggesting the dysregulation of nucleoporins may be cell type- and context-specific.
In this study, NUP62 rs9523A>G was associated with worse response to EGFR-TKIs, PFS, and OS. The luciferase assay showed that rs9523A-to-G change in 3ʹUTR of NUP62 led to altered binding efficiency of miR-1914, causing decreased NUP62 mRNA expression. Consistently, NUP62 rs9523A>G was significantly associated with decreased NUP62 expression level in lung tumor tissues. Interestingly, the decreased NUP62 expression in resected tumor samples is correlated with poor OS after surgery, collectively suggesting a potential tumor suppressor role of NUP62. Because export of mRNAs from the nucleus to the cytoplasm through NPC is a key regulatory step in protein expression, NUP62, a component of NPC, may play a role in regulating protein expression involved in survival, proliferation, metastases, and invasion of cancer cells in response to aberrant EGFR signals in lung cancer with activating EGFR mutation. Therefore, altered expression of NUP62 may modulate the effect of EGFR-TKI. Based on our results, it can be speculated that decreased NUP62 expression may have a negative impact on the effect of EGFR-TKI. Previous studies suggested a potential mechanism for the association between NPC and resistance to EGFR-TKIs. It was reported that EGFR translocates from the cell surface to the nucleus through NPCs in response to EGF,33,34 and that nuclear localized EGFR was associated with increased resistance to anti-EGFR therapies.35 Wang et al showed that down-regulation of NUP62 expression inhibited EGF-dependent EGFR translocation,34 suggesting that decreased NUP62 expression may reduce resistance to EGFR-TKI mediated by nuclear translocation of EGFR, which seems to conflict with our results. Therefore, the molecular mechanism of the potential role of NUP62 in the resistance to EGFR-TKIs is required to be further evaluated in the future studies.
Although most EGFR-mutant tumors exhibit dramatic initial response to EGFR-TKIs, the magnitude and duration of the responses varies widely even among responders, leading to considerable variation in survival outcomes. The identification of patients who may experience early progression after the EGFR-TKI treatment is important for optimizing personalized therapeutic strategies. Possible mechanisms for these heterogeneous clinical outcomes include clinical characteristics,36 tumor heterogeneity,37 genetic variants,38–40 or various drug-resistance mechanisms.9–13 Studies reported that several genetic variants could predict clinical outcomes in patients treated with EGFR-TKIs, including polymorphisms in EGFR gene, TGF-β pathway genes, or BIM deletion polymorphism.13,38–40 Our result suggests that four genetic polymorphisms at miRNA binding sites, especially NUP62 rs9523A>G, may be useful in predicting the PFS and OS after EGFR-TKIs. Because the duration of response to EGFR-TKIs is not predictable for individual patients even if the median PFS of 12 months from clinical trials is often referred to, the bad genotypes of those variants may be used as minor resistance factors helping the clinicians to predict the therapeutic course and to make a closer monitoring plan for disease progression. For potential clinical applicability, further studies are required to validate our findings.
Several limitations should be considered in this study. First, the EGFR mutation status was not assessed for all enrolled subjects because EGFR mutation test was not widely adopted in the early part of the enrollment period. Therefore, patients with unknown EGFR mutation status who experienced treatment responses or stable disease for longer than 6 months with EGFR-TKI as a second-line therapy were enrolled.22 However, there was no difference in genotype distribution between those with mutant EGFR and those with unknown EGFR status (data not shown). Second, because osimertinib was not available to many patients upon resistance to EGFR-TKIs, we could not analyze the role of osimertinib in patients who experienced disease progression after EGFR-TKI treatment, which could have had significant impact on overall survival.41 Third, we did not conduct experiments to confirm the difference in the efficacy of EGFR-TKIs according to the genotypes. Additional experiments such as CRISPR-Cas9 to generate PC9 cells with rs9523AA and PC9 cells with rs9523GG genotype may help reveal the different efficacy of EGFR-TKIs between A and G alleles.
In conclusion, this study shows that four SNPs in miRNA binding sites, especially NUP62 rs9523A>G, may be useful for predicting the clinical outcomes of EGFR-mutant lung adenocarcinoma patients treated with EGFR-TKIs. Further studies including larger population with various ethnicity are required to validate our findings.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (NRF-2020R1A5A2017323), and by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2019R1I1A3A01061137).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi:10.1056/NEJMoa0810699
2. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised Phase 3 trial. Lancet Oncol. 2012;13:239–246. doi:10.1016/S1470-2045(11)70393-X
3. Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of Afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–3334. doi:10.1200/JCO.2012.44.2806
4. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388.
5. Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–222. doi:10.1016/S1470-2045(13)70604-1
6. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi:10.1056/NEJMoa044238
7. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi:10.1126/science.1141478
8. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi:10.1126/scitranslmed.3002003
9. Su KY, Chen HY, Li KC, et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012;30:433–440. doi:10.1200/JCO.2011.38.3224
10. Cappuzzo F, Janne PA, Skokan M, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol. 2009;20:298–304. doi:10.1093/annonc/mdn635
11. Sos ML, Koker M, Weir BA, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69:3256–3261. doi:10.1158/0008-5472.CAN-08-4055
12. Takeda M, Okamoto I, Fujita Y, et al. De novo resistance to epidermal growth factor receptor-tyrosine kinase inhibitors in EGFR mutation-positive patients with non-small cell lung cancer. J Thorac Oncol. 2010;5:399–400. doi:10.1097/JTO.0b013e3181cee47e
13. Ng KP, Hillmer AM, Chuah CT, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18:521–528. doi:10.1038/nm.2713
14. Griffiths-Jones S, Grocock RJ, van Dongen S, et al. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–144. doi:10.1093/nar/gkj112
15. Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi:10.1038/nrc1840
16. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi:10.1038/nrc1997
17. Chin LJ, Ratner E, Leng S, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3ʹ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–8540. doi:10.1158/0008-5472.CAN-08-2129
18. Teo MT, Landi D, Taylor CF, et al. The role of microRNA-binding site polymorphisms in DNA repair genes as risk factors for bladder cancer and breast cancer and their impact on radiotherapy outcomes. Carcinogenesis. 2012;33:581–586. doi:10.1093/carcin/bgr300
19. Xu J, Yin Z, Gao W, et al. Genetic variation in a microRNA-502 minding site in SET8 gene confers clinical outcome of non-small cell lung cancer in a Chinese population. PLoS One. 2013;8:e77024. doi:10.1371/journal.pone.0077024
20. Hong MJ, Lee SY, Choi JE, et al. A genetic variation in microRNA target site of ETS2 is associated with clinical outcomes of paclitaxel-cisplatin chemotherapy in non-small cell lung cancer. Oncotarget. 2016;7:15948–15958. doi:10.18632/oncotarget.7433
21. Helwak A, Kudla G, Dudnakova T, et al. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. doi:10.1016/j.cell.2013.03.043
22. Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28:357–360. doi:10.1200/JCO.2009.24.7049
23. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–247. doi:10.1016/j.ejca.2008.10.026
24. Bhattacharya A, Ziebarth JD, Cui Y. PolymiRTS Database 3.0: linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014;42:D86–91. doi:10.1093/nar/gkt1028
25. Higgins ME, Claremont M, Major JE, et al. CancerGenes: a gene selection resource for cancer genome projects. Nucleic Acids Res. 2007;35:D721–726. doi:10.1093/nar/gkl811
26. Lee JK, Shin JY, Kim S, et al. Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non-small-cell lung cancer harboring TKI-sensitive EGFR mutations: an exploratory study. Ann Oncol. 2013;24:2080–2087. doi:10.1093/annonc/mdt127
27. Simon DN, Rout MP. Cancer and the nuclear pore complex. Adv Exp Med Biol. 2014;773:285–307.
28. Borden KLB. The Nuclear Pore Complex and mRNA Export in Cancer. Cancers. 2020;13:521.
29. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. cell. 2004;116:281–297. doi:10.1016/S0092-8674(04)00045-5
30. Hazawa M, Lin DC, Kobayashi A, et al. ROCK-dependent phosphorylation of NUP62 regulates p63 nuclear transport and squamous cell carcinoma proliferation. EMBO Rep. 2018;19:73–88. doi:10.15252/embr.201744523
31. Karacosta LG, Kuroski LA, Hofmann WA, et al. Nucleoporin 62 and Ca(2+)/calmodulin dependent kinase kinase 2 regulate androgen receptor activity in castrate resistant prostate cancer cells. Prostate. 2016;76:294–306. doi:10.1002/pros.23121
32. Kinoshita Y, Kalir T, Rahaman J, et al. Alterations in nuclear pore architecture allow cancer cell entry into or exit from drug-resistant dormancy. Am J Pathol. 2012;180:375–389. doi:10.1016/j.ajpath.2011.09.024
33. Lo H-W, Ali-Seyed M, Wu Y, et al. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin β1 and CRM1. J Cell Biochem. 2006;98:1570–1583. doi:10.1002/jcb.20876
34. Wang Y-N, Yamaguchi H, Huo L, et al. The translocon Sec61β localized in the inner nuclear membrane transports membrane-embedded EGF receptor to the nucleus. J Biol Chem. 2010;285:38720–38729. doi:10.1074/jbc.M110.158659
35. Brand TM, Iida M, Luthar N, et al. Nuclear EGFR as a molecular target in cancer. Radiother Oncol. 2013;108:370–377. doi:10.1016/j.radonc.2013.06.010
36. Lin J-H, Lin D, Xu L, et al. The association between clinical prognostic factors and epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) efficacy in advanced non-small-cell lung cancer patients: a retrospective assessment of 94 cases with EGFR mutations. Oncotarget. 2017;8:3412. doi:10.18632/oncotarget.13787
37. Taniguchi K, Okami J, Kodama K, et al. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci. 2008;99:929–935. doi:10.1111/j.1349-7006.2008.00782.x
38. Chang IS, Jiang SS, Yang JC, et al. Genetic modifiers of progression-free survival in never-smoking lung adenocarcinoma patients treated with first-line tyrosine kinase inhibitors. Am J Respir Crit Care Med. 2017;195:663–673. doi:10.1164/rccm.201602-0300OC
39. Zhang L, Li QX, Wu HL, et al. SNPs in the transforming growth factor-beta pathway as predictors of outcome in advanced lung adenocarcinoma with EGFR mutations treated with gefitinib. Ann Oncol. 2014;25:1584–1590. doi:10.1093/annonc/mdu172
40. Jung M, Cho BC, Lee CH, et al. EGFR polymorphism as a predictor of clinical outcome in advanced lung cancer patients treated with EGFR-TKI. Yonsei Med J. 2012;53:1128–1135. doi:10.3349/ymj.2012.53.6.1128
41. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2017;376:629–640. doi:10.1056/NEJMoa1612674
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.