Back to Journals » International Journal of Nanomedicine » Volume 16
Novel Strategies for Disrupting Cancer-Cell Functions with Mitochondria-Targeted Antitumor Drug–Loaded Nanoformulations
Authors Allemailem KS ![]() , Almatroudi A
, Almatroudi A ![]() , Alsahli MA
, Alsahli MA ![]() , Aljaghwani A, M El-Kady A
, Aljaghwani A, M El-Kady A ![]() , Rahmani AH, Khan AA
, Rahmani AH, Khan AA
Received 26 January 2021
Accepted for publication 24 April 2021
Published 9 June 2021 Volume 2021:16 Pages 3907—3936
DOI https://doi.org/10.2147/IJN.S303832
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 8
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Khaled S Allemailem,1,2 Ahmad Almatroudi,1 Mohammed A Alsahli,1 Aseel Aljaghwani,1 Asmaa M El-Kady,3 Arshad Husain Rahmani,1 Amjad Ali Khan2
1Department of Medical Laboratories, College of Applied Medical Sciences, Qassim University, Buraydah, Saudi Arabia; 2Department of Basic Health Sciences, College of Applied Medical Sciences, Qassim University, Buraydah, Saudi Arabia; 3Department of Medical Parasitology, Faculty of Medicine, South Valley University, Qena, Egypt
Correspondence: Amjad Ali Khan
Department of Basic Health Sciences, College of Applied Medical Sciences, Qassim University, PO Box 6699, Buraydah, 51452, Saudi Arabia
Tel +966-16-380-0050 Ext 4199
Fax +96616 3801628
Email [email protected]
Abstract: Any variation in normal cellular function results in mitochondrial dysregulation that occurs in several diseases, including cancer. Such processes as oxidative stress, metabolism, signaling, and biogenesis play significant roles in cancer initiation and progression. Due to their central role in cellular metabolism, mitochondria are favorable therapeutic targets for the prevention and treatment of conditions like neurodegenerative diseases, diabetes, and cancer. Subcellular mitochondria-specific theranostic nanoformulations for simultaneous targeting, drug delivery, and imaging of these organelles are of immense interest in cancer therapy. It is a challenging task to cross multiple barriers to target mitochondria in diseased cells. To overcome these multiple barriers, several mitochondriotropic nanoformulations have been engineered for the transportation of mitochondria-specific drugs. These nanoformulations include liposomes, dendrimers, carbon nanotubes, polymeric nanoparticles (NPs), and inorganic NPs. These nanoformulations are made mitochondriotropic by conjugating them with moieties like dequalinium, Mito-Porter, triphenylphosphonium, and Mitochondria-penetrating peptides. Most of these nanoformulations are meticulously tailored to control their size, charge, shape, mitochondriotropic drug loading, and specific cell-membrane interactions. Recently, some novel mitochondria-selective antitumor compounds known as mitocans have shown high toxicity against cancer cells. These selective compounds form vicious oxidative stress and reactive oxygen species cycles within cancer cells and ultimately push them to cell death. Nanoformulations approved by the FDA and EMA for clinical applications in cancer patients include Doxil, NK105, and Abraxane. The novel use of these NPs still faces tremendous challenges and an immense amount of research is needed to understand the proper mechanisms of cancer progression and control by these NPs. Here in this review, we summarize current advancements and novel strategies of delivering different anticancer therapeutic agents to mitochondria with the help of various nanoformulations.
Keywords: cancer, antitumor drugs, mitochondria targeting, theranostic nanoparticles, mitochondriopathies
Introduction
Mitochondria are multifunctional organelles found in most eukaryotic cells that form a comprehensive intracellular network controlled by a proper balance among fusion, fission, biogenesis, and mitophagy.1,2 These organelles are acknowledged for storing and harvesting energy, released by oxidative phosphorylation. Mitochondria are maternally inherited organelles, and most of their proteins are nuclear-encoded. However, these organelles retain a small DNA genome of about 16 kb mitochondrial DNA (mtDNA), which encodes rRNAs and tRNAs and proteins required for respiration. Eukaryotic cells generally contain hundreds of mitochondria with their mtDNA either free of mutations (wild type), mutated mtDNA, or a mixed population known as heteroplasmy.3
These organelles are significant junctions for intracellular interactions with other organelles. They interact with nuclei, the endoplasmic reticulum (ER), and peroxisomes through membrane contact, vesicle transport, and signal transduction to regulate biosynthesis, energy metabolism, immunoresponse, and cell turnover. However, when this normal communication among mitochondria and other organelles fails or when mitochondria are dysfunctional, it most often induces different diseases and especially tumorigenesis.4 Mitochondrial diseases are well known to be devastating, and can affect many organs like muscles, the heart, and the nervous system. These diseases can either be of maternal inheritance or by nuclear inheritance of loss-of-function mutations in some essential mitochondrial genes.5
Most cancers retain mitochondrial functions, including respiration, and even some tumors show enhanced oxidative phosphorylation.6 The energy-harvesting functions of mitochondria are at least as important for cancer progression as ATP generation.7 Cancer cells survive easily during hypoxic milieus by recycling NADH to NAD+ through plasma-membrane electron transport and lactate dehydrogenase to continue glycolytic ATP synthesis.8–10 The precise role of mitochondria during different phases of cancer has been recently reviewed. This review discusses the altered functions of mitochondria during cancer, changes in mitochondrial dynamics, and targeting mitochondria with specific mitochondriophilic biomolecules at multiple sites during cancer progression. In addition, therapeutic strategies, including the use of novel NPs (NPs) conjugated with mitochondriophilic biomolecules, to combat cancer progression are discussed.
Mitochondria as Eukaryotic Cell Organelles
The fundamental role of mitochondria as eukaryotic cell organelles was verified over a century ago.11 During a healthy state, these organelles regulate some vital cellular functions through the tricarboxylic acid cycle (TCA), oxidative phosphorylation, fatty-acid oxidation, and calcium homeostasis. Mitochondria also regulate heme biosynthesis, ketogenesis, urea cycle, gluconeogenesis, and iron–sulfur cluster formation.12 These organelles possess their own DNA (mtDNA), which controls some important functions and can get mutated or partially deleted. Mitochondria are enclosed by a lipid-bilayer double-membrane system — inner mitochondrial membrane (IMM) and outer mitochondrial membrane (OMM) — which are separated by intermembrane space. Much of the inner space of the mitochondrial matrix is occupied by cristae, which are formed by extensive IMM folds. Each mitochondrial component plays specific roles that controls many of the overall cellular activities.13
There are several proteins and enzymes that regulate mitochondrial function, fission, fusion, interaction, and cross talk with other organelles. Mitochondrial fission is promoted by mitochondrial fission 1 protein (FIS1) and mitochondrial fission factor (MFF) present in OMM. Mitochondrial fission controls important functions like autophagy, apoptosis, and cell death.14 On the other hand, mitochondrial fusion is achieved by the outer-membrane fusion proteins, Mfn1 and Mfn2 and the inner-membrane fusion protein OPAC1.15 Mitochondrial fusion controls mitochondrial membrane potential (MMP), cell growth, and electron-transport chain (ETC) functions.16
For the normal functioning of a cell, proper crosstalk between mitochondria and other organelles is very important. Any impairment in this connection may lead to alteration of the cellular environment, which can activate certain oncogenes and mitochondrial genome mutation.4 Precise control exists between the nucleus and mitochondria for the stability of these organelles. Any cross-talk dysfunction between these two organelles can lead to DNA damage in both, abnormal activation of growth factors, Ca2+ overload, and metabolic disorders, which are prominent hallmarks of cancer.17,18
An active and strong interaction exists between the ER and mitochondria for the coordination of important cellular biological functions like Ca2+ signaling, ER stress response, regulation of apoptosis, phospholipid biosynthesis, and translocation from the ER to mitochondria. In addition, proper interaction between mitochondria and peroxisomes is very important for reactive oxygen species (ROS) and lipid balance.
Role of Mitochondria in Autophagy, Apoptosis, and Senescence
In addition to the regulation of biosynthetic precursor balance, energy production, and cytosolic Ca2+ levels, mitochondria can modulate redox status and ROS generation and initiate apoptosis through the activation of mtPTP.19 Mitochondria are the major sources of ROS (ie, hydroxyl radicals, hydrogen peroxide, and superoxide anions), and these reflect the imbalance between antioxidant defense and ROS production. The most pronounced ROS-depended damage includes vascular tonus impairment and platelet adhesion and alterations in gene transcription and metabolism.20 This usually occurs due to hydrogen peroxide, which besides acting as intracellular messenger, controls autophagy, senescence, and apoptosis.20 These processes are closely related to one another, the relationship appears rather complex, and the boundaries are difficult to delineate.
In some circumstances, autophagy can lead to cell death while apoptosis is inhibited, so acting as a backup mechanism for death progression.21 During some failure conditions, to activate autophagy as a cell-survival mechanism during nutrient starvation, it leads to cell death by apoptosis with the involvement of BAX/BAK proteins. These are BCL2-family proteins that are required for caspase activation or mitochondrial outer-membrane permeabilization,22 which is the point of no return in different forms of apoptotic cell death, as it initiates proapoptotic, enzyme-mediated proteolytic cascades and damages mitochondrial functions.23 The cross-regulation among autophagy, senescence, and apoptosis is a complex phenomenon and still far from being understood. The role played by mitochondria in the onset of these process is briefly discussed in many sections of this article.
Mitochondrial Participation in Cancer Development
Mitochondria may play a significant role in the development of cancer phenotypes through at least five mechanisms. First, it has been commonly demonstrated that DNA mutations affect mitochondria and lead to many diseases, mainly due to alterations in ETC subunits.24 Second, ROS are mainly produced from mitochondria (mtROS), which mediates oxidative stress (OS) and is the principal cause of cancer generation and progression.25 mtROS can be generated either in the ETC or during the TCA cycle.26 Enhanced levels of ROS are usually found in cancer cells, due to altered antioxidant potential.27 Third, the mitochondria have a direct role in cell-death regulation, including but not limited to necrosis and apoptosis.28 For the induction of apoptosis, BCL2 proteins interact with mitochondria through binding with voltage-dependent anion channels (VDACs) to enhance the release of cytochrome c (cyt c).29 Mitochondria also control necroptosis, which is a regulated form of necrosis that requires mitochondrial permeability transition and mtROS.30 Fourth, metabolic reprogramming also affects gene mutations encoding enzymes of the TCA cycle, which can promote cancer transformation.31 Fifth, telomerase reverse transcriptase shuttles from the nucleus to mitochondria during enhanced oxidative stress. It is used to preserve mitochondrial functions, decrease oxidative stress, and protect mtDNA and nuclear DNA from oxidative damage to avoid apoptosis.32
Mitochondrial Functional Aberrations During Cancer
Though cancer cells are highly diverse, all display some stereotypical traits or hallmarks, and mitochondria play an important role in such hallmarks.33 Mitochondria play a significant role in initiation of cancer, which can be due to dysregulated signaling, mtDNA mutations, oxidative stress, metabolism, bioenergetics, fission and fusion dynamics, or biogenesis and turnover. The distorted bioenergetics within cancer cells help them meet the required energy demands by ATP generation through the ETC.
During acute myeloid leukemia (AML), altered mitochondrial metabolism occurs, due to mutations in isocitrate dehydrogenase (IDH). An isoform of IDH, IDH3 catalyses the formation of α-ketoglutarate from isocitrate in the TCA cycle. In parallel, IDH1 and IDH2 catalyse the same reaction, but outside the TCA cycle.34 Aberrant mitochondrial metabolism during AML has opened the possibility of using several drug nanoformulations to rectify mutations. Various studies on in vivo and in vitro AML models have demonstrated the benefits of using mitochondria-targeted mitocans in combination therapies. In this regard, arsenic trioxide has been found to be a potential drug at the clinical level for acute promyelocytic leukemia patients. The use of arsenic trioxide has raised hopes of discovery in more aberrant mitochondria–targeted drug nanoformulations as a therapeutic strategy in treating AML.
The transformed mitochondrial metabolism supports the rapidly dividing cancer cells by providing building blocks. Mitochondria show good flexibility in supporting cancer-cell survival during adverse conditions, such as starvation and chemotherapy.9 Therefore, understanding the mechanisms of mitochondrial function during normal and cancerous states will be crucial to develop next-generation cancer therapeutics. Some altered mitochondrial functions during cancerous state are outlined in the following sections.
Upregulation of Oxidative Phosphorylation
Recent experimental data on mitochondria have overturned the belief that cancer cells quench their bioenergetic and anabolic requirements predominantly through aerobic glycolysis. It is now well acknowledged that mitochondrial metabolism plays a crucial role in cancer development and progression. These organelles play an important role in different steps of oncogenesis and response to treatment.35 This is further supported by the findings that different cancer cells depend primarily on oxidative phosphorylation for promotion of their tumorigenic potential.6,36 These observations are supported further by analysis of glioma cells, which are rescued by pyruvate and lactate, oxidative substrates produced during low-glucose conditions.37 In parallel, it has been observed that in MCF7 cells, oxidative metabolism produces 80% of ATP and increased glucose consumption is not necessarily linked with increased glycolysis.38 Furthermore, it has been found that some drug-resistant tumor cells are subjected to mitochondrial oxidative phosphorylation for their survival. The treatment of these cells with ETC complex I inhibitors prolongs survival and tumor burden in murine xenograft models.39
Reprograming of Metabolism
Mitochondria are indispensable for tumor cells, as they provide the major share of energy and process metabolic intermediates. The TCA cycle provides some major metabolic intermediates and building blocks for anabolism. Kreb’s cycle constitutes an epicenter in cellular metabolism, as numerous substrates can feed into it. As such altered TCA-cycle regulation and its continued feedback with dysregulated oxidative phosphorylation is crucial for the progression of cancer.
Several types of human cancers show that TCA cycle enzymes are often dysregulated and frequently mutated. Some of the most vulnerable enzymes are isocitrate dehydrogenase, aconitate hydratase, succinate dehydrogenase (SDH), fumarate hydratase (FH), and the α-ketodehydrogenase complex.40,41 In addition, some metabolites of the TCA cycle control mitochondrial chromatin modifications and post-translational modification in proteins. The role of Kreb’s cycle rewiring during hepatocellular carcinoma has been summarized by Todisco et al, as they found dysregulation of glutamine metabolism, citrate–pyruvate, and malate–aspartate shuttles. A link has also been observed between the transcription factor NFκB–HIF1 and TCA cycle reprogramming.42
Significant alterations have been observed in the abundance of some enzymes linked with aerobic glycolysis and Kreb’s cycle in gliomata of IDH1-mutant types.43 Enzymes involved in the metabolism of lactate, glutamate, and α-ketoglutarate are also significantly enhanced in such mutant gliomata. In addition, increased expression of SLC4AG, a bicarbonate transporter has been observed in such gliomata. This suggests a mechanisms that preventing the glycolysis mediated intracellular acidification is active in such cells. This special type of metabolic rewiring preserves the activity of TCA cycle in IDH1-mutant glioma types.43
Enhanced Oxidative Stress
Cancer cells display metabolic aberrations accompanied by accumulation of ROS, which is the main cause of biomolecular damage, and if it exceeds the limit leads to cell death.44 The ETC releases electrons that are captured by O2 to generate O2–, which leads to the formation of ROS.45 These entities lead to DNA damage and bind with intracellular and surface receptors and signaling molecules. All these changes lead to angiogenesis, proliferation, and apoptosis, significant aspects of cancer progression.46,47 The extensive DNA damage by ROS can promote carcinogenesis and the malignant transformation of normal cells. Excessive production of ROS also leads to Cyt. c release and can trigger programmed cell death.48 Enhanced levels of ROS pose a severe threat to mitochondria and even cell viability.
Altered Dynamics
Normal cells exhibits continued mitochondrial fission and fusion cycles to maintain proper function. Fission- and fusion-machinery proteins also regulate intrinsic apoptotic pathways.49 In cancer cells, the genes responsible for mitochondrial dynamics regulation are amplified and inhibition of DRP1, a fission promoting GTPase known to induce apoptosis.50 DRP1 knockout in pancreatic cancer cells has been witnessed with diminished oxygen consumption and minimal ATP production, which results in reduced growth.51 In comparison to normal cells, lung adenocarcinoma has been reported to express lower levels of Mfn2 and higher levels of DRP1. Different phases of the cell cycle are also controlled by DRP1 dysregulation, which helps to maintain cell proliferation. DRP1 also inhibits p53 and promotes progression of the cell cycle.52 DRP1 expression has been found to be in phase with cell-cycle gene expression in ovarian cancer cells, as 55% of these genes regulate mitotic transition.53 Forced inhibition of DRP1 in these cells causes replication stress and delayed G2–M transition. This replication stress leads to hyperfused mitochondrial structures and improper cyclin E expression during the G2 phase.54 In addition to this, silencing of DRP1 in breast cancer cells leads to suppressed metastatic abilities.55 Furthermore, inhibition of upregulated expression of DRP1 in hypoxic glioblastoma U251 cells attenuates hypoxia-induced mitochondrial migration and fission. All these findings support the view that DRP1 can be a potential therapeutic target in cancer cells.56 In addition, proteasomal degradation of Mfn2 induced by chemotherapy leads to mitochondrial fragmentation, which results in apoptotic cell death.57,58
Altered Mitochondrial DNA
In addition to mitochondrial aberrations in different cancers, high levels of mtDNA mutations have been reported in several cases.59 The different mtDNA mutations in diverse cancers include deletion, inversion, point mutation, and variation in copy number. These mutations potentially arise from the clonal expansion of cells containing mtDNA mutations. mtDNA mutations have been reported in a large percentage of lung cancer, colorectal cancer, head and neck cancer, pancreatic cancer, urinary bladder cancer, ovarian carcinoma, gastric cancer, breast cancer, and several other tumors.60–66 Whether the enhanced frequency of mutated mtDNA in cancer cells is an outcome of their uncontrolled division or whether mtDNA mutations provide a selective advantage to these transformed cells that contributes to cancer initiation and progression remains an important open question.67
Some recent discoveries have shown intergenomic cross talk between mitochondria and the nucleus and the role of mtDNA variation in disturbing this signaling and thus indirectly targeting nuclear genes involved in tumerogenic and invasive phenotypes. Therefore, mitochondrial dysregulation is currently regarded as an important hallmark of carcinogenesis and a promising target for antitumor therapy.68 Moreover, the advancement of mtDNA editing tools is expected to improve strategies to characterize, track, and repair oncogenic mitochondria, which will further boost the understanding of mitochondrial epigenetics in cancer and therapeutic strategies.
Elevated Heme Levels
Heme (protoporphyrin IX), an iron-containing molecule, is synthesized by human cells at the basal level.69 Heme plays a significant role in mitochondrial respiratory-chain complexes and different enzymes and proteins involved in oxygen metabolism like cytochromes, peroxidase, and catalase. Different types of cancers have been reported with elevated heme levels, and this elevation may contribute to the maintenance and proliferation of cancer.70 Oxygen consumption and heme biosynthesis are significantly intensified in lung cancer cells. In addition, protein levels in heme synthesis and uptake are increased in lung cancer. It has been found that the inhibition of heme and mitochondrial functions suppresses the cancer-cell proliferation and migration.70,71 Furthermore, some epidemiological studies have suggested that increased heme intake via red meat is associated with greater risks of breast, lung, pancreatic, oesophageal, and colorectal cancer. A study based on almost 500,000 individuals revealed that the consumption of processed meat leads to a 16% increased risk of lung cancer. Important sites of altered mitochondrial metabolism in cancer as potential targets for therapy are shown in Figure 1.
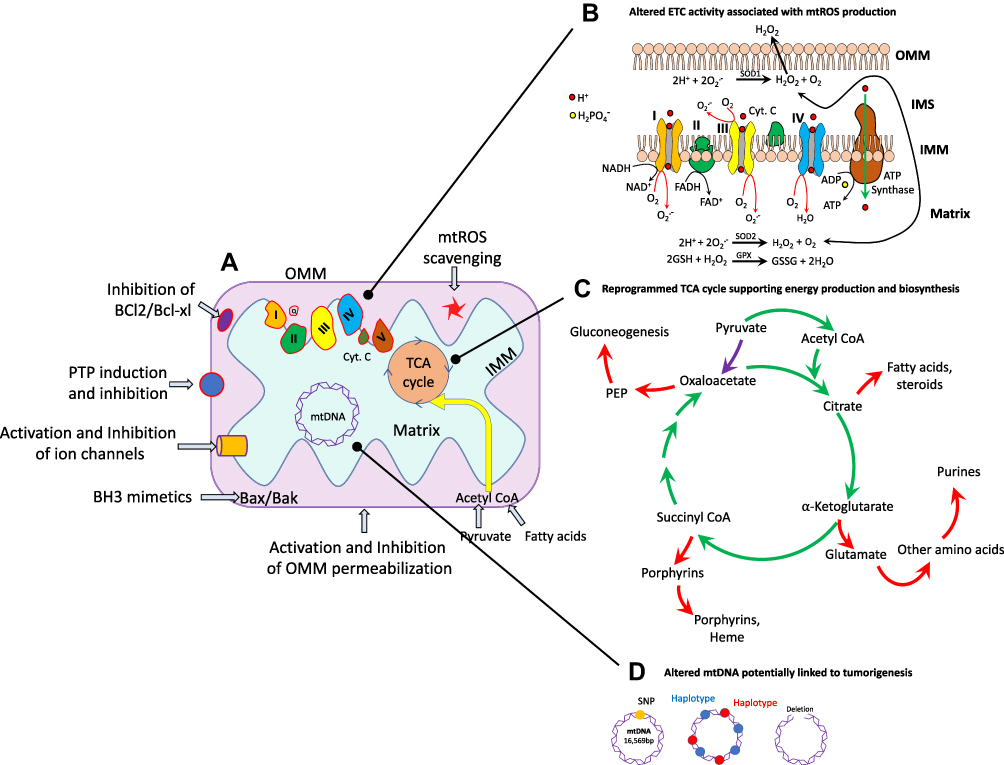 |
Figure 1 Some important locations of mitochondria that can be potential targets of anticancer-drug nanoformulations. (A) Mitochondria in normal and in cancer cells. (B) Highly metabolically active or hypoxic cancer cells generate superoxide (O2–), which is immediately dismutated to H2O2. (C) in cancer cells, the TCA cycle produces reducing equivalents to fuel the ETC (green arrows), and also generates intermediates necessary for cell proliferation (red arrows). The most important anaplerotic reaction produces oxaloacetate directly from pyruvate (purple arrow). (D) Mitochondrial DNA (mtDNA) variations, including single-nucleotide polymorphisms (SNPs), maternally inherited haplotypes, and deletions, have been studied for their associations with cancer. |
Novel Strategies for Targeting Mitochondria at Different Sites
Novel anticancer drugs have been synthesized that can selectively disrupt cancerous mitochondria at different function targets by inhibiting glycolysis, disrupting the ETC and oxidative phosphorylation and depolarizing membrane potential. Here, we elucidate different locations of mitochondria in cancerous cells that can be novel targets to hit these types of cells.
Targeting Oxidative Phosphorylation
Appropriate functioning of the ETC is very important to support oxidative phosphorylation and ATP synthesis, essential for tumorigenesis. Several ETC inhibitors like tamoxifen, α-tocopheryl succinate, metformin, and 3-bromopyruvate have been used to disrupt the proper functioning of ETC respiratory complexes. These inhibitors lead to the induction of enhanced ROS generation and ultimately kill some cancerous cells.72 Proper use of these drugs specific to mitochondria of cancerous cells is a novel approach of drug targeting and requires deeper investigations for future cancer therapy.
Some novel mitochondria-targeted therapeutic agents like MitoTam, a derivative of tamoxifen, have been found to inhibit ETC complex I and lead to increased ROS synthesis. This drug has been used in breast cancer cells to induce their death.73 In parallel, another mitochondria-specific drug — MitoVes, an analogue of vitamin E succinate — inhibits ETC complex II and minimizes tumor growth by triggering apoptotic cell death in colorectal, breast, and lung cancers.74 In addition, several signaling pathways and ETC complex I have been targeted by ME44, which induces cell death by interfering with mitochondrial permeability in colorectal cancer.75 Mitochondria were further targeted by ME143 and ME344, which significantly inhibit oxidation of NADH by complex I, thus preventing electron flux through other oxidative phosphorylation complexes. ROS are generated by ME344-mediated inhibition of complex I, thus leading to BAX translocation to the OMM. This translocation leads to mitochondrial permeability transition, which results in the discharge of proapoptotic molecules.76
Targeting the TCA Cycle and Glutamine Metabolism
As the TCA cycle is the bioenergetic hub of metabolism and redox-state balance, it also serves as an important location of biosynthesis of different compounds. This hub is considered a novel location for different therapeutic strategies for the prevention of cancer. It involves mutations of isocitrate dehydrogenase genes, which have been reported in cancers like AML and glioblastoma.41,77 Novel therapeutic agents against these mutated gene products are now being engineered for the treatment of AML and other cancers.78 Some mutations have also been reported in FH and SDH in association with certain cancers, and any loss of these enzymes increases the vulnerability of a cancerous cell to a therapeutic agent.79 In addition to TCA cycle enzymes, oncogenes like HIF, MYC, RAS, and P53 are known to regulate the metabolic phenotype of tumor cells.80 As such, these additional pathways are now being explored as new therapeutic targets against cancer cells.81
Many cancer cells use glutamine as a fuel to supply essential nutrients and precursors for their constant growth. Inhibition of glutaminolysis can be an innovative therapeutic strategy for the treatment of various cancers. Some glutamine analogues (azaserine and 6-diazo-5-oxo-l-norleucine, azotomycin) have been tried as a treatment strategy, but this therapeutic protocol was not continued, as these analogues induced severe toxicity.82 These analogues can be tried again if transported specifically to cancer cells through mitochondria-targeted NPs.
Targeting Mitochondrial Dynamics and Trafficking
Mitochondrial dynamics include morphology, distribution, fusion, and fission, which regulate different biological activities within the cell, including energy production. Most cancers show a prominent hallmark of increased fission compared to its fusion ratio.83 Some studies have targeted DRP1 with mitochondrial division inhibitor 1, a mitochondrial fission protein inhibitor, to reduce the tumorigenic activities of cancer stem cells.84 In parallel, an effective therapeutic strategy has been devised by using miR125a to inhibit Mfn2, which augments the mitochondrial fission in pancreatic tumor cells.85 The cell cycle is regulated by LATS2, which senses DNA-damage response. Overexpression of this gene activates mitochondrial fission and promotes mitochondrial stress in lung cancer cells, leading to their apoptosis. Targeting of LATS2 and its associated signaling can be an efficient therapeutic approach.86 The regulation of mitochondrial morphology through mitofusins and DRP1 is also regulated by E3 ubiquitin ligase (MARCH5). This protein is associated with breast cancer, and could serve as a potential therapeutic target.87
Cancer therapy can also be shortlisted by focusing on mitochondrial membrane transport mechanisms and associated proteins. This includes translocase of inner mitochondrial membrane 50 (TIMM50), involved in ERK–P90RSK signaling-pathway regulation. It prevents E-cadherin expression, which promotes cancer proliferation in NSCLC cells, so can be an efficient therapeutic target in such cells.88 Furthermore, mitochondria-mediated apoptosis is also regulated by VDAC1, which is found at the OMM. Specific targeting of this complex may serve as a novel therapeutic approach for cancer treatment.89 Some important examples of mitochondria-specific antitumor drugs (mitocans) and treatment strategies with these are listed in Table 1.
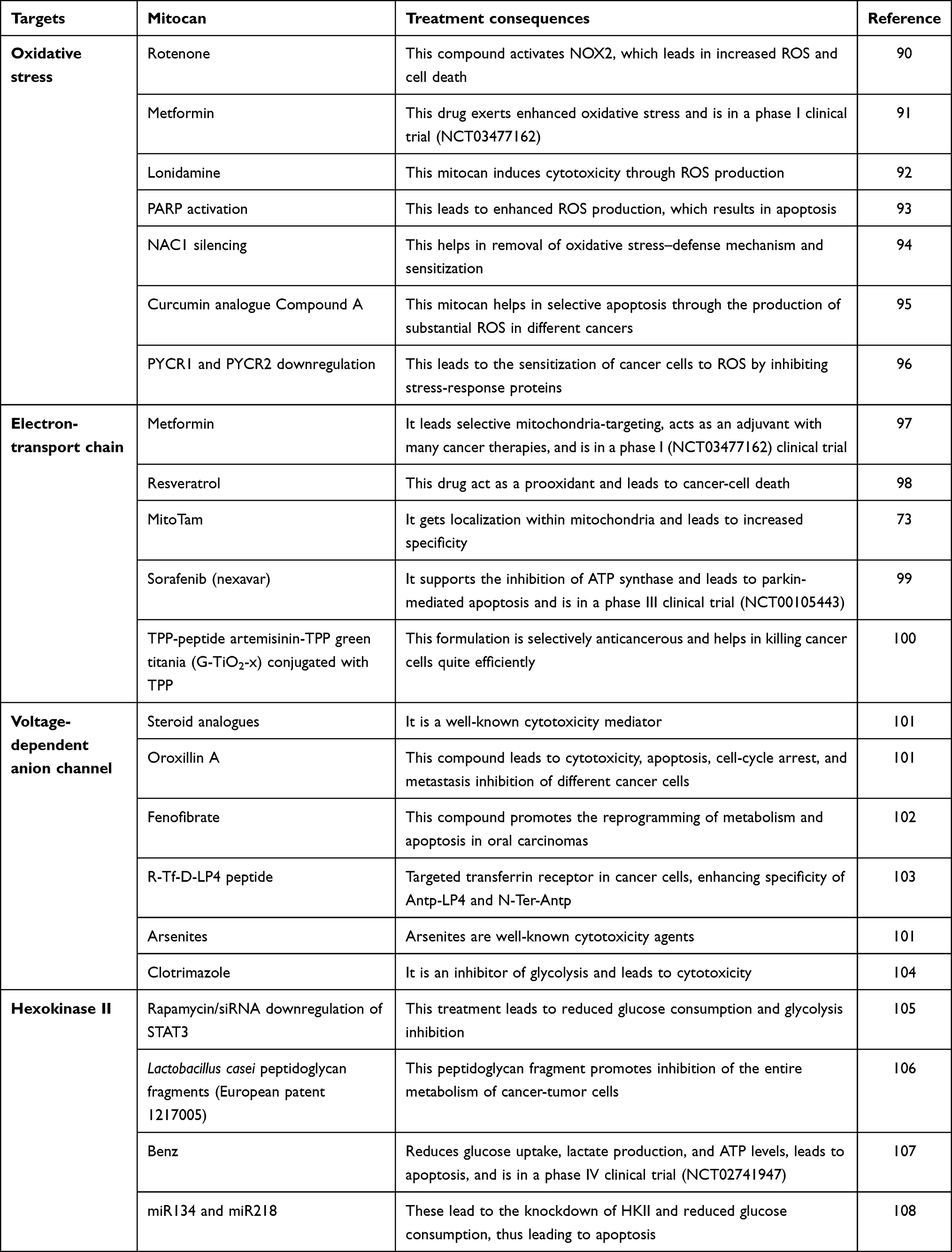 |
Table 1 Mitochondrial function target sites for different mitocans and their possible treatment effects |
Mitochondria-Specific Targeting Ligands and Accumulation Criteria
A number of mitochondria-specific targeting ligands have been discovered that have tremendously improved therapeutic efficacy and greatly reduced the side effects of conjugated drugs. The direct targeting of these moieties to mitochondria has resulted in rapid response to the attached drugs.109 The most widely used mitochondria-targeting ligands are triphenylphosphonium (TPP), dequalinium (DQA), short peptide, rhodamine 19, and rhodamine 123, pyridinium, guanidine, (E)-4-(1H-indol-3-ylvinyl)-N-methylpyridinium iodide (F16), and 2,3-dimethylbenzothiazolium iodide. The direct conjugation of these mitochondria-specific moieties with anticancer drugs, sensors, and antioxidants by different bonds and spacers has resulted in enhanced cytotoxicity, sensing activity, and antioxidizing activity, respectively.110 Transformed and cancer-cell mitochondria display amplified transmembrane potential compared to normal cells.111 This difference has been utilized to develop mitochondria-targeting moieties that preferentially accumulate within cancer-cell mitochondria. Most mitochondria-targeting moieties are delocalized lipophilic cations, and their chemical structure is shown in Figure 2. They are also elaborated upon below in the following sections.
 |
Figure 2 Chemical structure of some important mitochondria-targeted lipophilic cations. |
Triphenylphosphonium
TPP is a delocalized cationic lipid that readily penetrates through the mitochondrial membrane because of highly negative membrane potential. TPP is a well-known mitochondria-orienting moiety with cationic phosphorus bonded with three hydrophobic phenyl groups.112 This compound has been used significantly by conjugating with anticancer drugs like doxorubicin, porphyrin, coumarin, and chlorambucil to enhance mitochondria targeting within cancer cells.113 Accumulation of TPP is directly proportional to the negative charge of the mitochondrial membrane, and for every 60 mV negative membrane potential, TPP accumulation is increased by one order of magnitude. It has been reported that MMP is −180 mV, which can facilitate up to 1,000-fold buildup of TPP inside mitochondria.114 TPP has also been conjugated with chlorambucil (DNA-damaging anticancer agent) and used against breast cancer cell lines, resulting in an almost 12-fold reduction in IC50 compared to free drug.115 In a parallel study, vitamin E succinate has been conjugated with TPP, and this conjugate (MitoVES) presented enhanced mitochondrial accumulation.74
1,1′-Decamethylene-bis-(4-Aminoquinaldinium Chloride)
1,1′-Decamethylene-bis-(4-aminoquinaldinium chloride) also named dequalinium (DQA) is a well-known lipophilic dication composed of two quinolinium moieties bonded with each other through an alkyl chain of ten carbons. This compound displays antiproliferative potential against different in vitro cancer cell lines and also shows in vivo antitumor properties.116 In aqueous medium, DQA molecules (single-chain bola-amphiphile) self-assemble and form vesicles known as DQAsomes.117 These vesicles are used to deliver pDNA within the mitochondria without any off-target leakage.118 In another study, DQA-conjugated, peptide-conjugated, and F16-conjugated anticancer drugs were used against different cancers.119
Mitochondria-Penetrating Peptides
Mitochondria-penetrating peptides (MPPs) have repeating lipophilic and cationic residues, eg, (L-cyclohexyl alanine-D-arginine)3 (Figure 2). These peptides show mitochondrial buildup with low toxicity against human tumor cells.120 Doxorubicin has been conjugated with MPP via succinate linkage. Another examples of mitochondria-specific peptides is Szeto–Schiller (SS) peptides. These tetrapeptides denote a special class of novel chemical entities that precisely target mitochondrial cardiolipin, improve the plasticity of mitochondria, and recondition bioenergetics. They are water-miscible tetrapeptides composed of Tyr-dimethyltyrosine (Dmt)-Arg-Phe-Lys residues. These peptides get selectively built up within the IMM and scavenge ROS. Further, these peptides block the opening of mitochondrial permeability transition pores, thus stopping the release of cytochrome c (cyt c).121 Different types of SS peptides have been engineered (SS1–S31), and SS01 (Tyr-D-Arg-Phe-Lys-NH2) is shown in Figure 2.
The smart character of SS peptides lies in their mutual lipophilicity and positive charge, which is essential for easy passage through the cell membrane and mitochondrial membranes. To target mitochondria more specifically, a key strategy is to ensure alternate basic and aromatic amino residues. This approach has been used to design SS31 (D-Arg-Dmt-Lys-Phe-NH2), an efficient ROS scavenger through inhibiting lipid peroxidation. The safety and efficacy of SS31 led to a phase II clinical trial on microvascular and ischemia–reperfusion injuries, in patients with acute myocardial infarction. This trial also involved the treatment of hypertension-mediated renal microvascular dysfunction and kidney injuries. This drug has also been used for the treatment of diabetic macular edema and heart failure.122
In addition, an amphipathic molecule, α-helical D-(KLAKLAK)2, has been used for targeting the IMM, as this drug has improved anticancer potency.123 Keeping the chemistry of these drugs in mind in terms of their alternate hydrophobic and cationic nature, it has attracted to design similar MPPs. This led to the design of P11LRR, an arginine-modified amphiphilic peptide that comprises polyproline scaffolds and has a helical structure. It has been reported that accumulation of P11LRR within mitochondria is basically driven by its transmembrane potential. Its mitochondria-targeting impact is further enhanced by the amphipathic α-helical structure, as this is crucial for the import of some peptide sequences up to mitochondria.124
Guanidium and Biguanidium Moieties
Guanidinium and bigaunidinium moieties possess delocalized positive charges, and have been conjugated with hydrophobic porphyrins as photosensitizers and phototoxic agents to enhance mitochondrial accumulation. Guanidine and biguanidines have been reported to possess enhanced lipophilicity. These amphiphilic porphyrins have been bonded with different moieties to enhanced mitochondria targeting. These conjugates have been found to possess high membrane potential across the IMM and have been used for the treatment of cancer.125 Cellular uptake of these conjugates and their subcellular localization studies have revealed that guanidine-porphyrins are readily engulfed by cells and get accumulated in mitochondria more quickly than bigaunidine moieties. These conjugates possess enhanced mitochondria targeting and improved phototoxicity against certain cancer cells. The guanidine-porphyrin conjugates represent 1.8 fold enhanced phototoxicity than biguanidine–porphyrins.126 The presence of guanidinium shows a proton-sponge effect within lysosomes and promotes lysosomal membrane rupture and escape capacity, even when conjugated with porphyrins. Metformin is a good example of biguanide, which acts as an antihyperglycemic agent and suppresses mitochondrial respiration, as it inhibits respiratory complex I.127
(E)-4-(1H-Indol-3-ylvinyl)-N-Methylpyridinium Iodide
(E)-4-(1H-Indol-3-ylvinyl)-N-methylpyridinium iodide (F16) is a delocalized cation that accumulates within the mitochondrial matrix. The accumulation of this cation within this organelle lies in its higher MMP (Δψm) capability. This accumulation also causes depolarization of the membrane, disrupting the integrity of mitochondria, and opens mitochondrial permeability transition pores. These events lead to cytochrome c (cyt c) release, cell arrest, and ultimately cell death.128 The antiproliferative potential of F16 has been reported in a variety of human breast cancer cell lines and mouse mammary tumors.
Unlike other apoptosis inducers, F16 acts in mitochondria at the junction of the apoptotic and necrotic pathways. This compound results in the induction of permeability transition and changes the functional integrity essential for cell survival. It has been observed that cell death in F16-treated overexpressing BCL2 clones is prevented under conditions of higher concentration of ATP maintenance to neutralize superoxide anions. This indicates that overexpressing BCL2 cells show necrotic death that coincides with F16-mediated mitochondrial dysregulation.129
Rhodamine
Rhodamine has a mitochondria-targeting nature due to its lipophilic and cationic properties. These properties are the basis of crossing the double mitochondrial membrane ands accumulation within the negatively charged mitochondrial matrix.130,131 Rhodamine is also efficient at mitochondria targeting and damaging the ETC when bound to mitochondria. Both rhodamine 19 and rhodamine 123 have potential in targeting mitochondria.132 The mitochondria-targeting capacity of rhodamine 19 has been confirmed by substituting TPP. In brief, rhodamine 19 is a potential mitochondria-targeting cationic uncoupler, and shows its protonophorous uncoupling potential and maintains equilibration across the mitochondrial membranes in a Nernstian style. Some examples of important mitochondria-targeting moieties liganded with different drugs through specific bonds or spacers and their respective responses are listed in Table 2.
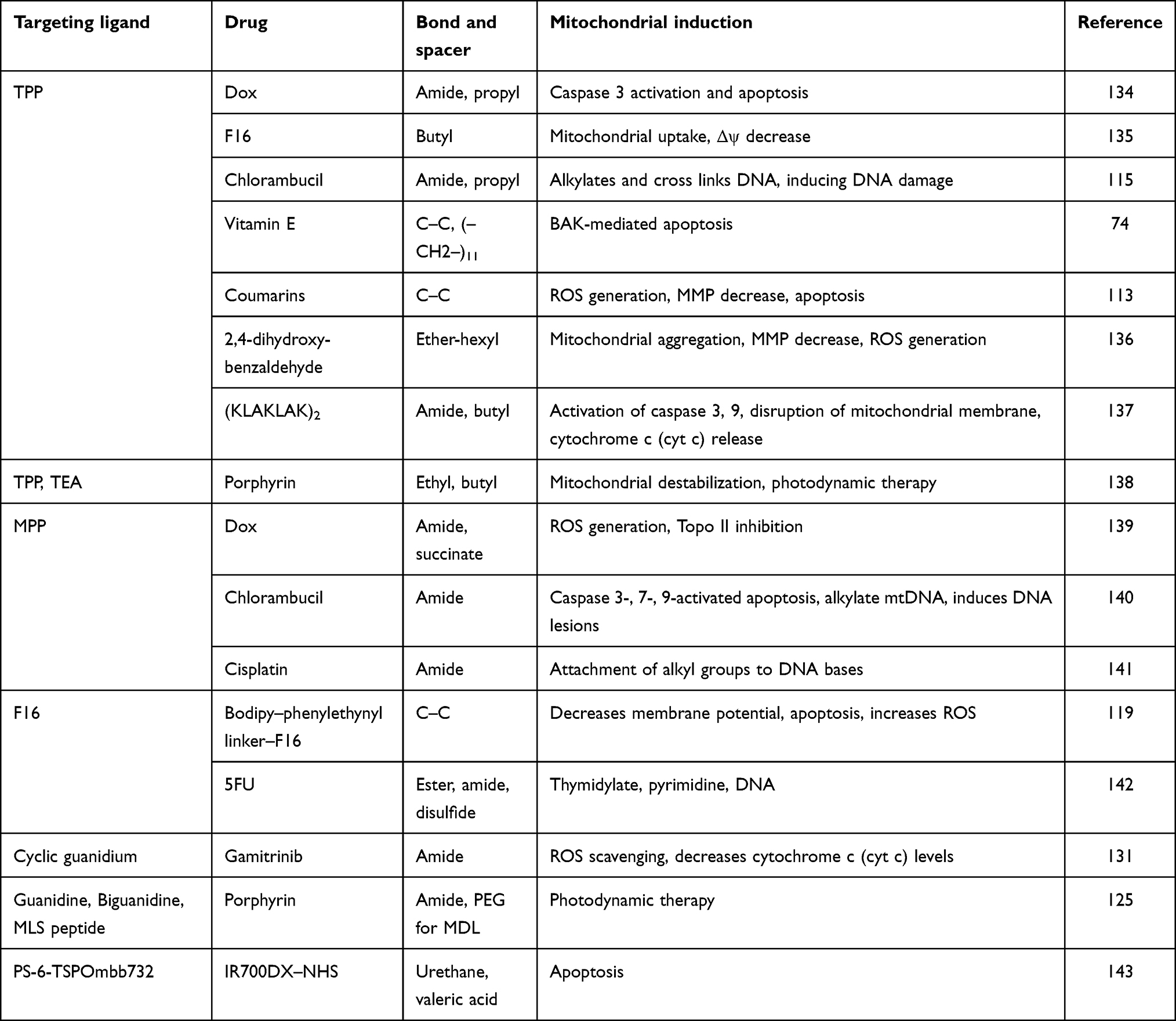 |
Table 2 Mitochondria-targeting moieties conjugated with various drugs through a specific bond or spacer, provoking different reactions and mediating important changes |
Mitochondria Targeting with the Aid of Ligand-Conjugated Nanoformulations
In clinical practice, the use of single-unit nanoformulations in therapeutics and diagnostics (theranostics) is a novel approach to drug delivery.145 Theranostic NPs exhibit several advantages over the conventional systemic administration of native drugs. These include overcoming the problems of limited solubility, inactivation, biodegradation, and minimal off-target toxicity. Other benefits include extended circulation time, higher concentration at tumor site, multiple synergistic drugs, diagnostic system delivery,146 controlled drug release at tumor sites through stimulus-sensitive delivery systems, eg, pH, temperature, enzyme-sensitive nanoformulation, overcoming multidrug resistance and enhanced therapeutic efficacy. The approach of this drug-delivery system even up to the organelle level (third-level drug targeting) with the aid of different nanoformulations has revolutionized the therapeutic approach to different diseases, including cancer.
Mitochondria-related diseases can be best addressed by the novel strategy of using nanoformulations, which can also prove to be a valuable tool to overcome the current limitations of treating mitochondrial diseases. These nanoformulations can be powerful targeted drug-delivery systems to mitochondria.147 Moreover, they can drastically improve the pharmacokinetic and biodistribution properties of various therapeutic drugs. The uptake of NPs loaded with chemotherapeutic agents by mitochondria stimulates ROS generation and Cyt. c release, sequentially activates the downregulation of caspase 3/9 precursors, and ultimately induces mitochondrial permeability. These responses result in mitochondrial edema and cause substantial damage. Moreover, NPs dysregulate membrane potential and promote mitochondrial death pathways, inducing the elevation of apoptotic events within cancer cells.148
Delivery systems based on NPs must be meticulously designed with proper size, shape, charge, lipophilic surface, and specific density to achieve center-point targeting within mitochondrial locations. NPs need to have spatiotemporal control over the release of their drug payloads at different mitochondrial compartments.149 NP size impacts drastically on cellular uptake by influencing adhesion strength with cellular receptors. Optimal cellular uptake with ligand-coated NPs has been found to be met at almost 50 nm diameter.150 Similarly, the highest uptake of spherical mesoporous silica NPs by HeLa cells is at 50 nm.151 In addition, targeted AuNPs have been reported to possess the highest cellular uptake by SKBR3 cells at 40–50 nm in size.152 Currently, NPs/nanoformulations are conjugated with different mitochondria-specific compounds to achieve best organelle targeting. Some common examples of NPs used against mitochondria of different cancer cell lines are presented in Figure 3.
 |
Figure 3 (A) Schematic representation of mitochondria-targeted nanoformulations loaded with hydrophilic and hydrophobic drugs. These NPs can also be loaded with Mitochondria-targeted genes. (B) Approaches for drug-loaded NP entry within a target cell. (C) of Mito-Porter approach for targeting of cancer-cell mitochondria. (D) Membrane fusion of Mito-Porter with OMM and IMM and the delivery of mitochondria-specific drugs and genes. |
Liposomes
Liposomes are spherical vesicles composed of one or more concentric lipid bilayers and are routinely used as drug-delivery vehicles. The physicochemical properties of these vesicles differ considerably on size, composition, surface charge, and even method of preparation.153 New modifications of conventional liposomes to achieve efficient mitochondria targeting are ongoing, as these entities need to be cheaper, atoxic, and biodegradable. This has led to the formation of TPP-modified liposomes coloaded with a photothermal near-infrared (NIR) imaging agent, IR780 iodide, and a photosensitizer known as chlorin e6. These novel liposomes show enhanced toxicity to HeLa cells and some tumor vessels in vitro compared to untargeted ones. In addition, this technique has led to easy and controlled release of drugs and imaging agents to achieve antitumor angiogenesis and photothermal therapy.154 In a parallel strategy, stearyl residues have been conjugated with TPP and incorporated as STPP within lipid bilayers.155 These STPP-modified liposomes were further loaded with ceramide, which showed significantly reduced tumor volume in BALB/c mice.
Liposome-based drug formulations face some aggregation and instability issues in blood. This complication has been resolved by using hybrid cerasomes based on the Si–O–Si framework and liposomes.156 The cerasomes were conjugated with TPP using 3-aminopropyl triethoxysilane, which acts as a linker. These TPP-modified cerasomes were loaded with doxorubicin (TPP–CER–Dox) through self-assembly process and formed phospholipid bilayer vesicles covering the cerasomes. These possessed extraordinary stability, biocompatibility, sustained drug release, and efficient drug accumulation within mitochondria.157
Recently, a novel liposome (Mito-Porter) has been designed with phosphatidic acid or sphingomyelin nd its surface modified with octaarginine (R8) and GALA, a membrane fusogenic peptide. This special type of liposome can introduce specific cargoes into mitochondria using the advantage of membrane fusion. The presence of highly dense R8 enables the liposomes to achieve micropinocytosis-mediated cell-membrane internalization. The presence of GALA helps the liposomes escape endosome formation. Mito-Porter helps to fuse successfully with both the OMM and IMM158,159 (Figure 3).
Mito-Porters have also been designed for targeting nucleic acids specific to the mitochondrial genome. The presence of phosphatidic acid or sphingomyelin in this formulation facilitates enhanced mitochondrial membrane binding and special cargoes are released within the mitochondrial compartment. For intracellular trafficking of Mito-Porter, R8 plays a crucial role, as its higher density leads to internalization by micropinocytosis and its lower-density vehicles being taken up by clathrin-mediated endocytosis and degraded by lysosomes.158,160
Mito-Porters have also been used to transport fluorescent dyes like propidium iodide for staining nuclear DNA.161 Advancement in the same study led to the discovery of a dual-function Mito-Porter system that penetrates the endosomal and mitochondrial membranes by phase-wise membrane fusion.162 A study was based on comparison of the effective dose for the two types of nanocarriers, and the results showed that the dual-function Mito-Porter was 15-fold higher in efficiency than conventional Mito-Porter for mitochondrial delivery.163
Furthermore, instead of R8, mitochondrial signal targeting signal peptide (MTS) with sequence NH2-MVSGSSGLAAARLLSRTFLLQQNGIRHGSYC was used to form MTS-Mito-Porter; however, this system showed labile aggregation, eventhough it was highly efficient for mitochondrial delivery compared to R8-Mito-Porter.164 In further research, S2 peptides modified with stearyl-Dmt-D-Arg-FK-Dmt-DArg-FK-NH2 were used to decorate the dual-function Mito-Porter, which provoked lower toxicity than the DF-R8-Mito-Porter.165 The mechanism of Mito-Porter uptake by cells and its fusion with mitochondria for the delivery of loaded drug is illustrated in Figure 3.
DQAsomes
DQAsomes are well known mitochondriotropic “bola-lipid”–based vesicles composed of dequalinium (DQA;1,1′-decamethylene-bis-[4-aminoquinaldinium chloride]), a dicationic amphiphilic molecule. These vesicles were designed for the transportation of drugs and DNA specific for mitochondria.166 Studies have now demonstrated that DQAsomes induce necrotic and apoptotic activities, as these nanovehicles induce mitochondrial dysregulation. DQAsomes cause mitochondrial membrane–potential reduction, excess ROS production, ATP depletion, activation of the protein kinase–signaling cascade, and induction of apoptosis by mitochondria-dependent pathways.167
A novel formulation of curcumin encapsulated by DQAsomes has been prepared with average hydrodynamic diameter about 185 nm, drug-loading capacity up to 61%, and encapsulation capacity up to 90%. These DQAsomes possessed enhanced antioxidant activity compared to free curcumin. These vesicles are potential mitochondria-targeting vehicles, thus representing a promising formulation and improved stability for mitochondria-targeting strategies.168
Some specifically modified DQAsomes have been engineered to deliver plasmid DNA to mitochondria as “DQAplexes”, a hybrid of DNA and DQAsomes.169 (Figure 3). The application of DQAsomes has been extended further to deliver mitochondria-specific chemotherapeutic drugs. This includes the use of the anticancer drug paclitaxel, which induces apoptosis and ultimately cell death.170 This technique has revolutionized mitochondrial gene-therapy protocols, as the preparation of DQAsome–DNA complexes is quite efficient and simple.169 Plasmid DNA is first coupled with mitochondrial homing sequences for mitochondrial delivery only.171 It has been reported that after harvested mitochondrial contact with DQAplexes, DNA gets released quickly and escapes endosomes.172
Polymeric Nanoparticles
Polymeric NPs are formed from poly(glycolic acid), polylactic acid, polycaprolactone, or polylactic-co-glycolic acid (PLGA). These NPs are efficient biocompatible and biodegradable polymers and promising drug carriers.173 They can encapsulate both hydrophilic and hydrophobic drugs.173 A special type of polymeric NP prepared from polycaprolactone modified with PEG and TPP and self-assembled into micelles with a diameter 38–60 nm showed CoQ10-loading efficiency of almost 9.5%. These micelles were efficiently loaded and accumulated in mitochondria.
In another study, TPP-modified PLGA-PEG and PLGA-COOH NPs were prepared and their size, potential, and stability optimized. Those with diameter <100 nm and potential >22 mV were efficiently taken up by mitochondria. For clinical application, they were loaded with four drugs: 2,4-dinitrophenol (mitochondrial decoupler), curcumin (amyloid-β protein inhibitor for Alzheimer’s disease), α-tocopheryl succinate (tumor targeting drug), and lodamine (mitochondrial glycolysis inhibitor).174 These NPs improved the therapeutic activity of 2,4-dinitrophenol and decreased the amyloid-β–mediated cytotoxicity.175
In other research, thioketal linker-modified camptothecin (Cpt) was conjugated with PEGylated TPP to form (TL-Cpt-PEG1K-TPP) and blended with DSPEPEG-NH2. This led to the synthesis of photodynamic and chemosensitive dual-function NPs loaded with the photosensitizer molecule as zinc phthalocyanine (ZnPc). These nanoformulations were irradiated by 633 nm laser to produce ROS by thioketal linker rupture, thus releasing Cpt. This led to increased antitumor efficiency against lung cancer. This demonstrates that TL-Cpt-PEG1K-TPP guided the development of mitochondria-targeting in malignant cells with sixfold the cytotoxic activity of free ZnPc and Cpt in NCI-H460 cells.176
Another study used chitosan NPs functionalized with TPP and loaded with Dox. These NPs showed increased antitumor efficiency in A549 and HeLa cells.177 In parallel, PEG-TPP was linked with a disulfide bond. This polymer self-assembled and led to the formation of a hydrophobic TPP core and a hydrophilic PEG shell. Dox was loaded within these NPs and endocytosed by specific cells. Within the cells, glutathione broke the disulfide bond between mPEG and TPP, so removing the mPEG shell, exposing TPP, and driving Dox directly to mitochondria. These results further demonstrated that Dox-encapsulated mPEG-TPP NPs possessed enhanced mitochondria-targeting efficacy and improved therapeutic activity compared to other nonbioreducible NPs.178
Dendrimers
The new investigational drugs for the treatment of an increasing number of hematological cancers still have a poor record. Healthcare professionals and researchers are working intensively to find an effective therapy against chronic lymphocytic leukemia.179 Currently, personalized and targeted therapies with active compounds in nanoformulations capable of center-point targeting of cancer cells are the most favorable trends in oncology.180 To date, among the different studies on NPs, dendrimers have demonstrated strong potential in pharmacological applications, and look to become a milestone achievement in oncology and nanomedicine.181,182
Dendrimers are synthetic, hyperbranched macromolecules possessing three components, ie, a central core, repeated branches, and a surface with a controlled number of available groups to load multiple functionalities. The core and branched space is used for biomolecular entrapment, and surface functionality is used to integrate different moieties. These special properties brand dendrimers as multipurpose pharmaceutical nanocarriers.183 They have potential in biomedical applications, and are used as drug carriers184 and gene-transfection vectors,185 as well as in magnetic resonance imaging (MRI) detection.186 The nanometric size of these NPs facilitates their specific and effective interaction with cellular components like proteins, nucleic acids, membranes, and organelles.187 Dendrimers with a generation number greater than five and higher positive charges due to lipophilic cationic molecules like TPP and rhodamine can be engineered. These NPs have the potential for endosomal escape and can deliver chemotherapeutic drugs directly to mitochondria.
Some of the most widely used dendrimers include polypropyleneimine (PPI) and polyamidoamine (PAMAM). These dendrimers are toxic, owing to their positively charged surfaces.188 Proper surface modification is the best way to minimize their toxicity. PPI dendrimers have been modified with maltose and maltotriose sugar residues, and these semi-modified open-shell (OS) PPI dendrimers (PPI-G4-OS) are lethal to selected cancer cells like CEM-SS, MEC1, and U87.189 In comparison to this, their fully modified dense-shell (DS) counterparts (PPI-G4-DS) show relatively weaker or no such effects. Furthermore, neutral DS and cationic OS PPI glycodendrimers have been utilized as stabilization and transfection agents for different particles.190 Third-generation cationic PPI glycodendrimers with open maltotriose shells (PPI-Mal-IIIG3) have been used for the transfection of AuNP conjugated with turbo green fluorescent protein (mitoTGFP) against selective mitochondria targeting of JIMT1 cancer cells. This facilitation of AuNPs by PPI dendrimers led to mitochondrial rupture, triggering apoptosis.191
PAMAM dendrimers are significantly used as a platform for the delivery of genomic materials and drugs.192 PAMAM-based G(5)-D-Ac-TPP dendrimers have been designed for mitochondria targeting in drug delivery.193 For monitoring intracellular localization, these NPs are labeled with a fluorescent dye, and less cytotoxicity has been reported with these nanocarriers. A parallel strategy has been used to deliver the luciferase gene and EGFP within the COS7 and HeLa cells by utilizing TPP-conjugated PAMAM dendrimers (G5-TPP).194 Under the transfection condition, these dendrimers have been reported to be atoxic. The G5-TPP dendrimer platform demonstrates efficient DNA packing and unpacking, endosomal escape, and efficient Mitochondria-targeting genome and drug delivery.
Carbon Nanotubes
Multiwalled carbon nanotubes (MWCNTs) have been used as anticancer delivery vehicles by surface functionalization with mitochondria-specific ligands. A novel mitochondria-targeted peptide sequence (MTS) with a primary structure of KMSVLTPLLLRGLTGSARRLPVPRAKC has been tagged on MWCNT surfaces to attain efficient mitochondria-specific drug delivery. With the help of confocal microscopy, these nanocarriers have been found to accumulate extensively in HeLa cells and macrophage mitochondria. Mitochondria targeting of these NPs has been further confirmed by transmission electron microscopy (TEM). Further, these NPs have not been reported to possess any significant toxicity, so are potential candidates as effective mitochondria-targeted drug-delivery systems.195 In this vista, mitochondrial-targeting has also been achieved by cationic rhodamine- 110 (MWCNT-ρ) and fluorescein (MWCNT-Fluo) used as an untargeted control.196 MWCNT-ρ has also been used to entrap a platinum prodrug (PtBz), which presented enhanced potency and efficient mitochondrial localization.
Inorganic Nanoparticles
Inorganic NPs cover a broad range of substances, including elemental metals, metal oxides, and metal salts. Inorganic NPs have been utilized as mitochondria-targeting agents, as these form uniform and smaller NPs. Among these, hydroxyapatite (HAp; Ca10(PO4)6(OH)2), displays outstanding drug-loading capacity and biocompatibility. It has been reported that HApNPs enter tumor-cell mitochondria and induce apoptosis by disturbing MMP, causing leakage of cytochrome c (cyt c).197 Rod-shaped HApNPs have been engineered with about 50 nm length and almost 10 nm width, and are engulfed by caveolate-mediated endocytosis by normal bronchial epithelial cells (16HBE) and lung cancer cells (A549). Interestingly, it has been further reported that A549 lung cancer cells engulf more NPs, causing sustained rise in Ca2+ concentration compared to 16HBE normal cells. This property of specific cell and mitochondria targeting causes increased Ca2+ concentration, resulted in almost 40% cancer-growth inhibition even without a drug, in lung cancer in nude mice.198 Anticancer efficacy was furthered with Dox-loaded HApNPs and coating with hyaluronic acid (HA). This nanoformulation specifically targets CD44-overexpressing cancer cells and overcomes the burst drug release. It has been found that Dox-loaded HAp-HA NPs exhibit almost four- to sevenfold the cytochrome c (cyt c) release of free Dox under similar conditions.199
Metallic NPs are emerging as innovative drug carriers and contrast agents for the treatment of different cancers. Metallic NPs are routinely used as site-specific targeting, drug delivery, and imaging of different tumor cells.200 Metal and metal oxide NPs can be precisely synthesized and modified with different functional groups. The novel functionalization of these metallic NPs helps in conjugating them with various mitochondria-specific moieties for use in specific cancer treatment. Some common metallic NPs are explained in the following sections to understand their significance for mitochondria targeting and cancer management.
Gold Nanoparticles
In addition to other metallic NPs, gold NPs (AuNPs) have been demonstrated to accumulate within mitochondria and trigger apoptosis after internalization by cells.201 AuNPs have been conjugated with GFP and tagged at the amino terminus with a mitochondrial localization sequence of the IMM protein COX8. To overcome the aggregation of AuNPs, these nanoformulations were altered with cationic maltotriose–amended polypropyleneimine dendrimers to coat mitTGFP-AuNPs. However, for proper transfection, this nanoformulation (mitoTGFP-AuNPs) required a cationic glycodendrimer (PPI-Mal-III G3) for traversing the plasma membrane. These NPs quite successfully escaped early endosome formation, efficiently ruptured the OMM, and finally got localized within the IMM. This resulted in cytochrome c (cyt c) release that triggered apoptosis.202 In a similar fashion, multilayered polypeptides were used to surround the AuNPs. The first layer used was a CALNN-based peptide to avoid the aggregation of AuNPs. The second layer was tetrameric streptavidin, a linker to join biotinylated molecules. The outermost layer was a biotinylated peptide (KLA:(KLAKLA)2), which possessed both the mitochondriotropic agent and cytotoxic peptide to kill the cancer cells. These KLA-tagged AuNPs possessed thousands of times the antitumor activity of free KLA peptide. KLA peptide is well recognized for its efficiency in cell entry and mitochondrial specificity.203
Titanium Dioxide Nanoparticles
TiO2NPs have stronger catalytic activity and have been widely used for different applications.204 These raise some concerns about adverse health effects, as they are smaller particles with larger surface area.205 Significant associations have been found between metabolic stress, inflammatory response, and ROS production and treatment with TiO2NPs in brains of mice.206
TiO2NPs can concentrate in the brain after crossing the BBB, thereby resulting in infiltration of inflammatory cells and apoptosis of hippocampus cells. This leads to a decrease in cognitive brain functioning.207 ROS generation damages the cell membrane, which further facilitates the entry of TiO2NPs, activating signaling pathways involved in oxidative stress. To check oxidative stress, expression ofNRF2 is very important. The association between oxidative stress and p38, JNK, and MAPK cascade is well established.206 In addition to this, TiO2NPs induce apoptosis, alter the immune system, and works as a secondary messenger for some intracellular signaling cascades. TiO2NP-mediated enhanced ROS production may also be related to p38–NRF2 signaling pathways during brain injury.208
Silver Nanoparticles
Silver NPs (AgNPs) have been widely used in chemical, antimicrobial, household, and medical applications.209 AgNP composition, size, shape, charge, and solubility affect their ability to bind with biological sites. The cytotoxicity of AgNPs is mainly related to cell-membrane destruction, which leads to mitochondrial destruction.210 These NPs usually induce oxidative stress, which is the major reason for their toxicity.211 AgNPs also deplete the antioxidant defense system, leading to enhanced ROS accumulation, which initiates the inflammatory response, and the destruction of mitochondria.212,213 The perturbation of mitochondria also leads to cytochrome c (cyt c) release and apoptosis as the final outcome. AgNPs also exhibit toxicity toward mammalian and HEPG2 cells by reducing MMP, DNA damage, and mediating apoptosis.214 In addition, this perturbation also leads to changes in the mitochondrial respiratory chain, dynamics, biogenesis, and autophagy control.215
Zinc Oxide Nanoparticles
ZnONPs are used in biomedical imaging and, fungicides and as anticancer drugs and antimicrobial agents.216 The toxicity of these NPs has been mainly related to the production of ROS, which leads to oxidative stress, inflammation, and DNA and protein modifications. The oxidative stress also leads to lipid peroxidation and apoptosis through the p38 and p53 pathways.217 The ROS production also leads to the activation of MAPK pathway, which regulates different cellular pathways.218
In one study, ZnONPs at 14–20 µg/mL exposed to HEPG2 cells for 12 hours induced apoptosis-mediated reduced cell viability. The cell-viability decline was due to oxidative stress–mediated DNA damage and decreased MMP. Furthermore, these NPs increased the ratio of BAX:Bcl2, which led to the induction of apoptotic pathways. ZnONPs also activated p38 and JNK pathways and induced the phosphorylation of p53 Ser15 residues.219
Various investigations support the role of ZnONPs in mitochondria-mediated toxicity induction in experimental animal studies and in vitro models.220 These NPs trigger excessive ROS production in zebrafish embryos by reducing MMP and inducing mitochondria-mediated apoptosis.221 Further, they decrease mitochondrial density by disrupting biogenesis, inhibit the PGC1α pathway, and interfere with mtDNA number control. PGC1α plays a significant role in mitochondrial biogenesis regulation by interaction with downstream targets like TFAM, which helps in transcription of some genes. This factor also plays an important role in controlling mitochondrial oxidative stress by the activation of manganese superoxide dismutase.222
Iron Oxide Nanoparticles
FeONPs have been used for cell labeling, gene delivery, and drug targeting and as hyperthermia-therapy agents. These NPs are also good contrast agents in magnetic resonance imaging.223 FeONPs induce such cellular responses as cell activation, ROS production, and cell death.224 In addition, FeONPs cause mitochondrial damage, though these NPs are not targeted for this organelle.225 Mitochondria are the principal source of ROS generation, and prolonged action initiates oxidative stress, which leads to activation of transcription factors and some inflammation-responsible genes like AP1 and NFKB.
Magnetic composite NPs for dual modal photothermal therapy and photodynamic therapy have been used to enhanced cancer therapeutic effect by mitochondria targeting. These composite NPs have the capacity to generate heat and ROS simultaneously upon NIR-laser irradiation. After surface modification of targeting ligands, they have been selectively delivered to mitochondria to amplify the cancer-cell apoptosis promoted by hyperthermia and cytotoxic ROS.226
Lung cancer cells have been reported to increase their ROS production after exposure to FeONPs. This increased production is blocked by N-acetyl cysteine (NAC), which results in significantly decreased cell death. In addition, this exposure also leads to decreased conversion of LC3-I to LC3-II within cancer cells pretreated with f-NAC. Therefore, FeONPs likely induce ROS production and autophagy-mediated cell death. These lethal consequences are likely due to mitochondrial damage caused by FeONPs, as MMP is significantly reduced as well.227
FeONPs have been probed with P13/Akt, and it was observed that they activated the classical AMPK–mTOR–Akt signaling cascades in lung cancer cells. The involvement of AMPK phosphorylation during autophagic cell death has been fully confirmed by pretreating the cells with the AMPK-phosphorylation inhibitor compound C.228 However, the direct role of FeONPs on AMPK- and mTOR-mediated autophagy and cell death with the help of certain inhibitors is still under observation. It has been observed that FeONPs possess autophagic potential by activating the classical pathway for autophagy induction.229 Some more examples of drug-loaded nanocarriers targeting mitochondria with the aid of varied targeting moieties are briefly listed in Table 3.
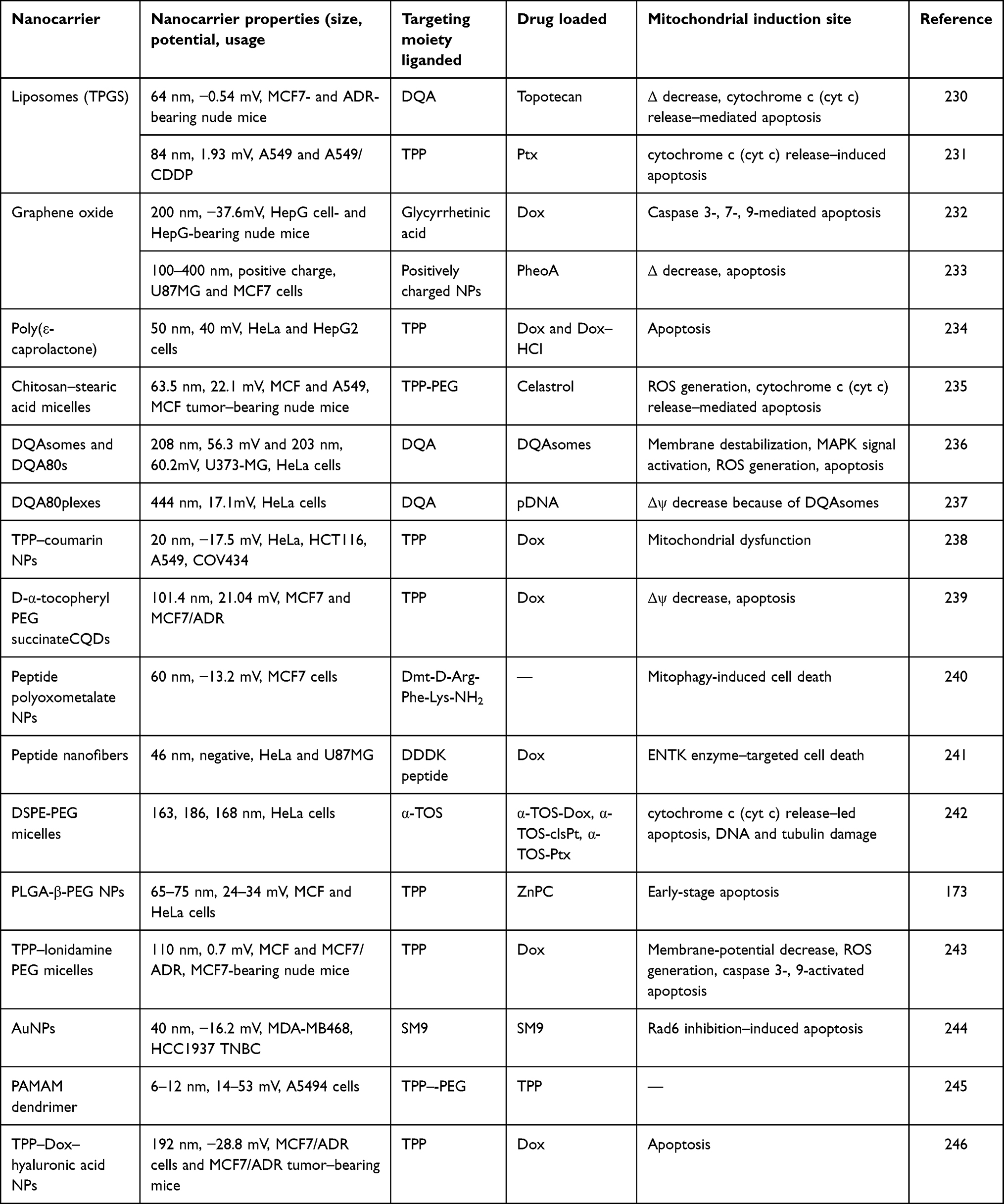 |
Table 3 Drug-loaded nanocarriers tagged with various mitochondriophilic ligands, inducing different mitochondrial functional irregularities |
Limitations of Native and Nanoformulation-Based Drugs
Using native drugs poses a number of challenges before reaching the final target of action. Healthcare researchers are working hard to design the drugs that can be specifically transported to the site of action while minimizing unwanted buildup in untargeted normal tissue. NPs can have different routes of administration like respiratory tract, skin, and parenteral administration to reach the actual target.247 Properties that can give rise to unexpected toxicities should be equally noted.248 In the blood, some drug nanoformulations can lead to the formation of protein corona while in contact with plasma proteins. The protein corona may consist of dozens to hundreds of proteins that can alter the physicochemical properties of NPs like morphology, size, aggregation, and -potential.249
The toxicity potential of cationic NPs like polystyrene and AuNPs can lead to clotting and hemolysis, whereas the toxicity potential of anionic NPs is considerably less.250 NP use leads to some basal toxicities, due to disruption of host homeostasis that leads to ROS production.251 Enhanced ROS can promote genome damage and micronuclei formation. AgNPs of 15 nm and amorphous TiO2 NPs of 30 nm in size induce the highest ROS generation. The possible engulfment of quantum dots and AgNPs by macrophages can lead to the expression of inflammatory mediators like IL1β, TNFα, MIP2, irrespective of their size.252 The chronic inflammation by ROS producing NPs can lead to the development of pulmonary diseases, atherosclerosis, or even cancer. Other NPs can affect calcium homeostasis, thus affecting cellular metabolism, signal transduction, and gene expression. Dissociated ions from metallic NPs can even prove to be more toxic, so NPs of biodegradable polymers can be beneficial to use.253,254 Single- or multiwalled CNTs induce the aggregation of platelets, while their building-block C60 fullerenes do not. All NPs tend to accumulate in the liver, and the mechanism of their elimination from the body needs to be investigated.247
Clinical Applications, Trials, and Phases of Mitochondria-Targeted Nanomedicine
NPs have been extensively studied in theranostic clinical applications, and several formulations have been approved by the US Food and Drug Administration and European Medicines Agency for clinical applications in patients with cancer.255 The formulation of cancer nanomedicines is mainly based on liposomes (eg, Doxil, Vyxeos, and Onivyde), polymeric micelles (eg, NK105, Genexol, and NC6004), albumin (eg, Abraxane), or inorganic NPs (eg, NBTXR3 and NanoTherm). Although most current cancer nanomedicines are administered intravenously for systemic delivery to tumors, some nanomedicine formulations (eg, NanoTherm and NBTXR3) have been designed for intratumoral administration.256 A brief overview of some major approaches of nanomedicine aiming to modulate the TCA cycle, ETC, anaplerosis, mtROS, and mitochondria-driven apoptosis in cancer cells is presented in Table 4. The table also highlights clinical phase stages and the clinical identifier numbers of molecular targets and the drugs used against these targets.
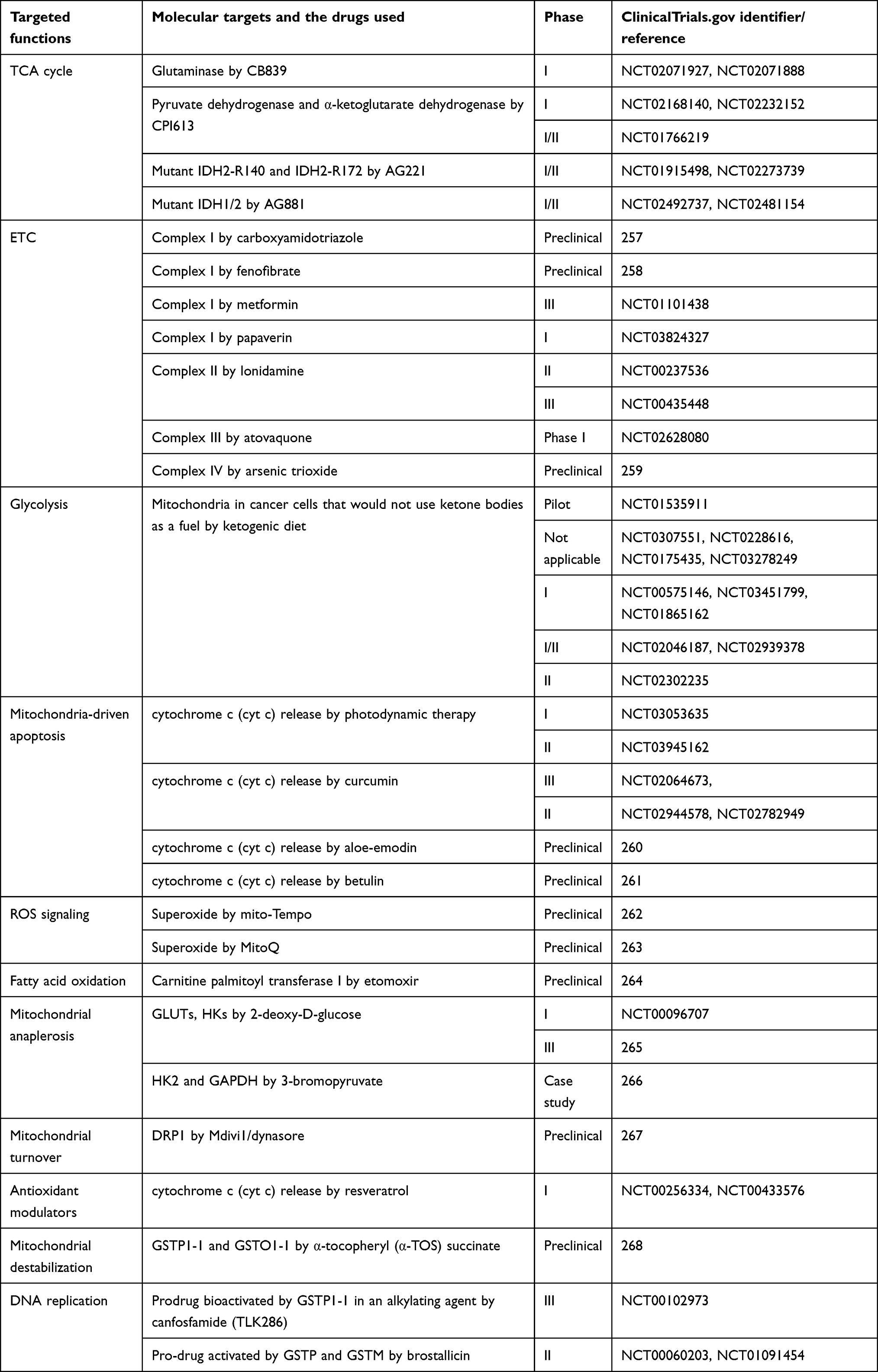 |
Table 4 Clinical perspectives (phases and trials) of some Mitochondria-targeted therapeutic strategies for cancer management |
Prospects of Mitochondria Targeting and Cancer Management
The strategy of direct therapeutic action by targeting mitochondria will dramatically decrease the side effects of a particular drug at aspecific locations, and is the ultimate goal of future therapeutics. Nanomedicine faces tremendous challenges due to the diverse nature of biological systems. The advantage of engineering multidimensional features within NPs for specific targeting to diseased cells and enhanced accumulation in particular organelles has drastically revolutionized therapeutic strategies, where mitochondrial dysfunction plays a central role.
NPs like liposomes, micelles, CNTs, and dendrimers tagged with specific mitochondria-targeting moieties have demonstrated their existence as novel delivery means. However, more comprehensive research is necessary to properly understand the safety aspects of drug nanoformulations when used in human subjects. For successful mitochondrial targeting, the characteristics of these theranostic NPs should be highly efficient to achieve their goals. Some novel characteristics include tumor-cell and tissue specificity, long circulation time in blood, and large accumulation within cancer-cell mitochondria.
Some types of currently used mitochondria-targeting nanoformulations suffer from certain drawbacks. For example, delocalized lipophilic cations accumulate efficiently in mitochondria because of negative mitochondrial membrane potential; however, they mediate intrinsic toxicity, which limits their clinical applications. In addition, other targeting ligands like peptides have bulky structures, solubility issues, poor membrane permeability, and very low stability in serum. To overcome these limitations, in-depth research is necessary to engineer such targeting ligands properly to make them clinically more useful as drug-loaded mitochondria-targeting agents.
It is a very challenging task to engineer nanoformulations that can perfectly target mitochondrial abnormalities in tumor cells without toxicity to nearby normal cells. To solve this challenge, different physicochemical factors of nanoformulations have been considered, which include shape, size, charge, membrane potential, tumor-cell specificity, andcombinations thereof.
The endosomal escape ability of theranostic nanoformulations is of utmost importance in enhancing their mitochondria-targeting abilities. For the production of effective mitochondria-targeting NPs, these nanoformulations should be equipped with endosomolytic features. Furthermore, as NIR photosensitizers enable the imaging of NIR fluorescence, photothermal signals, and photoacoustic signals, the corresponding diagnostic materials should be considered for other imaging tools like CT, PET, MRI, and SPECT.
Despite the success of these novel nanoformulations in in vitro studies, thorough and logical preclinical and clinical studies are obligatory to achieve their potential use in clinical settings. At present, there is a big gap in understanding the safety aspects unique to specific nanoformulations when used in varied systems. These of nanoformulations need to be properly addressed when targeting mitochondria of diseased cells only. Enormous efforts are required for the development of targeted nanoformulations. It is extremely difficult to use nanoformulations unless full understanding and characterization of them are achieved.
Conclusion
The direct mitochondria-targeting approach within cancer cells is a current focused to enhance therapeutic strategies, and has gained momentum in the last decade. The goal is to design nanoformulations that can show minimum off-target and side effects. Mitochondria of cancer cells have unique features, which are novel targets of different theranostic NPs as a therapeutic strategy. These novel targets include oxidative phosphorylation site, TCA cycle, glutamine metabolism, and mitochondrial dynamics and trafficking. The direct conjugation of anticancer drugs with mitochondria-targeting ligands (eg, TPP, DQA, and MPP) has been able to solve some complications like use of larger doses and drug resistance. This problem has been solved to some extent by the use of nanoformulations (eg, liposomes, Mito-Porter, and CNTs), but still there are so many other challenges like toxicity complications and center-point targeting that need to be sorted out. Despite the primary success of these nanoformulations, systematic preclinical and clinical investigations are obligatory before their actual use in clinical settings. At present, there is a lack of thorough understanding regarding safety concerns, which limits their use as nanomedicine. The safety aspects of these nanoformulations need to be properly addressed through appropriate safeguards. The prerequisite of thorough understanding of the mitochondrial role in cancer progression, employment of proper regulatory procedures, and advancements in nanoformulation technology will definitely boost cancer treatment in the near future.
Acknowledgment
We are thankful to all the researchers who have contributed immensely to the field of cancer biology by using mitochondria-specific drug nanoformulations as a strategy in cancer therapy.
Funding
The researchers would like to thank the Deanship of Scientific Research, Qassim University for funding the publication of this project.
Disclosure
The authors declare no conflicts of interest.
References
1. Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi:10.1146/annurev-genet-110410-132529
2. Hoitzing H, Johnston IG, Jones NS. What is the function of mitochondrial networks? A theoretical assessment of hypotheses and proposal for future research. Bioessays. 2015;37(6):687–700. doi:10.1002/bies.201400188
3. Rai PK, Russell OM, Lightowlers RN, Turnbull DM. Potential compounds for the treatment of mitochondrial disease. Br Med Bull. 2015;116:5–18. doi:10.1093/bmb/ldv046
4. Xia MF, Zhang YZ, Jin K, Lu ZT, Zeng Z, Xiong W. Communication between mitochondria and other organelles: a brand‑new perspective on mitochondria in cancer. Cell Biosci. 2019;9:27. doi:10.1186/s13578-019-0289-8
5. Gorman GS, Chinnery PF, DiMauro S, et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2(1):1–22. doi:10.1038/nrdp.2016.80
6. Solaini G, Sgarbi G, Baracca A. Oxidative phosphorylation in cancer cells. Biochim Biophys Acta. 2011;1807(6):534–542. doi:10.1016/j.bbabio.2010.09.003
7. Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015;3(1):1–10. doi:10.1186/s40170-015-0128-2
8. Herst P, Berridge M. Plasma membrane electron transport: a new target for cancer drug development. Curr Mol Med. 2006;6:895–904. doi:10.2174/156652406779010777
9. Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166:555–566. doi:10.1016/j.cell.2016.07.002
10. Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell. 2016;61(5):667–676. doi:10.1016/j.molcel.2016.02.011
11. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–530. doi:10.1085/jgp.8.6.519
12. Reichert AS, Neupert W. Mitochondriomics or what makes us breathe. Trends Genet. 2004;20(11):555–562. doi:10.1016/j.tig.2004.08.012
13. Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698. doi:10.1038/nrc3365
14. Hasnat M, Yuan Z, Naveed M, et al. Drp1-associated mitochondrial dysfunction and mitochondrial autophagy: a novel mechanism in triptolide-induced hepatotoxicity. Cell Biol Toxicol. 2019;35:267–280. doi:10.1007/s10565-018-9447-8
15. Cipolat S, de Brito M, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101:15927–15932. doi:10.1073/pnas.0407043101
16. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi:10.1074/jbc.M503062200
17. Guha M, Avadhani NG. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion. 2013;13:577–591. doi:10.1016/j.mito.2013.08.007
18. Tu C, Zeng Z, Qi P, et al. Identification of genomic alterations in nasopharyngeal carcinoma and nasopharyngeal carcinoma-derived Epstein-Barr virus by whole-genome sequencing. Carcinogenesis. 2018;39:1517–1528. doi:10.1093/carcin/bgy108
19. Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013;8:2003–2014. doi:10.3969/j.issn.1673-5374.2013.21.009
20. Rigoulet M, Yoboue ED, Devin A. Mitochondrial ROS generation and its regulation: mechanisms involved in H2O2 signaling. Antioxid Redox Signal. 2010;14:459–468. doi:10.1089/ars.2010.3363
21. Shimizu S, Kanaseki T, Mizushima N, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi:10.1038/ncb1192
22. Maiuri MC, Zalckvar E, Kimchi A, et al. Selfeating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi:10.1038/nrm2239
23. Dewson RM, Kluck RM. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci. 2009;122:2801–2808. doi:10.1242/jcs.038166
24. Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283(5407):1482–1488. doi:10.1126/science.283.5407.1482
25. Kumari S, Badana AK, G MM, et al. Reactive oxygen species: a key constituent in cancer survival. Biomark Insights. 2018;13:1177271918755391. doi:10.1177/1177271918755391
26. Galvan DL, Green NH, Danesh FR. The hallmarks of mito-chondrial dysfunction in chronic kidney disease. Kidney Int. 2017;92(5):1051–1057. doi:10.1016/j.kint.2017.05.034
27. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48(2):158–167. doi:10.1016/j.molcel.2012.09.025
28. Arakawa S, Nakanomyo I, Kudo-Sakamoto Y, Akazawa H, Komuro I, Shimizu S. Identification of a novel compound that inhibits both mitochondria-mediated necrosis and apoptosis. Biochem Biophys Res Commun. 2015;467(4):1006–1011. doi:10.1016/j.bbrc.2015.10.022
29. Shimizu S, Narita M, Tsujimoto Y, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochon-drial channel VDAC. Nature. 1999;399(6735):483–487. doi:10.1038/20959
30. Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22(2):263–268. doi:10.1016/j.ceb.2009.12.003
31. Gaude E, Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014;2:1–9. doi:10.1186/2049-3002-2-10
32. Muzza M, Colombo C, Cirello V, Perrino M, Vicentini L, Fugazzola L. Oxidative stress and the subcellular localization of the te-lomerase reverse transcriptase (TERT) in papillary thyroid cancer. Mol Cell Endocrinol. 2016;431:54–61. doi:10.1016/j.mce.2016.05.005
33. Giampazolias E, Tait SW. Mitochondria and the hallmarks of cancer. Fed Eur Biochem Soc J. 2016;283:803–814.
34. Basak NP, Banerjee S. Mitochondrial dependency in progression of acute myeloid leukemia. Mitochondrion. 2015;21:41–48. doi:10.1016/j.mito.2015.01.006
35. Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res. 2018;28:265–280. doi:10.1038/cr.2017.155
36. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. 2018;24:2482–2490. doi:10.1158/1078-0432.CCR-17-3070
37. Griguer CE, Oliva CR, Gillespie GY. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J Neuro Oncol. 2005;74:123–133. doi:10.1007/s11060-004-6404-6
38. Guppy M, Leedman P, Zu XL, Russell V. Contribution by dierent fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J. 2002;364:309–315. doi:10.1042/bj3640309
39. Hirpara J, Eu JQ, Tan JKM, et al. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 2019;25:101076. doi:10.1016/j.redox.2018.101076
40. Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3:193–202. doi:10.1038/nrc1013
41. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi:10.1056/NEJMoa0808710
42. Todisco S, Convertini P, Iacobazzi V, Infantino V. TCA cycle rewiring as emerging metabolic signature of hepatocellular carcinoma. Cancers. 2019;12:68. doi:10.3390/cancers12010068
43. Dekker LJM, Wu S, Jurriens C, et al. Metabolic changes related to the idh1 mutation in gliomas preserve TCA-cycle activity: an investigation at the protein level. Fed Am Soc Exp Biol J. 2020;3646–3657.
44. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi:10.1038/nrd4002
45. Bezawork-Geleta A, Rohlena J, Dong L, Pacak K, Neuzil J. Mitochondrial complex II: at the crossroads. Trends Biochem Sci. 2017;42:312–325. doi:10.1016/j.tibs.2017.01.003
46. Hecht F, Pessoa CF, Gentile LB, Rosenthal D, Carvalho DP, Fortunato RS. The role of oxidative stress on breast cancer development and therapy. Tumour Biol. 2016;37:4281–4291. doi:10.1007/s13277-016-4873-9
47. Valko M, Rhodes CJ, Moncol JM, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40.
48. Zhang J, Wang X, Vikash V, et al. ROS and ROS-mediated cellular signaling. Oxidative Med Cell Longev. 2016;2016:4350965. doi:10.1155/2016/4350965
49. Grandemange S, Herzig S, Martinou JC. Mitochondrial dynamics and cancer. Semin Cancer Biol. 2009;19:50–56. doi:10.1016/j.semcancer.2008.12.001
50. Anderson GR, Wardell SE, Cakir M, et al. Dysregulation of mitochondrial dynamics proteins are a targetable feature of human tumors. Nat Commun. 2018;9:1677. doi:10.1038/s41467-018-04033-x
51. Yu MF, Nguyen ND, Huang YQ, et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. J Clin Investig Insight. 2019;4:e126915.
52. Simula L, Nazio F, Campello S. The mitochondrial dynamics in cancer and immune-surveillance. Semin Cancer Biol. 2017;47:29–42. doi:10.1016/j.semcancer.2017.06.007
53. Tanwar DK, Parker DJ, Gupta P, et al. Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget. 2016;7:60021–60037. doi:10.18632/oncotarget.11047
54. Qian W, Choi S, Gibson GA, Watkins SC, Bakkenist CJ, Van Houten B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes atm-dependent g2/m arrest and aneuploidy through DNA replication stress. J Cell Sci. 2012;125:5745–5757. doi:10.1242/jcs.109769
55. Zhao J, Zhang J, Yu M, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32:4814–4824. doi:10.1038/onc.2012.494
56. Wan YY, Zhang JF, Yang ZJ, et al. Involvement of Drp1 in hypoxia-induced migration of human glioblastoma u251 cells. Oncol Rep. 2014;32:619–626.
57. Maycotte P, Marin-Hernandez A, Goyri-Aguirre M, Anaya-Ruiz M, Reyes-Leyva J, Cortes-Hernandez P. Mitochondrial dynamics and cancer. Tumour Biol. 2017;39:1010428317698391. doi:10.1177/1010428317698391
58. Leboucher GP, Tsai YC, Yang M, et al. Stress-induced phosphorylation and proteasomal degradation of Mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell. 2012;47:547–557. doi:10.1016/j.molcel.2012.05.041
59. Gammage PA, Frezza C. Mitochondrial DNA: the overlooked oncogenome? BMC Biol. 2019;17(1). doi:10.1186/s12915-019-0668-y
60. Jakupciak JP, Maragh S, Markowitz ME, et al. Performance of mitochondrial DNA mutations detecting early stage cancer. BMC Cancer. 2008;8:285. doi:10.1186/1471-2407-8-285
61. Habano W, Nakamura S, Sugai T. Microsatellite instability in the mitochondrial DNA of colorectal carcinomas: evidence for mismatch repair systems in mitochondrial genome. Oncogene. 1998;17(15):1931–1937. doi:10.1038/sj.onc.1202112
62. Mithani SK, Smith IM, Zhou S, et al. Mitochondrial resequencing arrays detect tumor-specific mutations in salivary rinses of patients with head and neck cancer. Clin Cancer Res. 2007;13(24):7335–7340. doi:10.1158/1078-0432.CCR-07-0220
63. Jones JB, Song JJ, Hempen PM, Parmigiani G, Hruban RH, Kern SE. Detection of mitochondrial DNA mutations in pancreatic cancer offers a “mass”-ive advantage over detection of nuclear DNA mutations. Cancer Res. 2001;61(4):1299–1304.
64. Liu VW, Shi HH, Cheung AN, et al. High incidence of somatic mitochondrial DNA mutations in human ovarian carcinomas. Cancer Res. 2001;61(16):5998–6001.
65. Habano W, Sugai T, Nakamura SI, Uesugi N, Yoshida T, Sasou S. Microsatellite instability and mutation of mitochondrial and nuclear DNA in gastric carcinoma. Gastroenterology. 2000;118(5):835–841. doi:10.1016/S0016-5085(00)70169-7
66. Parrella P, Xiao Y, Fliss M, et al. Detection of mitochondrial DNA mutations in primary breast cancer and fine-needle aspirates. Cancer Res. 2001;61(20):7623–7626.
67. Moraes CT, DiMauro S, Zeviani M, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegian and Kearns-Sayre syndrome. N Engl J Med. 1989;320(20):1293–1299. doi:10.1056/NEJM198905183202001
68. Errichiello E, Venesio T. Mitochondrial DNA variations in tumors: drivers or passengers? Mitochondrial DNA New Insights. 2018.
69. Zhang L. Heme Biology: Heme Acts as a Versatile Signaling Molecule Regulating Diverse Biological Processes.
70. Hooda J, Cadinu D, Alam MM, et al. Enhanced heme function and mitochondrial respiration promote the progression of lung cancer cells. PLoS One. 2013;8(5):e63402. doi:10.1371/journal.pone.0063402
71. Hooda J, Shah A, Zhang L. Heme, an essential nutrient from dietary proteins, critically impacts diverse physiological and pathological processes. Nutrients. 2014;6:1080–1102. doi:10.3390/nu6031080
72. Dong L, Neuzil J. Targeting mitochondria as an anticancer strategy. Cancer Commun (Lond Engl). 2019;39:63. doi:10.1186/s40880-019-0412-6
73. Rohlenova K, Sachaphibulkij K, Stursa J, et al. Selective disruption of respiratory supercomplexes as a new strategy to suppress her2high breast cancer. Antioxid Redox Signal. 2017;26:84–103. doi:10.1089/ars.2016.6677
74. Dong LF, Jameson VJ, Tilly D, et al. Mitochondrial targeting of alpha-tocopheryl succinate enhances its pro-apoptotic ecacy: a new paradigm for eective cancer therapy. Free Radic Biol Med. 2011;50:1546–1555. doi:10.1016/j.freeradbiomed.2011.02.032
75. Huang Q, Chen Z, Cheng P, et al. LYRM2 directly regulates complex I activity to support tumor growth in colorectal cancer by oxidative phosphorylation. Cancer Lett. 2019;455:36–47. doi:10.1016/j.canlet.2019.04.021
76. Lim SC, Carey KT, McKenzie M. Anti-cancer analogues me-143 and me-344 exert toxicity by directly inhibiting mitochondrial NADH: ubiquinone oxidoreductase (complex I). Am J Cancer Res. 2015;5:689–701.
77. DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345–19350. doi:10.1073/pnas.0709747104
78. Yen K, Travins J, Wang F, et al. Ag-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7:478–493. doi:10.1158/2159-8290.CD-16-1034
79. Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol. 2017;24:1161–1180.
80. Anderson NM, Mucka P, Kern JG, Feng H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. 2018;9:216–237. doi:10.1007/s13238-017-0451-1
81. Han H, Jain AD, Truica MI, et al. Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell. 2019;36:483–497.e15. doi:10.1016/j.ccell.2019.10.001
82. O’Dwyer PJ, Alonso MT, Leyland-Jones B. Acivicin: a new glutamine antagonist in clinical trials. J Clin Oncol. 1984;2:1064–1071. doi:10.1200/JCO.1984.2.9.1064
83. Senft D, Ronai ZA. Regulators of mitochondrial dynamics in cancer. Curr Opin Cell Biol. 2016;39:43–52. doi:10.1016/j.ceb.2016.02.001
84. Peiris-Pages M, Bonuccelli G, Sotgia F, Lisanti MP. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (csc) signalling. Oncotarget. 2018;9:13254–13275. doi:10.18632/oncotarget.24285
85. Pan L, Zhou L, Yin W, Bai J, Liu R. miR-125a induces apoptosis, metabolism disorder and migration impairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission. Int J Oncol. 2018;53:124–136. doi:10.3892/ijo.2018.4380
86. Pan L, Zhou L, Yin W, Bai J, Liu R, Pan L. LATS2 promotes apoptosis in non-small cell lung cancer A549 cells via triggering m-dependent mitochondrial fission and activating the JNK signaling pathway. Biomed Pharmacother. 2019;109:679–689. doi:10.1016/j.biopha.2018.10.097
87. Tang H, Peng S, Dong Y, et al. MARCH5 overexpression contributes to tumor growth and metastasis and associates with poor survival in breast cancer. Cancer Manag Res. 2019;11:201–215. doi:10.2147/CMAR.S190694
88. Zhang X, Han S, Zhou H, et al. TIMM50 promotes tumor progression via ERK signaling and predicts poor prognosis of non-small cell lung cancer patients. Mol Carcinog. 2019;58:767–776. doi:10.1002/mc.22969
89. Liu D, Angelova A, Liu J, et al. Self-assembly of mitochondria-specific peptide amphiphiles amplifying lung cancer cell death through targeting the VDAC1-hexokinase-II complex. J Mater Chem B. 2019;7:4706–4716. doi:10.1039/C9TB00629J
90. Hu W, Tian H, Yue W, et al. Rotenone induces apoptosis in human lung cancer cells by regulating autophagic flux. IUBMB Life. 2016;68:388–393. doi:10.1002/iub.1493
91. DeWaal D, Nogueira V, Terry AR, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. 2018;9:
92. Nath K, Guo L, Nancolas B, et al. Mechanism of antineoplastic activity of lonidamine. Biochim Biophys Acta. 2016;1866:151–162. doi:10.1016/j.bbcan.2016.08.001
93. Hocsak E, Szabo V, Kalman N, et al. PARP inhibition protects mitochondria and reduces ROS production via PARP-1-ATF4-MKP-1-MAPK retrograde pathway. Free Radic Biol Med. 2017;108:770–784. doi:10.1016/j.freeradbiomed.2017.04.018
94. Ren YJ, Wang XH, Ji C, et al. Silencing of NAC1 expression induces cancer cells oxidative stress in hypoxia and potentiates the therapeutic activity of elesclomol. Front Pharm. 2017;8:804. doi:10.3389/fphar.2017.00804
95. Pignanelli C, Ma D, Noel M, et al. Selective targeting of cancer cells by oxidative vulnerabilities with novel curcumin analogs. Sci Rep. 2017;7:1105. doi:10.1038/s41598-017-01230-4
96. Kuo ML, Lee MB, Tang M, et al. PYCR1 and PYCR2 interact and collaborate with RRM2B to protect cells from overt oxidative stress. Sci Rep. 2016;6:18846. doi:10.1038/srep18846
97. Queiroz EA, Puukila S, Eichler R, et al. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One. 2014;9:e98207. doi:10.1371/journal.pone.0098207
98. Sassi N, Mattarei A, Azzolini M, et al. Mitochondria-targeted resveratrol derivatives act as cytotoxic pro-oxidants. Curr Pharm Des. 2014;20:172–179. doi:10.2174/13816128113199990034
99. Zhang C, Liu Z, Bunker E, et al. Sorafenib targets the mitochondrial electron transport chain complexes and ATP synthase to activate the PINK1-Parkin pathway and modulate cellular drug response. J Biol Chem. 2017;292:15105–15120. doi:10.1074/jbc.M117.783175
100. Mou J, Lin T, Huang F, Shi J, Chen H. A new green titania with enhanced NIR absorption for mitochondria-targeted cancer therapy. Theranostics. 2017;7:1531–1542. doi:10.7150/thno.17247
101. Belzacq AS, El Hamel C, Vieira HL, et al. Adenine nucleotide translocator mediates the mitochondrial membrane permeabilization induced by lonidamine, arsenite and CD437. Oncogene. 2001;20:7579–7587. doi:10.1038/sj.onc.1204953
102. Jan CI, Tsai MH, Chiu CF, Huang YP, Liu CJ, Chang NW. Fenofibrate suppresses oral tumorigenesis via reprogramming metabolic processes: potential drug repurposing for oral cancer. Int J Biol Sci. 2016;12:786–798. doi:10.7150/ijbs.13851
103. Shteinfer-Kuzmine A, Amsalem Z, Arif T, Zooravlov A, Shoshan-Barmatz V. Selective induction of cancer cell death by VDAC1-based peptides and their potential use in cancer therapy. Mol Oncol. 2018;12:1077–1103. doi:10.1002/1878-0261.12313
104. Kadavakollu S, Stailey C, Kunapareddy CS, White S. Clotrimazole as a cancer drug: a short review. Med Chem. 2014;4:722–724.
105. Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi:10.1016/S0092-8674(00)81959-5
106. Fichera GA, Fichera M, Milone G. Antitumoural activity of a cytotoxic peptide of Lactobacillus casei peptidoglycan and its interaction with mitochondrial-bound hexokinase. Anti Cancer Drugs. 2016;27:609–619. doi:10.1097/CAD.0000000000000367
107. Li W, Zheng M, Wu S, et al. Benserazide, a dopadecarboxylase inhibitor, suppresses tumor growth by targeting hexokinase 2. J Exp Clin Cancer Res. 2017;36:58. doi:10.1186/s13046-017-0530-4
108. Sun X, Zhang L. MicroRNA-143 suppresses oral squamous cell carcinoma cell growth, invasion and glucose metabolism through targeting hexokinase 2. Biosci Rep. 2017;37:BSR20160404. doi:10.1042/BSR20160404
109. Lu P, Bruno BJ, Rabenau M, Lim CS. Delivery of drugs and macromolecules to the mitochondria for cancer therapy. J Control Release. 2016;240:38–51. doi:10.1016/j.jconrel.2015.10.023
110. Khan AA, Allemailem KS, Almatroudi A, Almatroodi SA, Alsahli MA, Rahmani AH. Novel strategies of third level (organelle-specific) drug targeting: an innovative approach of modern therapeutics. J Drug Deliv Sci Technol. 2020;102315.
111. Dairkee SH, Hackett AJ. Differentialretentionofrhodamine123by breast carcinoma and normal human mammary tissue. Breast Cancer Res Treat. 1991;18:57–61. doi:10.1007/BF01975444
112. Cortes LA, Castro L, Pesce B, et al. Novel gallate triphenylphosphonium derivatives with potent antichagasic activity. PLoS One. 2015;10:e0136852. doi:10.1371/journal.pone.0136852
113. Wang H, Xu W. Mito-methyl coumarin, a novel mitochondria-targeted drug with great antitumor potential was synthesized. Biochem Biophys Res Commun. 2017;489:1–7. doi:10.1016/j.bbrc.2017.05.116
114. Zielonka J, Joseph J, Sikora A, et al. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev. 2017;117(15):10043–10120. doi:10.1021/acs.chemrev.7b00042
115. Millard M, Gallagher JD, Olenyuk BZ, Neamati N. A selective mitochondrial-targeted chlorambucil with remarkable cytotoxicity in breast and pancreatic cancers. J Med Chem. 2013;56:9170–9179. doi:10.1021/jm4012438
116. Christman JE, Miller DS, Coward P, Smith LH, Teng NN. Study of the selective cytotoxic properties of cationic, lipophilic mitochondrial-specific compounds in gynecologic malignancies. Gynecol Oncol. 1990;39:72–79. doi:10.1016/0090-8258(90)90402-7
117. Weissig V, Lasch J, Erdos G, Meyer HW, Rowe TC, Hughes J. DQAsomes: a novel potential drug and gene delivery system made from dequalinium TM. Pharm Res. 1998;15:334–337. doi:10.1023/A:1011991307631
118. Song YF, Liu DZ, Cheng Y, et al. Dual subcellular compartment delivery of doxorubicin to overcome drug resistant and enhance antitumor activity. Sci Rep. 2015;5:16125. doi:10.1038/srep16125
119. He H, Li DW, Yang LY, et al. A novel bifunctional mitochondria-targeted anticancer agent with high selectivity for cancer cells. Sci Rep. 2015;5:13543. doi:10.1038/srep13543
120. Horton KL, Stewart KM, Fonseca SB, Guo Q, Kelley SO. Mitochondria-penetrating peptides. Chem Biol. 2008;15:375–382. doi:10.1016/j.chembiol.2008.03.015
121. Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:601–619. doi:10.1089/ars.2007.1892
122. Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther. 2014;96(6):672–683. doi:10.1038/clpt.2014.174
123. Jiang L, Li L, He X, et al. Overcoming drug-resistant lung cancer by paclitaxel loaded dual-functional liposomes with mitochondria targeting and pH-response. Biomaterials. 2015;52:126–139. doi:10.1016/j.biomaterials.2015.02.004
124. Li L, Geisler I, Chmielewski J, et al. Cationic amphiphilic polyproline helix P11LRR targets intracellular mitochondria. J Control Release. 2010;142(2):259–266. doi:10.1016/j.jconrel.2009.10.012
125. Sibrian-Vazquez M, Nesterova IV, Jensen TJ, Vicente MGH. Mitochondria targeting by guanidine-and biguanidine-porphyrin photosensitizers. Bioconjug Chem. 2008;19:705–713. doi:10.1021/bc700393u
126. Battogtokh G, Choi YS, Kang DS, et al. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: current strategies and future perspectives. Acta Pharmaceutica Sinica B. 2018;8(6):862–880. doi:10.1016/j.apsb.2018.05.006
127. Luengo A, Sullivan LB, Vander Heiden MG. Understanding the complex-I-ty of metformin action: limiting mitochondrial respiration to improve cancer therapy. BMC Biol. 2014;12(1):1–4. doi:10.1186/s12915-014-0082-4
128. Fantin VR, Berardi MJ, Scorrano L, Korsmeyer SJ, Leder P. A novel mitochondrio-toxic small molecule that selectively inhibits tumor cell growth. Cancer Cell. 2002;2:29–42. doi:10.1016/S1535-6108(02)00082-X
129. Tee TT, Cheah YH, Hawariah LPA. F16, a fraction from Eurycoma longifolia jack extract, induces apoptosis via a caspase-9-independent manner in MCF-7 cells. Anticancer Res. 2007;27(5A):3425–3430.
130. Lampidis TJ, Hasin Y, Weiss MJ, Chen LB. Selective killing of carcinoma cells “in vitro” bylipophilic-cationic compounds: a cellular basis. Biomed Pharmacother. 1985;39:220–226.
131. Baracca A, Sgarbi G, Solaini G, Lenaz G. Rhodamine 123 as a probe of mitochondrial membrane potential: evaluation of proton flux through F(0) during ATP synthesis. Biochim Biophys Acta. 2003;1606:137–146. doi:10.1016/S0005-2728(03)00110-5
132. Antonenko YN, Avetisyan AV, Cherepanov DA, et al. Derivatives of rhodamine 19 as mild mitochondria-targeted cationic uncouplers. J Biol Chem. 2011;286:17831–17840. doi:10.1074/jbc.M110.212837
133. Antonenko YN, Avetisyan AV, Bakeeva LE, et al. Mitochondria-targeted plastoquinone derivatives a stools to interrupt execution of the aging program.1. Cationic plastoquinone derivatives: synthesis and in vitro studies. Biochem (Mosc). 2008;73:1273–1287. doi:10.1134/S0006297908120018
134. Han M, Vakili MR, Soleymani Abyaneh H, Molavi O, Lai R, Lavasanifar A. Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells. Mol Pharm. 2014;11:2640–2649. doi:10.1021/mp500038g
135. Wu S, Cao Q, Wang X, Cheng K, Cheng Z. Design, synthesis and biological evaluation of mitochondria targeting theranostic agents. Chem Commun (Camb). 2014a;50:8919–8922. doi:10.1039/C4CC03296A
136. Hu QL, Gao M, Feng GX, Liu B. Mitochondria-targeted cancer therapy using a light-up probe with aggregation-induced-emission characteristics. Angew Chem Int Edt. 2014;53:14225–14229. doi:10.1002/anie.201408897
137. Chen WH, Xu XD, Luo GF, et al. Dual-targeting pro-apoptotic peptide for programmed cancer cell death via specific mitochondria damage. Sci Rep. 2013;3:3468. doi:10.1038/srep03468
138. Lei W, Xie J, Hou Y, et al. Mitochondriatargeting properties and photodynamic activities of porphyrin derivatives bearing cationic pendant. J Photochem Photobiol B. 2010;98:167–171. doi:10.1016/j.jphotobiol.2009.12.003
139. Chamberlain GR, Tulumello DV, Kelley SO. Targeted delivery of doxorubicin to mitochondria. ACS Chem Biol. 2013;8:1389–1395. doi:10.1021/cb400095v
140. Fonseca SB, Pereira MP, Mourtada R, et al. Rerouting chlorambucil to mitochondria combats drug deactivation and resistance in cancer cells. Chem Biol. 2011;18:445–453. doi:10.1016/j.chembiol.2011.02.010
141. Wisnovsky SP, Wilson JJ, Radford RJ, et al. Targeting mitochondrial DNA with a platinum-based anticancer agent. Chem Biol. 2013;20:1323–1328. doi:10.1016/j.chembiol.2013.08.010
142. Agemy L, Friedmann-Morvinski D, Kotamraju VR, et al. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc Natl Acad Sci USA. 2011;108:17450–17455. doi:10.1073/pnas.1114518108
143. Zhang S, Yang L, Ling X, et al. Tumor mitochondria-targeted photodynamic therapy with a translocator protein (TSPO)-specific photosensitizer. Acta Biomater. 2015;28:160–170. doi:10.1016/j.actbio.2015.09.033
144. Li J, Lu J, Zhou Y. Mitochondrial-targeted molecular imaging in cardiac disease. BioMed Res Int. 2017;2017.
145. Chen F, Ehlerding EB, Cai W. Theranostic nanoparticles. J Nucl Med. 2014;55(12):1919–1922. doi:10.2967/jnumed.114.146019
146. Wongrakpanich A, Geary SM, Joiner ML, Anderson ME, Salem AK. Mitochondria-targeting particles. Nanomedicine. 2014;9(16):2531–2543. doi:10.2217/nnm.14.161
147. Pathak RK, Kolishetti N, Dhar S. Targeted nanoparticles in mitochondrial medicine. Wiley Interdisciplin Rev. 2015;7(3):315–329.
148. Liu CG, Han YH, Kankala RK, Wang SB, Chen AZ. Subcellular performance of nanoparticles in cancer therapy. Int J Nanomed. 2020;15:675. doi:10.2147/IJN.S226186
149. Oladimeji O, Akinyelu J, Singh M. Nanomedicines for subcellular targeting: the mitochondrial perspective. Curr Med Chem. 2020;27(33):5480–5509. doi:10.2174/0929867326666191125092111
150. Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S. Sizedependent endocytosis of nanoparticles. Adv Mater. 2009;21:419–424. doi:10.1002/adma.200801393
151. Lu F, Wu SH, Hung Y, Mou CY. Size effect on cell uptake in well-suspended, uniform mesoporous silica nanoparticles. Small. 2009;5:1408–1413. doi:10.1002/smll.200900005
152. Jiang W, Kim BYS, Rutka JT, Chan WCW. Nanoparticle mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145–150. doi:10.1038/nnano.2008.30
153. Khan AA, Allemailem KS, Almatroodi SA, Almatroudi A, Rahmani AH. Recent strategies towards the surface modification of liposomes: an innovative approach for different clinical applications. 3 Biotech. 2020c;10(4):1–15. doi:10.1007/s13205-020-2144-3
154. Guo F, Yu M, Wang J, et al. The mitochondria-targeted and IR780-regulated theranosomes for imaging and enhanced photodynamic/photothermal therapy. RSC Adv. 2016;6(14):11070–11076. doi:10.1039/C5RA19521G
155. Boddapati SV, D’Souza GG, Erdogan S, et al. Organelle- targeted nanocarriers: specific delivery of liposomal ceramide to mitochondria enhances its cytotoxicity in vitro and in vivo. Nano Lett. 2008;8(8):2559–2563. doi:10.1021/nl801908y
156. Katagiri K, Ariga K, Kikuchi J-I. Preparation of organic- inorganic hybrid vesicle “cerasome” derived from artificial lipid with alkoxysilyl head. Chem Lett. 1999;28(7):661–662. doi:10.1246/cl.1999.661
157. Wang Y, Wang B, Liao H, et al. Liposomal nanohybrid cerasomes for mitochondria-targeted drug delivery. J Mater Chem B. 2015;3(36):7291–7299. doi:10.1039/C5TB01197C
158. Yamada Y, Akita H, Kamiya H, et al. MITO-Porter: a liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim Biophys Acta. 2008;1778(2):423–432. doi:10.1016/j.bbamem.2007.11.002
159. Yamada Y, Nakamura K, Abe J, et al. Mitochondrial delivery of Coenzyme Q10 via systemic administration using a MITO-Porter prevents ischemia/reperfusion injury in the mouse liver. J Control Release. 2015;213:86–95. doi:10.1016/j.jconrel.2015.06.037
160. Khalil IA, Kogure K, Futaki S, et al. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J Biol Chem. 2006;281(6):3544–3551. doi:10.1074/jbc.M503202200
161. Yasuzaki Y, Yamada Y, Harashima H. Mitochondrial matrix delivery using MITO-Porter, a liposome-based carrier that specifies fusion with mitochondrial membranes. Biochem Biophys Res Commun. 2010;397(2):181–186. doi:10.1016/j.bbrc.2010.05.070
162. Yamada Y, Furukawa R, Yasuzaki Y, et al. Dual function MITO-Porter, a nano carrier integrating both efficient cytoplasmic delivery and mitochondrial macromolecule delivery. Mol Ther. 2011;19(8):1449–1456. doi:10.1038/mt.2011.99
163. Yamada Y, Harashima H. Delivery of bioactive molecules to the mitochondrial genome using a membrane-fusing, liposome-based carrier, DF-MITO-Porter. Biomaterials. 2012;33(5):1589–1595. doi:10.1016/j.biomaterials.2011.10.082
164. Yamada Y, Harashima H. Enhancement in selective mitochondrial association by direct modification of a mitochondrial targeting signal peptide on a liposomal based nanocarrier. Mitochondrion. 2013;13(5):526–532. doi:10.1016/j.mito.2012.09.001
165. Kawamura E, Yamada Y, Harashima H. Mitochondrial targeting functional peptides as potential devices for the mitochondrial delivery of a DF-MITO-Porter. Mitochondrion. 2013;13(6):610–614. doi:10.1016/j.mito.2013.08.010
166. Wang Z, Guo W, Kuang X, Hou S, Liu H. Nanopreparations for mitochondria targeting drug delivery system: current strategies and future prospective. Asian J Pharm Sci. 2017;12(6):498–508. doi:10.1016/j.ajps.2017.05.006
167. Garcia-Perez EG, Sancho P, Sancho P, Sancho P. Dequalinium induces human leukemia cell death by affecting the redox balance. Leuk Res. 2011;35:1395–1401. doi:10.1016/j.leukres.2011.03.012
168. Zupancic S, Kocbek P, Zariwala MG, et al. Design and development of novel mitochondrial targeted nanocarriers, DQAsomes for curcumin inhalation. Mol Pharm. 2014;11(7):2334–2345. doi:10.1021/mp500003q
169. D’Souza GGM, Rammohan R, Cheng S-M, et al. DQAsome mediated delivery of plasmid DNA toward mitochondria in living cells. J Control Release. 2003;92(1–2):189–197. doi:10.1016/S0168-3659(03)00297-9
170. Vaidya B, Paliwal R, Rai S, et al. Cell-selective mitochondrial targeting: a new approach for cancer therapy. Cancer Ther. 2009;7:141–148.
171. D’Souza GG, Boddapati SV, Weissig V. Mitochondrial leader sequence–plasmid DNA conjugates delivered into mammalian cells by DQAsomes co-localize with mitochondria. Mitochondrion. 2005;5(5):352–358. doi:10.1016/j.mito.2005.07.001
172. Weissig V, Lizano C, Torchilin VP. Selective DNA release from DQAsome/DNA complexes at mitochondria-like membranes. Drug Deliv. 2000;7(1):1–5. doi:10.1080/107175400266722
173. Kamaly N, Xiao Z, Valencia PM, et al. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010.
174. Mulik RS, Monkkonen J, Juvonen RO, et al. ApoE3 mediated polymeric nanoparticles containing curcumin: apoptosis induced in vitro anticancer activity against neuroblastoma cells. Int J Pharm. 2012;437(1–2):29–41. doi:10.1016/j.ijpharm.2012.07.062
175. Marrache S, Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc Natl Acad Sci USA. 2012;109:16288–16293. doi:10.1073/pnas.1210096109
176. Yue C, Yang Y, Zhang C, et al. ROS-responsive mitochondria targeting blended nanoparticles: chemo- and photodynamic synergistic therapy for lung cancer with on-demand drug release upon irradiation with a single light source. Theranostics. 2016;6(13):2352–2366. doi:10.7150/thno.15433
177. Hou J, Yu X, Shen Y, et al. Triphenyl phosphine functionalized chitosan nanoparticles enhanced antitumor efficiency through targeted delivery of doxorubicin to mitochondria. Nanoscale Res Lett. 2017;12(1):158. doi:10.1186/s11671-017-1931-1
178. Khatun Z, Choi YS, Kim YG, et al. A bioreducible poly(ethylene glycol)-triphenylphosphonium conjugate as a bioactivable mitochondria-targeting nano-carrier. Biomacromolecules. 2017;18(4):1074–1085. doi:10.1021/acs.biomac.6b01324
179. Mignani S, El Brahmi N, Eloy L, et al. Anticancer copper(II) phosphorus dendrimers are potent proapoptotic Bax activators. Eur J Med Chem. 2017;132:142–156. doi:10.1016/j.ejmech.2017.03.035
180. Lopez-Lazaro M. How many times should we screen a chemical library to discover an anticancer drug? Drug Discov Today. 2015;20:167–169. doi:10.1016/j.drudis.2014.12.006
181. Gorzkiewicz M, Klajnert-Maculewicz B. Dendrimers as nanocarriers for nucleoside analogues. Eur J Pharm Biopharm. 2017;114:43–56. doi:10.1016/j.ejpb.2016.12.030
182. Franiak-Pietryga I, Ziemba B, Sikorska H, et al. Maltotriose-modified poly (propylene imine) Glycodendrimers as a potential novel platform in the treatment of chronic lymphocytic Leukemia. A proof-of-concept pilot study in the animal model of CLL. Toxicol Appl Pharmacol. 2020;403:115139. doi:10.1016/j.taap.2020.115139
183. Tomalia DA, Reyna LA, Svenson S. Dendrimers as multi-purpose nanodevices for oncology drug delivery and diagnostic imaging. Biochem Soc Trans. 2007;35:61–67. doi:10.1042/BST0350061
184. Shao N, Su Y, Hu J, Zhang J, Zhang H, Cheng Y. Comparison of generation 3 polyamidoamine dendrimer and generation 4 polypropylenimine dendrimer on drug loading, complex structure, release behavior, and cytotoxicity. Int J Nanomed. 2011;6:3361–3372. doi:10.2147/IJN.S27028
185. Pedziwiatr-Werbicka E, Ferenc M, Zaborski M, Gabara B, Klajnert B, Bryszewska M. Characterization of complexes formed by polypropylene imine dendrimers and anti-HIV oligonucleotides. Colloids Surf B Biointerfaces. 2011;83:360–366. doi:10.1016/j.colsurfb.2010.12.008
186. Bumb A, Brechbiel MW, Choyke P. Macromolecular and dendrimer-based magnetic resonance contrast agents. Acta Radiol. 2010;51:751–767. doi:10.3109/02841851.2010.491091
187. Franiak-Pietryga I, Ziolkowska E, Ziemba B, et al. Glycodendrimer PPI as a potential drug in chronic lymphocytic leukaemia. The influence of glycodendrimer on apoptosis in in vitro B-CLL cells defined by microarrays. Anti Cancer Agents Med Chem. 2017;17(1):102–114.
188. Mukherjee SP, Lyng FM, Garcia A, Davoren M, Byrne HJ. Mechanistic studies of in vitro cytotoxicity of poly(amidoamine) dendrimers in mammalian cells. Toxicol Appl Pharmacol. 2010;248:259–268. doi:10.1016/j.taap.2010.08.016
189. Franiak-Pietryga I, Ziółkowska E, Ziemba B, et al. The influence of maltotriose-modified poly (propylene imine) dendrimers on the chronic lymphocytic leukemia cells in vitro: dense shell G4 PPI. Mol Pharm. 2013;10(6):2490–2501. doi:10.1021/mp400142p
190. Kubeil M, Stephan H, Pietzsch H-J, et al. Sugar-decorated dendritic nanocarriers: encapsulation and release of the octahedral rhenium cluster complex [Re6S8(OH)6]4−. Chem Asian J. 2010;5:2507–2514. doi:10.1002/asia.201000284
191. Xiong Z, Shen M, Shi X. Dendrimer-based strategies for cancer therapy: recent advances and future perspectives. Sci China Mater. 2018;61(11):1387–1403. doi:10.1007/s40843-018-9271-4
192. Choi YS, Cho TS, Kim JM, Han SW, Kim SK. Amine terminated G-6 PAMAM dendrimer and its interaction with DNA probed by Hoechst 33258. Biophys Chem. 2006;121:142–149. doi:10.1016/j.bpc.2006.01.005
193. Biswas S, Dodwadkar NS, Piroyan A, Torchilin VP. Surface conjugation of triphenylphosphonium to target poly(amidoamine) dendrimers to mitochondria. Biomaterials. 2012;33:4773–4782. doi:10.1016/j.biomaterials.2012.03.032
194. Wang X, Shao N, Zhang Q, Cheng Y. Mitochondrial targeting dendrimer allows efficient and safe gene delivery. J Mater Chem B. 2014;2:2546–2553. doi:10.1039/C3TB21348J
195. Battigelli A, Russier J, Venturelli E, et al. Peptide-based carbon nanotubes for mitochondrial targeting. Nanoscale. 2013;5:9110–9117. doi:10.1039/c3nr02694a
196. Yoong SL, Wong BS, Zhou QL, et al. Enhanced cytotoxicity to cancer cells by mitochondria-targeting MWCNTs containing platinum(IV) prodrug of cisplatin. Biomaterials. 2014;35:748–759. doi:10.1016/j.biomaterials.2013.09.036
197. Xu J, Xu P, Li Z, et al. Oxidative stress and apoptosis induced by hydroxyapatite nanoparticles in C6 cells. J Biomed Mater Res A. 2012;100A(3):738–745. doi:10.1002/jbm.a.33270
198. Sun Y, Chen Y, Ma X, et al. Mitochondria-targeted hydroxyapatite nanoparticles for selective growth inhibition of lung cancer in vitro and in vivo. ACS Appl Mater Interfaces. 2016;8(39):25680–25690. doi:10.1021/acsami.6b06094
199. Xiong H, Du S, Ni J, et al. Mitochondria and nuclei dualtargeted heterogeneous hydroxyapatite nanoparticles for enhancing therapeutic efficacy of doxorubicin. Biomaterials. 2016;94:70–83. doi:10.1016/j.biomaterials.2016.04.004
200. Sharma A, Goyal AK, Rath G. Recent advances in metal nanoparticles in cancer therapy. J Drug Target. 2018;26(8):617–632. doi:10.1080/1061186X.2017.1400553
201. Zhang X, Chibli H, Mielke R, et al. Ultrasmall golddoxorubicin conjugates rapidly kill apoptosis-resistant cancer cells. Bioconjug Chem. 2011;22(2):235. doi:10.1021/bc100374p
202. Mkandawire MM, Lakatos M, Springer A, et al. Induction of apoptosis in human cancer cells by targeting mitochondria with gold nanoparticles. Nanoscale. 2015;7(24):10634–10640. doi:10.1039/C5NR01483B
203. Ma X, Wang X, Zhou M, et al. A mitochondria-targeting gold peptide nanoassembly for enhanced cancer-cell killing. Adv Healthc Mater. 2013;2(12):1638–1643. doi:10.1002/adhm.201300037
204. Federici G, Shaw BJ, Handy RD. Toxicity of titanium dioxide nanoparticles to rainbow trout (Oncorhynchus mykiss): gill injury, oxidative stress, and other physiological effects. Aquat Toxicol. 2007;84(4):415–430. doi:10.1016/j.aquatox.2007.07.009
205. Periasamy VS, Athinarayanan J, Al-Hadi AM, Juhaimi FA, Mahmoud MH, Alshatwi AA. Identification of titanium dioxide nanoparticles in food products: induce intracellular oxidative stress mediated by TNF and CYP1A genes in human lung fibroblast cells. Environ Toxicol Pharmacol. 2015;39(1):176–186. doi:10.1016/j.etap.2014.11.021
206. Wu J, Sun J, Xue Y. Involvement of JNK and P53 activation in G2/M cell cycle arrest and apoptosis induced by titanium dioxide nanoparticles in neuron cells. Toxicol Lett. 2010;199(3):269–276. doi:10.1016/j.toxlet.2010.09.009
207. Cui Y, Gong X, Duan Y, et al. Hepatocyte apoptosis and its molecular mechanisms in mice caused by titanium dioxide nanoparticles. J Hazard Mater. 2010;183(1–3):874–880. doi:10.1016/j.jhazmat.2010.07.109
208. Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 2008;83(5):761–769. doi:10.1038/sj.clpt.6100400
209. Ahamed M, Posgai R, Gorey TJ, Nielsen M, Hussain SM, Rowe JJ. Silver nanoparticles induced heat shock protein 70, oxidative stress and apoptosis in Drosophila melanogaster. Toxicol Appl Pharmacol. 2010;242(3):263–269. doi:10.1016/j.taap.2009.10.016
210. Kim HR, Kim MJ, Lee SY, Oh SM, Chung KH. Genotoxic effects of silver nanoparticles stimulated by oxidative stress in human normal bronchial epithelial (BEAS-2B) cells. Mutat Res. 2011;726(2):129–135. doi:10.1016/j.mrgentox.2011.08.008
211. Piao MJ, Kang KA, Lee IK, et al. Silver nanoparticles induce oxidative cell damage in human liver cells through inhibition of reduced glutathione and induction of mitochondria-involved apoptosis. Toxicol Lett. 2011;201(1):92–100. doi:10.1016/j.toxlet.2010.12.010
212. Ranjbar A, Ataie Z, Khajavi F, Ghasemi H. Effects of silver nanoparticle (Ag NP) on oxidative stress biomarkers in rat. Nanomed J. 2014;1(3):205–210.
213. Berk M, Kapczinski F, Andreazza AC, et al. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev. 2011;35(3):804–817. doi:10.1016/j.neubiorev.2010.10.001
214. Xue Y, Wang J, Huang Y, et al. Comparative cytotoxicity and apoptotic pathways induced by nanosilver in human liver HepG2 and L02 cells. Human Exp Toxicol. 2018;960327118769718.
215. Li J, Zhang B, Chang X, et al. Silver nanoparticles modulate mitochondrial dynamics and biogenesis in HepG2 cells. Environ Pollut. 2020;256:113430. doi:10.1016/j.envpol.2019.113430
216. Sharma V, Shukla RK, Saxena N, Parmar D, Das M, Dhawan A. DNA damaging potential of zinc oxide nanoparticles in human epidermal cells. Toxicol Lett. 2009;185(3):211–218. doi:10.1016/j.toxlet.2009.01.008
217. Sharma V, Anderson D, Dhawan A. Zinc oxide nanoparticles induce oxidative stress and genotoxicity in human liver cells (HepG2). J Biomed Nanotechnol. 2011;7(1):98–99. doi:10.1166/jbn.2011.1220
218. Ahamed M, Akhtar MJ, Raja M, et al. ZnO nanorod-induced apoptosis in human alveolar adenocarcinoma cells via p53, survivin and bax/bcl-2 pathways: role of oxidative stress. Nanomedicine. 2011;7(6):904–913. doi:10.1016/j.nano.2011.04.011
219. Sharma V, Anderson D, Dhawan A. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria mediated apoptosis in human liver cells (HepG2). Apoptosis. 2012;17(8):852–870. doi:10.1007/s10495-012-0705-6
220. Park YH, Bae HC, Kim J, Jeong SH, Yang SI, Son SW. Zinc oxide nanoparticles induce HIF-1alpha protein stabilization through increased reactive oxygen species generation from electron transfer chain complex III of mitochondria. J Dermatol Sci. 2018;91(1):104–107. doi:10.1016/j.jdermsci.2018.03.010
221. Zhao X, Ren X, Zhu R, Luo Z, Ren B. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria-mediated apoptosis in zebrafish embryos. Aquat Toxicol. 2016;180:56–70. doi:10.1016/j.aquatox.2016.09.013
222. Li Y, Li F, Zhang L, et al. Zinc oxide nanoparticles induce mitochondrial biogenesis impairment and cardiac dysfunction in human iPSC-derived cardiomyocytes. Int J Nanomed. 2020;15:2669. doi:10.2147/IJN.S249912
223. Rahman MM, Khan SB, Aslam Jamal MF, Aisiri AM. Iron oxide nanoparticles. In: Rahman M, editor. Nanomaterials. England: InTech; 2011.
224. Naqvi S, Samim M, Abdin M, et al. Concentration-dependent toxicity of iron oxide nanoparticles mediated by increased oxidative stress. Int J Nanomedicine. 2010;5:983–989. doi:10.2147/IJN.S13244
225. Zhu MT, Wang Y, Feng WY, et al. Oxidative stress and apoptosis induced by iron oxide nanoparticles in cultured human umbilical endothelial cells. J Nanosci Nanotechnol. 2010;10(12):8584–8590. doi:10.1166/jnn.2010.2488
226. Guo R, Peng H, Tian Y, Shen S, Yang W. Mitochondria‐targeting magnetic composite nanoparticles for enhanced phototherapy of cancer. Small. 2016;12(33):4541–4552. doi:10.1002/smll.201601094
227. Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777e90. doi:10.1089/ars.2008.2270
228. Jin J, Mullen TD, Hou Q, et al. AMPK inhibitor compound C stimulates ceramide production and promotes Bax redistribution and apoptosis in MCF-7 breast carcinoma cells. J Lipid Res. 2009;50:2389e97. doi:10.1194/jlr.M900119-JLR200
229. Mahmoudi M, Simchi A, Vali H, et al. Cytotoxicity and cell cycle effects of bare and polyvinyl alcohol coated iron oxide nanoparticles in mouse fibroblasts. Adv Eng Mater. 2009;11:B243e50. doi:10.1002/adem.200990035
230. Yu Y, Wang ZH, Zhang L, et al. Mitochondrial targeting topotecan-loaded liposomes for treating drug-resistant breast cancer and inhibiting invasive metastases of melanoma. Biomaterials. 2012;33:1808–1820. doi:10.1016/j.biomaterials.2011.10.085
231. Zhou J, Zhao WY, Ma X, et al. The anticancer efficacy of paclitaxel liposomes modified with mitochondrial targeting conjugate in resistant lung cancer. Biomaterials. 2013;34:3626–3638. doi:10.1016/j.biomaterials.2013.01.078
232. Zhang C, Liu Z, Zheng Y, et al. Glycyrrhetinic acid functionalized graphene oxide for mitochondria targeting and cancer treatment in vivo. Small. 2018;14:1703306. doi:10.1002/smll.201703306
233. Wei Y, Zhou F, Zhang D, Chen Q, Xing D. A graphene oxide based smart drug delivery system for tumor mitochondria-targeting photodynamic therapy. Nanoscale. 2016;8:3530–3538. doi:10.1039/C5NR07785K
234. Cho DY, Cho H, Kwon K, et al. Triphenylphosphonium-conjugated poly(+-caprolactone)- based self-assembled nanostructures as nanosized drugs and drug delivery carriers for mitochondria-targeting synergistic anticancer drug delivery. Adv Funct Mater. 2015;25:5479–5491. doi:10.1002/adfm.201501422
235. Tan Y, Zhu Y, Zhao Y, et al. Mitochondrial alkaline pH-responsive drug release mediated by celastrol loaded glycolipidlike micelles for cancer therapy. Biomaterials. 2018;154:169–181. doi:10.1016/j.biomaterials.2017.07.036
236. Bae Y, Jung MK, Lee S, et al. Dequalinium-based functional nanosomes show increased mitochondria targeting and anticancer effect. Eur J Pharm Biopharm. 2018;124:104–115. doi:10.1016/j.ejpb.2017.12.013
237. Bae Y, Jung MK, Song SJ, et al. Functional nanosome for enhanced mitochondria-targeted gene delivery and expression. Mitochondrion. 2017;37:27–40. doi:10.1016/j.mito.2017.06.005
238. Lee JH, Kim KY, Jin H, et al. Self-assembled coumarin nanoparticle in aqueous solution as selective mitochondrial-targeting drug delivery system. ACS Appl Mater Interfaces. 2018;10:3380–3391. doi:10.1021/acsami.7b17711
239. Zhang Y, Zhang C, Chen J, et al. Trackable mitochondria-targeting nanomicellar loaded with doxorubicin for overcoming drug resistance. ACS Appl Mater Interfaces. 2017;9:25152–25163. doi:10.1021/acsami.7b07219
240. Zhang Z, Zhou L, Zhou Y, et al. Mitophagy induced by nanoparticle-peptide conjugates enabling an alternative intracellular trafficking route. Biomaterials. 2015b;65:56–65. doi:10.1016/j.biomaterials.2015.06.029
241. He H, Wang J, Wang H, et al. Enzymatic cleavage of branched peptides for targeting mitochondria. J Am Chem Soc. 2018;140:1215–1218. doi:10.1021/jacs.7b11582
242. Mallick A, More P, Ghosh S, et al. Dual drug conjugated nanoparticle for simultaneous targeting of mitochondria and nucleus in cancer cells. ACS Appl Mater Interfaces. 2015;7:7584–7598. doi:10.1021/am5090226
243. Liu Y, Zhang X, Zhou M, Nan X, Chen X, Zhang X. Mitochondrial-targeting lonidamine-doxorubicin nanoparticles for synergistic chemotherapy to conquer drug resistance. ACS Appl Mater Interfaces. 2017;9:43498–43507. doi:10.1021/acsami.7b14577
244. Haynes B, Zhang Y, Liu F, et al. Gold nanoparticle conjugated Rad6 inhibitor induces cell death in triple negative breast cancer cells by inducing mitochondrial dysfunction and PARP-1 hyperactivation: synthesis and characterization. Nanomedicine. 2016;12:745–757. doi:10.1016/j.nano.2015.10.010
245. Young K, Jin H, Park J, et al. Mitochondria-targeting self-assembled nanoparticles derived from triphenylphosphonium-conjugated cyanostilbene enable site-specific imaging and anticancer drug delivery. Nano Res. 2018;11:1082. doi:10.1007/s12274-017-1728-7
246. Liu HN, Guo NN, Wang TT, et al. Mitochondrial targeted doxorubicin-triphenylphosphonium delivered by hyaluronic acid modified and pH responsive nanocarriers to breast tumor: in vitro and in vivo studies. Mol Pharm. 2018;15:882–891. doi:10.1021/acs.molpharmaceut.7b00793
247. Parveen S, Misra R, Sahoo SK. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine. 2012;8(2):147–166. doi:10.1016/j.nano.2011.05.016
248. Gnach A, Lipinski T, Bednarkiewicz A, Rybka J, Capobianco JA. Upconverting nanoparticles: assessing the toxicity. Chem Soc Rev. 2015;44(6):1561–1584. doi:10.1039/C4CS00177J
249. Glancy D, Zhang Y, Wu JL, Ouyang B, Ohta S, Chan WC. Characterizing the protein corona of sub-10 nm nanoparticles. J Control Release. 2019;304:102–110. doi:10.1016/j.jconrel.2019.04.023
250. De Jong WH, Hagens WI, Krystek P, et al. Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomaterials. 2008;29(12):1912–1919. doi:10.1016/j.biomaterials.2007.12.037
251. Khanna P, Ong C, Bay BH, Baeg GH. Nanotoxicity: an interplay of oxidative stress, inflammation and cell death. Nanomaterials. 2015;5(3):1163–1180. doi:10.3390/nano5031163
252. Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14:1–6. doi:10.1146/annurev-bioeng-071811-150124
253. Tomsa AM, Alexa AL, Junie ML, et al. Oxidative stress as a potential target in acute kidney injury. PeerJ. 2019;7:e8046. doi:10.7717/peerj.8046
254. Zuberek M, Grzelak A. Nanoparticles-caused oxidative imbalance. Adv Exp Med Biol. 2018;1048:85–98.
255. Hare JI, Lammers T, Ashford MB, et al. Challenges and strategies in anti-cancer nanomedicine development: an industry perspective. Adv Drug Deliv Rev. 2017;108:25–38. doi:10.1016/j.addr.2016.04.025
256. Bobo D, Robinson KJ, Islam J, et al. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res. 2016;33:2373–2387. doi:10.1007/s11095-016-1958-5
257. Ju R, Guo L, Li J, et al. Carboxyamidotriazole inhibits oxidative phosphorylation in cancer cells and exerts synergistic anti-cancer effect with glycolysis inhibition. Cancer Lett. 2016;370(2):232–241. doi:10.1016/j.canlet.2015.10.025
258. Wilk A, Wyczechowska D, Zapata A, et al. Molecular mechanisms of fenofibrate-induced metabolic ca-tastrophe and glioblastoma cell death. Mol Cell Biol. 2015;35(1):182–198. doi:10.1128/MCB.00562-14
259. Jordan BF, Sonveaux P. Targeting tumor perfusion and oxygenation to improve the outcome of anticancer therapy. Front Pharmacol. 2012;3:94. doi:10.3389/fphar.2012.00094
260. Levitsky DO, Dembitsky VM. Anti-breast cancer agents derived from plants. Nat Prod Bioprospect. 2014. doi:10.1007/s13659-014-0048-9
261. Zhou Z, Zhu C, Cai Z, et al. Betulin induces cytochrome c release and apoptosis in colon cancer cells via NOXA. Oncol Lett. 2018;15(5):7319–7327. doi:10.3892/ol.2018.8183
262. Porporato PE, Payen VL, Perez-Escuredo J, et al. A mitochondrial switch pro-motes tumor metastasis. Cell Rep. 2014;8(3):754–766. doi:10.1016/j.celrep.2014.06.043
263. Liou G-Y, Döppler H, DelGiorno Kathleen E, et al. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016;14(10):2325–2336. doi:10.1016/j.celrep.2016.02.029
264. Dheeraj A, Agarwal C, Schlaepfer IR, et al. A novel approach to target hypoxic cancer cells via combining beta-oxidation inhibitor etomoxir with radiation. Hypoxia. 2018;6:23–33. doi:10.2147/HP.S163115
265. Venkataramanaa NK, Venkatesh PK, Dwarakanath BS, Vani S. Protective effect on normal brain tissue during a combination-al therapy of 2-deoxy-d-glucose and hypofractionated irradiation in malignant gliomas. Asian J Neurosurg. 2013;8(1):9–14. doi:10.4103/1793-5482.110274
266. El Sayed SM, Mohamed WG, Seddik MA, et al. Safety and outcome of treatment of metastatic melanoma using 3-bromopyruvate: a concise literature review and case study. Chin J Cancer. 2014;33(7):356–364. doi:10.5732/cjc.013.10111
267. Zhong B, Shi D, Wu F, et al. Dynasore suppresses cell proliferation, migration, and invasion and enhances the antitumor capacity of cis-platin via STAT3 pathway in osteosarcoma. Cell Death Dis. 2019;10(10):687. doi:10.1038/s41419-019-1917-2
268. Angulo-Molina A, Reyes-Leyva J, Lopez-Malo A, Hernandez J. The role of alpha tocopheryl succinate (alpha-TOS) as a poten-tial anticancer agent. Nutr Cancer. 2014;66(2):167–176. doi:10.1080/01635581.2014.863367
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.