")
Back to Journals » Lung Cancer: Targets and Therapy » Volume 12
Novel Therapies for Metastatic Non-Small Cell Lung Cancer with MET Exon 14 Alterations: A Spotlight on Capmatinib
Authors Tan AC , Loh TJ, Kwang XL, Tan GS, Lim KH, Tan DSW
Received 8 September 2020
Accepted for publication 20 November 2020
Published 18 March 2021 Volume 2021:12 Pages 11—20
DOI https://doi.org/10.2147/LCTT.S263610
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ignatius Ou
Aaron C Tan,1 Tracy J Loh,2 Xue Lin Kwang,1 Gek San Tan,2 Kiat Hon Lim,2 Daniel SW Tan1
1Division of Medical Oncology, National Cancer Centre Singapore, Singapore, 169610, Singapore; 2Department of Pathology, Singapore General Hospital, Singapore, 169608, Singapore
Correspondence: Daniel SW Tan
National Cancer Centre Singapore, 11 Hospital Crescent, Singapore, 169610, Singapore
Tel +65 6436 8000
Email [email protected]
Abstract: MET exon 14 (METex14) alterations are now an established therapeutically tractable target in non-small cell lung cancer (NSCLC). Recently reported trials of several MET tyrosine kinase inhibitors (TKI) in this patient population have demonstrated promising efficacy data in both the treatment naïve and pre-treated settings and have led to regulatory approvals. This review will focus on practical diagnostic considerations for METex14 alterations, the trial evidence for capmatinib in this molecular subset including dosing and toxicity management, and the future therapeutic landscape of METex14 altered NSCLC.
Keywords: capmatinib, MET exon 14 skipping, non-small cell lung cancer, targeted therapy
Introduction
Non-small cell lung cancer (NSCLC) remains a leading cause of cancer death globally.1 With greater understanding of the molecular alterations that underpin lung cancer pathogenesis, there are now multiple tyrosine kinase inhibitors (TKI) approved for a myriad of targets including activating mutations in BRAF and EGFR as well as fusions involving ALK, NTRK, RET and ROS1.2 Additionally, there is emerging data for therapies targeting oncogenic drivers such as HER2 mutations, KRAS G12C mutations, MET exon 14 (METex14) alterations and NRG1 fusions.3–6 Targeting the MET signaling pathway to date has yielded disappointing results in several randomized trials of MET TKIs.7–9 However, these studies were conducted in either MET unselected populations or patients with MET overexpression, and there has since been a growing body of literature characterizing METex14 alterations as therapeutically tractable. This review will focus on METex14 alterations, including practical diagnostic considerations, the promising efficacy data for capmatinib in early trials and future directions for the therapeutic landscape of METex14 altered NSCLC.
Biology of METex14 Alterations in NSCLC
In cancer, activation of the MET pathway is involved in cellular transformation, vasculogenesis, tumor motility and invasion.10 The MET gene is located on chromosome 7 and is translated as a precursor protein, which is then split into an α-chain (extracellular) and a β-chain (transmembrane) after cleavage to form the mature protein.11 The intracellular component contains a juxtamembrane domain consisting of the binding site for an E3 ubiquitin-protein ligase, c-CBL, including Y1003 which is encoded by exon 14. In the 1980s, a TPR-MET oncogenic fusion was first discovered in a chemically transformed osteosarcoma cell line.12 Subsequent findings illustrated that activation of the MET receptor results in activation of downstream signaling pathways such as RAS/ERK/MAPK, PI3K/Akt and STAT (Figure 1). The importance of MET exon 14 in the regulation of MET activation was also an important finding.13 A MET splice variant resulting in an in-frame deletion of the 47 amino acids composing the juxtamembrane domain was first detected in mice.14 It was also then found that mutations in Y1003 prevented binding of c-CBL to MET, disrupted c-CBL mediated degradation and resulted in MET oncogenic activity.15 In NSCLC, splice site mutations in MET were first detected in 2005, and further characterization revealed the resulting skipping of MET exon 14.16,17 The incidence of METex14 alterations in NSCLC has been estimated at 2–4%, and lung adenocarcinoma is the predominant tumor type harboring METex14 skipping alterations.18,19 Numerous case series demonstrating response to MET TKI in patients with METex14 altered NSCLC then emerged20–23 and has reinvigorated interest in the development of MET targeted therapies in lung cancer.
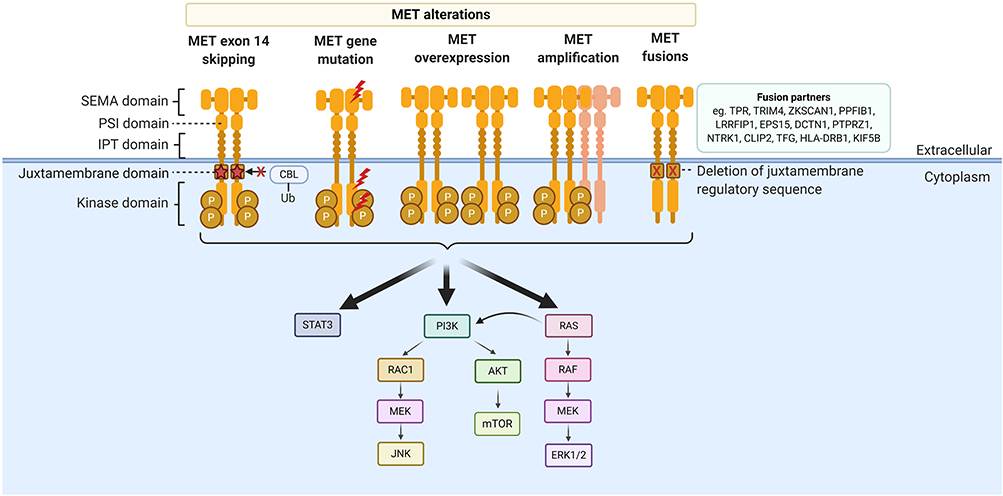 |
Figure 1 Structure of the MET receptor and MET alterations. MET alterations include MET exon 14 skipping mutations, other MET gene mutations, MET overexpression, MET amplification and MET fusions. |
Molecular Diagnostic Approaches for METex14 Skipping Alterations
METex14 alterations are diverse and can be challenging to detect.18 Immunohistochemical (IHC) studies, while relatively inexpensive, have not been proven useful thus far in detection of METex14 alterations. IHC is only able to detect MET overexpression, which may occur due to not only METex14 alterations but also increased gene copy number and gene amplification.24 Several studies have shown MET IHC overexpression poorly predicts for the presence of METex14 alterations.25–27 Furthermore, there may be a high degree of inter-observer variability in the interpretation of IHC.28 As such, most of the assays for METex14 involve molecular techniques (Figure 2). These assays can be divided into two main groups based on their starting nucleic acid substrate, i.e. DNA or RNA and their respective advantages and disadvantages will be discussed subsequently.
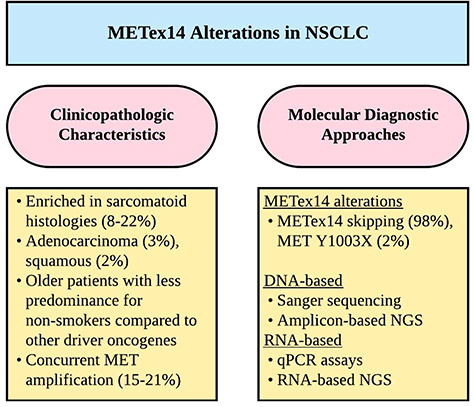 |
Figure 2 Diagnostic considerations for METex14 alterations in NSCLC. Abbreviations: METex14, MET exon 14; NGS, next-generation sequencing; NSCLC, non-small cell lung cancer; qPCR, quantitative PCR. |
DNA-based assays range from Sanger sequencing of single genes to next-generation sequencing (NGS) panels which can simultaneously analyze multiple regions of multiple genes. In either scenario, primer design is vital, as genomic deletion of sequences within the primer binding site impairs primer binding and can prevent amplification of the mutant allele, leading to a false-negative result.29,30 Sanger sequencing has a high specificity but has a relatively lower sensitivity compared to NGS panels.31 This, in combination with the scarcity of diagnostic tissue and the increasing approval of targeted therapies has led to the increasing implementation of NGS in clinical practice.
NGS panels can be further subdivided into hybrid capture-based panels including whole-exome panels and amplicon-based panels which tend to target clinically significant genes.32 With appropriate probe/bait design to cover the region of interest and algorithms to detect large deletions, hybrid capture mediated target enrichment tends to produce fewer false-negative results.18,33,34 However, hybrid capture-based panels have historically required more tumor DNA compared to amplicon-based panels and were often not implemented clinically.33 The diversity of METex14 alterations, however, could be important, and the ability to detect large insertions or deletions may provide advantages to hybrid capture-based panels.29
Amplicon-based NGS panels are not standardized and detection of specific alterations depends heavily on the primers within the panel.35 To highlight the importance of primer design, an earlier in silico analysis study of 8 amplicon-based NGS panels by Poirot et al30 highlighted limitations in accuracy, with only 63% of the literature reported cases of METex14 alterations being detected. However, as more is known about METex14 alterations, NGS panels can be optimized for greater accuracy and in a more recent study conducted by Pruis et al,36 in silico analysis of their customized NGS panel was able to detect 96% of reported METex14 alterations.
RNA-based methods of detecting METex14 skipping mutations such as quantitative PCR assays24,37 are based on the detection of a fusion transcript, which in this case, is between MET exon 13 and 15. As such, the interpretation of this assay is more straightforward than that of DNA-based assays for screening purposes.37 The main weakness of PCR-based assays is that for the purposes of primer design, knowledge of the fusion partner is required which makes the detection of novel fusions difficult. In contrast, RNA-based NGS panels for example the ArcherDX FusionPlex Solid Tumor Assay allow for fusion gene detection without a priori knowledge of the fusion partner.29,38
Another possible advantage of RNA-based methods is that the over-expression of MET can theoretically yield higher proportional concentrations of altered MET RNA transcripts available for analysis - especially if further enriched through micro dissection. This is as compared to extracted genomic DNA which is often derived not only from tumor cells but also from the admixed non-neoplastic inflammatory and stromal cells. As such, in some cases with a low proportion of tumor cells, the wild-type DNA can reduce the proportion of mutant DNA and lead to false-negative results.31 A study by Davies et al29 compared a DNA-based approach (Illumina TruSight Tumor 26 assay) and RNA-based detection (ArcherDX FusionPlex Solid Tumor Assay) of METex14 skipping events in lung cancer, and albeit not unexpectedly, found that RNA-based detection detected a higher proportion of METex14 skipping alteration cases as compared to DNA-based detection. Other studies have also demonstrated the detection of METex14 alterations using RNA-based NGS that were not detected using DNA-based assays.39 The main disadvantage of RNA-based testing is that RNA is less stable than DNA and in clinical samples, which are predominantly formalin fixed and paraffin-embedded (FFPE) tissues, poor quality RNA can lead to uninformative results on testing.29
Liquid biopsy, with the detection of alterations in plasma cell-free DNA (cfDNA) or circulating tumor cells (CTCs), is also emerging as a feasible diagnostic approach.40 METex14 alterations and MET amplification are detectable on numerous commercially available assays with high sensitivity, although are currently restricted to DNA-based assays.41,42 These diagnostic assays have already been incorporated in many of the clinical trials evaluating MET inhibitors in METex14 altered NSCLC.43–45
Pharmacology of Capmatinib and Early Phase Data
MET TKI can be broadly categorized based on the binding site and mechanism.46 Type I inhibitors are ATP-competitive and bind to Y1230 in the MET activation loop. Type Ia inhibitors such as crizotinib also interact with the solvent front glycine residue G1163, resulting in greater off-target effects, whereas type Ib inhibitors such as capmatinib, tepotinib and savolitinib have stronger interactions with Y1230 without interaction with G1163.10,47 Type II inhibitors, such as cabozantinib bind to the ATP adenine binding site extending to the hydrophobic back pocket, meaning potency depends on the activation state of the MET protein.10,47
Capmatinib (INC280), a type Ib inhibitor, is a potent highly selective oral MET inhibitor. In pre-clinical studies, it has been demonstrated to block MET phosphorylation and activation of key downstream effectors in MET-dependent cell lines.48 Furthermore, pleiotropic effects on other signaling pathways such as EGFR and HER3 were also seen. In the dose-escalation part of the Phase 1 trial (NCT01324479), 38 patients were treated with capmatinib, starting at 100mg bid in capsule formulation.49 There were no NSCLC patients in this cohort. Dose-limiting toxicities (DLT) occurred at dose levels 200mg bid (grade 3 fatigue), 250mg bid (grade 3 bilirubin increase) and 450mg bid capsules (grade 3 fatigue); however, the maximum tolerated dose (MTD) was not reached. There were no DLTs at the 600mg bid capsule formulation dose level, and additionally, 400mg bid tablets were found to have comparable tolerability and exposure. As this dose level was expected to achieve and maintain inhibition of MET, this became the recommended Phase 2 dose (RP2D). Overall, capmatinib was well tolerated, with nausea (32%), decreased appetite (29%), vomiting (29%) and fatigue (26%), the most frequent all grade capmatinib-related adverse events (AE). Fatigue (8%), ALT increase (5%) and hypophagia (5%) were the most frequent grade 3 or 4 capmatinib-related AEs.
There were several dose-expansion cohorts included in the trial, including two cohorts for advanced NSCLC patients.50 The first consisted of MET dysregulated NSCLC, defined as MET overexpression and amplification. The second cohort consisted of EGFR wild-type NSCLC with MET overexpression by IHC. A post hoc analysis further evaluated MET status using gene copy number (GCN) and amplification by fluorescence in-situ hybridization (FISH) and MET mutation by NGS. A total of 55 patients were enrolled (26 in the first cohort and 29 in the second cohort). The overall response rate was 20% (95% CI 10.4–33.0), with a response rate of 47% in patients with MET GCN ≥6 (n=15). Importantly however, all four patients with METex14 alterations achieved tumor response, including one complete response (CR). Most frequent all grade capmatinib-related AEs included nausea (42%), peripheral edema (33%) and vomiting (31%), with no grade 3 or 4 capmatinib-related AEs occurring in >10% of patients.
The Pivotal GEOMETRY Mono-1 Trial
GEOMETRY mono-1 (NCT02414139) is an ongoing phase II single-arm, multi-centre, multi-cohort trial of capmatinib tablets at 400mg bid in advanced EGFR and ALK wild-type NSCLC. There are seven cohorts, each analysed separately, based on centrally prescreened MET status and prior therapy. Enrollment in cohorts 1b, 2 and 3 (pre-treated patients with MET amplification <10 GCN) has been stopped early due to futility at a pre-planned interim analysis. Preliminary results have been reported for METex14 altered (regardless of GCN status) NSCLC patients in cohort 4 (1–2 prior lines of therapy, n=69) and cohort 5b (treatment naïve, n=28).6 The primary endpoint was objective response rate (ORR) by Blinded Independent Review Committee (BIRC), with key secondary endpoint of duration of response (DOR) by BIRC (Table 1). In pre-treated patients (cohort 4), the ORR was 41% (95% CI 27.6–51.6), with a median DOR of 9.7 months (95% CI 5.5–13.0). In treatment naïve patients (cohort 5b), the ORR was 68% (95% CI 47.6–84.1), with a median DOR of 12.6 months (95% CI 5.5–25.3). There were a small number of patients with brain metastases included in cohorts 4 and 5b, with intracranial response in 54% (7/13) of patients, including several cases of complete intracranial response.
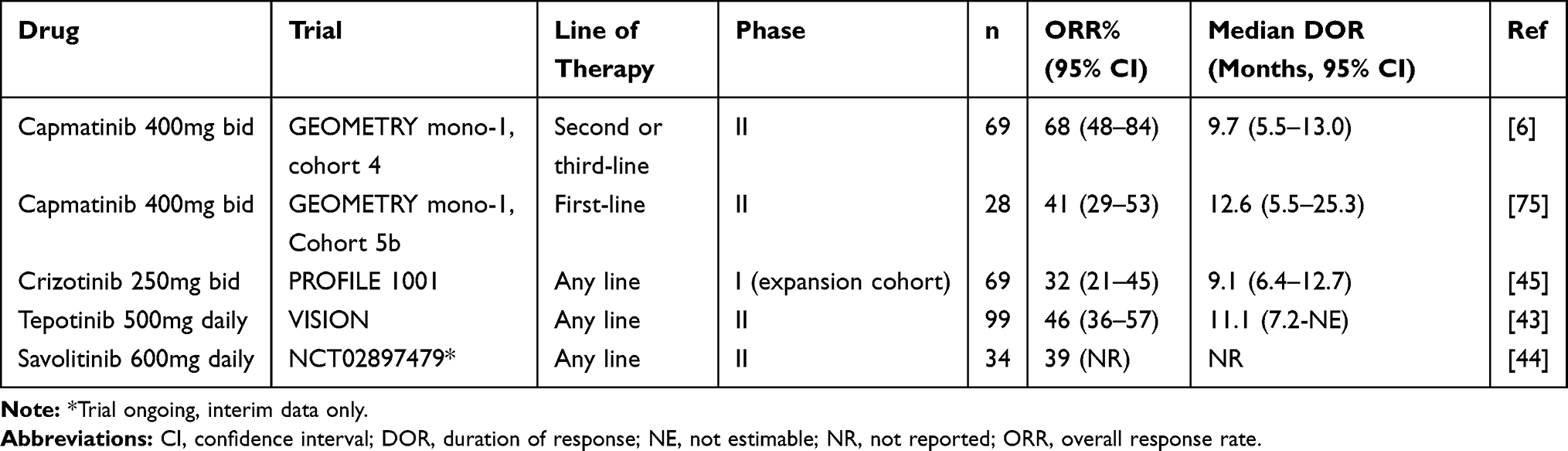 |
Table 1 Clinical Trials of MET Inhibitors in METex14 Altered NSCLC |
In the overall study population (n=334), the safety profile was consistent with the earlier trials of capmatinib.6,51 The most common all grade capmatinib-related AEs were peripheral edema (42%), nausea (33%), creatinine increase (20%), vomiting (19%), fatigue (14%), decreased appetite (13%) and diarrhea (11%) – the majority of which were grade 1 or 2. Pneumonitis was seen in 4.5% of patients, with grade 3 pneumonitis in 1.8%, and one death (0.3%). Treatment was discontinued in 8 (2.4%) patients due to pneumonitis. Hepatotoxicity with AST/ALT elevation was seen in 13% of patients, with grade 3 or 4 elevation in 6% and treatment discontinued in 3 (0.9%) patients.
Based on this data, the United States (US) Food and Drug Administration (FDA) granted accelerated approval for capmatinib (TabrectaTM) in NSCLC patients with a METex14 alteration detected by a companion diagnostic (FoundationOne CDx assay).52
Practical Clinical Considerations Including Dosing and Toxicity Management
Capmatinib is approved in tablet formulation at a starting dose of 400mg bid with or without food, coming in tablets with strengths of 150mg or 200mg. In patients that experience adverse reactions, dose reductions to 300mg bid and subsequently 200mg bid may be considered. Permanent discontinuation is recommended in patients that do not tolerate the 200mg bid dose level.
Clinically significant adverse reactions include pneumonitis and hepatotoxicity.51 In GEOMETRY mono-1, the median time to onset was 1.4 months (range 0.2–14.4) for grade 3 or higher pneumonitis, and 1.4 months (range 0.5–4.1) for grade 3 or higher AST/ALT elevation. Permanent treatment discontinuation is recommended for any grade pneumonitis and grade 4 AST/ALT elevation, or grade 2 or higher AST/ALT elevation in the presence of bilirubin increase >2 times upper limit of normal (ULN). For Grade 3 AST/ALT elevation without increase in bilirubin, dose interruption with subsequent dose reduction if recovery to baseline AST/ALT takes longer than 7 days is suggested.
Common toxicities, as described previously, include peripheral edema, nausea, fatigue, vomiting, dyspnea and decreased appetite. Dose interruptions were required in 54% of patients in GEOMETRY mono-1, with dose reductions in 23% of patients.6,51 Dose reductions were most commonly due to peripheral edema, increased ALT, increased blood creatinine and nausea. Peripheral edema, in particular, has also been seen in other agents targeting the MET pathway and may require additional management with limb elevation, compression stockings and occasionally diuretics.53,54
Therapeutic Resistance to Capmatinib
Despite promising efficacy data, drug resistance inevitably ensues, and the mechanisms of resistance to capmatinib in METex14 NSCLC is yet to be well characterized. In EGFR mutant and ALK rearranged NSCLC, for which resistance to TKI therapy is better understood, a diverse range of mechanisms may occur.55–57 This includes acquisition of secondary resistance mutations or alterations, activation of bypass signaling pathways or phenotypic change such as small cell transformation.
Secondary resistance MET mutations in D1228 and Y1230 to type I MET inhibitors have been demonstrated in in vitro drug screens and mutagenesis assays.58,59 Numerous case studies have described these resistance mutations to crizotinib therapy in METex14 altered NSCLC.60–63 In vitro studies of MET-amplified cell lines treated with capmatinib, also suggest activation of EGFR signaling and downstream effectors such as PIK3CA may mediate resistance.64 The EGFR pathway has further been implicated in resistance to MET kinase inhibition in other studies.58,65 MET-directed therapy in EGFR TKI resistant EGFR mutant NSCLC is an area of ongoing investigation, including clinical trials with capmatinib.66 The cross-talk between the MET and EGFR signaling pathways in NSCLC therefore has potential significance in resistance.67 Similarly, there is pre-clinical data suggesting KRAS signaling may be upregulated in METex14 tumors, and expression of mutant KRAS may induce resistance to MET-directed therapy.68 The PI3K pathway is also implicated in resistance, and potentially primary resistance.69 In a series of 20 MET TKI treated patients with post-treatment NGS testing, acquired MET resistance mutations, MET exon 14 mutant allele amplification, KRAS mutations and KRAS, EGFR, HER3 and BRAF amplifications were seen.70 There were two patients treated with capmatinib, with post-resistance NGS demonstrating acquisition of MET D1228N mutation, EGFR and HER3 amplification in the first patient, and EGFR amplification and HER3 gain in the second patient.
A large series of 298 patients with METex14 altered NSCLC indicated rates of concurrent MDM2 amplification in 35%, CDK4 amplification in 21%, EGFR amplification in 6% and KRAS mutation in 3%.34 Additionally, concurrent MET amplification was seen in 15%, and this was also associated with higher tumor mutation burden (TMB). Another series of 289 patients suggested co-occurring RAS-MAPK pathway gene alterations such as KRAS and NF1, may be associated with decreased response to MET TKI.71 Ultimately though, any potential implication of these concurrent alterations on response and resistance to capmatinib is yet to be validated.
Patterns of clinical progression to capmatinib are also important to evaluate. In particular, brain metastases occur with greater frequency in oncogenic driven NSCLC.72 However, the prevalence of brain metastases in METex14 altered NSCLC and indeed the overall natural history of disease remains to be completely elucidated. In one series of 34 patients with METex14 altered NSCLC, brain metastases were seen in 21% of patients and were the second most common site of metastases after bone.73 In another series of 71 patients, the incidence of brain metastases was 37%.74 As described previously, only a small number of patients with brain metastases were included on GEOMETRY mono-1, although intracranial responses were seen in over half,75 suggesting reasonable intracranial activity for capmatinib.
Therapeutic Landscape of METex14 Altered NSCLC
Capmatinib is the first FDA-approved therapy for patients with METex14 altered NSCLC (Figure 3). However, there are other MET TKIs which have also been studied in this patient population (Table 1). Crizotinib was evaluated in an expansion cohort of the PROFILE 1001 trial (NCT00585195), in patients with METex14 altered NSCLC.45 A total of 69 patients were enrolled, with 62% having received at least one prior line of therapy, and 96% had METex14 alterations detected by NGS. Among 65 evaluable patients, ORR was 32% (95% CI 21–45) with median progression-free survival (PFS) of 7.3 months (95% CI 5.4–9.1) and median DOR of 9.1 months (95% CI 6.4–12.7). Subsequently, crizotinib was granted FDA breakthrough therapy designation in 2018, and has also been incorporated into the National Comprehensive Cancer Network (NCCN) guidelines for NSCLC.76
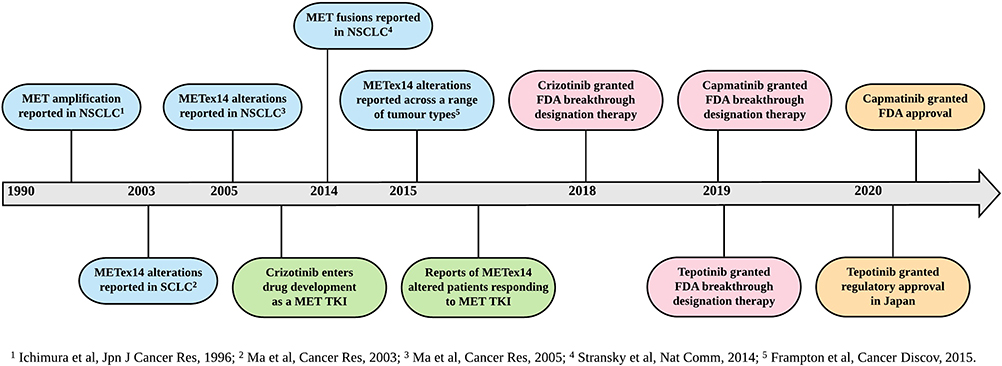 |
Figure 3 Timeline of key events in the development of METex14 altered NSCLC targeted therapies. Abbreviations: FDA, Food and Drug Administration; METex14, MET exon 14; NSCLC, non-small cell lung cancer; TKI, tyrosine kinase inhibitor. |
Tepotinib was evaluated in the phase II VISION trial (NCT02864992), in patients with METex14 altered NSCLC.43 A total of 152 patients were treated (safety population), with 99 having follow-up of at least 9 months (efficacy population). In the efficacy population, METex14 alterations were detected using NGS on liquid biopsy (66%) and/or tissue biopsy (60%), and 43% of patients were treatment naïve. The ORR was 46% (95% CI 36–57) with median PFS of 8.5 months (95% CI 6.7–11.0) and median DOR of 11.1 months (95% CI 7.2-NE). Outcomes were similar comparing METex14 alterations detected on liquid versus tissue biopsy. Tepotinib was also granted FDA breakthrough therapy designation in 2019 and has been fully approved in Japan with a companion diagnostic (ArcherMET CDx) assay.77 For tepotinib, in contrast to capmatinib, response rates and PFS was no different comparing patients treated as first-line versus subsequent lines of therapy.6,43
Finally, savolitinib is being evaluated in an ongoing phase II trial (NCT02897479) in China.44 Patients may be treatment naïve or pre-treated. Preliminary data demonstrated an ORR of 39% in 34 patients treated on the trial to date.
The therapeutic landscape in METex14 altered NSCLC is evolving, with numerous aforementioned TKIs showing promising early efficacy data leading to regulatory approvals. A confirmatory phase III trial for capmatinib (NCT04427072) in pre-treated METex14 altered NSCLC patients versus docetaxel chemotherapy is due to commence recruitment. Crizotinib continues to be evaluated in basket trials for METex14 altered NSCLC such as the National Lung Matrix trial (NCT02664935). Other MET TKI in development for METex14 altered NSCLC include cabozantinib, glesatinib and merestinib.47,78 With confirmation of METex14 alterations as a bona fide target in NSCLC, careful consideration on sequencing and combining therapies becomes crucial. Accordingly, there are also ongoing trials to address these questions. Phase II trials of capmatinib after resistance to prior MET TKI (NCT02750215) and capmatinib in combination with immunotherapy with spartalizumab (NCT04323436) are examples of currently recruiting or planned trials.
Conclusion
METex14 alterations in advanced NSCLC are now established as a therapeutic target in both the treatment naïve and pre-treated settings. It joins a growing list of biomarkers in NSCLC, further emphasizing the importance of molecular profiling and diagnosis in this patient population. Consequently, this also brings greater complexity in the appropriate selection and sequencing of therapies. Ongoing clinical trials and translational studies will aid in determining the role for capmatinib and other MET-directed therapies in METex14 altered NSCLC.
Acknowledgments
A.C.T is the recipient of an International Association for the Study of Lung Cancer (IASLC) Fellowship 2018–2020. D.S.W.T is the recipient of the NMRC clinician-scientist award (NMRC/CSA/007/2016).
Disclosure
Aaron C. Tan reports honoraria from ThermoFisher and personal fees from Thermo Fisher, outside the submitted work. Kiat Hon Lim reports honoraria from AstraZeneca and consulting and advisory role for MSD. Daniel S.W. Tan reports research grant advisory for and consultancy honoraria from Novartis, Pfizer, and Astra Zeneca, research grant from and advisory/consultancy for Bayer, advisory for and consultancy honoraria from Boehringer Ingelheim and Takeda, honoraria from Bristol-Myers Squibb, Takeda Pharmaceuticals, Novartis, Roche, Pfizer, and Merck; consulting or advisory role for Eli-Lilly, Merrimack, Novartis, Merck, Loxo Oncology, AstraZeneca, Roche, and Pfizer, and research funding from Novartis (Inst), GlaxoSmithKline (Inst), AstraZeneca (Inst). The authors report no other potential conflicts of interest for this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492.
2. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446. doi:10.1038/nature25183
3. Tsurutani J, Iwata H, Krop I, et al. Targeting HER2 with trastuzumab deruxtecan: a dose-expansion, phase I study in multiple advanced solid tumors. Cancer Discov. 2020;10:688–701. doi:10.1158/2159-8290.CD-19-1014.
4. Fakih M, O’Neil B, Price TJ, et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS G12C inhibitor, in advanced solid tumors. J Clin Oncol. 2019;37(15_suppl):3003. doi:10.1200/JCO.2019.37.15_suppl.3003.
5. Schram AM, O’Reilly EM, Somwar R, et al. Abstract PR02: clinical proof of concept for MCLA-128, a bispecific HER2/3 antibody therapy, in NRG1 fusion-positive cancers. Mol Cancer Ther. 2019;18:PR02. doi:10.1158/1535-7163.TARG-19-PR02.
6. Wolf J, Seto T, Han J-Y, et al. Capmatinib in MET exon 14–mutated or MET -amplified non–small-cell lung cancer. N Engl J Med. 2020;383(10):944–957. doi:10.1056/NEJMoa2002787.
7. Scagliotti G, von Pawel J, Novello S, et al. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol. 2015;33(24):2667–2674. doi:10.1200/JCO.2014.60.7317.
8. Yoshioka H, Azuma K, Yamamoto N, et al. A randomized, double-blind, placebo-controlled, phase III trial of erlotinib with or without a c-Met inhibitor tivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall-cell lung cancer harboring wild-type epidermal growth factor receptor (ATTENTION study). Ann Oncol. 2015;26:2066–2072. doi:10.1093/annonc/mdv288.
9. Spigel DR, Edelman MJ, O’Byrne K, et al. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV non-small-cell lung cancer: mETLung. J Clin Oncol. 2017;35(4):412–420. doi:10.1200/JCO.2016.69.2160.
10. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. doi:10.1038/nrc3205.
11. Komada M, Hatsuzawa K, Shibamoto S, Ito F, Nakayama K, Kitamura N. Proteolytic processing of the hepatocyte growth factor/scatter factor receptor by furin. FEBS Lett. 1993;328(1–2):25–29. doi:10.1016/0014-5793(93)80958-w.
12. Cooper CS, Park M, Blair DG, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311(5981):29–33. doi:10.1038/311029a0.
13. Vigna E, Gramaglia D, Longati P, Bardelli A, Comoglio PM. Loss of the exon encoding the juxtamembrane domain is essential for the oncogenic activation of TPR-MET. Oncogene. 1999;18(29):4275–4281. doi:10.1038/sj.onc.1202791.
14. Lee CC, Yamada KM. Identification of a novel type of alternative splicing of a tyrosine kinase receptor. Juxtamembrane deletion of the c-met protein kinase C serine phosphorylation regulatory site. J Biol Chem. 1994;269:19457–19461.
15. Peschard P, Fournier TM, Lamorte L, et al. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell. 2001;8(5):995–1004. doi:10.1016/S1097-2765(01)00378-1
16. Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65(4):1479–1488. doi:10.1158/0008-5472.CAN-04-2650.
17. Kong-Beltran M, Seshagiri S, Zha J, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66(1):283–289. doi:10.1158/0008-5472.CAN-05-2749.
18. Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–859. doi:10.1158/2159-8290.CD-15-0285.
19. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi:10.1038/nature13385..
20. Paik PK, Drilon A, Fan P-D, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5(8):842–849. doi:10.1158/2159-8290.CD-14-1467.
21. Lee C, Usenko D, Frampton GM, McMahon C, Ali SM, Weiss J. MET 14 deletion in sarcomatoid non-small-cell lung cancer detected by next-generation sequencing and successfully treated with a MET inhibitor. J Thorac Oncol. 2015;10(12):e113–114. doi:10.1097/JTO.0000000000000645.
22. Waqar SN, Morgensztern D, Sehn J. MET mutation associated with responsiveness to crizotinib. J Thorac Oncol. 2015;10(5):e29–31. doi:10.1097/JTO.0000000000000478.
23. Mendenhall MA, Goldman JW. MET-mutated NSCLC with major response to crizotinib. J Thorac Oncol. 2015;10(5):e33–4. doi:10.1097/JTO.0000000000000491.
24. O’Brien O, Wright MC, O’Brien C, et al. Cost-efficient and easy to perform PCR-based assay to identify met exon 14 skipping in formalin-fixed paraffin-embedded (FFPE) non-small cell lung cancer (NSCLC) samples. Diagnostics (Basel). 2019;9. doi:10.3390/diagnostics9010013.
25. Mignard X, Ruppert A-M, Antoine M, et al. c-MET overexpression as a poor predictor of MET amplifications or exon 14 mutations in lung sarcomatoid carcinomas. J Thorac Oncol. 2018;13(12):1962–1967. doi:10.1016/j.jtho.2018.08.008.
26. Guo R, Berry LD, Aisner DL, et al. MET IHC is a poor screen for MET amplification or MET exon 14 mutations in lung adenocarcinomas: data from a tri-institutional cohort of the lung cancer mutation consortium. J Thorac Oncol. 2019;14(9):1666–1671. doi:10.1016/j.jtho.2019.06.009.
27. Baldacci S, Figeac M, Antoine M, et al. High MET overexpression does not predict the presence of MET exon 14 splice mutations in NSCLC: results from the IFCT PREDICT. J Thorac Oncol. 2020;15(1):120–124. doi:10.1016/j.jtho.2019.09.196.
28. Bubendorf L, Dafni U, Schöbel M, et al. Prevalence and clinical association of MET gene overexpression and amplification in patients with NSCLC: results from the European thoracic oncology platform (ETOP) lungscape project. Lung Cancer. 2017;111:143–149. doi:10.1016/j.lungcan.2017.07.021.
29. Davies KD, Lomboy A, Lawrence CA, et al. DNA-based versus RNA-based detection of MET exon 14 skipping events in lung cancer. J Thorac Oncol. 2019;14(4):737–741. doi:10.1016/j.jtho.2018.12.020.
30. Poirot B, Doucet L, Benhenda S, Champ J, Meignin V, Lehmann-Che J. MET exon 14 alterations and new resistance mutations to tyrosine kinase inhibitors: risk of inadequate detection with current amplicon-based NGS panels. J Thorac Oncol. 2017;12(10):1582–1587. doi:10.1016/j.jtho.2017.07.026.
31. Kim EK, Kim KA, Lee CY, et al. Molecular diagnostic assays and clinicopathologic implications of MET exon 14 skipping mutation in non-small-cell lung cancer. Clin Lung Cancer. 2019;20(1):e123–32. doi:10.1016/j.cllc.2018.10.004.
32. Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome Med. 2015;7(1):80. doi:10.1186/s13073-015-0203-x.
33. Awad MM, Oxnard GR, Jackman DM, et al. MET exon 14 mutations in non–small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression. J Clin Oncol. 2016;34(7):721–730. doi:10.1200/JCO.2015.63.4600.
34. Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol. 2016;11(9):1493–1502. doi:10.1016/j.jtho.2016.06.004.
35. Drilon A, Cappuzzo F, Ou S-HI, Camidge DR. Targeting MET in lung cancer: will expectations finally be MET? J Thorac Oncol. 2017;12(1):15–26. doi:10.1016/j.jtho.2016.10.014.
36. Pruis MA, Geurts-Giele WRR, von der TJH. Highly accurate DNA-based detection and treatment results of MET exon 14 skipping mutations in lung cancer. Lung Cancer. 2020;140:46–54. doi:10.1016/j.lungcan.2019.11.010.
37. Lung J, Hung M-S, Lin Y-C, et al. MET exon 14 skipping mutations and gene amplification in a Taiwanese lung cancer population. PLoS One. 2019;14(8):e0220670. doi:10.1371/journal.pone.0220670.
38. Davies KD, Ng TL, Estrada-Bernal A, et al. Dramatic response to crizotinib in a patient with lung cancer positive for an HLA-DRB1-MET gene fusion. JCO Precis Oncol. 2017;2017. doi:10.1200/PO.17.00117.
39. Benayed R, Offin M, Mullaney K, et al. High yield of RNA sequencing for targetable kinase fusions in lung adenocarcinomas with no mitogenic driver alteration detected by DNA sequencing and low tumor mutation burden. Clin Cancer Res. 2019;25(15):4712–4722. doi:10.1158/1078-0432.CCR-19-0225.
40. Rolfo C, Mack PC, Scagliotti GV, et al. Liquid biopsy for advanced non-small cell lung cancer (NSCLC): a statement paper from the IASLC. J Thorac Oncol. 2018;13(9):1248–1268. doi:10.1016/j.jtho.2018.05.030.
41. Mondelo-Macía P, Rodríguez-López C, Valiña L, et al. Detection of MET alterations using cell free DNA and circulating tumor cells from cancer patients. Cells. 2020;9(2):522. doi:10.3390/cells9020522.
42. Salgia R, Sattler M, Scheele J, Stroh C, Felip E. The promise of selective MET inhibitors in non-small cell lung cancer with MET exon 14 skipping. Cancer Treat Rev. 2020;87:102022. doi:10.1016/j.ctrv.2020.102022.
43. Paik PK, Felip E, Veillon R, et al. Tepotinib in non–small-cell lung cancer with MET exon 14 skipping mutations. N Engl J Med. 2020;383(10):931–943. doi:10.1056/NEJMoa2004407.
44. Lu S, Fang J, Cao L, et al. Abstract CT031: preliminary efficacy and safety results of savolitinib treating patients with pulmonary sarcomatoid carcinoma (PSC) and other types of non-small cell lung cancer (NSCLC) harboring MET exon 14 skipping mutations. Cancer Res. 2019;79:CT031–CT031. doi:10.1158/1538-7445.AM2019-CT031.
45. Drilon A, Clark JW, Weiss J, et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med. 2020;26(1):47–51. doi:10.1038/s41591-019-0716-8.
46. Backes A, Zech B, Felber B, Klebl B, Müller G. Small-molecule inhibitors binding to protein kinase. Part II: the novel pharmacophore approach of type II and type III inhibition. Expert Opin Drug Discov. 2008;3(12):1427–1449. doi:10.1517/17460440802580106.
47. Reungwetwattana T, Liang Y, Zhu V, Ou S-HI. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: the why, the how, the who, the unknown, and the inevitable. Lung Cancer. 2017;103:27–37. doi:10.1016/j.lungcan.2016.11.011.
48. Liu X, Wang Q, Yang G, et al. A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin Cancer Res. 2011;17(22):7127–7138. doi:10.1158/1078-0432.CCR-11-1157.
49. Bang Y-J, Su W-C, Schuler M, et al. Phase 1 study of capmatinib in MET-positive solid tumor patients: dose escalation and expansion of selected cohorts. Cancer Sci. 2020;111(2):536–547. doi:10.1111/cas.14254.
50. Schuler M, Berardi R, Lim W-T, et al. Molecular correlates of response to capmatinib in advanced non-small-cell lung cancer: clinical and biomarker results from a phase I trial. Ann Oncol. 2020;31(6):789–797. doi:10.1016/j.annonc.2020.03.293.
51. Food and Drug Administration. Full Prescribing Information for Tabrecta. 2020.
52. Food and Drug Administration. FDA Grants Accelerated Approval to Capmatinib for Metastatic Non-Small Cell Lung Cancer. 2020.
53. Garajová I, Giovannetti E, Biasco G, Peters GJ. c-Met as a target for personalized therapy. Transl Oncogenomics. 2015;7:13–31. doi:10.4137/TOG.S30534.
54. Rothenstein JM, Letarte N. Managing treatment-related adverse events associated with Alk inhibitors. Curr Oncol. 2014;21(1):19–26. doi:10.3747/co.21.1740.
55. Oxnard GR, Hu Y, Mileham KF, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M–positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 2018;4(11):1527–1534. doi:10.1001/jamaoncol.2018.2969.
56. Yu HA, Suzawa K, Jordan E, et al. Concurrent alterations in EGFR-mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of MTOR as a mediator of resistance. Clin Cancer Res. 2018;24(13):3108–3118. doi:10.1158/1078-0432.CCR-17-2961.
57. Lin JJ, Zhu VW, Yoda S, et al. Impact of EML4-ALK variant on resistance mechanisms and clinical outcomes in ALK-positive lung cancer. JCO. 2018;36(12):1199–1206. doi:10.1200/JCO.2017.76.2294.
58. Qi J, McTigue MA, Rogers A, et al. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res. 2011;71(3):1081–1091. doi:10.1158/0008-5472.CAN-10-1623.
59. Tiedt R, Degenkolbe E, Furet P, et al. A drug resistance screen using a selective MET inhibitor reveals a spectrum of mutations that partially overlap with activating mutations found in cancer patients. Cancer Res. 2011;71(15):5255–5264. doi:10.1158/0008-5472.CAN-10-4433.
60. Heist RS, Sequist LV, Borger D, et al. Acquired resistance to crizotinib in NSCLC with MET exon 14 skipping. J Thorac Oncol. 2016;11(8):1242–1245. doi:10.1016/j.jtho.2016.06.013.
61. Jin W, Shan B, Liu H, et al. Acquired mechanism of crizotinib resistance in NSCLC with MET exon 14 skipping. J Thorac Oncol. 2019;14(7):e137–9. doi:10.1016/j.jtho.2019.04.021.
62. Zhang Y, Yin J, Peng F. Acquired resistance to crizotinib in advanced lung adenocarcinoma with MET exon 14 skipping. Lung Cancer. 2017;113:69–71. doi:10.1016/j.lungcan.2017.09.006.
63. Ou S-HI, Young L, Schrock AB, et al. Emergence of preexisting MET Y1230C mutation as a resistance mechanism to crizotinib in NSCLC with MET exon 14 skipping. J Thorac Oncol. 2017;12(1):137–140. doi:10.1016/j.jtho.2016.09.119.
64. Kim S, Kim TM, Kim D-W, et al. Acquired resistance of MET-amplified non-small cell lung cancer cells to the MET inhibitor capmatinib. Cancer Res Treat. 2019;51(3):951–962. doi:10.4143/crt.2018.052.
65. McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010;70(4):1625–1634. doi:10.1158/0008-5472.CAN-09-3620.
66. Wu Y-L, Zhang L, Kim D-W, et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J Clin Oncol. 2018;36:3101–3109. doi:10.1200/JCO.2018.77.7326.
67. Puri N, Salgia R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J Carcinog. 2008;7(1):9. doi:10.4103/1477-3163.44372.
68. Suzawa K, Offin M, Lu D, et al. Activation of KRAS mediates resistance to targeted therapy in MET exon 14-mutant non-small cell lung cancer. Clin Cancer Res. 2019;25(4):1248–1260. doi:10.1158/1078-0432.CCR-18-1640.
69. Jamme P, Fernandes M, Copin M-C, et al. Alterations in the PI3K pathway drive resistance to MET inhibitors in NSCLC harboring MET exon 14 skipping mutations. J Thorac Oncol. 2020;15(5):741–751. doi:10.1016/j.jtho.2020.01.027.
70. Recondo G, Bahcall M, Spurr LF, et al. Molecular mechanisms of acquired resistance to MET tyrosine kinase inhibitors in patients with MET exon 14-mutant NSCLC. Clin Cancer Res. 2020;26(5981):2615–2625. doi:10.1158/1078-0432.CCR-19-3608.
71. Rotow JK, Gui P, Wu W, et al. Co-occurring alterations in the RAS-MAPK pathway limit response to MET inhibitor treatment in MET exon 14 skipping mutation-positive lung cancer. Clin Cancer Res. 2020;26(2):439–449. doi:10.1158/1078-0432.CCR-19-1667.
72. Remon J, Besse B. Brain metastases in oncogene-addicted non-small cell lung cancer patients: incidence and treatment. Front Oncol. 2018;8:88. doi:10.3389/fonc.2018.00088.
73. Digumarthy SR, Mendoza DP, Zhang EW, Lennerz JK, Heist RS. Clinicopathologic and imaging features of non-small-cell lung cancer with MET exon 14 skipping mutations. Cancers (Basel). 2019;11. doi:10.3390/cancers11122033.
74. Awad MM, Leonardi GC, Kravets S, et al. Impact of MET inhibitors on survival among patients with non-small cell lung cancer harboring MET exon 14 mutations: a retrospective analysis. Lung Cancer. 2019;133(22):96–102. doi:10.1016/j.lungcan.2019.05.011.
75. Wolf J, Seto T, Han J-Y, et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): efficacy data from the phase II GEOMETRY mono-1 study. JCO. 2019;37(15_suppl):9004. doi:10.1200/JCO.2019.37.15_suppl.9004.
76. Ettinger DS, Aisner DL, Wood DE, et al. NCCN guidelines insights: non-small cell lung cancer, version 5.2018. J Natl Compr Canc Netw. 2018;16(7):807–821. doi:10.6004/jnccn.2018.0062.
77. Markham A. Tepotinib: first approval. Drugs. 2020;80(8):829–833. doi:10.1007/s40265-020-01317-9.
78. Rybkin II, Kio EA, Masood A, et al. Amethyst NSCLC trial: phase 2, parallel-arm study of receptor tyrosine kinase (RTK) inhibitor, MGCD265, in patients (pts) with advanced or metastatic non-small cell lung cancer (NSCLC) with activating genetic alterations in mesenchymal-epithelial transition factor (MET). JCO. 2016;34:TPS9099–TPS9099. doi:10.1200/JCO.2016.34.15_suppl.TPS9099.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.