")
Back to Journals » OncoTargets and Therapy » Volume 13
Novel Therapeutic Strategies for CDK4/6 Inhibitors in Metastatic Castrate-Resistant Prostate Cancer
Authors Kase AM , Copland III JA, Tan W
Received 4 June 2020
Accepted for publication 18 September 2020
Published 15 October 2020 Volume 2020:13 Pages 10499—10513
DOI https://doi.org/10.2147/OTT.S266085
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Adam M Kase,1 John A Copland III,2 Winston Tan1
1Mayo Clinic Florida Division of Hematology Oncology, Jacksonville, FL 32224, USA; 2Mayo Clinic Florida Department of Cancer Biology, Jacksonville, FL 32224, USA
Correspondence: Adam M Kase Tel +1 904-953-0315
Fax +1 905-953-2315
Email [email protected]
Abstract: The majority of patients with castrate-resistant prostate cancer will have metastatic disease at the time of diagnosis. Investigative efforts on new therapeutics for this patient population have improved with the development of androgen signaling inhibitors, such as abiraterone and enzalutamide, and PARP inhibitors, such as rucaparib and olaparib, to accompany the previously FDA-approved docetaxel, cabazitaxel, sipuleucel-T, and Radium 223. However, new therapeutic strategies are necessary to prolong survival as progression after these agents is inevitable. CDK4/6 inhibitors have advanced the field of estrogen receptor positive breast cancer treatment and are being investigated in prostate cancer given the role of androgen receptor signaling effects on the cell cycle. Response to CDK4/6 inhibitors may be predicted by the tumors’ genomic profile and may provide insight into combinatory therapy with CDK4/6 inhibitors in order to delay resistance or provide synergistic effects. Here, we review the use of CDK4/6 inhibitors in prostate cancer and potential combinations based on known resistance mechanisms to CDK4/6 inhibitors, prostate cancer regulatory pathways, and prostate-cancer-specific genomic alterations.
Keywords: metastatic castrate-resistant prostate cancer, CDK4/6 inhibitors, genomics, combination therapy
Introduction
In 1853, surgeon J. Adams once described prostate cancer (PCa) as a very rare disease. Fast forward over 160 years, PCa is now the second leading cause of cancer deaths among men in the United States.1 Despite androgen deprivation therapy (ADT), PCa will eventually develop resistance known as castrate-resistant prostate cancer (CRPC) and thus, progress to end-stage disease. There are currently eight FDA-approved therapies indicated for metastatic castrate-resistant prostate cancer (mCRPC) that include: docetaxel, abiraterone, enzalutamide, cabazitaxel, sipuleucel-T, radium-223, rucaparib, and olaparib. Unfortunately, the overall survival benefit from these therapies ranges from 2.4 to 4.8 months, demonstrating the importance of continued development of new therapeutics.2
The cell cycle contains key regulatory factors to control cellular growth. These factors include enzymes such as cyclin-dependent kinases (CDK).3 In cancer, this pathway is universally disrupted resulting in tumorigenesis and over the past decade has become a focus of cancer therapeutics leading to the development of CDK4/6 inhibitors (CDK4/6i).4 CDK4/6i’s first gained approval in 2015 when palbociclib showed encouraging activity in breast cancer with clinical trials showing a double in median progression-free survival (mPFS) and also an overall survival (OS) benefit. Estrogen receptors, as well as androgen receptors, are key regulators of the cell cycle and instrumental in the regulation of transcription genes that allow for G1 to S transition.5,6 In PCa, this hormone pathway can be targeted with ADT; however, PCa eventually becomes androgen resistant which provides opportunity for the use of CDK4/6i’s to disrupt AR signaling. In patients with hormone sensitive prostate cancer (HSPC), a Phase II clinical trial (NCT02059213) investigating ADT ± palbociclib showed tolerability and multiple other trials are underway with combining androgen signaling inhibitors with CDK4/6i’s. Understanding the cell cycle as well as associated regulatory pathways and PCa specific genomic mutations (Table 1) will provide insight to potential combinatorial therapies with CDK4/6i’s. Table 1 shows mutated genes in mCRPC and will be referred throughout this review in guiding novel combinatorial therapeutic strategies.
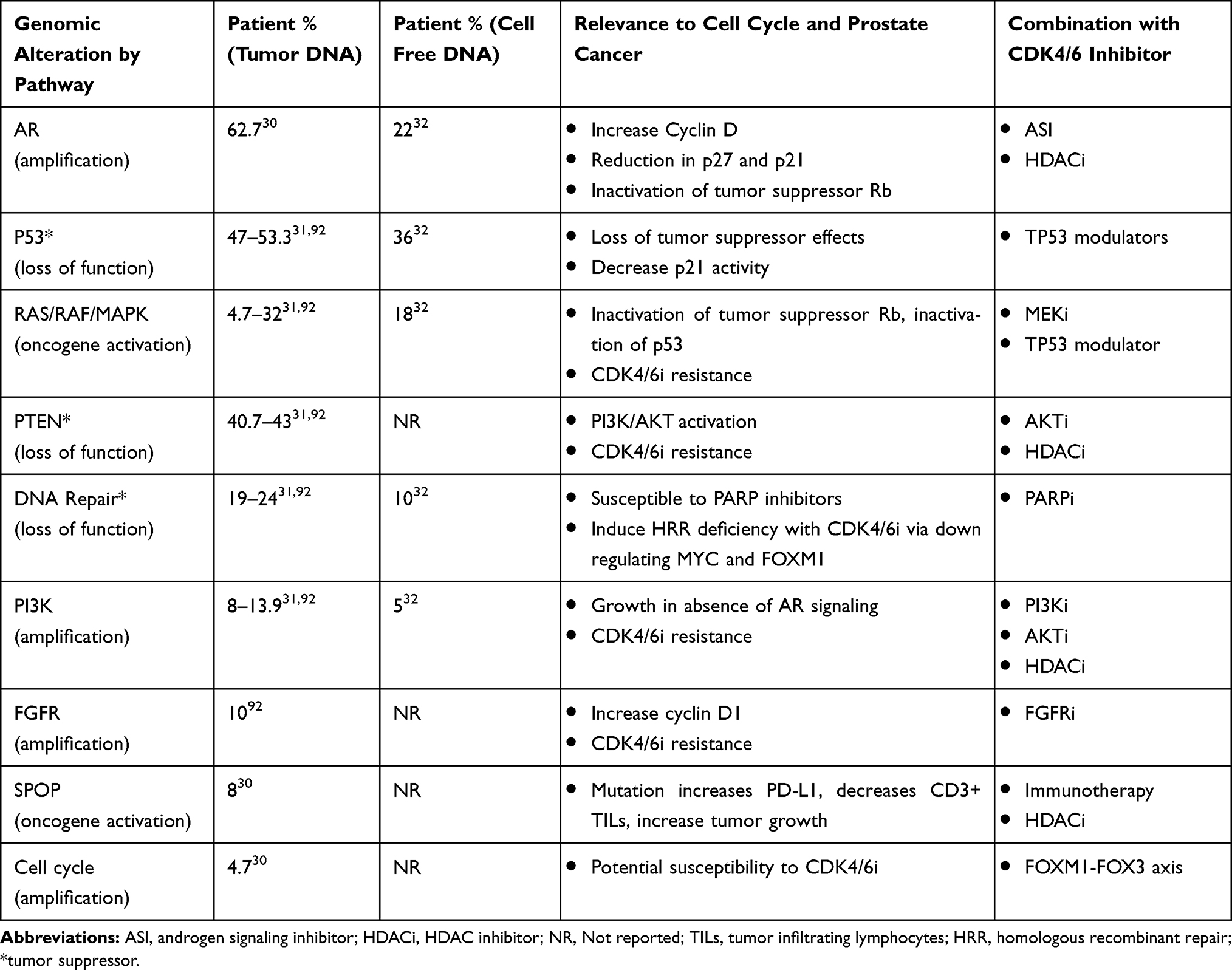 |
Table 1 Subset of Mutations Found in mCRPC and the Percent Occurrence as Measured in Patient Tumor Tissue or from Blood Sequencing Cell Free DNA |
Cell Cycle Pathway and CDK4/6 Inhibitors in Cancer
Cellular proliferation begins with a stimulus allowing cells to enter G1 phase from the quiescent G0 phase. In order for a cell to continue through the cell cycle, it must pass checkpoints controlled by regulatory proteins. The regulatory proteins involved in the transition from G1 phase (growth/metabolism) to S phase (replication) include: cyclin D, CDK4, CDK6, Retinoblastoma gene product (Rb) and E2 transcription factor (E2F).7 Cyclin D is induced by growth factors and binds to CDK4 and CDK6 converting them to an active state. The cyclin-D-CDK4/6 complex phosphorylates the active tumor suppressor, Rb, sending it in to its hyper-phosphorylated inactive state. When Rb is in the active state it binds and inhibits the activating E2F transcription factors. However, when Rb is in the inactive hyper-phosphorylated state, Rb dissociates from the activating E2F transcription factor and drives E2F targeted genes involved in DNA replication allowing the cell to enter S phase.8 The transition from G1 to S is highly regulated by the CDK pathway involving cyclin D-CDK4/6-p16-Rb making CDK4/6 inhibitors prime agents for disrupting the cell cycle. There are multiple CDK4/6i’s that are FDA approved as anti-neoplastic agents including palbociclib, ribociclib, and abemaciclib for metastatic breast cancer.
Palbociclib (PD 0332991, Ibrance) was the first CDK4/6i to gain FDA approval in 2015. It was approved for combination therapy with letrozole in postmenopausal women with locally advanced or metastatic HER2-negative, estrogen receptor positive breast cancer.9 In PALMOA-1 and PALMOA-2, letrozole with and without palbociclib was given as first-line treatment to patients with ER-positive/HER2 negative advanced breast cancer and showed a mPFS prolongation of 10 months (20.2 months vs 10.2 months) and 10.3 months (24.8 months vs 14.5 months), respectively.10,11 The most common grade 3 and 4 adverse events seen were neutropenia (66%), leukopenia (24.8%), anemia and fatigue.11 Palbociclib is given on a 28-day cycle with daily dosing on days 1–21 and off on days 22–28. Clinical trials which are currently recruiting or active for PCa include: palbociclib in patients with mCRPC (NCT02905318) and a Phase II study of ADT with or without palbociclib in Rb-positive (ie, wild type) metastatic PCa (NCT02059213).
Ribociclib (LEE011, Kisqali) gained its first FDA approval in 2017 for treatment of postmenopausal hormone receptor (HR) positive HER2 negative metastatic breast cancer in combination with an aromatase inhibitor. MONALEESA-2 was a Phase III randomized placebo controlled trial investigating letrozole ± ribociclib in post-menopausal patients with HR positive and HER2 negative advanced breast cancer. The ribociclib group had an improved 18 month PFS, 63% vs 42.2% (Hazard Ratio 0.56), leading to its approval.12 The dosing regimen is the same as palbociclib with once-daily dosing for 21 days followed by 7 days off. Adverse effects are similar to palbociclib and may also cause prolong the QT interval (1–6%), limiting its use in patients with cardiac comorbidities.
Abemaciclib (LY2835219, Verzenio) was first approved in 2017 in women with HR positive, HER2 negative advanced breast cancer after recurrence following endocrine therapy. The phase III MONARCH-2, investigated abemaciclib or placebo with fulvestrant (anti-estrogen) in patients who are HR positive HER2 negative and progressed following endocrine therapy. The mPFS was 16.4 months compared to 9.3 months in the placebo group, leading to its approval. Due to its lower toxicity profile, dosing for abemaciclib is continuous, unlike the other two approved CDK4/6i’s. Common adverse effects of abemaciclib include: neutropenia (lower rate compared to palbociclib and ribociclib), diarrhea, nausea and fatigue. Tolerability of CDK 4/6 inhibitors and the benefit of oral administration provides an exceptional alternative to standard chemotherapy and is now first-line treatment for estrogen receptor (ER) positive metastatic breast cancer.
Androgen Receptor Influence on Cell Cycle in Prostate Cancer
ADT has been the gold standard for PCa therapy due to androgen receptor (AR) activation causing PCa cell proliferation and survival. When androgens (ie, testosterone, dihydrotestosterone) enter the cell and bind to cytoplasmic AR, this leads to AR dimerization and subsequent nuclear translocation for genomic signaling. Activated AR dimers bind to DNA androgen response elements in promoter regions of genes to induce gene transcription such as prostate-specific antigen (PSA) and transmembrane protease serine 2 (TMPRSS2).13 This genomic AR signaling pathway influences prostate cancer proliferation, invasion and survival. Rapid non-genomic AR signaling by way of ligand-transformed AR associating with molecular substrates in the cytoplasm and inner leaflet of the cell membrane activating kinase cascades also occurs resulting in enhancement of cell proliferation and survival with many of these pathways regulating the cell cycle (Figure 1).14 This non-genomic AR signaling by the Src, Ras/Raf, protein kinase C, and AKT/PI3K pathways activate mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) to cause increase cell proliferation.15,16 At low androgen concentrations (0.01 −10 nM), AR N-terminal domain binds to Src homology domain 3 (SH3) which results in Src unfolding and autophosphorylation. This activates Src and causes enhanced cell proliferation via MAPK/ERK.17 As prostate cancer progresses, aberrant Src activation occurs, independent of androgens, as a result of increased Src expression or stimulation via growth factors and interleukins.18,19 In breast cancer, c-Src suppression resulted in down regulation of cyclin D1 and increase p27kip1 (intrinsic CDK4/6 inhibitor) suggesting the Src pathway regulates cell cycle progression.20 AR has also been found to activate the PKC pathway, which is regulated by modulation of intracellular calcium leading to activation of MAPK/ERK.21 While the activation of MAPK/ERK by PI3K/AKT pathway relies on AR’s activation of the p85α subunit of PI3K and AR’s interactions with Src leading to activation of AKT and subsequent MAPK/ERK activation.22 Non-genomic AR signaling independent of MAPK/ERK also occurs leading to increase cell proliferation. This is seen with AR causing increase intracellular calcium which not only activates PKC, but also activates protein kinase A which interacts with transcription factors promoting cell proliferation.21 Another AR signaling effect not reliant on MAPK/ERK includes AR interacting with PI3K/AKT causing phosphorylation of the tumor suppressor transcription factor Forkhead box O1 (FOXO1) causing it to be retained in the cytoplasm for degradation preventing its pro-apoptotic effects,23 in addition, PI3K/AKT phosphorylation of mTOR also causes increase cell proliferation.
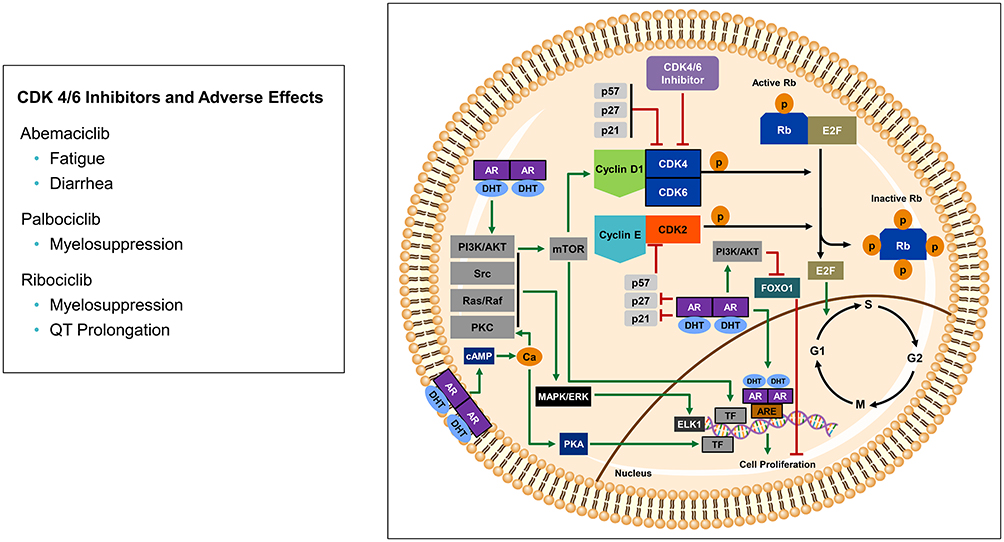 |
Figure 1 Androgen regulation of cell cycle and CDK4/6 pathway. Genomic AR signaling activates androgen response elements (ARE) to promote cell proliferation. Non-genomic AR signaling acts via membrane bound AR and activated cytosolic AR by activating PI3K/AKT, Src, Ras/Raf, and PKC to regulate MAPK/ERK activating ELK1 transcription factor to promote cell proliferation. Non-MAPK/ERK-dependent AR signaling includes membrane bound AR increasing intracellular calcium activating PKA and transcription factors (TF) to also promote cell proliferation. PI3K/AKT activates mTOR which leads to increase cyclin D1, in addition, PI3K/AKT phosphorylates the transcription factor FOXO1 which causes cytosolic degradation preventing FOXO1’s tumor suppressor effects. Cytosolic AR also down-regulates p21 and p27, cyclin-dependent kinase inhibitors that negatively regulate cell cycle progression. Cyclin D1 activates CDK4/6 which leads to phosphorylation of the hypo-phosphorylated active Rb, generating a hyper-phosphorylated inactive Rb state. When Rb is hyper-phosphorylated, the activating transcription factor E2F is released and enters the nucleus to promote transition from G1 (pre-DNA synthesis) to S phase (DNA synthesis). CDK4/6 inhibitors block this pathway by preventing phosphorylation of Rb, thereby keeping Rb in its active tumor suppressor state, and not allowing the activating E2F to be released to enter the nucleus. |
Through all these pathways mentioned above, AR has been found to be a key regulator of transcription of genes that allow for G1 to S transition.5 Further investigation of AR’s role in the cell cycle has revealed its effects on cyclin D1, a substrate for activating CDK4 and CDK6. The mechanism for increasing cyclin D1 is through the activation of MAPK and Akt24,25 as well as via mammalian target of rapamycin (mTOR) and subsequent upregulation of protein translation.26 Androgens, via activated AR, also have effects on the intrinsic CDK inhibitors p21, p27 and p16. Androgens have been shown to transcriptionally downregulate the CDK inhibitors p21 and p27.27 Therefore, these pathways via MAPK, Akt, and mTOR increasing cyclin D1 and reduction of intrinsic CDK4/6i’s (p21 and p27), all promote inactivation of Rb tumor suppressor allowing the cell to progress from G1 to S phase. Using ADT will influence these pathways, unfortunately, these androgen sensitive PCa cells will eventually become androgen-independent by way of AR mutations, AR amplification and aberrant activation of AR.28 To circumvent this resistance, CDK 4/6 inhibitors have been investigated to disrupt these AR signaling pathways, in turn, decreasing their cancer cell promoting effects. Preclinical models using palbociclib revealed CDK 4/6 inhibitors can be used in the treatment of PCa. Early models showed this agent limited proliferation in HSPC and CRPC cells in vitro, as well as xenografts and primary human tumors ex vivo.29
In the evolving paradigm of precision medicine, genomic analysis of metastatic PCa has shown many alterations that utilize the cell cycle to promote survival and growth.30–32 Therefore, patients with these alterations, such as amplification of cyclin D1 gene (ie, CCND1), found in 4.7% of mCRPC patients (Table 1), should be more responsive to CDK4/6i’s. However, it is important to note that some genomic alterations found in PCa may make these agents ineffective. For example, patients with Rb gene (ie, RB) loss, p16INKa high, cycle E1/E2 amplification or E2F amplification would likely be resistant.6 In patients with mCRPC, RB loss is found in 9–17% of patients.30,31 Therefore, RB expression and potentially genomic analysis should be monitored to predict response, especially in patients who have CRPC.
Current Clinical Trials with CDK 4/6 Inhibitors in Prostate Cancer
There are currently five clinical trials (Table 2) involving CDK 4/6 inhibitors in PCa, excluding two mutation-specific, tumor agnostic “basket” trials using palbociclib. A phase II trial (NCT03706365) is evaluating the safety and effectiveness of abiraterone with and without abemaciclib in patients with mCRPC. Ribociclib is being evaluated in PCa in two separate trials. A phase IB/II trial (NCT02555189) is investigating enzalutamide with and without ribociclib in patients with mCRPC who are chemotherapy-naïve and retain Rb expression. The second phase II trial (NCT02494921) is evaluating ribociclib with docetaxel in patients with mCRPC. There are two clinical trials testing palbociclib’s use in PCa. A phase II trial (NCT02905318), using palbociclib in patients with PCa, is recruiting to evaluate side effects, but also determine markers that could predict response to palbociclib. The only CDK4/6i trial with results to date is a phase II trial (NCT02059213) evaluating ADT with and without palbociclib in patients with mHSPC who are RB-positive.33 Twenty patients were randomized to ADT alone and 40 to ADT plus palbociclib. The primary outcome was proportion of patients who achieved a PSA < 4 ng/mL after seven months of treatment. The primary PSA endpoint was met in 80% of patients in both arms with a p-value of 0.87 for superiority. All-cause mortality in both groups was 0%. Serious adverse events occurred in 25% of patients with ADT alone and 17.5% with ADT plus palbociclib. In the palbociclib group, Grade 3 or 4 neutropenia was seen in 33% of patients. The clinical PFS is not mature, but 12-month biochemical PFS in ADT was 69% vs 74% in palbociclib.33 In this study 97% of patients were RB-positive. Further investigation is warranted in PCa in order to find the appropriate patient population based on genomic alterations and potential synergistic combinations to make these agents more efficacious.
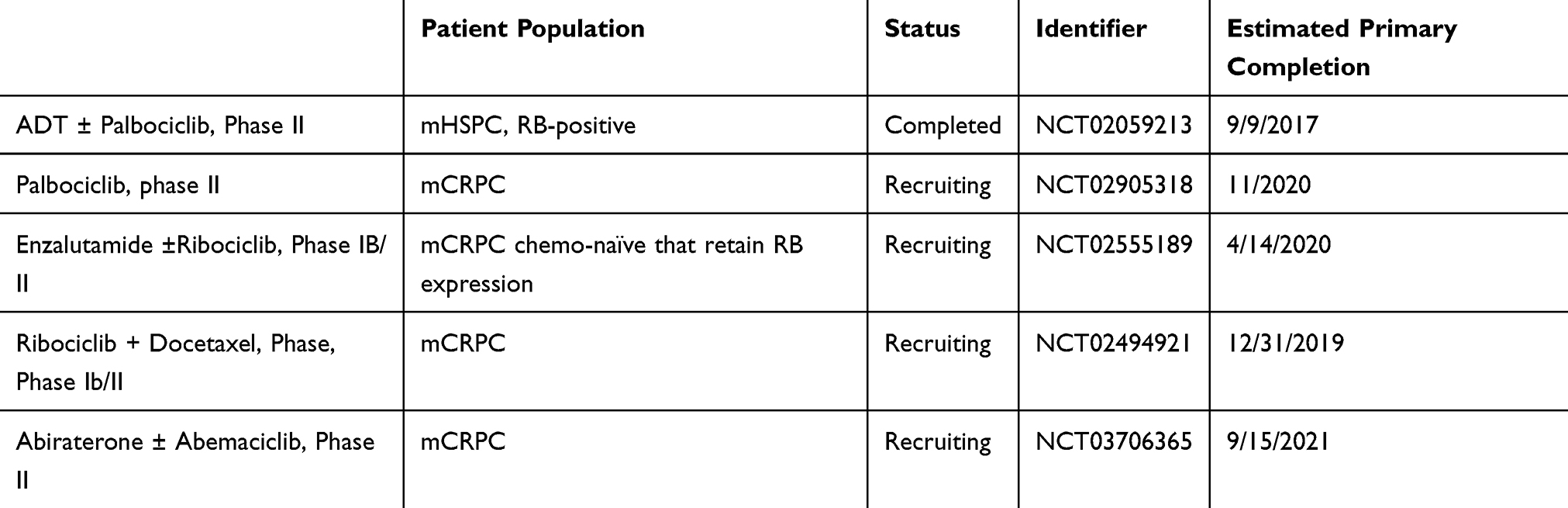 |
Table 2 CDK4/6 Inhibitor with Androgen Signaling Inhibitor Combination Trials |
Potential Combinations with CDK 4/6 Inhibitors
As with most cancer therapeutics, resistance develops with time; therefore, it is important to understand these mechanisms as they provide insight into which patients will respond and potential combination therapies to slow or overcome resistance. Multiple reviews have been published outlining CDK4/6i mechanisms of resistance in breast cancer.34 Potential mechanism of resistance include loss of Rb, upregulated CCNE1, activation of E2F, Cyclin E-CDK2 axis, driver mutations in PIK3CA, fibroblast growth factor axis and increasing 3-phosphoinositide-dependent protein kinase 1 (PDK1) and mitogen-activated protein kinase (MAPK) activation.34,35 In addition to mechanism of resistance, mechanism of action of CDK4/6 inhibitors are also important to guide combination therapy. CDK4/6 inhibitors’ mechanism of action is largely dependent on the Rb pathway; however, Rb independent effects are also present. These Rb independent pathways include CDK4/6 inhibitor effects on FOXM1 and SPOP;36 however, the impact of these Rb independent pathways is unclear since most patients with Rb loss have shown little activity to CDK4/6 inhibitors and is one of the main mechanisms of resistance.34 Guided by key pathways within PCa, PCa specific genomics and resistance profiles of CDK4/6i’s, we will review how CDK4/6i’s can be combined with agents from the following categories: chemotherapy, immunotherapy, DNA repair pathway, PI3K/AKT, FOXO3-FOXM1 axis, FGF-FGFR axis, Ras/Raf/MEK/ERK axis, and TP53 modulators.
Chemotherapy
In 2004, the US Food and Drug Administration (FDA) approved a taxane, docetaxel, for mCRPC based on its survival benefit.37 Subsequently, semisynthetic taxane cabazitaxel was first approved in 2010 for second-line treatment for mCRPC.38 Taxane’s mechanism of action includes inhibition of microtubular depolymerization arresting cells in G2/M phase of the cell cycle and attenuation of bcl-2 and bcl-xL gene expression promoting apoptosis.39 The ability to use cabazitaxel after docetaxel resistance is due to its poor affinity for P-glycoprotein efflux pumps which allows cabazitaxel to remain in the cell. Given the effects of taxanes on the cell cycle, these agents have been combined with CDK4/6i’s in various malignancies. However, the scheduling of these agents appears to affect the response. In preclinical breast cancer models, when the taxane paclitaxel was given concurrently with palbociclib it showed antagonism, but showed synergy when given sequentially via intermittent dosing.40,41 This finding can be explained by their mechanisms of action. When a CDK4/6i is administered it arrests cells in G1 which prevents cancer cells from eventually entering M phase, thereby, protecting cells from paclitaxel-induced cell death in M phase. When given sequentially it is hypothesized that G1 synchronization will occur after CDK4/6i is held, allowing more cells to enter M phase and enable cell death from paclitaxel.41 A Phase I trial with intermittent dosing in advanced breast cancer was performed and showed safety and tolerability.41 In PCa, a phase Ib/2 clinical trial is investigating ribociclib with docetaxel and prednisone in mCRPC patients with androgen signaling inhibitor resistance and no prior chemotherapy.42 Preliminary results of 14 patients showed PSA decrease by 50% or more in 29% of patients with neutropenia as the most commonly observed adverse event. Further trials in PCa with combining chemotherapy could be conducted, but dosing schedule should be considered.
Immunotherapy
Over the past decade, immunotherapy has taken the field of oncology by storm; however, in PCa the only FDA-approved autologous cellular immunotherapy is sipuleucel-T (Provenge).43 PCa has been unresponsive to immunotherapy which could be explained by its cold tumor environment as evident by a compromised cellular immunity and highly immune suppressive tumor microenvironment.44 CDK4/6i’s have been shown to enhance T-cell activation, increase T-cell tumor infiltration, and increase tumor expression of programmed death-ligand 1 (PD-L1) (Figure 2). 45,46 These changes support the potential synergistic effects with combining CDK4/6i’s and immunotherapy given the increased amount of immune cells in the tumor microenvironment and ability to block the immune suppressive effects of PD-L1. Inhibition of CDK4/6 increases PD-L1 protein by preventing cyclin D1-CDK4 phosphorylation of speckle-type POZ protein (SPOP), which without phosphorylation, compromises ubiquitination of PD-L1 that leads to PD-L1 protein degradation.46 High PD-L1 results in suppression of the host’s immune response to the cancer; however, this may make the tumor more vulnerable to immunotherapy. In PCa, cancer-derived SPOP mutations, seen in 8–15% of patients (Table 1),30,31,47,48 also increases PD-L1, but can cause decreased CD3+ tumor infiltrating lymphocytes and thus, leading to a cold tumor environment. This mutation resulted in increased growth of PCa xenografts compared to wild-type SPOP. Therefore, patients with SPOP mutations treated with CDK4/6i’s may be responsive to immunotherapy. In triple negative breast cancer mouse xenografts, combinations of CDK4/6i with a PI3K inhibitor and a PD-1 inhibitor induced complete and durable regressions.49 A clinical trial using CDK4/6i’s with immunotherapy is ongoing in various cancers. For example, abemaciclib plus pembrolizumab is currently being investigated in patients with metastatic breast cancer,50 and also in glioblastoma (NCT04118036), head and neck (NCT03938337), metastatic gastroesophageal (NCT03997448), and lung cancer (NCT02079636). There are no ongoing clinical trials in PCa using these class combinations; however, over 1000 clinical trials in PCa are investigating vaccines, immune checkpoint inhibitors, immunomodulators, adoptive cell transfer, and oncolytic virus-mediated immune response.44
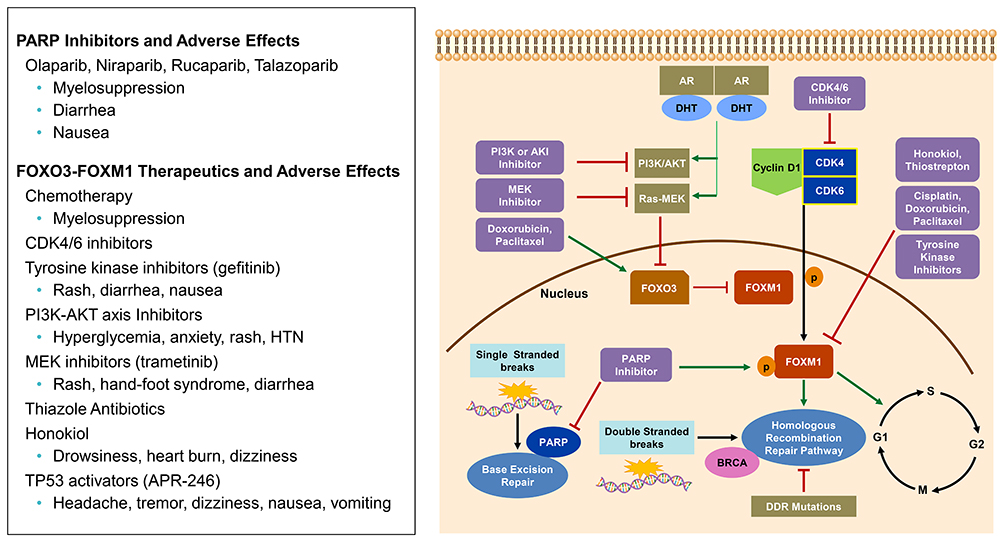 |
Figure 2 CDK4/6 pathway and FOXO3-FOXM1 axis: Cyclin D-CDK4/6 complex phosphorylates FOXM1 transcription factor which is involved in expression of G1/S phase genes. FOXM1 also up-regulates genes involved in the homologous recombination Repair (HRR) pathway. FOXO3, when acting as a tumor suppressor gene, inactivates FOXM1. The tumor suppressor activity is regulated by PI3K/AKT and Ras-MEK. Multiple therapeutics have been shown to have activity in the FOXO3-FOXM1 axis. PARP inhibitors can increase FOXM1 expression and nuclear localization. PARP is involved with base excision repair (BER), however, if PARP is inhibited single stranded breaks accumulate and double stranded breaks occur making the cell rely on HRR. If HRR is deficient, by mutation (i.e BRCA), then synthetic lethality occurs in the presence of PARP inhibition. |
DNA Repair Pathway
When DNA is damaged it relies on various pathways to undergo repair including: direct repair, mismatch repair (MMR), base excision repair (BER), nucleotide excision repair (NER) and double strand break (DSB) via non-homologous end joining and homologous recombination repair (HRR).51 Poly (adenosine diphosphate) [ADP]-ribose polymerases (PARP) are DNA repair enzymes involved in single stranded breaks and BER.52 When PARP is inhibited, single stranded breaks accumulate and lead to double stranded breaks which are repaired via HRR. However, aberrations within the HRR pathway, such as BRCA1/2 mutations, can result in synthetic lethality in the presence of PARP inhibitors (PARPi) (Figure 3).53 Therefore, patients with DNA repair aberrations (germline or somatic) are most sensitive to PARPi. DNA repair aberrations are seen in approximately 24% of patients with mCRPC.30 Specifically, BRCA2 loss of function alteration is seen between 5–13% of this cohort.30–32 In PCa xenografts, PARP-1 activity has been associated with progression to CRPC and PARP inhibition leads to delayed progression to CRPC, diminished androgen receptor function and reduced CRPC growth.54 In the TOPARP trial, a PARP inhibitor, olaparib, was used as a single agent in patients with heavily pretreated mCRPC and showed antitumor activity in patients with DNA damage response (DDR) gene aberrations, especially in BRCA1/2 alterations.55 Over 592 patients were screened for this trial and 27% of patients had DDR aberrations (7% BRCA2, 7% ATM, 6% CDK12, 7% other).55 The response evaluation criteria in solid tumors (RECIST) objective response rate based on these aberrations was 52.4%, 8.3%, and 0%, respectively; in addition, PSA was decreased by 50% or more in 76.7%, 5.3% and 0%, respectively.55 Of the 33 evaluated patients in the 400 mg olaparib cohort, a radiologic response was seen in 24.2%.55 Since this trial, two PARPi are now approved for use in mCRPC including rucaparib and Olaparib. Rucaparib gained FDA approval in 2020 for mCRPC patients with germline or somatic BRCA mutations who progressed on ADT and taxane-based chemotherapy. The TRITON2 trial showed rucaparib had an objective response rate (ORR) of 44% (11 of 25 patients) with 56% of these patients having a duration of response of ≥6 months.56 Also in 2020, olaparib gained approval for HRR gene-mutated mCRPC. In the PROfound trial,57 radiologic progression-free survival (rPFS) for olaparib was 7.4 months vs 3.6 months for patients treated with enzalutamide or abiraterone; median overall survival (mOS) was also improved, 19.1 months vs 14.7 months. The ORR was 33% vs 2%. These trials demonstrated that single agent PARP inhibitor can be used in PCa patients with HRR gene aberrations, but could combination therapy be used to improve responses? Multiple clinical trials are evaluating PARP inhibitors in combination with other agents such as androgen signaling inhibitors (NCT04179396) and immunotherapy (NCT03572478) in PCa patients with DNA damage alterations. Combination therapy with PARP inhibitors is also being investigated in patients with a proficient homologous recombination pathway. In vitro and in vivo ovarian cancer models, combining PARPi with CDK4/6i’s showed synergistic effects with palbociclib by inducing homologous recombination repair deficiency through downregulation of MYC regulated HR pathway genes leading to synthetic lethality with olaparib.58 Additionally, high MYC expression determined sensitivity to combination therapy.58 Since MYC amplifications occur in 20% of patients with mCRPC,30,32 combining these agents is a promising combination. PARP inhibitors also demonstrated effects on the FOXM1 transcription factor axis by inducing expression and nuclear localization of FOXM1 which in turn up regulates expression of genes involved in the homologous recombination (HR) pathway.59 When FOXM1 inhibited cells were treated with olaparib it resulted in increased DNA damage and PARP trapping. It has been demonstrated that CDK4/6i’s can decrease FOXM1,60 which could potentially circumvent the increase FOXM1 effects of PARPi. In clinical trials, olaparib, palbociclib and fulvestrant combination is being investigated in patients with BRCA-mutation-associated metastatic breast cancer (NCT03685331). No clinical trials are investigating PARPi +CDK4/6i in PCa.
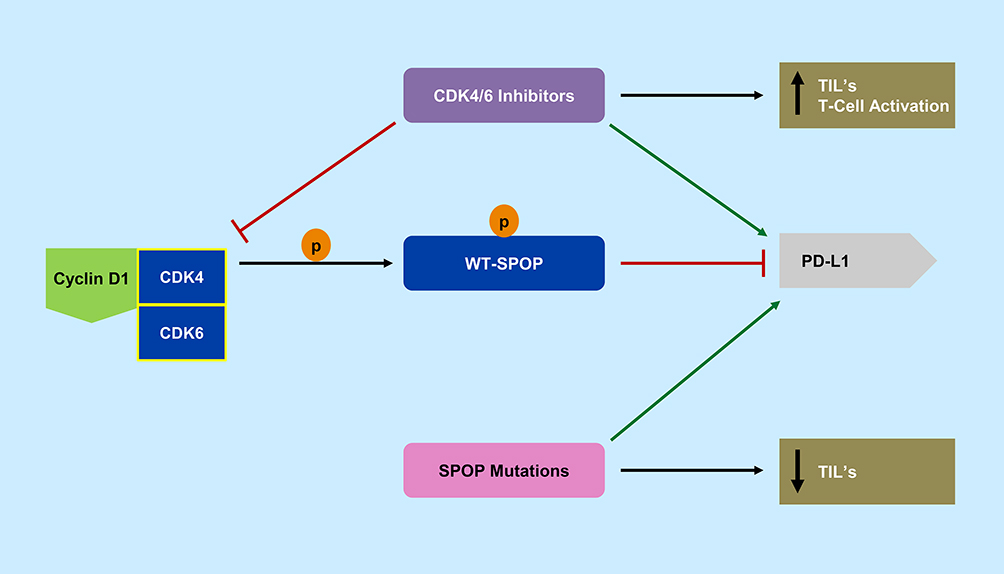 |
Figure 3 Speckle-type POZ protein (SPOP) and CDK4/6 pathway. Cyclin D-CDK4/6 complex phosphorylates wild type-SPOP (WT-SPOP) which results in ubiquitination and destruction of programmed death ligand 1 (PD-L1). CDK4/6 inhibitors are able to prevent this ubiquitination and also increase T-cell activation and tumor infiltrating lymphocytes (TIL’s). Cancer mutated SPOP allows for increased PD-L1 expression of tumor cells and decreases tumor infiltrating lymphocytes. |
FOXO3-FOXM1 Axis
Forkhead box O3 (FOXO3) and forkhead box M1 (FOXM1) axis contains important transcription factors for cell proliferation, differentiation, cell survival, senescence, and DNA damage repair.61 FOXO3 has been shown to act as a tumor suppressor gene in many cancers including PCa. In transgenic adenocarcinoma of the mouse prostate (TRAMP) mice, prostate progression was increased when FOXO3a activity was blocked.62 FOXO3a is regulated by the phosphorylation by PI3K-AKT, RAS-ERK, serum glucocorticoid inducible kinases (SGK), and IkB kinase beta (IKKB) which prevents translocation into the nucleus.61,63,64 When FOXO3 is able to translocate to the nucleus it inhibits the oncogene FOXM1 (Figure 3). Similarly to CDK4/6, FOXM1 is involved in cell proliferation and cell cycle progression by promoting entry into S-phase and M-phase; thereby, leading to tumorigenesis.65,66 FOXM1 has been identified as a critical phosphorylation target of CDK4/6 allowing expression of G1/S phase genes which protects cells from senescence. It was also demonstrated that FOXM1 had involvement with regulation of genes in cell cycle DNA replication (cycle E2, MYB, MCM2, MCM10, CDT1) as well as DNA repair (XRCC2, SFRS4).60 Since FOXM1 is overexpressed in many solid tumors including ovarian, breast, prostate, melanoma, hepatoma, angiosarcoma, colorectal cancer, lung cancer and gastric cancer,67 targeting the FOXO3-FOXM1 axis could be a synergistic approach to further prevent tumorigenesis by cell cycle mechanisms. When CDK4/6 is inhibited, using palbociclib, it decreased FOXM1 protein levels by 70%, indicating blockade of CDK4/6 can down-regulate FOXM1.60 Multiple therapeutics have been shown to have effects on this axis to either increase the activity of FOXO3 or inhibit FOXM1 including: chemotherapy (paclitaxel,68 doxorubicin,69 cisplatin),70 CDK4/6i’s (ribociclib,71 palbociclib),60 tyrosine kinase inhibitors (gefitinib),72 PI3K-AKT inhibitors,61 endocrine receptor modulators,73 aurora kinase inhibitors,74 thiazole antibiotics (thiostrepton),75,76 natural compounds like Honokiol,77 and potentially TP 53 activators.78 Combining these agents with CDK4/6i’s may provide synergistic activity against tumorigenesis given FOXO3-FOXM1 role in cell proliferation in mCRPC.
PI3K/Akt Axis
In mCRPC, phosphatidylinositol 3-kinase (PI3K) alterations are seen in almost 50% of patients, with loss of PTEN tumor suppressor occurring in approximately 40% of these patients (Table 1).30 When PTEN loss occurs, it allows PI3K/AKT activation to promote PCa growth in the absence of AR signaling and is associated with worse outcomes.79–81 Single agents targeting PI3K, AKT and mTOR have failed to progress in PCa as a result of toxicity, inadequate target inhibition, and limited efficacy.79 However, combination therapy has shown some activity. AKT inhibitor, ipatasertib, was combined with abiraterone in a randomized phase II study and showed superior antitumor activity with improved OS and time to PSA progression, although not statistically significant, compared to abiraterone alone in patients with mCRPC and PTEN loss.82 Since the PI3K axis can be a means of developing CDK4/6i resistance, combining these agents or sequential treatment is under investigation. In breast cancer, targeting CDK4/6 and PI3K in vitro and in patient-derived tumor xenografts (PDTX) resulted in tumor regression.83 As a result, clinical trials with this combination are ongoing in breast cancer. As more studies are being completed investigators are developing a better understanding of resistance for these agents. It has been shown that chronic exposure to CDK4/6i’s upregulates cyclin D and promotes emergence of PIK3CA driver mutations.34 In breast cancer, PTEN loss has been shown to cause resistance to CDK4/6i’s or PI3K inhibitors such as alpelisib.84,85 This CDK4/6 resistance develops as a result of increased AKT activation. However, selective AKT inhibitors have been shown to restore sensitivity in vitro and in vivo.84 Since PTEN loss is frequent in mCRPC (40%), using AKT inhibitors with CDK4/6i’s, instead of PI3K inhibitors, may result in better efficacy and synergy. There are no clinical trials investigating PI3K or AKT inhibitors with CDK4/6i’s in PCa.
Although PI3K/AKT inhibitors remain a viable option, negative feedback loops are present between AR and PI3K/AKT in which inhibition of one activates the other;81,86 therefore, other targets in this pathway have been investigated. Histone deacetylase 3 (HDAC3), class I, known to be upregulated in PCa, has roles in S phase progression, DNA damage control, maintenance of genomic stability and T cell development.87 Blocking HDAC3 has inhibitory effects on both, AKT and AR, through inhibition of AKT phosphorylation and histone deacetylation and condensed chromatin, respectively (Figure 4).86 It was hypothesized that patients with loss of PTEN or mutated SPOP, which results in aberrant activation of AR and AKT, would respond to HDAC3 inhibitors. This was shown with a selective HDAC3 inhibitor, RGFP966, when it inhibited growth in PTEN or SPOP mutated PCa cells in culture and PDTX models.87 Given the inhibitory effects as a single agent, combining HDAC inhibitors with CDK4/6i’s could decrease PI3K associated CDK4/6i resistance. HDAC inhibitors have also been shown to upregulate FOXO1 mRNA and protein levels which have effects on the cell cycle. FOXO1 is a tumor suppressor that induces cell cycle arrest at G1 by modulating p27kip1, p21, Rb protein, cyclin D1/D2 and induces cell cycle arrest at G2 via GADD45.88 Therefore, this HDAC inhibitor effect may add additional inhibition to the cell cycle when added to CDK4/6i’s. Combining CDK4/6i’s with HDAC inhibitors was investigated in preclinical breast cancer models when abemaciclib and vorinostat were combined and revealed synergy with significant diminished tumor growth.89 Otherwise, this combination has not been studied extensively. Further studies with this combination in PCa are warranted.
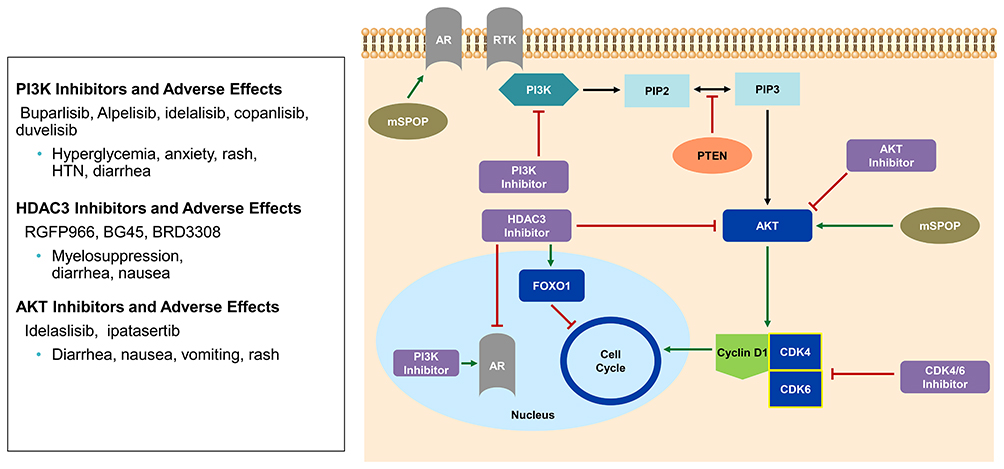 |
Figure 4 PI3K-AKT axis and CDK4/6 relationship. PI3K allows for conversion of phosphatidylinositol-4,5 biphosphate (PIP2) to phosphatidylinositol-3,4,5 triphosphate (PIP3) resulting in activation of AKT via phosphorylation and increase cyclin D1. PTEN tumor suppressor regulates this axis by inhibiting PIP2 conversion to PIP3. Mutated SPOP (mSPOP) also regulates this axis by causing aberrant activation of AR and AKT. HDAC3 inhibitors inhibit AKT phosphorylation and AR mediated transcriptional activity. PI3K inhibitors have been shown to increase transcriptional activity of AR. |
FGF-FGFR Axis
Fibroblast growth factor pathway aberrancy has been described in many cancers including bladder, breast, endometrial, lung, rhabdomyosarcoma, melanoma, head and neck, brain and PCa.90 FGF and fibroblast growth factor receptor (FGFR) have been shown to be dysregulated in the development of prostate intraepithelial neoplasia, epithelial-mesenchymal transition, angiogenesis, promotion of bone metastases and emergence of CRPC.91 In a cohort of 101 patients with mCRPC, FGFR1 was amplified in 10% of patients.92 The FGF-FGFR signaling pathway leads to activation of STAT, PI3K-AKT-mTOR and RAS-RAF-MEK pathway (Figure 5). Each of these pathways have downstream effects on the cell cycle by increasing cyclin D1.93,94 In breast cancer, the FGF-FGFR axis, specifically FGFR1 over expression, is a way for breast cancer cells to become resistant to CDK4/6i’s and have resulted in shorter progression-free survival.95 As a result of this, combining a CDK4/6i with an FGFR inhibitor has been pursued. The resistance was overcome in breast cancer xenograft models when FGFR1 amplified tumors were given FGFR tyrosine kinase inhibitor, lucitanib and complete responses were seen in xenografts when the FGFR inhibitor, erdafitinib, was given with CDK4/6i and endocrine therapy.95 This triple therapy is being used in a phase Ib trial in patients with metastatic ER+/HER2-/FGFR amplified breast cancer (NCT03238196). In PCa, erdafitinib is being pursued in combination with androgen signaling inhibitors in patients who are androgen receptor negative and no neuroendocrine differentiation (NCT03999515). Dovitinib, a pan class inhibitor targeting FGFR along with PDGFR and VEGF, was used in patients with mCRPC and showed modest activity with PFS of 3.67 months and mOS 13.7 months.96 However, no other clinical trials are ongoing in PCa with this agent. Other FGFR inhibitors under investigation include futibatinib (FGFR 1–4 inhibitor) in a mutation-specific and solid tumor agnostic basket trial (NCT04189445). Further investigation is warranted by combining CDK4/6i’s with FGFR inhibitors in PCa.
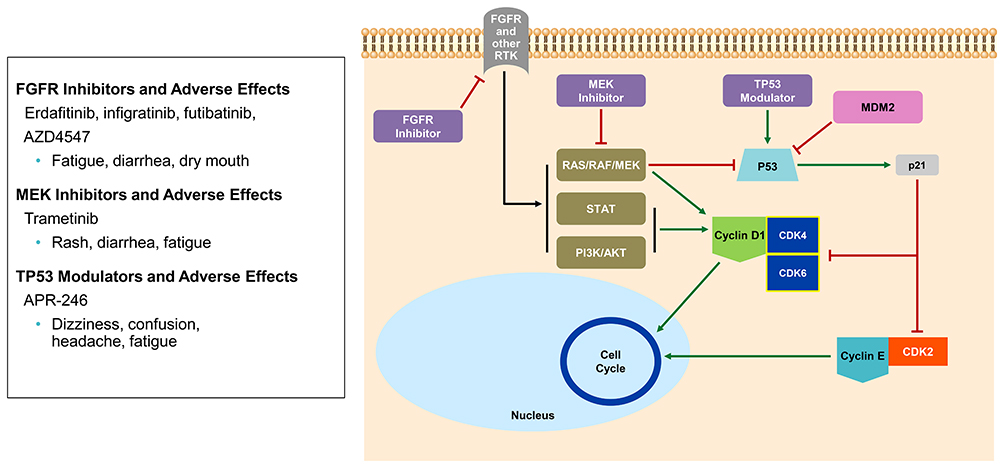 |
Figure 5 FGFR axis, Ras-Raf-MEK and TP53 relationship to CDK4/6; FGFR results in activation of PI3K, Ras-Raf-MEK, STAT which increases cyclin D1, thereby promoting progression through the cell cycle. The Ras-Raf-MEK pathway is needed for inactivation of tumor suppressor TP53 pathway. In addition, MDM2 also regulates p53 by proteasomal degradation. However, when the tumor suppressor, p53, is active it allows for activation of p21 which regulates the cell cycle via CDK4/6 and CDK2 causing cell cycle arrest. RTK, receptor tyrosine kinase. |
Ras-Raf-MEK-ERK Axis
Ras/Raf/MEK/ERK is an important signaling pathway involving MAPKs that are involved in regulating proliferation, differentiation and apoptosis.97 Amplification of members within the MAPK pathway is as high as 32% in patients with mCRPC.92 Phosphorylated ERK1/2 is associated with biochemical recurrence, rapid progression to CRPC and reduced disease-specific survival.92,98 The MAPK pathway influence on the cell cycle has been investigated in multiple studies. It has been suggested that Ras signaling is required for inactivation of tumor suppressor pRb within the cell cycle when Ras neutralizing antibodies caused G1 arrest.99 Ras signaling is also essential in inactivation of p53-mediated induction of p21Cip1 (Figure 5). This was demonstrated when loss of Ras resulted in transcriptional activation of p53.100 If p53 is activated, then this tumor suppressor leads to increase p21Cip1 and G1/S cell cycle arrest.100 p21 causes this arrest by suppressing activity of cyclin A/CDK2, cyclin A/CDK1, and decreasing transcription of E2F1, STAT3, and MYC.101 Paradoxically, p21 can bind CDK4/6 to increase kinase activity to promote progression through G1.101 The MAPK pathway has also been a means of CDK4/6i resistance. Resistant cells have been shown to have increased MAPK activation leading to CDK4/6-Rb bypass to induce aggressive phenotypes and metastasis.35 In PCa, these CDK4/6 resistant cells were sensitized to MEK inhibitors.35 Therefore, this suggests that MEK inhibitors could be used in combination with CDK4/6i’s to delay resistance or used sequentially after resistance occurs. Combining MEK inhibitors with CDK4/6i’s is being investigated in multiple tumor types including: KRAS mutant non-small cell lung cancer (NCT03170206),102 KRAS mutant colorectal cancer,103 pancreatic cancer,104 and melanoma.105 The MEK inhibitor, trametinib, is being investigated as single agent in mCRPC (NCT02881242); however, no combination trials are underway providing an opportunity to combine MEKi with CDK4/6i.
TP53 Axis
The guardian of the genome, tumor suppressor p53, has many roles essential for protecting against oncogenic transformation including: cell cycle arrest, DNA repair, senescence and cell death, modulation of autophagy, and cancer metabolism.106 G1 cell cycle arrest by p53 is mainly the result of transcriptional activation of p21 which binds and inhibits cyclin E/CDK2 and cyclin D/CDK4, thereby preventing Rb phosphorylation (Figure 5).107 The main regulator of p53 is a ubiquitin ligase, mouse double minute 2 (MDM2), which results in proteasome ubiquitination of the tumor suppressor.108 As mentioned earlier, Ras signaling is also essential in inactivation of p53-mediated induction of p21Cip1.100 Therapeutic targets within this pathway are needed since TP53 is mutated in over 50% of cancers and in mCRPC, an aberration causing loss of function is found in 36–53% of patients (Table 1).30–32 Mutated TP53 results in loss of function of wild-type p53 and gain of function in other aspects that lead to oncogenic activity.109 Patients with these mutations are found to have worse outcomes, progression and high rate of recurrence.110,111 Treatment strategies targeting TP53 mutated cancers are focused on restoration of wild-type p53 function (APR-246, MIRA-1, JNJ-26854165, Calcein AM, NSC59984), direct attack on p53 deficient cells, enhancement of normal p53 function (MDM2 inhibitors Nutlin-3a, RITA) and mimicking DNA damage with a virus.109 APR-246 is being investigated in multiple clinical trials in hematologic malignancies along with solid tumors including: ovarian (NCT02098343) and prostate (NCT00900614). In patients with mCRPC, a phase I trial with APR-246 was shown to be safe and tolerable when given to 7 patients intravenously for 4 consecutive days.112 Adverse effects included fatigue, dizziness, headache and confusion. Combining therapeutics that target p53 and CDK4/6 has been investigated in sarcoma cancer cells when MDM2 antagonists were combined with CDK4/6i’s in the preclinical setting.113 It resulted in reduced MDM2 antagonistic activity and diminished RNA polymerase II recruitment and decreased transcription of p53 target genes (MDM2 and p21).113 These effects led to antagonistic cytotoxic effects suggesting combining these agents may not be beneficial. This suggests CDK4-cyclin D1 complex has a positive impact on p53 as more checks and balances are needed when cells are entering the cell cycle. However, a separate study with liposarcoma xenografts showed synergy when MDM2 and CDK4/6 targeting agents were combined.114 There are no clinical trials ongoing combining APR-246 with CDK4/6i’s. Given the varying results, additional studies are warranted in combining these agents.
Conclusion
The benefits of CDK4/6i’s in PCa can be optimized by fully understanding pathways that are involved with the cell cycle, resistance patterns of CDK4/6i’s and by utilizing therapies that target driver mutations in mCRPC. Additional research is needed in these key areas in order to provide more insightful reasoning to combinatorial therapies with CDK4/6i’s to maximize efficacy and durability of response. The best opportunity for synergistic success occurs when agents are combined based on interrelated mechanisms, resistance profiles, and genomics, however, it is extremely important to consider the adverse effects of the combined agents. Precision medicine should not only aid with improving effectiveness of treatment, but also protect and identify patients who would not benefit from therapy thus avoiding toxicity and decreasing morbidity. Novel strategic combinatorial therapies for mCRPC, with a CDK4/6i as the common backbone, have the potential to improve overall survival and quality of life in a patient population with few therapeutic options.
Acknowledgments
Julie Nielsen, Mayo Clinic senior production designer.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Key Statistics for Prostate Cancer. American Cancer Society.
2. Overview of the treatment of castration-resistant prostate cancer (CRPC). UpToDate, 2018. (
3. Karp G. Cell and Molecular Biology: Concepts and Experiments.
4. Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24(11):1770–1783. doi:10.1200/JCO.2005.03.7689
5. Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nucl Recept Signal. 2008;6(1):e001. doi:10.1621/nrs.06001
6. Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer. 2017;3(1):39–55. doi:10.1016/j.trecan.2016.11.006
7. Michael Lieberman ADM. Marks’ Basic Medical Biochemistry: A Clinical Approach.
8. Topacio BR, Zatulovskiy E, Cristea S, et al. Cyclin D-Cdk4,6 drives cell-cycle progression via the retinoblastoma protein’s C-Terminal Helix. Mol Cell. 2019;74(4):758–70.e4. doi:10.1016/j.molcel.2019.03.020
9. First CDK. 4/6 inhibitor heads to market. Cancer Discov. 2015;5:339–340.
10. Finn RS, Crown JP, Ettl J, et al. Efficacy and safety of palbociclib in combination with letrozole as first-line treatment of ER-positive, HER2-negative, advanced breast cancer: expanded analyses of subgroups from the randomized pivotal trial PALOMA-1/TRIO-18. Breast Cancer Res. 2016;18(1):67. doi:10.1186/s13058-016-0721-5
11. Finn RS, Martin M, Rugo HS, et al. Palbociclib and Letrozole in advanced breast cancer. N Eng J Med. 2016;375(20):1925–1936. doi:10.1056/NEJMoa1607303
12. Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first-line therapy for HR-Positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738–1748. doi:10.1056/NEJMoa1609709
13. Tan MH, Li J, Xu HE, Melcher K, Yong EL. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin. 2015;36:3–23.
14. Leung JK, Sadar MD. Non-genomic actions of the androgen receptor in prostate cancer. Front Endocrinol (Lausanne). 2017;8.
15. Peterziel H, Mink S, Schonert A, Becker M, Klocker H, Cato AC. Rapid signalling by androgen receptor in prostate cancer cells. Oncogene. 1999;18(46):6322–6329. doi:10.1038/sj.onc.1203032
16. Migliaccio A, Castoria G, Auricchio F. Analysis of androgen receptor rapid actions in cellular signaling pathways: receptor/Src association. Methods Mol Biol. 2011;776:361–370.
17. Migliaccio A, Castoria G, Di Domenico M, et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000;19(20):5406–5417. doi:10.1093/emboj/19.20.5406
18. Asim M, Siddiqui IA, Hafeez BB, Baniahmad A, Mukhtar H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene. 2008;27(25):3596–3604. doi:10.1038/sj.onc.1211016
19. Varkaris A, Katsiampoura AD, Araujo JC, Gallick GE, Corn PG. Src signaling pathways in prostate cancer. Cancer Metastasis Rev. 2014;33(2–3):595–606. doi:10.1007/s10555-013-9481-1
20. Liu X, Du L, Feng R. c-Src regulates cell cycle proteins expression through protein kinase B/glycogen synthase kinase 3 beta and extracellular signal-regulated kinases 1/2 pathways in MCF-7 cells. Acta Biochim Biophys Sin (Shanghai). 2013;45(7):586–592. doi:10.1093/abbs/gmt042
21. Liao RS, Ma S, Miao L, et al. Androgen receptor-mediated non-genomic regulation of prostate cancer cell proliferation. Future Oncol. 2013;2:187–196.
22. Sun M, Yang L, Feldman RI, et al. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J Biol Chem. 2003;278(44):42992–43000. doi:10.1074/jbc.M306295200
23. Huang H, Tindall DJ. FOXO factors: a matter of life and death. Future Oncol. 2006;2(1):83–89. doi:10.2217/14796694.2.1.83
24. Di Donato M, Giovannelli P, Cernera G, et al. Non-genomic androgen action regulates proliferative/migratory signaling in stromal cells. Front Endocrinol (Lausanne). 2015;5:225. doi:10.3389/fendo.2014.00225
25. Castoria G, Giovannelli P, Di Donato M, et al. Role of non-genomic androgen signalling in suppressing proliferation of fibroblasts and fibrosarcoma cells. Cell Death Dis. 2014;5(12):e1548–e. doi:10.1038/cddis.2014.497
26. Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66(15):7783–7792. doi:10.1158/0008-5472.CAN-05-4472
27. Fang Z, Zhang T, Dizeyi N, et al. Androgen receptor enhances p27 degradation in prostate cancer cells through rapid and selective TORC2 activation. J Biol Chem. 2012;287(3):2090–2098. doi:10.1074/jbc.M111.323303
28. Tilki D, Schaeffer EM, Evans CP. Understanding mechanisms of resistance in metastatic castration-resistant prostate cancer: the role of the androgen receptor. Eur Urol Focus. 2016;2(5):499–505. doi:10.1016/j.euf.2016.11.013
29. Comstock CES, Augello MA, Goodwin JF, et al. Targeting cell cycle and hormone receptor pathways in cancer. Oncogene. 2013;32(48):5481–5491. doi:10.1038/onc.2013.83
30. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. doi:10.1016/j.cell.2015.05.001
31. Frank S, Nelson P, Vasioukhin V. Recent advances in prostate cancer research: large-scale genomic analyses reveal novel driver mutations and DNA repair defects. F1000 Res. 2018;7.
32. Sonpavde G, Agarwal N, Pond GR, et al. Circulating tumor DNA alterations in patients with metastatic castration-resistant prostate cancer. Cancer. 2019;125(9):1459–1469. doi:10.1002/cncr.31959
33. Palmbos PL, Tomlins SA, Agarwal N, et al. Cotargeting AR signaling and cell cycle: a randomized phase II study of androgen deprivation therapy with or without palbociclib in RB-positive metastatic hormone sensitive prostate cancer (mHSPC). J Clin Oncol. 2018;36(6_suppl):251. doi:10.1200/JCO.2018.36.6_suppl.251
34. McCartney A, Migliaccio I, Bonechi M, et al. Mechanisms of resistance to CDK4/6 inhibitors: potential implications and biomarkers for clinical practice. Front Oncol. 2019;9:666. doi:10.3389/fonc.2019.00666
35. de Leeuw R, McNair C, Schiewer MJ, et al. MAPK reliance via acquired CDK4/6 inhibitor resistance in cancer. Clin Cancer Res. 2018;24(17):4201–4214. doi:10.1158/1078-0432.CCR-18-0410
36. Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell. 2018;34(1):9–20. doi:10.1016/j.ccell.2018.03.023
37. Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502–1512. doi:10.1056/NEJMoa040720
38. Paller CJ, Antonarakis ES. Cabazitaxel: a novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des Devel Ther. 2011;5:117–124.
39. Pienta KJ. Preclinical mechanisms of action of docetaxel and docetaxel combinations in prostate cancer. Semin Oncol. 2001;28:3–7. doi:10.1016/S0093-7754(01)90148-4
40. Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem. 2012;287(34):29075–29087. doi:10.1074/jbc.M112.365494
41. Clark AS, McAndrew NP, Troxel A, et al. Combination Paclitaxel and Palbociclib: results of a Phase I trial in advanced breast cancer. Clin Cancer Res. 2019;25(7):2072–2079. doi:10.1158/1078-0432.CCR-18-0790
42. Lewis C, Smith DC, Carneiro BA, et al. c15-149: A phase 1b study of the oral CDK4/6 inhibitor ribociclib in combination with docetaxel plus prednisone in metastatic castration resistant prostate cancer (mCRPC) – a prostate cancer clinical trials consortium study. J Clin Oncol. 2018;36(15_suppl):e17028–e. doi:10.1200/JCO.2018.36.15_suppl.e17028
43. Slovin SF. Sipuleucel-T: when and for whom to recommend it. Oncology. 2017;31:900–901.
44. Boettcher AN, Usman A, Morgans A, VanderWeele DJ, Sosman J, Wu JD. Past, current, and future of immunotherapies for prostate cancer. Front Oncol. 2019;9:9. doi:10.3389/fonc.2019.00009
45. Deng J, Wang ES, Jenkins RW, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018;8:216–233.
46. Zhang J, Bu X, Wang H, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–95.
47. Shi Q, Zhu Y, Ma J, et al. Prostate cancer-associated SPOP mutations enhance cancer cell survival and docetaxel resistance by upregulating Caprin1-dependent stress granule assembly. Mol Cancer. 2019;18(1):170. doi:10.1186/s12943-019-1096-x
48. Abeshouse A, Ahn J, Akbani R, et al. The molecular taxonomy of primary prostate cancer. Cell. 2015;163(4):1011–1025. doi:10.1016/j.cell.2015.10.025
49. Teo ZL, Versaci S, Dushyanthen S, et al. Combined CDK4/6 and PI3Kα inhibition is synergistic and immunogenic in triple-negative breast cancer. Cancer Res. 2017;77(22):6340–6352. doi:10.1158/0008-5472.CAN-17-2210
50. Tolaney SM, Kabos P, Dickler MN, et al. Updated efficacy, safety, & PD-L1 status of patients with HR+, HER2- metastatic breast cancer administered abemaciclib plus pembrolizumab. J Clin Oncol. 2018;36(15_suppl):1059. doi:10.1200/JCO.2018.36.15_suppl.1059
51. Plummer R. Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clin Cancer Res. 2010;16(18):4527–4531. doi:10.1158/1078-0432.CCR-10-0984
52. Dziadkowiec KN, Gąsiorowska E, Nowak-Markwitz E, Jankowska A. PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Prz Menopauzalny. 2016;15:215–219.
53. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly (ADP-Ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi:10.1056/NEJMoa0900212
54. Schiewer MJ, Goodwin JF, Han S, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2(12):1134–1149. doi:10.1158/2159-8290.CD-12-0120
55. Mateo J, Porta N, Bianchini D, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, Phase 2 trial. Lancet Oncol. 2020;21(1):162–174. doi:10.1016/S1470-2045(19)30684-9
56. FDA grants accelerated approval to rucaparib for BRCA-mutated metastatic castration-resistant prostate cancer. 2020. (
57. de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Eng J Med. 2020;382(22):2091–2102. doi:10.1056/NEJMoa1911440
58. Yi J, Liu C, Tao Z, et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine. 2019;43:225–237. doi:10.1016/j.ebiom.2019.03.027
59. Fang P, Madden JA, Neums L, Moulder RK, Forrest ML, Chien J. Olaparib-induced adaptive response is disrupted by FOXM1 targeting that enhances sensitivity to PARP inhibition. Mol Cancer Res. 2018;16(6):961–973. doi:10.1158/1541-7786.MCR-17-0607
60. Anders L, Ke N, Hydbring P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20(5):620–634. doi:10.1016/j.ccr.2011.10.001
61. Yao S, Fan LY-N, Lam EW-F. The FOXO3-FOXM1 axis: a key cancer drug target and a modulator of cancer drug resistance. Semin Cancer Biol. 2018;50:77–89. doi:10.1016/j.semcancer.2017.11.018
62. Shukla S, Bhaskaran N, Maclennan GT, Gupta S. Deregulation of FoxO3a accelerates prostate cancer progression in TRAMP mice. Prostate. 2013;73(14):1507–1517. doi:10.1002/pros.22698
63. Yamaguchi H, Hsu JL, Hung MC. Regulation of ubiquitination-mediated protein degradation by survival kinases in cancer. Front Oncol. 2012;2:15. doi:10.3389/fonc.2012.00015
64. Liu Y, Ao X, Ding W, et al. Critical role of FOXO3a in carcinogenesis. Mol Cancer. 2018;17(1):104. doi:10.1186/s12943-018-0856-3
65. Wierstra I. FOXM1 (Forkhead box M1) in tumorigenesis: overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor-suppressive properties, and target of anticancer therapy. Adv Cancer Res. 2013;119:191–419.
66. Wang X, Kiyokawa H, Dennewitz MB, Costa RH The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration.
67. Liao GB, Li XZ, Zeng S, et al. Regulation of the master regulator FOXM1 in cancer. Cell Commun Signal. 2018;16(1):57. doi:10.1186/s12964-018-0266-6
68. Khongkow P, Gomes AR, Gong C, et al. Paclitaxel targets FOXM1 to regulate KIF20A in mitotic catastrophe and breast cancer paclitaxel resistance. Oncogene. 2016;35(8):990–1002. doi:10.1038/onc.2015.152
69. Ho KK, McGuire VA, Koo CY, et al. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. J Biol Chem. 2012;287(2):1545–1555. doi:10.1074/jbc.M111.284224
70. Fernandez de Mattos S, Villalonga P, Clardy J, Lam EW-F. FOXO3a mediates the cytotoxic effects of cisplatin in colon cancer cells. Mol Cancer Ther. 2008;7(10):3237–3246. doi:10.1158/1535-7163.MCT-08-0398
71. Rader J, Russell MR, Hart LS, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013;19(22):6173–6182. doi:10.1158/1078-0432.CCR-13-1675
72. McGovern UB, Francis RE, Peck B, et al. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol Cancer Ther. 2009;8(3):582–591. doi:10.1158/1535-7163.MCT-08-0805
73. Millour J, Constantinidou D, Stavropoulou AV, et al. FOXM1 is a transcriptional target of ERalpha and has a critical role in breast cancer endocrine sensitivity and resistance. Oncogene. 2010;29:2983–2995.
74. Yang N, Wang C, Wang Z, et al. FOXM1 recruits nuclear Aurora kinase A to participate in a positive feedback loop essential for the self-renewal of breast cancer stem cells. Oncogene. 2017;36(24):3428–3440. doi:10.1038/onc.2016.490
75. Kwok JM, Myatt SS, Marson CM, Coombes RC, Constantinidou D, Lam EW. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol Cancer Ther. 2008;7:2022–2032.
76. Pandit B, Gartel AL. New potential anti-cancer agents synergize with bortezomib and ABT-737 against prostate cancer. Prostate. 2010;70(8):825–833. doi:10.1002/pros.21116
77. Halasi M, Hitchinson B, Shah BN, et al. Honokiol is a FOXM1 antagonist. Cell Death Dis. 2018;9(2):84. doi:10.1038/s41419-017-0156-7
78. Millour J, de Olano N, Horimoto Y, et al. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol Cancer Ther. 2011;10(6):1046–1058. doi:10.1158/1535-7163.MCT-11-0024
79. Crumbaker M, Khoja L, Joshua AM. AR signaling and the PI3K pathway in prostate cancer. Cancers. 2017;9(12):34. doi:10.3390/cancers9040034
80. Mahajan NP, Liu Y, Majumder S, et al. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation.
81. Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19(5):575–586. doi:10.1016/j.ccr.2011.04.008
82. de Bono JS, De Giorgi U, Rodrigues DN, et al. Randomized Phase II study evaluating Akt blockade with Ipatasertib, in combination with Abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin Cancer Res. 2019;25(3):928–936. doi:10.1158/1078-0432.CCR-18-0981
83. Herrera-Abreu MT, Palafox M, Asghar U, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76(8):2301–2313. doi:10.1158/0008-5472.CAN-15-0728
84. Costa C, Wang Y, Ly A, et al. PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kalpha inhibitors in breast cancer. Cancer Discov. 2020;10(1):72–85. doi:10.1158/2159-8290.CD-18-0830
85. Juric D, Castel P, Griffith M, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature. 2015;518:240–244.
86. Zoubeidi A, Gleave ME. Co-targeting driver pathways in prostate cancer: two birds with one stone. EMBO Mol Med. 2018;10(4):e8928. doi:10.15252/emmm.201808928
87. Yan Y, An J, Yang Y, et al. Dual inhibition of AKT-m TOR and AR signaling by targeting HDAC 3 in PTEN – or SPOP-mutated prostate cancer. EMBO Mol Med. 2018;10(4):e8478. doi:10.15252/emmm.201708478
88. Yuan C, Wang L, Zhou L, Fu Z. The function of FOXO1 in the late phases of the cell cycle is suppressed by PLK1-mediated phosphorylation. Cell Cycle. 2014;13:807–819.
89. Huang Z, Zhou W, Li Y, et al. Novel hybrid molecule overcomes the limited response of solid tumours to HDAC inhibitors via suppressing JAK1-STAT3-BCL2 signalling. Theranostics. 2018;8:4995–5011.
90. Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437:199–213.
91. Corn PG, Wang F, McKeehan WL, Navone N. Targeting fibroblast growth factor pathways in prostate cancer. Clin Cancer Res. 2013;19:5856–5866.
92. Nickols NG, Nazarian R, Zhao SG, et al. MEK-ERK signaling is a therapeutic target in metastatic castration resistant prostate cancer. Prostate Cancer Prostatic Dis. 2019;22:531–538.
93. Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci. 2008;121:3853–3857.
94. Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl). 2016;94:1313–1326.
95. Formisano L, Lu Y, Servetto A, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019;10:1373.
96. Choi YJ, Kim HS, Park SH, et al. Phase II study of Dovitinib in patients with castration-resistant prostate cancer (KCSG-GU11-05). Cancer Res Treat. 2018;50:1252–1259.
97. Huynh H, Nguyen TT, Chow KH, Tan PH, Soo KC, Tran E. Over-expression of the mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK in hepatocellular carcinoma: its role in tumor progression and apoptosis. BMC Gastroenterol. 2003;3:19.
98. Mukherjee R, McGuinness DH, McCall P, et al. Upregulation of MAPK pathway is associated with survival in castrate-resistant prostate cancer. Br J Cancer. 2011;104:1920–1928.
99. Mittnacht S, Paterson H, Olson MF, Marshall CJ. Ras signalling is required for inactivation of the tumour suppressor pRb cell-cycle control protein. Curr Biol. 1997;7:219–221.
100. Drosten M, Sum EYM, Lechuga CG, et al. Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway.
101. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414.
102. Pacheco J, Schenk E. CDK4/6 inhibition alone and in combination for non-small cell lung cancer. Oncotarget. 2019;10(6):618–619. doi:10.18632/oncotarget.26545
103. Lee MS, Helms TL, Feng N, et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget. 2016;7(26):39595–39608. doi:10.18632/oncotarget.9153
104. Maust JD, Frankowski-McGregor CL, Bankhead A, Simeone DM, Sebolt-Leopold JS. Cyclooxygenase-2 influences response to cotargeting of MEK and CDK4/6 in a subpopulation of pancreatic cancers. Mol Cancer Ther. 2018;17(12):2495–2506. doi:10.1158/1535-7163.MCT-18-0082
105. Romano G, Chen P-L, Song P, et al. A preexisting rare PIK3CAE545K subpopulation confers clinical resistance to MEK plus CDK4/6 inhibition in NRAS melanoma and is dependent on S6K1 signaling. Cancer Discov. 2018;8:556–567.
106. Zhou X, Hao Q, Lu H. Mutant p53 in cancer therapy – the barrier or the path. J Mol Cell Biol. 2018;11(4):293–305. doi:10.1093/jmcb/mjy072
107. The Cell-Cycle CJ. Arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6(3):a026104–a. doi:10.1101/cshperspect.a026104
108. Pitolli C, Wang Y, Candi E, Shi Y, Melino G, Amelio I. p53-mediated tumor suppression: DNA-damage response and alternative mechanisms. Cancers. 2019;11.
109. Tanaka T, Watanabe M, Yamashita K. Potential therapeutic targets of TP53 gene in the context of its classically canonical functions and its latest non-canonical functions in human cancer. Oncotarget. 2018;9(22):16234–16247. doi:10.18632/oncotarget.24611
110. De Laere B, Oeyen S, Mayrhofer M, et al. TP53 outperforms other androgen receptor biomarkers to predict abiraterone or enzalutamide outcome in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2019;25(6):1766–1773. doi:10.1158/1078-0432.CCR-18-1943
111. Ecke TH, Schlechte HH, Schiemenz K, et al. TP53 gene mutations in prostate cancer progression. Anticancer Res. 2010;30:1579–1586.
112. Lehmann S, Bykov VJN, Ali D, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol. 2012;30(29):3633–3639. doi:10.1200/JCO.2011.40.7783
113. Sriraman A, Dickmanns A, Najafova Z, Johnsen SA, Dobbelstein M. CDK4 inhibition diminishes p53 activation by MDM2 antagonists. Cell Death Dis. 2018;9(9):918. doi:10.1038/s41419-018-0968-0
114. Laroche-Clary A, Chaire V, Algeo M-P, Derieppe M-A, Loarer FL, Italiano A. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol Oncol. 2017;10(1):123. doi:10.1186/s13045-017-0482-3
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.