Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Nrf2 expression is increased in peripheral blood mononuclear cells derived from mild–moderate ex-smoker COPD patients with persistent oxidative stress
Authors Fratta Pasini AM, Ferrari M, Stranieri C, Vallerio P, Mozzini C, Garbin U, Zambon G, Cominacini L
Received 10 December 2015
Accepted for publication 5 May 2016
Published 28 July 2016 Volume 2016:11(1) Pages 1733—1743
DOI https://doi.org/10.2147/COPD.S102218
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Anna Maria Fratta Pasini,1 Marcello Ferrari,2 Chiara Stranieri,1 Paola Vallerio,1 Chiara Mozzini,1 Ulisse Garbin,1 Giorgia Zambon,1 Luciano Cominacini1
1Department of Medicine, Section of Internal Medicine, 2Department of Medicine, Unit of Respiratory Diseases, University of Verona, Verona, Italy
Abstract: Inadequacy of antioxidant nuclear factor-E2-related factor 2 (Nrf2) and endoplasmic reticulum stress-mediated unfolded protein response has been implicated in severe chronic obstructive pulmonary disease (COPD) and cigarette smoking-induced emphysema. As evidence suggests that the ability to upregulate Nrf2 expression may influence the progression of COPD and no data exist up to now in ex-smokers with mild–moderate COPD, this study was first aimed to evaluate Nrf2 and unfolded protein response expression in peripheral blood mononuclear cells (PBMC) of mild–moderate ex-smokers with COPD compared to smoking habit-matched non-COPD subjects. Then, we tested whether oxidative stress persists after cigarette smoking cessation and whether the concentrations of oxidized phospholipids (oxidation products of the phospholipid 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine [oxPAPC]) in the PBMC of the same subjects may have a causative role in determining the upregulation of Nrf2. The expression (mRNA and protein) of Nrf2 and of its related gene heme oxygenase-1 was significantly increased in COPD group without differences in the unfolded protein response. Plasma malondialdehyde, the circulating marker of oxidative stress, and oxPAPC in PBMC were significantly higher in COPD than in non-COPD subjects. The fact that the expression of p47phox, a subunit of NADPH oxidase, was increased in PBMC of COPD patients and that it was directly correlated with oxPAPC may indicate that oxPAPC may be one of the determinants of oxidative stress-induced Nrf2 upregulation. Finally, we also demonstrated that lung function inversely correlated with plasma malondialdehyde and with Nrf2 and heme oxygenase-1 mRNA expression in all subjects. Our results indicate that mild–moderate ex-smokers with COPD may be able to counteract oxidative stress by increasing the expression of Nrf2/antioxidant-response elements. Because Nrf2 failure significantly contributes to the development of COPD, our findings suggest that the possibility to prevent Nrf2 reduction may open a new scenario in helping to prevent the oxidative stress-associated lung function decline.
Keywords: mild–moderate COPD, Nrf2/ARE, UPR, oxidative stress, cigarette smoking, peripheral blood mononuclear cells
Introduction
Chronic obstructive pulmonary disease (COPD) is a common disease affecting millions of people worldwide characterized by progressive airflow limitation, destruction of the lung parenchyma, and various systemic manifestations, which significantly impact mortality.1 Although cigarette smoking (CS) is the major risk factor for COPD,1 the fact that only a minority of smokers develop COPD2 and that CS cessation is ineffective at halting disease3 has led to the suggestion that other endogenous factors may also be driving the disease. Convincing evidence suggests that oxidative stress and inflammation play a key pathogenetic role in COPD onset and progression.4,5 In COPD patients, oxidative stress results from an increase of reactive oxygen species (ROS) present in CS per se, as well as generated by various inflammatory, immune, and epithelial cells of the airways.5 Increased oxidative stress, in turn, sustains pulmonary inflammation that induces recruitment and activation of immune cells into the lungs,5 producing inflammatory mediators that may spill over into the systemic circulation.1 It has been suggested that host ability to defend from oxidative stress by upregulating lung antioxidant defenses may be one of the critical events that determines the severity and progression of COPD.6 The nuclear factor-E2-related factor 2 (Nrf2) is a master transcription factor that regulates antioxidant-response element (ARE)-mediated expression of cytoprotective proteins and antioxidant enzymes, such as heme oxygenase-1 (HO-1). Under basal conditions, Nrf2-dependent transcription is repressed by its negative regulator Kelch-like ECH-associated protein 1; when cells are exposed to oxidative stress or electrophiles, Nrf2 accumulates in the nucleus and drives the expression of its target genes.7 There are significant data suggesting a critical role for Nrf2 in preventing lung disease; in this context, previous studies have shown an increased susceptibility to CS-induced emphysema in Nrf2-deficient mice8 and a decline of Nrf2 expression in pulmonary macrophages of current smokers and COPD patients.9 Recent evidence demonstrates that endoplasmic reticulum (ER) stress is a form of intracellular stress that occurs whenever the protein-folding capacity of the ER is overwhelmed.10 This accumulation of unfolded and misfolded proteins activates transcriptional and translational pathways, known as unfolded protein response (UPR). When UPR is induced, the three ER transmembrane sensors, protein kinase-like ER kinase (PERK), inositol-requiring kinase 1 (IRE1), and the transcriptional factor activating transcription factor 6 (ATF6), which are maintained in an inactive state through interaction with binding immunoglobulin protein (BiP) become activated to initiate adaptive responses.10 Once the UPR fails to control the level of unfolded and misfolded proteins in the ER, apoptotic signaling is induced through the expression of the death factor, CCAAT/enhancer-binding protein homologous protein (CHOP).11 Former research has shown that CS extract causes ER stress and apoptosis in human bronchial epithelial cells and in mice12 and chronic CS exposure induces UPR activation in lungs of smokers13 and patients with COPD.13,14 Furthermore, an increased ER stress, particularly an overexpression of apoptotic mediators, has been recently demonstrated in lung tissues of COPD patients with severe emphysema.15 Peripheral blood mononuclear cells (PBMC) are the progenitors of the alveolar macrophages that play a crucial role in maintaining and controlling the inflammatory process in COPD.16 We have previously shown that oxidation products of the phospholipid 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (oxPAPC) increased the generation of ROS in PBMC through the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase,17 and the increase of oxPAPC in PBMC was associated with the activation of the Nrf2/ARE pathway in mild smokers compared to nonsmokers, whereas in heavy smokers the Nrf2/ARE expression was similar to nonsmokers.17 COPD is typically diagnosed late in the course of disease when the symptoms become clinically evident18 and consequently very few studies have focused on oxidative stress in its early phases, potentially crucial for the subsequent evolution of airway damage. Moreover, it is uncertain whether systemic and cellular oxidative stress is still present after long-term CS cessation;3 no data exist up to now on Nrf2/ARE and UPR expression in mild–moderate ex-smokers with COPD. Therefore, this study was performed in ex-smokers with COPD with mild–moderate bronchial obstruction compared to age-, sex-, and smoking habit-matched non-COPD subjects and aimed to evaluate: 1) the expression of Nrf2, HO-1, and UPR in PBMC; 2) circulating markers of oxidative stress and inflammation; 3) whether cellular oxidative stress, and in particular oxPAPC, may contribute to PBMC gene expression; and 4) the possible correlations between lung function and circulating markers of oxidative stress and antioxidant gene expression.
Materials and methods
Ethics statement
The study was approved by the ethics committee of the Azienda Ospedaliera Universitaria Integrata Verona, in accordance with the standards of the Declaration of Helsinki, and written informed consent was obtained from all the patients before their enrollment.
Study population
In this study, we enrolled two groups of subjects: one group was composed of 30 consecutive, ex-smokers mild–moderate (16 mild and 14 moderate) COPD patients referring to Respiratory Medicine Outpatient Clinic of our Institution. The Global initiative for chronic Obstructive Lung Disease guideline was used to make the diagnosis and to grade COPD severity.19 The other group comprised age-, sex-, and smoking habit-matched non-COPD subjects randomly selected from the general population.20 Major requirements for the enrollment of both groups were absence of infectious or acute/chronic inflammatory diseases or known cardiovascular diseases, malignancy, and absence of acute/chronic renal failure and hepatic failure. No COPD subjects were using supplemental oxygen, glucocorticoids, antibiotic, and bronchodilator agents. Past smoking exposure was evaluated as years of smoking duration and pack-years, which is an indicator of a person’s cumulative cigarette consumption. Pack-years have been calculated by multiplying the number of packs of cigarettes smoked per day by the number of years the subject had smoked. All subjects reported that they had discontinued smoking for at least 1 year. All subjects underwent clinical evaluation and pulmonary function test.
Pulmonary function test
Forced expiratory volume in 1 second (FEV1) and FEV1/forced vital capacity (FVC) were measured using a water-sealed spirometer (Biomedin, Padua, Italy). Lung function values were expressed as a percentage of predicted values, and the lower limit of normal low limit of normality for the FEV1/FVC was calculated according to Quanjer.21
Blood samples and PBMC isolation
Venous blood samples were obtained from non-COPD and COPD subjects after 12 hours fasting. Blood was collected from each subject and drawn into pyrogen-free blood collection tubes. Multiple aliquots of plasma were placed into sterile 1 mL screw-capped polypropylene vials containing the phenolic antioxidant 2,6-di-tert-butyl-4-methylphenol (10 mM; Sigma-Aldrich Co., St Louis, MO, USA) to inhibit lipid peroxidation and stored at -80°C. The samples were frozen and thawed only once. PBMC were isolated as previously described.22 Blood samples were collected from subjects into sodium heparin-buffered Vacutainer® CPT™ tubes (from Becton Dickinson). The BD CPT™ tube is an evacuated tube containing anticoagulant, separation gel, and density gradient liquid. Whole blood is collected, centrifuged, and processed entirely within this tube. The CPT™ contains a gel barrier and Ficoll to allow for separation of PBMC and plasma from erythrocytes and granulocytes following a single centrifugation step. Immediately following blood collection with Vacutainer® CPT™, the tubes were inverted ten times and centrifuged at 1,500× g for 20 minutes at room temperature. After centrifugation, the PBMC layer was gently suspended in the plasma and transferred to 15 mL conical tubes and washed with phosphate-buffered saline by centrifugation at 300× g for 10 minutes. Monocyte purity was greater than 97% as assessed by flow cytometry. C-reactive protein (CRP) was measured using a commercially available high-sensitivity turbidimetric method (Syncron-PCR; Beckman Coulter, Brea, CA, USA).
Glutathione measurement in plasma
The detailed procedure for the measurement of plasma glutathione (GSH) has been previously described.23 Samples were derivatized with 7-fluorobenzo-2-oxa-1,3-diazol-4-sulfonic acid and quantitated using high-performance liquid chromatography with fluorescence detection. Fluorimetric detector was a Shimadzu RF-10 Axl and was set with λex =385 nm and λem =515 nm.
Malondialdehyde measurement in plasma
The detailed procedure for the measurement of plasma malondialdehyde (MDA) has been previously described.24 Briefly, 400 μL of phosphoric acid solution (44 mM) and 100 μL of thiobarbituric acid solution (42 mM) were added to 150 μL of plasma sample. Then, samples were heated at 100°C for 60 minutes, extracted with 250 μL of n-butanol; 20 μL of each sample was injected into the column. MDA was measured by high-performance liquid chromatography with fluorescence detection. Fluorimetric detector was a Shimadzu RF-10 Axl and was set with λex =520 nm and λem =542 nm.
oxPAPC measurement in PBMC
Among the different oxPAPC, 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphorylcholine (POVPC) and 1-palmitoyl-2-glutaroyl-sn-glycero-3-phosphorylcholine (PGPC) were taken into consideration in this study because they have been previously identified as the most bioactive components.25 Measurement of POVPC and PGPC was obtained on an Agilent 1100 (Agilent Technologies, Santa Clara, CA, USA) mass spectrometer equipped with an electrospray ion source, as previously described.25 Quantification of the peak areas was performed by single-ion monitoring in the elution time range of 10–20 minutes using appropriate software. Authentic 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine, POVPC, and PGPC were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL, USA).
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (PCR) analysis was performed as previously described.22 Total RNA was extracted from PBMC with an RNeasy Mini Kit (Qiagen, Milan, Italy) and reverse transcribed using an IScript cDNA Synthesis Kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). Real-time RT-PCR was conducted by iCycler themocycler (Bio-Rad Laboratories Inc.) using IQSYBR Green SuperMix (Bio-Rad Laboratories Inc.) and 300 pmol/mL each primer pair. Primer design was done with Beacon Design 4.0 software (PREMIER Biosoft International, Palo Alto, CA, USA): Nrf2, sense 5′-TTCAGCCAGCCCAGCACATC-3′ and antisense 5′-CGTAGCCGAAGAAACCTCATTGTC-3′; HO-1, sense 5′-GGTGACCCGAGACGGCTTC-3′ and antisense 5′-AGACTGGGCTCTCCTTGTTGC-3′; p47phox, sense 5′-CCCACAGACAACCAGACAAA-3′, antisense 5′-TTTGTCTGGTTGTCTGTGGG-3′; β-actin, sense 5′-ATCAAGATCATTGCTCCTCCTG-3′ and antisense 5′-GCAACTAAGTCATAGTCCGCC-3′. All primers were optimized to an equal annealing temperature of 60°C, a similar GC content, and supplied by MWG-Biotech AG (Ebersberg, Germany). Cycling conditions were: 3 minutes at 95°C, followed by 50 cycles during 10 seconds at 95°C, 30 seconds at 60°C, and 1 minute at 55°C. The relative expression levels of mRNA encoding UPR genes were performed using the QuantiTect Primer Assay and QuantiTect SYBR Green PCR Kit (Qiagen) on the MyiQ Thermal Cycler (Bio-Rad Laboratories Inc.). QuantiTect Hs-ACTB Assay (Qiagen) was used as a normalizer. UPR QuantiTect Primer Assays were purchased from Qiagen: BiP: QT00096404, PERK: QT00066003, IRE1: QT00025760, ATF6: QT00083370, CHOP: QT00082278, and β-actin: QT00095431. They are bioinformatically validated primer sets for use in SYBR Green-based real-time RT-PCR at an annealing temperature of 55°C, in accordance with the specifications provided by the supplier. Cycling conditions were: 15 minutes at 95°C, followed by 40 cycles during 15 seconds at 94°C, 30 seconds at 55°C, and 30 seconds at 72°C. Normalized gene expression levels are given as the ratio between the mean value for the target gene and β-actin in each sample.
Western blotting
Western blot analysis was performed as previously described.22 Nrf2, HO-1, BiP, PERK, IRE1, ATF6, CHOP, and p47phox were immunoprecipitated from 1 mg of each PBMC nuclear lysate or protein lysate with mouse monoclonal antibodies and anti-β-actin (Santa Cruz Biotechnology, Heidelberg, Germany). Immune complexes were captured with protein A/G–Sepharose beads (Pierce, Rockford, IL, USA) for 2 hours, and the beads were washed four times with 100 mM NaCl. Nrf2 (sc-722), HO-1 (sc-10789), BiP (sc-1050), PERK (sc-13073), IRE1 (sc-20790), ATF6 (sc-22799), CHOP (sc-7351), and p47phox (sc-14015) were detected by probing immunoprecipitates with rabbit polyclonal antibodies (Santa Cruz Biotechnology), followed by goat antirabbit horseradish peroxidase-conjugated secondary antibody (Bio-Rad Laboratories Inc.). Reactive antigens were visualized with Supersignal chemiluminescence substrate (Pierce) and quantified by densitometric analysis with ChemiDoc XRS (Bio-Rad Laboratories Inc.). Protein expression data were quantified with Quantity One Software (Bio-Rad Laboratories Inc.).
Statistical analysis
Data are expressed as mean ± standard deviation values. Differences between the groups were analyzed by a two-tailed unpaired Student’s t-test. The relationship between variables was assessed by linear regression. Statistical analysis of the data was conducted using SPSS version 20.0 (IBM Corporation, Armonk, NY, USA); P<0.05 was considered to be statistically significant.
Results
Lung function, clinical, and laboratory characteristics
Lung function and medical history of subjects participating in the study are reported in Table 1. According to the inclusion criteria, FEV1 and FEV1/FVC resulted significantly lower in COPD than non-COPD subjects (P≤0.01). Smoking habits, prevalence of hypertension, diabetes, and dyslipidemia were similar in COPD and non-COPD subjects. As shown in Table 2, also anthropometric characteristics, plasma glucose, triglycerides, high-density lipoprotein cholesterol, and diastolic blood pressure were comparable in both groups. On the contrary, COPD subjects presented lower values of systolic blood pressure and total and low-density lipoprotein cholesterol (all P=0.01) than non-COPD subjects. These results are probably due to the fact that treatment with antihypertensive drugs and statins was more frequent in patients with COPD, as reported in Table 1.
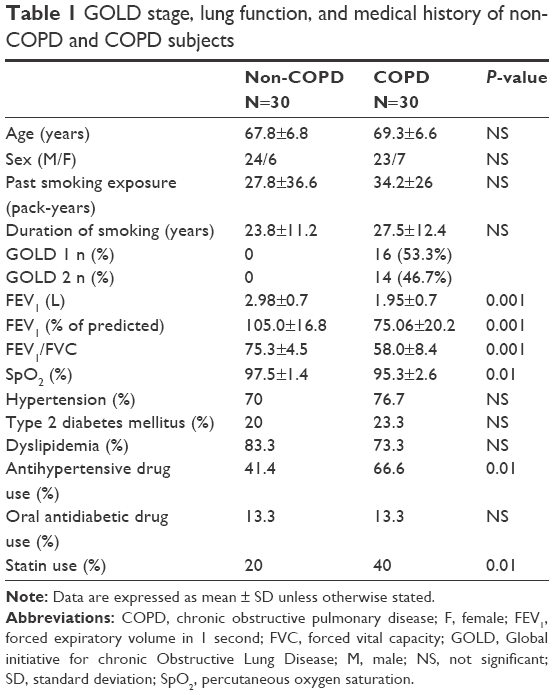 | Table 1 GOLD stage, lung function, and medical history of non-COPD and COPD subjects |
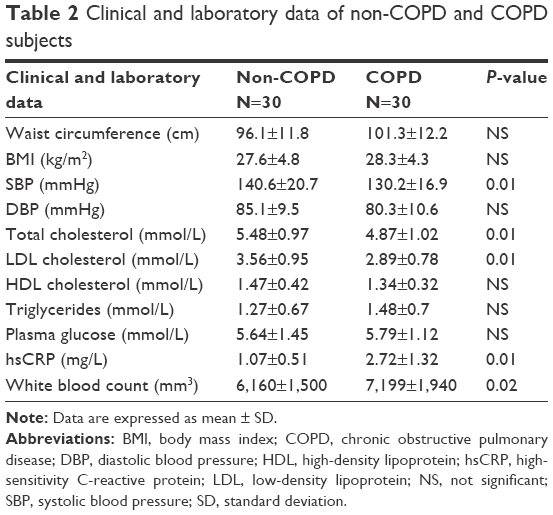 | Table 2 Clinical and laboratory data of non-COPD and COPD subjects |
Nrf2, HO-1, and UPR expression in PBMC derived from non-COPD and COPD subjects
We evaluated the expression of Nrf2, HO-1, and UPR (BiP, ATF6, PERK, IRE1, and CHOP) in PBMC derived from non-COPD and COPD subjects; our results show an increased mRNA and protein expression of both Nrf2 and HO-1 (P<0.001) in PBMC derived from COPD compared to non-COPD subjects (Figure 1A and B). On the contrary, UPR (BiP, ATF6, PERK, IRE1, and CHOP) mRNA and protein expression were similar in both groups (Figure 2A and B).
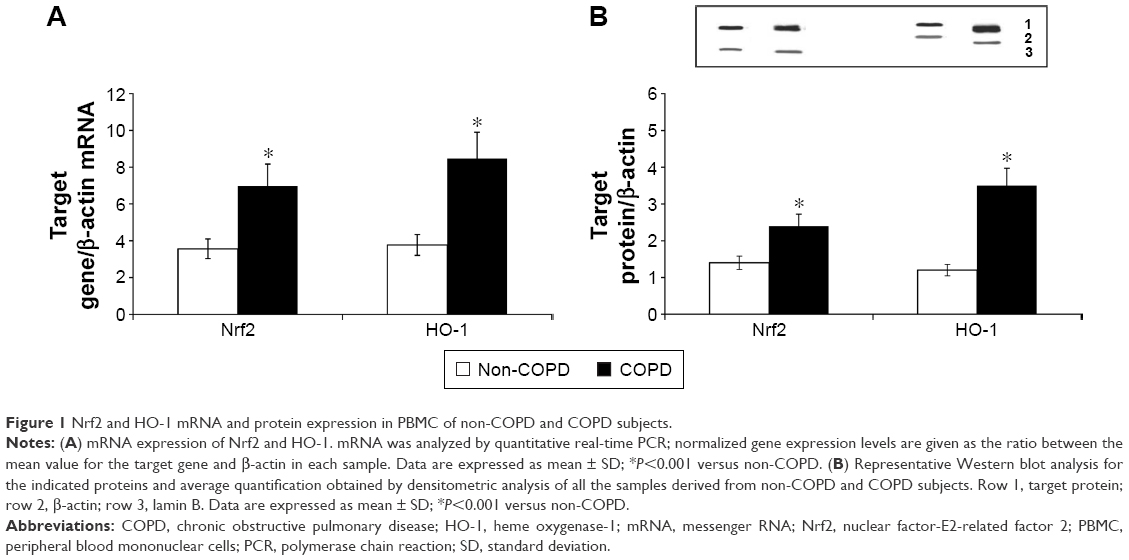 | Figure 1 Nrf2 and HO-1 mRNA and protein expression in PBMC of non-COPD and COPD subjects. |
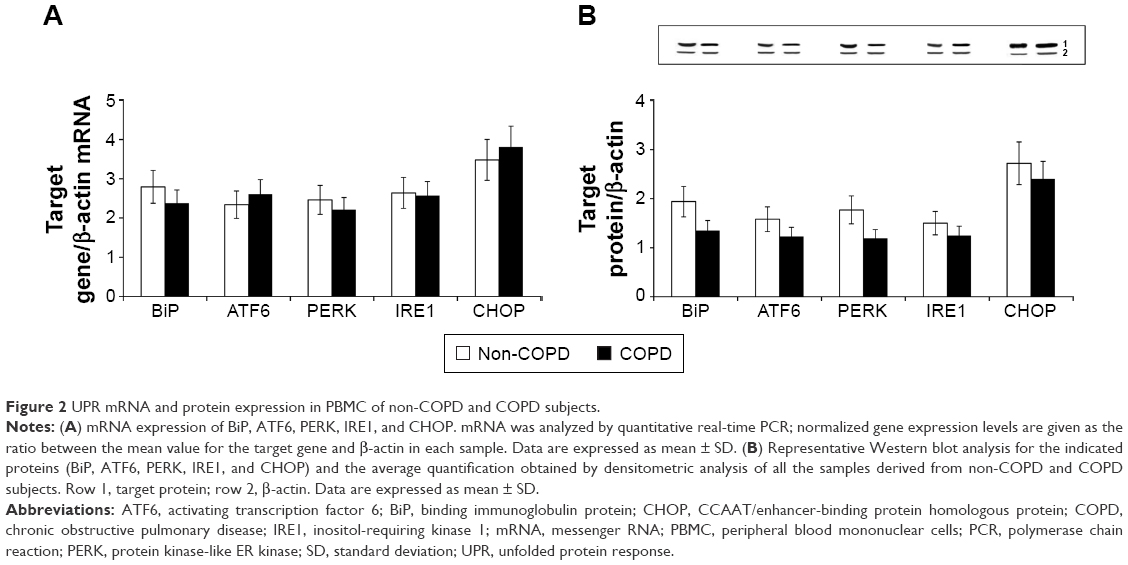 | Figure 2 UPR mRNA and protein expression in PBMC of non-COPD and COPD subjects. |
Circulating markers of oxidative stress and inflammation
The plasma concentrations of MDA resulted higher in COPD than in non-COPD subjects (P<0.01), whereas plasma concentrations of GSH were significantly higher in the latter than in the former (P<0.01) (Figure 3). Moreover, high-sensitivity CRP concentrations (P=0.01) and white blood count were significantly higher (P=0.02) in COPD than in non-COPD subjects (Table 2).
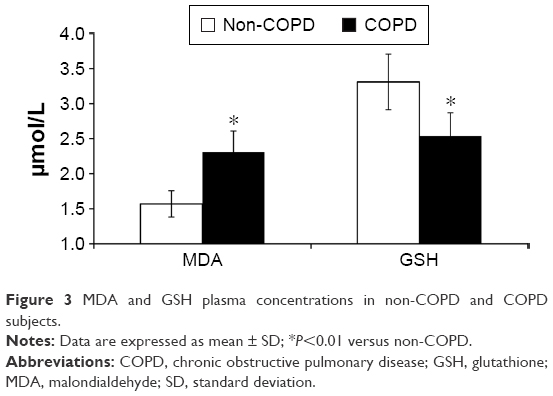 | Figure 3 MDA and GSH plasma concentrations in non-COPD and COPD subjects. |
Concentrations of oxPAPC and expression of p47phox in PBMC
In order to ascertain whether the increased expression of Nrf2 was related to the cellular oxidative stress, we also assessed the concentrations of oxPAPC in PBMC. Our results demonstrate that the concentrations of both PGPC and POVC, the oxPAPC considered in this study, were significantly higher (P<0.01) in PMBC derived from COPD than non-COPD subjects (Figure 4). Moreover, we found a significant direct correlation between plasma MDA and both PGPC (r=0.47, P<0.001) and POVPC (r=0.50, P<0.001) in PBMC of all subjects (data not shown). As oxPAPC has been shown to act as a second messenger in triggering NADPH oxidase activity,26 the expression of p47phox was also evaluated. Our results show an increased mRNA and protein expression (P<0.001) of p47phox in PBMC derived from COPD compared to non-COPD subjects (Figure 5A and B). Moreover, we found a positive correlation between the expression of p47phox and the concentrations of both PGPC (r=0.36, P=0.005) and POVPC (r=0.44, P<0.001) in PBMC of all subjects (Figure 5C and D), Finally, as shown in Figure 6, p47phox mRNA expression directly correlated with Nrf2 mRNA expression (r=0.52, P<0.001) in PBMC of all subjects.
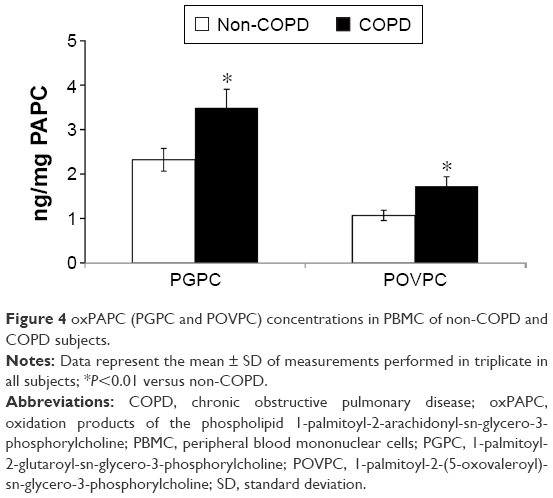 | Figure 4 oxPAPC (PGPC and POVPC) concentrations in PBMC of non-COPD and COPD subjects. |
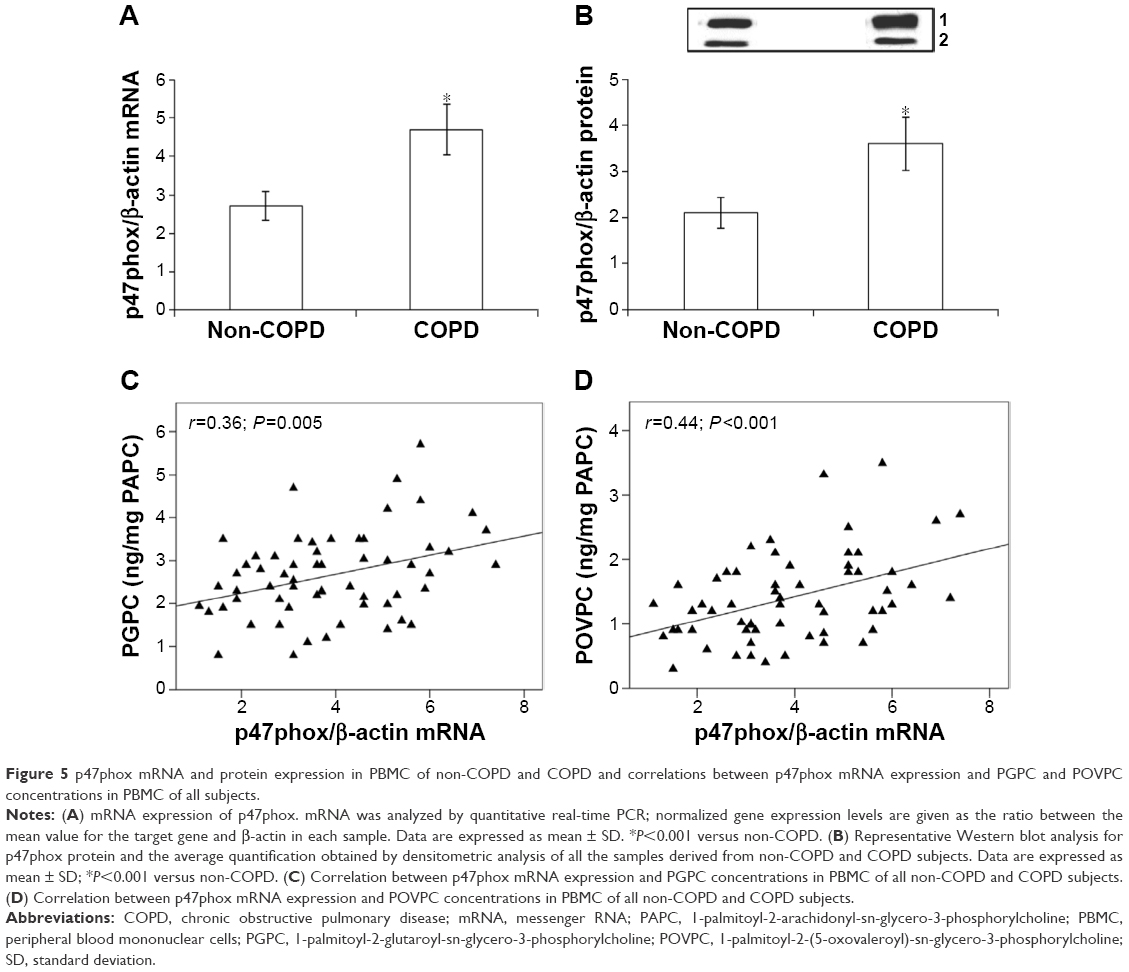 | Figure 5 p47phox mRNA and protein expression in PBMC of non-COPD and COPD and correlations between p47phox mRNA expression and PGPC and POVPC concentrations in PBMC of all subjects. |
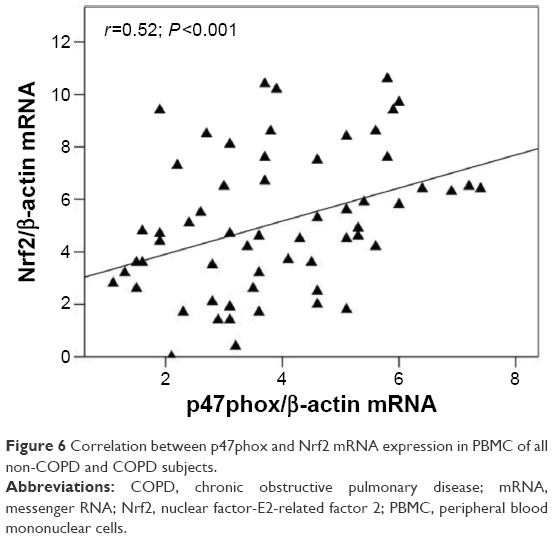 | Figure 6 Correlation between p47phox and Nrf2 mRNA expression in PBMC of all non-COPD and COPD subjects. |
Relationship between lung function and circulating markers of oxidative stress and antioxidant gene expression
Figure 7A and B shows that lung function, expressed as FEV1 (% predicted), directly correlated with plasma concentrations of GSH (r=0.61, P<0.001) and inversely with plasma concentrations of MDA (r=-0.46; P<0.001) in all the subjects participating in the study. Furthermore, our results also demonstrate an inverse relationship between FEV1 and both Nrf2 (r=-0.53, P<0.001) and HO-1 (r=-0.50, P<0.001) mRNA expression in PBMC (Figure 7C and D) of all the subjects.
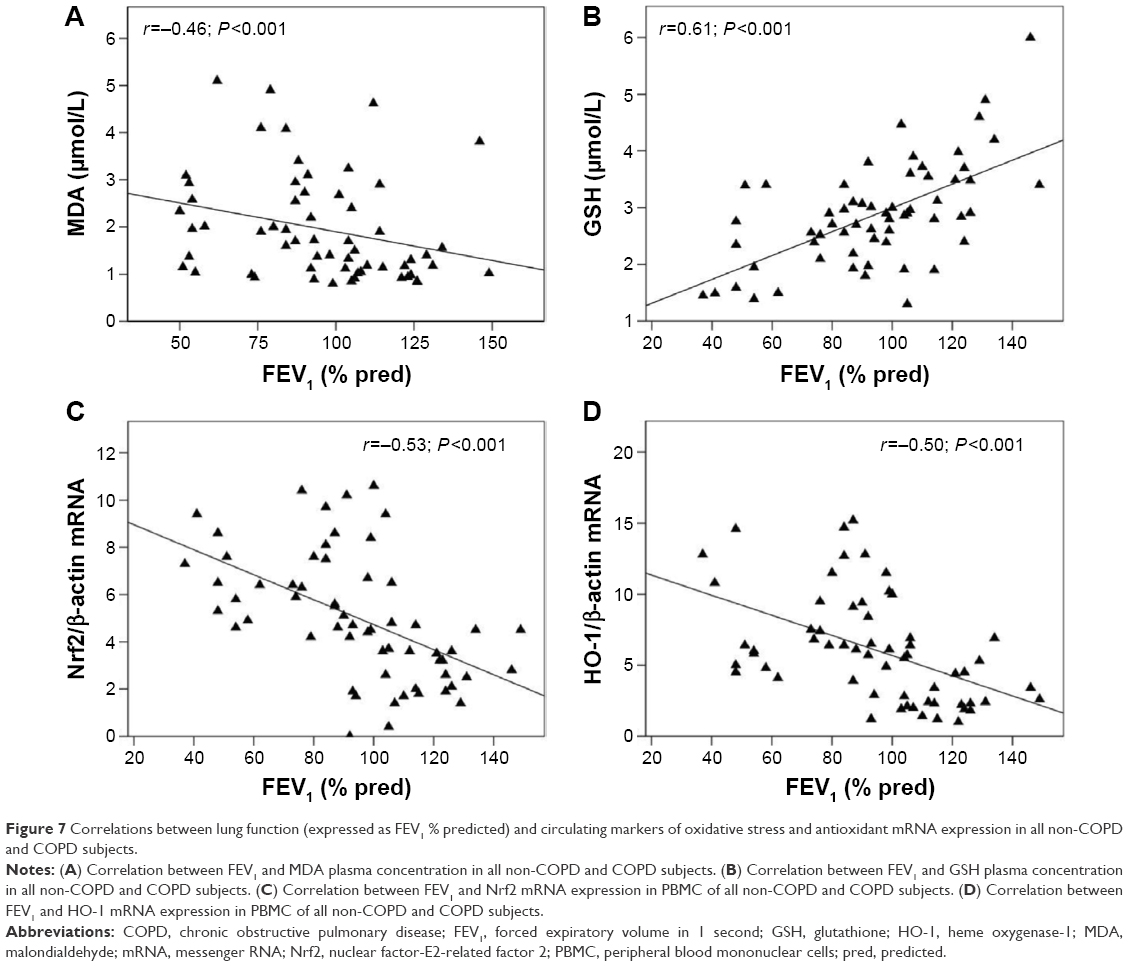 | Figure 7 Correlations between lung function (expressed as FEV1 % predicted) and circulating markers of oxidative stress and antioxidant mRNA expression in all non-COPD and COPD subjects. |
Discussion
COPD is characterized by a slowly progressive development of irreversible airflow limitation in which persistent CS-induced oxidative stress and inflammation are recognized as the major pathogenetic factors.4,5 Evidence indicates that CS cessation is effective in reducing mortality and delaying, but not halting, the rate of lung function decline in patients with mild COPD,27 so that it is possible to speculate that oxidative stress and inflammation may persist even after CS ending.3,28 The studies performed in the last decades on this issue have included mainly moderate–severe COPD patients with different CS exposure (current and ex-smokers, as well as subjects who had never smoked).3,29,30 On the contrary, very scarce data are available in ex-smoker COPD patients with a less severe airway obstruction. Therefore, a first important feature of this study is the peculiar setting of the subjects, that is, mild–moderate ex-smokers with COPD, compared with age- and sex-matched non-COPD ex-smoker subjects. Although recent studies suggest that PBMC gene expression may represent a promising noninvasive useful alternative to biopsy or invasive procedures especially at an early stage of the disease and a possible expression of systemic involvement of COPD,31,32 the majority of data available so far come from airway cells or lung tissues.9,14,15 In this study, we have first evaluated the expression of Nrf2 and of HO-1 (one of its main target genes) because several studies have shown that Nrf2 and its Nrf2 downstream genes have an essential protective role in the lung against oxidative stress.29,30,33–35 Rather unexpectedly, we demonstrated an increased expression of both Nrf2 and HO-1 in PBMC derived from ex-smokers with COPD compared with ex-smokers non-COPD. Although this finding confirms that PBMC may be a suitable noninvasive tool in mild–moderate COPD, it is not in line with prior reports showing a decline of Nrf2 expression in whole lung tissue,9,30 alveolar macrophages of current smokers,9 and patients with COPD.29,30 In these studies, the decrease of Nrf2 in alveolar macrophages and lung tissues of patients with emphysema was due to an increase of Bach-1 and Kelch-like ECH-associated protein 129 or a loss of Nrf2 protein stability.30 Similarly, it has been previously shown that the expression of HO-1 was diminished in pulmonary macrophages from patients with severe emphysema.29,33 Hence, at variance with previous studies performed in patients with different smoking exposure9,17 and/or COPD stages,29,30,36 our finding shows that in ex-smokers with mild–moderate bronchial obstruction, the antioxidant Nrf2/ARE is overexpressed in PBMC. Another peculiar result of this study is that UPR and CHOP expression in PBMC was similar in both groups of subjects. Recent studies have demonstrated the involvement of CS in UPR and, in particular, that CS caused a UPR response in the airway cells12 and human lungs, which is partially reversible after CS cessation.13 However, as far as COPD is concerned, the results are not completely in agreement; in fact, while some authors reported that UPR and apoptosis progressively increase in the lungs of ex- and current smokers with normal lungs, with mild and advanced COPD,14 an absence of ER stress in the lungs of patients with severe COPD has also been reported.37 Recent evidence indicates that there is a crosstalk between Nrf2 and UPR; it has been shown that impaired Nrf2 expression exaggerated ER stress and apoptosis in mice after exposure to CS8 and increased apoptosis in alveolar macrophages of smoker COPD subjects.38 Moreover, we have recently shown an increased ER stress with Nrf2 repression in PBMC derived from stable coronary artery disease patients.39 On the contrary, the results of a very recent in vitro study indicate that Nrf2 overexpression reduced CHOP and protected cells from apoptosis.40 From a mechanistic point of view, it has been recently demonstrated that a Nrf2-dependent regulation of 26S proteasome system exists,41 a major disposal pathway for degradation of misfolded proteins from ER.42 Taken together with the results of these previous studies, we are tempted to speculate that the activation of Nrf2 found in PBMC of our mild–moderate COPD subjects may have a role in preventing ER stress response in PBMC. Although recent studies suggest that a short period of CS cessation reduces, but not normalizes oxidative stress in airways43 and plasma44 of COPD patients, it is uncertain whether systemic oxidative stress is still present after long-term CS cessation. To our knowledge, this is the first demonstration of an oxidative–antioxidative imbalance, that is, increased plasma levels of MDA and decreased plasma levels of GSH after a long period (>1 year) of CS cessation in mild–moderate COPD compared with ex-smoker non-COPD subjects. Moreover, as past CS exposure and other cardiovascular risk factors were similar in both groups, we may speculate that this persistent redox imbalance found in our ex-smoker COPD subjects most likely arises from endogenous sources of ROS.4 Because oxidative stress is one of the most potent stimuli for Nrf2/ARE pathway activation,7 in order to ascertain whether the increased expression of Nrf2 was related to the cellular oxidative stress, we have evaluated the concentrations of oxPAPC in PBMC. Our results demonstrate that the concentrations of both PGPC and POVPC were significantly higher in PBMC derived from COPD than non-COPD subjects. Previous studies have indicated that ROS generation is dependent on NADPH oxidase in pulmonary epithelial cells45 and that PGPC and POVPC dose-dependently raise the generation of ROS in PBMC through the activation of NADPH oxidase.17 Consequently, we have evaluated the expression of p47phox that has been shown to significantly contribute to the activity of NADPH oxidase46 and found an increased expression of this enzyme subunit in PBMC derived from COPD subjects. The fact that the expression of p47phox directly correlated with both PGPC and POVPC in PBMC suggests that oxPAPC may play a role in inducing cellular oxidative stress. Moreover, we found a direct correlation between the expression of p47phox and Nrf2 in PBMC. Although correlations do not permit to distinguish between cause and effect, taken together, these results support the hypothesis that our mild–moderate COPD subjects can still increase Nrf2/ARE expression in response to the intracellular oxidative stress. In this context, we have already shown that a progressive increase of oxidative stress in PBMC was associated with the activation of the Nrf2/ARE pathway in mild young smokers, whereas in heavy smokers this pathway was repressed.17 Moreover, we have previously demonstrated that both PGPC17 and oxPAPC47 at low concentrations induced the expression of Nrf2 in PBMC while at higher concentrations the expression of Nrf2 was repressed. We can therefore hypothesize that the increased Nrf2 expression found in our ex-smokers CODP is likely due to the reduced burden of oxidative stress consequent to CS suspension. This ability to counteract oxidative stress by increasing Nrf2 pathway is a novel finding that could have important implications because the extensive studies performed in models of COPD and COPD patients clearly indicate that Nr2 is a key determinant of COPD susceptibility.29,30,33–35 In this study, we also show that plasma levels of high-sensitivity CRP and white blood cells were higher in COPD than in non-COPD subjects. Although an improvement in inflammation might be expected after CS cessation, the increase of systemic markers of inflammation found in our ex-smokers with COPD is in line with previous studies indicating that airway inflammation is similar in ex-smokers and smokers with COPD.48,49 We might therefore speculate that in our COPD subjects the persistent oxidative stress may have a role in these results as an imbalance in the oxidant–antioxidant system has been recognized as one of the first events that ultimately leads to inflammatory reactions in the lung.4 Population-based studies strongly reported that reduced lung function, as assessed by FEV1, independent of CS, is a significant risk factor for total mortality and in particular for cardiovascular morbidity.1,21 The results of this study show that FEV1 directly correlated with plasma concentrations of GSH and inversely with MDA in all subjects participating in the study suggesting that in ex-smokers also oxidant–antioxidant imbalance may be associated with lung function. These results agree with previous data showing, albeit in different COPD subjects, that an oxidative stress marker inversely correlated with lung function.5 Our finding of an inverse correlation between FEV1 and both Nrf2 and HO-1 expression in PBMC supports the hypothesis that ex-smokers with mild–moderate COPD can properly respond in terms of Nrf2 and HO-1 expression to lung function decline. Our data are in line with a former study showing that HO-1 expression in lung homogenates of severe COPD patients was inversely correlated with airway obstruction and increased macrophage expression of oxidative stress markers.29 At variance with these results, a direct correlation between lung function (FEV1:FVC) and mRNA expression of Nrf2 target antioxidant genes in lung of COPD subjects has been previously shown.30 Based on recent evidence suggesting that the pharmacological activation of Nrf2 protects mice against CS-induced emphysema,50 it has been proposed that the new Nrf2 inducers, also called “indirect antioxidants”,51 may be a useful tool for preventing and treating COPD. Thus, as there are currently no treatments that significantly reverse or slow the progression of COPD,4 the future possibility to counteract Nrf2 decline in COPD patients may help in reducing the negative effects of the oxidative stress-induced progression of the disease.
Acknowledgment
This work was supported in part by Ministero della Salute of Italy (project number RF-2009-1471235). The funding source had no involvement in study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the article for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J. 2009;33(5):1165–1185. | ||
Rennard SI, Vestbo J. COPD: The dangerous underestimate of 15%. Lancet. 2006;367(9518):1216–1219. | ||
Rutgers SR, Postma DS, ten Haken NH, et al. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax. 2000;55(1):12–18. | ||
Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. 2013;144(1):266–273. | ||
Zuo L, He F, Sergakis GG, et al. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am J Physiol Lung Cell Mol Physiol. 2014;307(3):L205–L218. | ||
Cho HY, Kleeberger SR. Nrf2 protects against airway disorders. Toxicol Appl Pharmacol. 2010;244(1):43–56. | ||
Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. | ||
Rangasamy T, Cho CY, Thimmulappa RK, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114(9):1248–1259. | ||
Suzuki M, Betsuyaku T, Ito Y, et al. Downregulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2008;39(6):673–682. | ||
Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110(10):1389–1398. | ||
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. | ||
Tagawa Y, Hiramatsu N, Kasai A, et al. Induction of apoptosis by cigarette smoke via ROS dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Radic Biol Med. 2008;5(1):50–59. | ||
Kelsen SG, Duan X, Ji R, Perez O, Liu C, Merali S. Cigarette smoke induces an unfolded protein response in the human lung: a proteomic approach. Am J Respir Cell Mol Biol. 2008;38(5):541–550. | ||
Malhotra D, Thimmulappa R, Vij N, et al. Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: the role of Nrf2-regulated proteasomal activity. Am J Respir Crit Care Med. 2009;180(12):1196–1207. | ||
Min T, Bodas M, Mazur S, Vij N. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J Mol Med. 2011;89(6):577–593. | ||
Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688. | ||
Garbin U, Fratta Pasini A, Stranieri C, et al. Cigarette smoking blocks the protective expression of Nrf2/ARE pathway in peripheral mononuclear cells of young heavy smokers favouring inflammation. PLoS One. 2009;4(12):e8225. | ||
Murtagh E, Heaney L, Gingles J, et al. Prevalence of obstructive lung disease in a general population sample: the NICECOPD study. Eur J Epidemiol. 2005;20(5):443–445. | ||
Fabbri L, Pauwels RA, Hurd SS. Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease: GOLD executive summary updated 2003. COPD. 2004;1(1):105–141. | ||
de Marco R, Accordini S, Antonicelli L, et al. The Gene-Environment Interactions in Respiratory Diseases (GEIRD) Project. Int Arch Allergy Immunol. 2010;152(3):255–263. | ||
Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF, Peslin R, Yernault JC. Lung volumes and forced ventilator flows. Eur Respir J. 1993;16:5–40. | ||
Fratta Pasini A, Anselmi M, Garbin U, et al. Enhanced levels of oxidized low-density lipoprotein prime monocytes to cytokine overproduction via upregulation of CD14 and toll-like receptor 4 in unstable angina. Arterioscler Thromb Vasc Biol. 2007;27(9):1991–1997. | ||
Ubbink JB, Vermaak WJH, Bissbort S. Rapid high performance liquid chromatographic assay for total homocysteine levels in human serum. J Chromatogr. 1991;565(1–2):441–446. | ||
Mao J, Zhang H, Luo J, et al. New method for HPLC separation and fluorescence detection of malonaldehyde in normal human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;832(1):103–108. | ||
Subbanagounder GN, Leitinger DC, Schwenke JW, et al. Determinants of bioactivity of oxidized phospholipids. Specific oxidized fatty acyl groups at the sn-2 position. Arterioscler Thromb Vasc Biol. 2000;20(10):2248–2254. | ||
Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86(5):494–501. | ||
Scanlon PD, Connett JE, Waller LA, et al. Smoking cessation and lung function in mild-to moderate chronic obstructive pulmonary disease. The lung health study. Am J Respir Crit Care Med. 2000;161(2 Pt 1):381–390. | ||
Rahman I, MacNee W. Role of transcription factors in inflammatory lung diseases. Thorax. 1998;53(7):601–612. | ||
Goven D, Boutten A, Lecon-Malas V, et al. Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax. 2008;63(10):916–924. | ||
Malhotra D, Thimmulappa R, Navas-Acien A, et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. 2008;178(6):592–604. | ||
Bhattacharya S, Tyagi S, Srisuma S, et al. Peripheral blood gene expression profiles in COPD subjects. J Clin Bioinforma. 2011;1(1):12. | ||
Poliska S, Csanky E, Szanto A, et al. Chronic obstructive pulmonary disease-specific gene expression signatures of alveolar macrophages as well as peripheral blood monocytes overlap and correlate with lung function. Respiration. 2011;81(6):499–510. | ||
Cho HY, Jedlicka AE, Reddy SP, et al. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26(2):175–182. | ||
Singh A, Ling G, Suhasini AN, et al. Nrf2-dependent sulfiredoxin-1 expression protects against cigarette smoke-induced oxidative stress in lungs. Free Radic Biol Med. 2009;46(3):376–386. | ||
Boutten A, Goven D, Boczkowski J, Bonay M. Oxidative stress targets in pulmonary emphysema: focus on the Nrf2 pathway. Expert Opin Ther Targets. 2010;14(3):329–346. | ||
Maestrelli P, Páska C, Saetta M, et al. Decreased haeme oxygenase-1 and increased inducible nitric oxide synthase in the lung of severe COPD patients. Eur Respir J. 2003;21(6):971–976. | ||
Korfei M, Ruppert C, Mahavadi P, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(8):838–846. | ||
Yamada K, Asai K, Nagayasu F, et al. Impaired nuclear factor erythroid 2-related factor 2 expression increases apoptosis of airway epithelial cells in patients with chronic obstructive pulmonary disease due to cigarette smoking. BMC Pulm Med. 2016;16(1):27. | ||
Mozzini C, Fratta Pasini A, Garbin U, et al. Increased endoplasmic reticulum stress and Nrf2 repression in peripheral blood mononuclear cells of patients with stable coronary artery disease. Free Radic Biol Med. 2014;68:178–185. | ||
Huang C, Wang JJ, Ma JH, Jin C, Yu Q, Zhang SX. Activation of the UPR protects against cigarette smoke-induced RPE apoptosis through up-regulation of Nrf2. J Biol Chem. 2015;290(9):5367–5380. | ||
Lee S, Hur EG, Ryoo IG, Jung KA, Kwak J, Kwak MK. Involvement of the Nrf2-proteasome pathway in the endoplasmic reticulum stress response in pancreatic beta-cells. Toxicol Appl Pharmacol. 2012;264(3):431–438. | ||
Poppek D, Grune T. Proteasomal defense of oxidative protein modifications. Antioxid Redox Signal. 2006;8(1–2):173–184. | ||
Louhelainen N, Rytilä P, Haahtela T, Kinnula VL, Djukanović R. Persistence of oxidant and protease burden in the airways after smoking cessation. BMC Pulm Med. 2009;9:25. | ||
Wofniak A, Górecki D, Szpinda M, Mila-Kierzenkowska C, Woźniak B. Oxidant-antioxidant balance in the blood of patients with chronic obstructive pulmonary disease after smoking cessation. Oxid Med Cell Longev. 2013(2013):897075. | ||
Papaiahgari S, Kleeberger SR, Cho HY, Kalvakolanu DV, Reddy SP. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J Biol Chem. 2004;279(40):42302–42312. | ||
Li JM, Mullen AM, Yun S, et al. Essential role of the NADPH oxidase subunit p47(phox) in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-alpha. Circ Res. 2002:90(2):143–150. | ||
Fratta Pasini A, Albiero A, Stranieri C, et al. Serum oxidative stress-induced repression of Nrf2 and GSH depletion: a mechanism potentially involved in endothelial dysfunction of young smokers. PLoS One. 2012;7(1):e30291. | ||
Willemsee BWM, ten Hacken NH, Rutgers B, et al. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J. 2005;26(5):835–845. | ||
Lapperre TS, Postma DS, Gosman MM, et al. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax. 2006;61(2):115–121. | ||
Sussan TE, Rangasamy T, Blake DJ, et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci U S A. 2009;106(1):250–255. | ||
Gao B, Doan A, Hybertson BM. The clinical potential of influencing Nrf2 signaling in degenerative and immunological disorders. Clin Pharmacol. 2014;6:19–34. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.