Back to Journals » OncoTargets and Therapy » Volume 11
Novel quinazoline-based sulfonamide derivative (3D) induces apoptosis in colorectal cancer by inhibiting JAK2–STAT3 pathway
Authors Al-Obeed O, Vaali-Mohammed MA, Eldehna WM ![]() , Al-Khayal K, Mahmood A, Abdel-Aziz HA
, Al-Khayal K, Mahmood A, Abdel-Aziz HA ![]() , Zubaidi A, Alafeefy A, Abdulla M, Ahmad R
, Zubaidi A, Alafeefy A, Abdulla M, Ahmad R
Received 2 August 2017
Accepted for publication 9 December 2017
Published 5 June 2018 Volume 2018:11 Pages 3313—3322
DOI https://doi.org/10.2147/OTT.S148108
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Omar Al-Obeed,1 Mansoor-Ali Vaali-Mohammed,1 Wagdy M Eldehna,2 Khayal Al-Khayal,1 Amer Mahmood,3 Hatem A Abdel-Aziz,4 Ahmed Zubaidi,1 Ahmed Alafeefy,5 Maha Abdulla,1 Rehan Ahmad1
1Colorectal Research Chair, Department of Surgery, King Khaled University Hospital, College of Medicine, King Saud University, Riyadh, Saudi Arabia; 2Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh, Egypt; 3Stem Cell Unit, Department of Anatomy, College of Medicine, King Saud University, Riyadh, Saudi Arabia; 4Department of Applied Organic Chemistry, National Research Center, Cairo, Egypt; 5Department of Chemistry, Kulliyyah of Science, International Islamic University, Kuantan, Malaysia
Introduction: Colorectal cancer (CRC) is a major worldwide health problem owing to its high prevalence and mortality rate. Developments in screening, prevention, biomarker, personalized therapies and chemotherapy have improved detection and treatment. However, despite these advances, many patients with advanced metastatic tumors still succumb to the disease. New anticancer agents are needed for treating advanced stage CRC as most of the deaths occur due to cancer metastasis. A recently developed novel sulfonamide derivative 4-((2-(4-(dimethylamino) phenyl)quinazolin-4-yl)amino)benzenesulfonamide (3D) has shown potent antitumor effect; however, the mechanism underlying the antitumor effect remains unknown.
Materials and methods: 3D-mediated inhibition on cell viability was evaluated by MTT and real-time cell proliferation was measured by xCelligence RTDP instrument. Western blotting was used to measure pro-apoptotic, anti-apoptotic proteins and JAK2-STAT3 phosphorylation. Flow cytometry was used to measure ROS production and apoptosis.
Results: Our study revealed that 3D treatment significantly reduced the viability of human CRC cells HT-29 and SW620. Furthermore, 3D treatment induced the generation of reactive oxygen species (ROS) in human CRC cells. Confirming our observation, N-acetylcysteine significantly inhibited apoptosis. This is further evidenced by the induction of p53 and Bax; release of cytochrome c; activation of caspase-9, caspase-7 and caspase-3; and cleavage of PARP in 3D-treated cells. This compound was found to have a significant effect on the inhibition of antiapoptotic proteins Bcl2 and BclxL. The results further demonstrate that 3D inhibits JAK2–STAT3 pathway by decreasing the constitutive and IL-6-induced phosphorylation of STAT3. 3D also decreases STAT3 target genes such as cyclin D1 and survivin. Furthermore, a combination study of 3D with doxorubicin (Dox) also showed more potent effects than single treatment of Dox in the inhibition of cell viability.
Conclusion: Taken together, these findings indicate that 3D induces ROS-mediated apoptosis and inhibits JAK2–STAT3 signaling in CRC.
Keywords: sulfonamide, apoptosis, colorectal cancer, STAT3 pathway, Bcl2 proteins, reactive oxygen species
Introduction
Colorectal cancer (CRC) has the third highest incidence rate of malignant tumors worldwide. CRC is associated with increased morbidity and mortality over the years.1 In the USA, it is estimated that 134,000 patients were diagnosed with CRC in 2016.2 Surgery is the mainstay of treatment for early CRC. CRC patients are diagnosed with an advanced stage of disease, often with distant metastasis. Gastrointestinal epithelial cells acquire genetic and epigenetic mutations in oncogenes and tumor suppressor genes, which confer them for selective advantage on proliferation and self-renewal.3,4 Aberrant inflammation of gastrointestinal tract by bacterial infections also leads to CRC development.5 Tumor growth and progression during tumor-associated inflammation involve multiple mechanisms including aberrant proliferation, antiapoptosis, angiogenesis, metastasis and tumor evasion.6,7 During CRC development, normal epithelial cells become hyperproliferative that leads to benign adenoma, which further evolves into carcinoma leading to CRC metastasis.8
Many inflammatory signals promote tumorigenesis by involving activation of NF-κB and JAK/STAT signaling pathways. The JAK–STAT pathway is known be involved in tumor-associated inflammation, proliferation, invasion and migration. The STAT3 proteins are transcription factors that participate in cellular signaling using cytokine and growth factors from the cell membrane into nucleus. Constitutive activation of STAT3 has been reported in various human malignancies such as breast cancer, pancreatic cancer and glioblastoma.9–12 Several previous reports have demonstrated that modulating JAK2–STAT3 pathway arrests growth of primary human cancer cells and promotes antitumor immunity.13 STAT3 as a drug target has been exploited in several cancers.14 Some studies have also reported that the inhibition of the JAK–STAT pathway induces apoptosis in CRC cells.15 During cancer pathogenesis, apoptosis deregulation has been widely recognized as a hallmark of cancer.16 Mainly, there are two major signaling pathways leading to cell death. The first is the death receptor-mediated extrinsic apoptotic pathway, which is activated from outside by proapoptotic ligands binding to the cell surface death receptor.17 The second is the mitochondrial apoptotic pathway (intrinsic apoptotic pathway), which is activated from inside the cell by Bcl2 family proteins.18 Both these pathways converge onto the activation of effector caspases, leading to apoptotic cell death program.
Recently, we published some derivatives of aminobenzenesulfonamide as potential antitumor agents and evaluated the mechanism of a novel antitumor agent (3c) and found that this particular derivative acts by inducing reactive oxygen species (ROS)-mediated apoptosis in CRC.19,20 In the present study, we evaluated the inhibitory effect of a novel quinazoline-based sulfonamide derivative, 4-((2-(4-(dimethylamino)phenyl)quinazolin-4-yl)amino)benzenesulfonamide (3D), in CRC. We demonstrated that compound 3D inhibits proliferation of CRC cell lines by inducing ROS-mediated apoptosis. This finding further indicated that 3D induces p53, Bax and the intrinsic mitochondrial apoptotic pathway and inhibits Bcl2 family protein. Furthermore, 3D inhibits constitutive and IL-6-induced STAT3 phosphorylation and target genes.
Materials and methods
Cell culture
Human colorectal carcinoma HT-29 and SW620 cell lines were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA) and maintained in the Roswell Park Memorial Institute medium (RPMI; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% heat-inactivated fetal bovine serum, 100 μg/mL streptomycin, 100 units/mL penicillin and 2 mmol/L L-glutamine.
Cell viability assay
Cell viability was determined using 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT).20 Briefly, after culturing cells in 96-well plates for 24 h, they were treated with various concentrations of 3D for 24 h. Freshly prepared 10 μL of 5 mM MTT solution was added to the cells and was further incubated for 2 h at 37°C in 5% CO2. In all, 100 μL of dimethyl sulfoxide (DMSO) was added and mixed in each well to dissolve the formazan crystal formed in the reaction of MTT at the time of incubation. The absorbance of the product was measured at 540 nm using a microplate reader. The experiments were performed in triplicates for each condition and the mean and standard deviation (SD) values of three independent experiments were provided.
Cytotoxicity assay using xCELLigence system
Optimal seeding concentration for proliferation of HT-29 cells was determined. Briefly, HT-29 cells (5,000 cells in 150 μL medium/well) were seeded in 16-well plates (E-plate 16; ACEA Biosciences Inc., San Diego, CA, USA) following the xCELLigence Real-Time Cell Analyzer (RTCA)-DP instrument manual as provided by the manufacturer’s protocol. After 24 h, 3D (5, 10, 20 μM) was added and incubation continued for another 48 h. Baseline cell indices were calculated for at least two measurements from three replicate experiments.
Apoptosis
Control and 3D-treated cells were cultured in six-well plates for 24 h. Cells were harvested and washed twice with ice-cold PBS. Cells were resuspended in Annexin-binding buffer and incubated with Annexin V-fluorescein isothiocyanate (5 μL) and propidium iodide (1 μL) at room temperature for 15 min and then analyzed by flow cytometry on FACSCalibur (BD Biosciences, San Jose, CA, USA).
Western blot
Whole cell lysates were prepared using radioimmunoprecipitation assay buffer (RIPA) lysis buffer as described.20 Total protein was extracted by RIPA lysis buffer (Boston Bio Products, Boston, MA, USA), and concentration was determined using Bradford protein reagent (Bio-Rad Laboratories Inc., Hercules, CA, USA). Equal amounts of soluble proteins were loaded and electrophoresed on 4%–20% of Mini-Protean TGX precast gels (Cat No 456-1094; Bio-Rad Laboratories Inc.) and subsequently transferred to trans-blot turbo 0.2 μm nitrocellulose transfer membrane using trans-blot turbo transfer system (Bio-Rad Laboratories Inc.). The membranes were blocked in 5% skimmed milk in PBS containing 0.1% Tween-20 (PBST) for 1 h at room temperature and were incubated overnight with the following primary antibodies at 4°C: anti-cytochrome c (1:200; Abcam, Cambridge, MA, USA), anti-poly-ADP-ribose polymerase (PARP, 1:200; BioVision, San Farncisco, CA, USA), anti-p53, anti-Bax, anti-Bcl2, anti-BclxL, anti-cyclin D1, anti-survivin, anti-JAK2, anti-STAT3 (1:1,000; Santa Cruz Biotechnology Inc., Dallas, TX, USA), anti-Phospho-JAK2 and anti-Phospho-STAT3 (1:1,000; Cell Signaling Technology, Danvers, MA, USA) and anti-β-actin (1:10,000; Sigma-Aldrich Co., St Louis, MO, USA). The membranes were washed twice with PBST and incubated with horseradish peroxisome-conjugated secondary antibodies (1:3,000; Santa Cruz Biotechnology Inc.) at room temperature for 1 h. After two PBST washes, reactivity was detected using chemiluminescence by Clarity Western ECL substrate (Bio-Rad Laboratories Inc.). Membranes were developed using C-DiGit Blot Scanner (LI-COR, Hamburg, Germany). β-actin was used as an internal loading control.
Cytochrome c measurement
Briefly, cells were treated with 3D for 24 h, and harvested cells were incubated in 1× cytosolic extraction buffer. The homogenate was centrifuged at 10,000× g for 10 min. The cytosolic extracts were immunoblotted for cytochrome c.
ROS measurement by flow cytometry
Cells were pretreated with different concentrations of compound 3D for 24 h. Cells were then treated with 2′,7′-dichlorodihydrofluorescein diacetate (c-H2DCFDA, 5 μM) for 15 min at 37°C to assess hydrogen peroxide (H2O2)-mediated oxidation to fluorescent compound DCF.20 Fluorescence of oxidized DCF was measured using flow cytometry (FACSCalibur) at an excitation wavelength of 480 nm and an emission wavelength of 525 nm.
Measurement of mitochondrial membrane potential
Cells were treated with 3D (10 μM) for 24 h and then were incubated with rhodamine 123 (25 ng/mL; Molecular Probes, Eugene, OR, USA) in PBS for 15 min at 37°C. Rhodamine 123-positive populations were monitored using flow cytometry.20
Caspase activity assay
Caspase activity assay was determined using Caspase Colorimetric Protease Assay Sampler Kit for measuring caspase-3, -6, -8 and -9 (KHZ1001; Thermo Fisher Scientific). In brief, control and treated cells were harvested and resuspended in 50 μL cold cell lysis buffer with incubation on ice for 10 min. Cytosolic fraction was extracted by centrifuging at 10,000× g for 1 min. In all, 50 μg of cytosolic extract was loaded into 96-well plates, followed by addition of 50 μL of reaction buffer containing 10 mM of dithiothreitol. A total of 5 μL of caspase substrate was added and incubated at 37°C for 2 h. Plate was read at 400 nm on the microplate reader.
Cell viability assay for combination studies
The effect of 3D and 3D/doxorubicin (Dox) combination on cell proliferation was measured by MTT assay. The cells were seeded in 96-well plates and incubated overnight. Cells were treated with different concentrations of Dox alone or in combination with 3D (10 μM) for 24 h. Freshly prepared 10 μL of 5 mM MTT solutions were added to the cells, and the cells were further incubated for 2 h at 37°C in 5% CO2 incubator. In all, 100 μL of DMSO was added in each well to dissolve formazan crystal formed in the reaction of MTT at the time of incubation. The absorbance of the product was measured at 540 nm using a microplate reader.
Statistical analysis
Data were presented as mean ± SD values. Statistical analysis was performed using Microsoft Excel (Microsoft Corporation, Redmond, WA, USA). The mean values between the control and treated groups were compared using Student’s t-test. P≤0.05 was considered to indicate a statistically significant difference.
Results
3D inhibits cell viability
Treatment of HT-29 cells with 3D (Figure 1A) for 24 h resulted in the inhibition of cell viability in a dose-dependent manner. The IC50 was found to be 10 μM in HT-29 cells (Figure 1B). Similar results were obtained in SW620, another human CRC cell-line with the IC50 calculated to be 6.25 μM (Figure 1C). These findings were further confirmed using the xCELLigence real-time cell proliferation system. 3D was found to inhibit the cell proliferation in a dose- and time-dependent manner (Figure 1D). These findings indicate that 3D significantly inhibited viability of CRC cells.
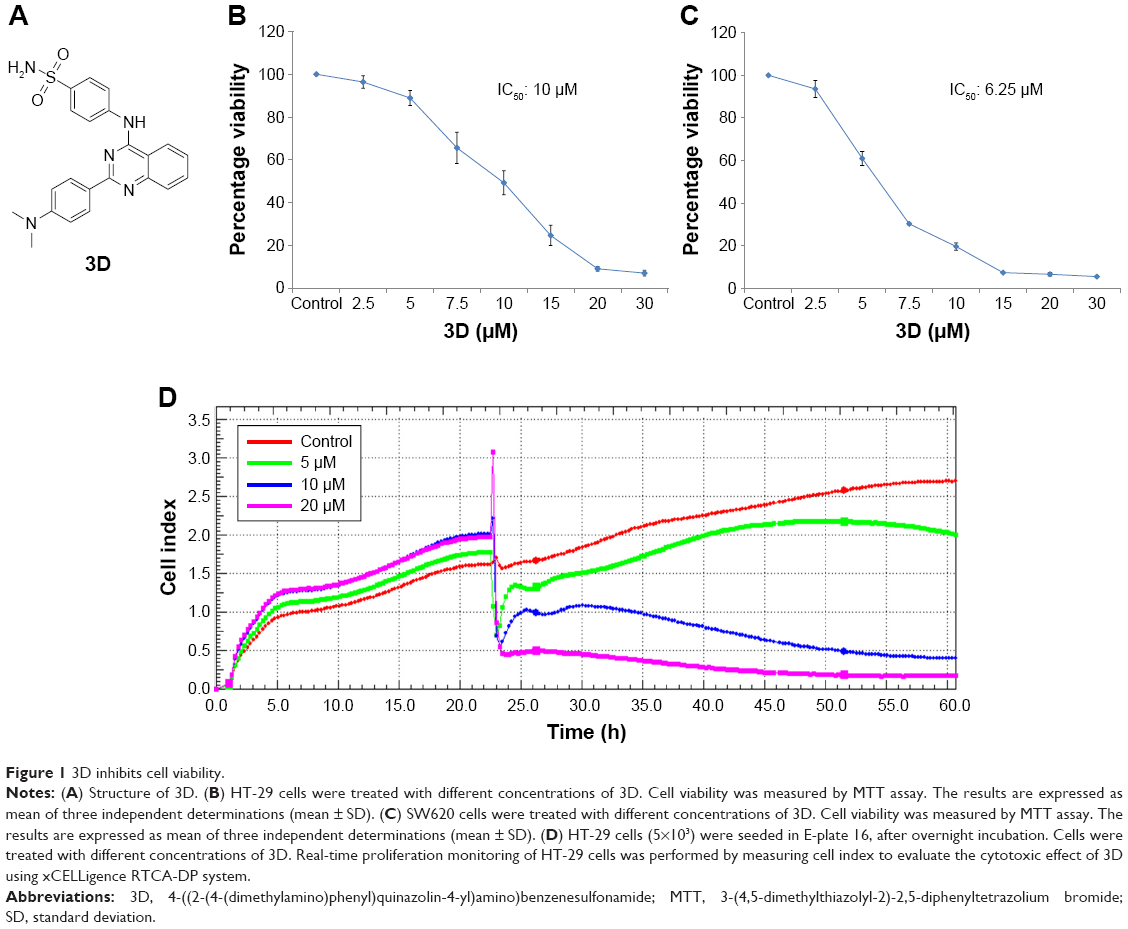 | Figure 1 3D inhibits cell viability. |
3D induces ROS-mediated apoptosis
Most of the inhibition in cell viability occurs due to apoptosis or necrosis. To understand whether 3D induces apoptosis or necrosis, SW620 cells treated with different concentrations of 3D resulted in a significant induction of apoptosis and lesser necrosis (Figure 2A). Chronic accumulation of intracellular ROS induces cell death. Preincubation with an antioxidant, N-acetylcysteine (NAC), decreased apoptosis indicating that 3D-induced apoptosis is ROS dependent (Figure 2B). A similar result was obtained in HT-29 cells (Figure 2C). We examined the effect of 3D on ROS production. Treatment of HT-29 with 3D resulted in enhanced production of ROS in a dose-dependent manner (Figure 2D). Similarly, 3D also induced ROS production in SW620 cells (Figure 2E). Most of the intracellular ROS generation takes place from mitochondria. Treatment of human CRC cell lines with 3D resulted in the decrease of mitochondrial membrane potential (Figure 2F, left). A similar result was obtained in SW620 cells (Figure 2F, right). These results demonstrated that 3D induces ROS production, inhibits mitochondrial membrane potential and enhances ROS-dependent apoptosis.
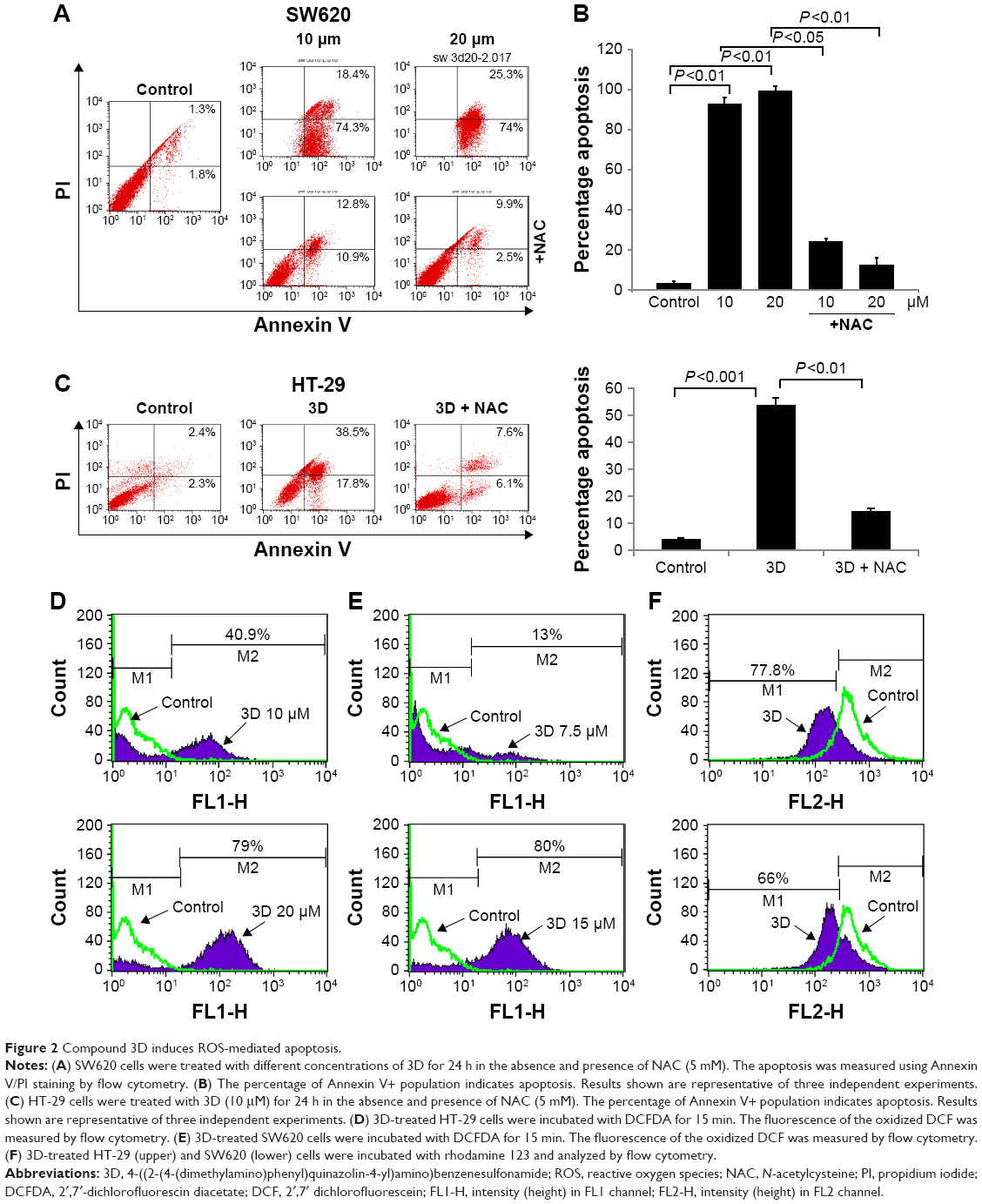 | Figure 2 Compound 3D induces ROS-mediated apoptosis. |
3D induces p53, modulates Bcl2 family proteins and induces cytochrome and PARP activation
p53 is a tumor suppressor gene and acts through several mechanisms for anticancer effect. It activates DNA repair proteins and induces growth arrest, apoptosis and senescence. 3D treatment resulted in the induction of p53 expression in HT-29 cells (Figure 3A). Bax is a p53 target gene, and as expected, treatment of HT-29 cells with different concentrations of 3D induced Bax expression (Figure 3A). Similar results were obtained in SW620 cells (Figure 3B). The balance between Bax and Bcl2/BclxL determines the cell fate under stress. Treatment with 3D resulted in the inhibition of Bcl2 and BclxL in HT-29 (Figure 3A) and SW620 (Figure 3B) cells. Thus, 3D was found to inhibit antiapoptotic proteins and induce proapoptotic proteins, thereby triggering the apoptotic pathway. Upon initiation of apoptosis signaling, activated Bax binds to the mitochondrial outer membrane and is known to induce the opening of the mitochondrial voltage-dependent anion channel (VDAC) that would lead to the release of cytochrome c from mitochondria into cytosol where it activates the caspase cascade. Indeed, treatment with 3D resulted in the release of cytochrome c in HT-29 cells (Figure 3C). Similar results were obtained in SW620 cell line (Figure 3D). PARP family of proteins is known to be involved in DNA repair and programmed cell death. Our results further demonstrated that treatment with 3D resulted in the cleavage of PARP as shown by an increase in cleaved PARP in HT-29 cells (Figure 3C). Similar results were obtained in SW620 cells (Figure 3D).
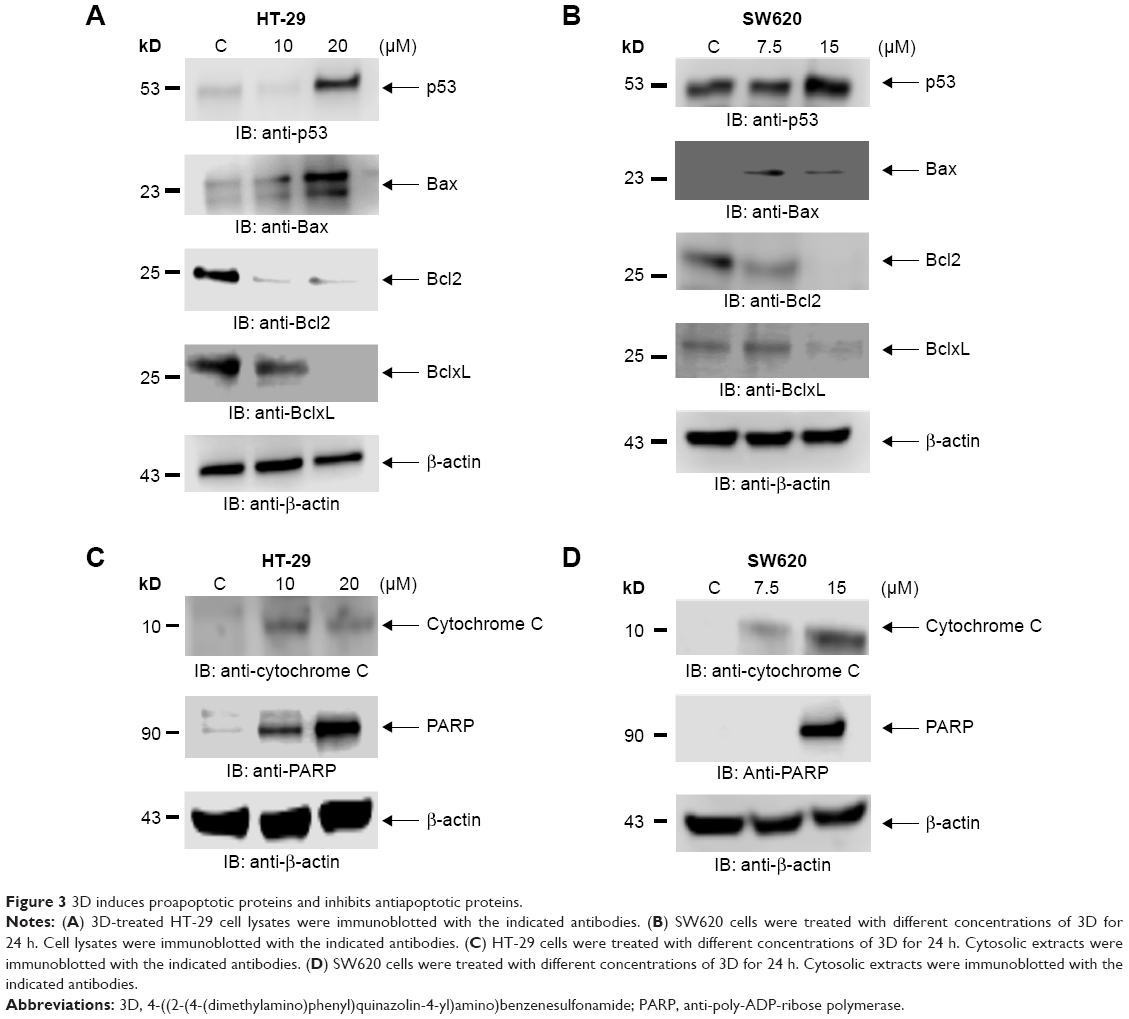 | Figure 3 3D induces proapoptotic proteins and inhibits antiapoptotic proteins. |
3D activates caspase cascade
Release of cytochrome c from mitochondria into cytosol leads to caspase activation that plays an essential role in apoptosis. To determine which caspases were involved in inducing apoptosis, 3D-treated HT-29 cells were incubated with various caspase substrates. 3D was found to activate caspase-9, caspase-6 and caspase-3 and to lesser extent caspase-8 as well (Figure 4A). Similar finding was obtained in SW620 cells (Figure 4B). Thus, these results indicate that 3D activated mostly those caspases that are involved in intrinsic apoptotic pathway. However, some induction of caspase-8 was also observed, which is important for extrinsic apoptotic pathway.
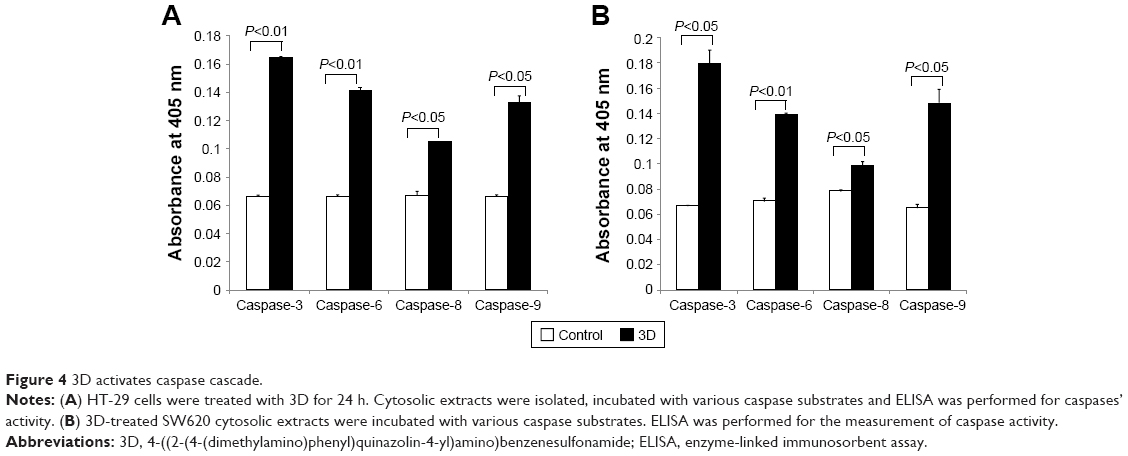 | Figure 4 3D activates caspase cascade. |
3D inhibits JAK2 and STAT3 phosphorylation
Phosphorylation of STAT3 at Tyr-705 is critical for its dimerization and nuclear translocation. STAT3 is constitutively activated in HT-29 cells. Treatment of HT-29 cells with 3D resulted in the inhibition of STAT3 phosphorylation (Figure 5A). Janus kinases are upstream kinases that are known to phosphorylate STAT3. 3D treatment of HT-29 cells inhibited JAK2 phosphorylation in a dose-dependent manner (Figure 5A). STAT3 phosphorylation can be induced by IL-6. SW620 cells, which do not express phosphorylated STAT3, were used to determine if 3D is capable of inhibiting IL-6-induced STAT3 phosphorylation. We found that 3D inhibited IL-6-induced phosphorylation of STAT3 in SW620 cell line (Figure 5B, left). STAT3 together with NF-κB is known to be involved in inflammation-induced cancer progression. We also determined if 3D may also block NF-κB activation; however, 3D was not found to have any inhibitory effect on NF-κB activation (Figure 5B, right). Binding of STAT3 to the binding site present in the promoters of the target genes induces the transcription of several proliferation genes. Cyclin D1 is a STAT3 target gene for inducing cell proliferation, and another STAT3 target gene survivin is also known to inhibit caspase activation and thereby to inhibit apoptosis. The expression of STAT3 target genes such as cyclin D1 and survivin was decreased after treatment with 3D in a dose-dependent manner in HT-29 cells (Figure 5C). Similar results were obtained in SW620 cells (Figure 5D). AG490 is a known STAT3 inhibitor; we asked if 3D could have synergistic effect with AG490. Indeed, 3D inhibited STAT3 phosphorylation along with cyclin D1 and survivin synergistically with AG490 (Figure 5E). These findings thus indicate that 3D inhibited the JAK–STAT pathway and downstream target genes in CRC cells.
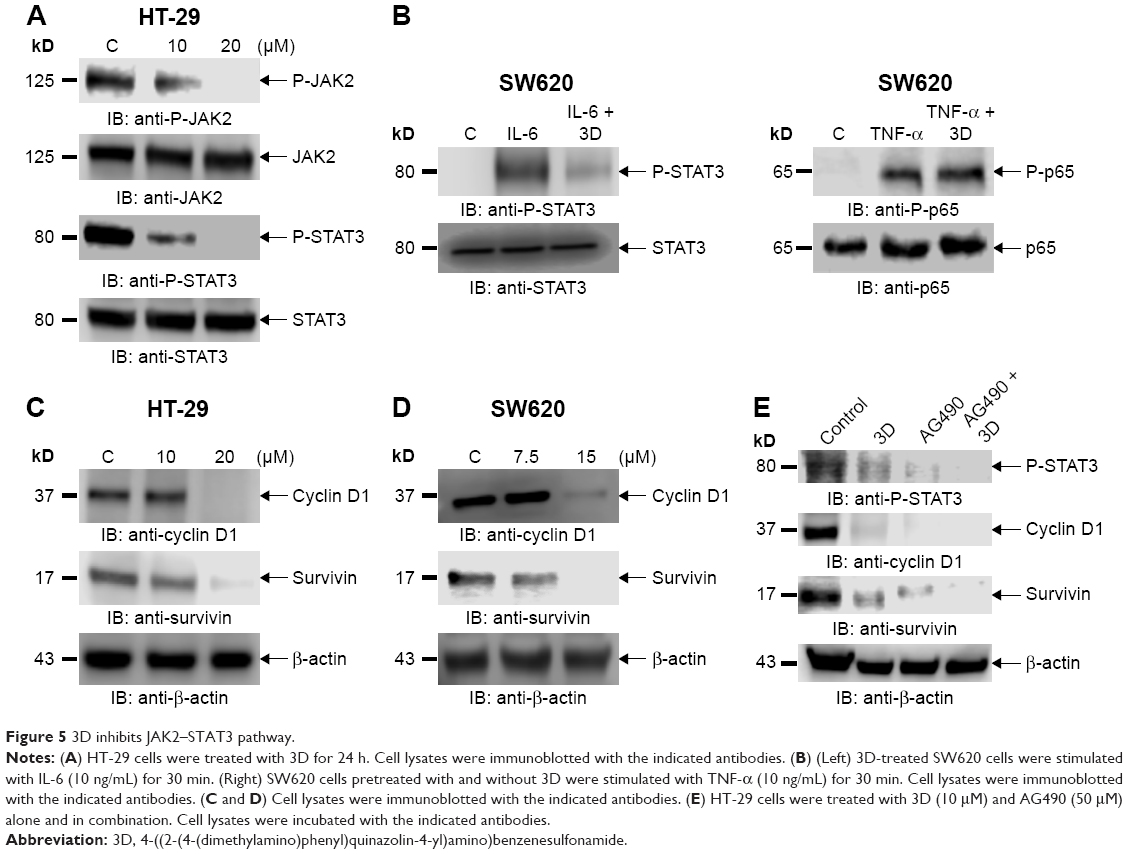 | Figure 5 3D inhibits JAK2–STAT3 pathway. |
3D sensitizes HT-29 to the Dox treatment
To compare the anticancer effect of 3D and potential synergy with a known standard drug, combination studies were performed with Dox. We investigated how 3D and Dox influence growth and proliferation of cancer cells in combination therapy. Treatment of HT-29 cells with Dox alone or in combination with 3D indicated that 3D potentiated the effect of Dox on inhibition of proliferation. Dox inhibited the cell viability of HT-29 in a dose-dependent manner; however, 3D significantly potentiated the effect of Dox (Figure 6A). Dox was found to be ineffective at lower concentration in inhibiting cell viability of metastatic CRC cell line SW620. However, in the combination approach, Dox with 3D significantly inhibited the cell viability (Figure 6B).
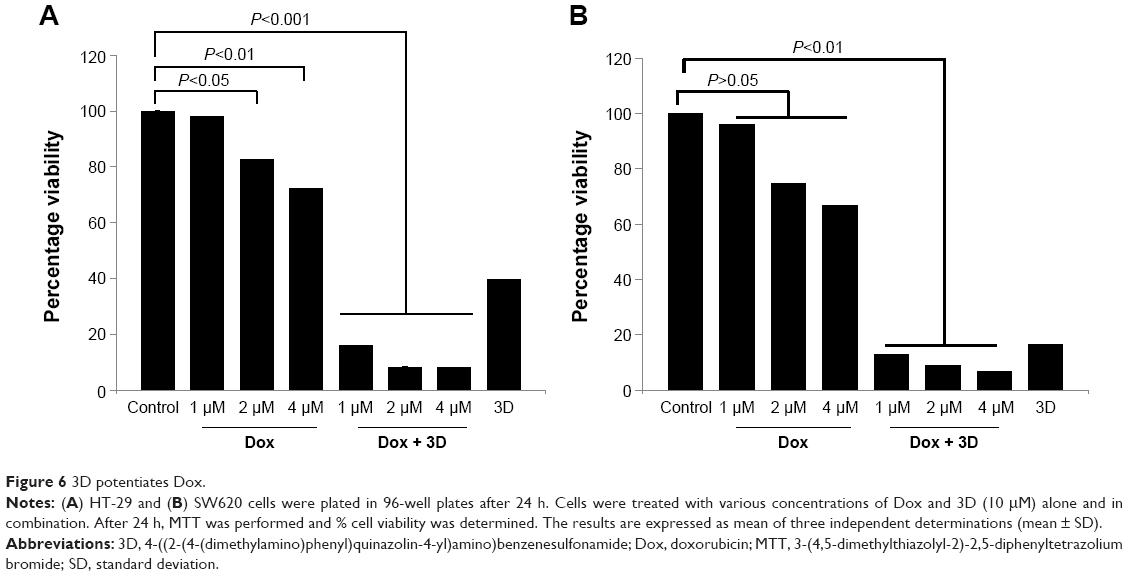 | Figure 6 3D potentiates Dox. |
Discussion
3D has been reported to exert anticancer effect.19 However, the biochemical basis of the anticancer effect of 3D remains unknown. In the present study, we found that 3D inhibited the cell viability of human CRC cell lines in a dose- and time-dependent manner. 3D was found to have more sensitive effect on metastatic CRC cell SW620 as compared to adenocarcinoma CRC cell HT-29. Most of the inhibition of cell viability and proliferation occurs due to apoptosis induction. Most anticancer agents target apoptosis as a major route to eradicate cancer. This novel quinazoline-based derivative of sulfonamide (3D) significantly induced apoptosis as demonstrated by flow cytometry. Various anticancer agents have been shown to exhibit their anticancer effect by altering the levels of ROS.20–22 ROS are byproducts of the oxygen metabolism during oxidative stress. ROS plays a vital role in the maintenance of homeostasis. 3D-induced apoptosis was found to be inhibited with addition of antioxidant NAC. These findings confirmed that 3D-induced apoptosis is ROS dependent. Excessive production of ROS leads to a loss of cellular integrity and cell death.23 Several therapeutic agents used to treat different malignant tumors have been shown to generate a high level of ROS.24 Our findings demonstrated that 3D robustly induces ROS generation in a dose-dependent manner in human CRC cells. These results thus demonstrate that the ROS production is an upstream event and is essential for the apoptosis induction in 3D-treated cells. These findings strongly support the idea that 3D-induced increase in ROS production may be an effective therapeutic strategy against CRC. Mitochondria are considered a prime location where cellular stress signals converge leading to the execution of apoptosis.25 Loss of mitochondrial membrane potential is an early event in the intrinsic apoptotic pathway. 3D treatment resulted in a significant loss of mitochondrial membrane potential.
P53 is a well-known tumor suppressor that plays important role in apoptosis and prevents cells from tumorigenic alterations.26 Our compound of interest induces p53 expression in a dose-dependent manner. The balance between the proapoptotic proteins (p53, Bax) and antiapoptotic proteins (Bcl2, BclxL) determines the cell fate. The induction of Bax expression is essential for apoptosis in human colon cancer cells.27 The overexpression of Bcl2 and BclxL inhibits apoptosis and promotes survival of cancer cells.28 The antiapoptotic Bcl2 protein is reported to be overexpressed in colon cancer. The Bcl2-mediated apoptosis inhibition restores the tumorigenicity of regressed colon cancer.29 In this study, we found that 3D induces the expression of proapoptotic proteins such as p53 and Bax; however, it inhibits the expression of Bcl2 and BclxL in human adenocarcinoma and metastatic CRC cells. Thus, the induction of Bax expression and the inhibition of Bcl2 and BclxL expression provide a mechanistic basis for 3D-induced apoptosis. Induction in Bax activation leads to opening of VDAC channel, thereby releasing cytochrome c. Cytochrome c release from mitochondria into cytosol resulted in activation of caspases. 3D was indeed found to enhance cytochrome c release from mitochondria into cytosol. 3D treatment also resulted in PARP cleavage. Caspase cascade begins with activation of initiator caspase-9, which in turn activated caspase-3 and -6, leading to mitochondria-mediated cell death pathway. Caspase-8 is involved in death receptor-mediated apoptosis pathway. 3D was found to activate caspase-9, -3 and -6 and to a little extent caspase-8 as well. These findings indicate that 3D induces mainly intrinsic apoptotic death pathway and to some extent extrinsic apoptotic pathway.
The essential role of STAT3 in cancer tumorigenesis makes STAT3 an important target for cancer therapeutics.15 We studied the effect of 3D on STAT3 signaling pathway in human CRC cell lines. STAT3 has been reported to be constitutively active in HT-29 cells. 3D was found to decrease phosphorylation of STAT3 at Tyr-705 in HT-29 cells. 3D also inhibited the IL-6-induced phosphorylation of STAT3 in metastatic CRC cell line SW620. Inhibition of STAT3 phosphorylation is an essential step in blocking STAT3 pathway as STAT3 phosphorylation leads to STAT3 dimerization, followed by translocation into nucleus and binding to the promoters of target genes. STAT3 is activated by the upstream kinases including JAK2.12 Our finding indicates that 3D inhibits JAK2 phosphorylation in human CRC cells. 3D-induced inhibition of JAK2 and STAT3 suggests that apoptosis induction by 3D is mediated by inhibition of JAK2-STAT3 signaling. 3D may affect other pathways as well. This was further demonstrated by 3D-induced inhibition of STAT3 target genes such as cyclin D1 and survivin. AG490 is known to inhibit JAK–STAT pathway; 3D treatment in combination with AG490 further inhibits the STAT3 signaling in CRC cells. The anticancer efficacies of current therapeutics are limited because of the high degree of cancer clonal heterogeneity and intratumor genetic variation. Therefore, use of combinations of molecular-targeted agents has been on the rise for better therapeutics. Our compound of interest 3D sensitizes the efficacy of standard anticancer drug Dox. Drug–drug interaction can pose serious side effects in patients. The interaction could be pharmacokinetic (PK) and pharmacodynamic (PD) in nature. The PK interactions result in altered distribution, absorption and elimination of the drug. Pharmacodynamic interactions cause alterations in the way a drug or compound affects a tissue or organ system. These interactions affect the action of a drug in a qualitative way by either enhancing (synergy or additive) or decreasing (antagonize) the therapeutic effect. The interaction between 3D and Dox may be synergistic or additive with resultant potentiation of cytotoxicity in combination therapy. Further studies are needed to study the interaction between 3D and other cytotoxic agents in combination therapy.
Conclusion
Based on these results, we conclude that this novel derivative of sulfonamide (3D) induces 1) ROS-mediated apoptosis; 2) induction of p53 and Bax; 3) inhibition of Bcl2 and BclxL; and 4) cytochrome c release, PARP cleavage and activation of caspases. Our findings further demonstrate that 3D inhibits the JAK2–STAT3 signaling pathway and target genes, which may provide a mechanistic basis for this compound. 3D-mediated inhibition of STAT3 activation also sensitizes Dox efficacy in CRC. Hence, 3D-mediated inhibition of STAT3 activation and induction of ROS-dependent apoptosis suggest that 3D is an attractive candidate for further investigation as a potential anticancer agent.
Acknowledgments
The authors are grateful to the Deanship of Scientific Research, King Saud University for funding through Vice Deanship of Scientific Research Chairs.
Disclosure
The authors report no conflicts of interest in this work.
References
De Rosa M, Pace U, Rega D, et al. Genetics, diagnosis and management of colorectal cancer. Oncol Rep. 2015;34(3):1087–1096. | ||
Strum BW. Colorectal adenomas. N Engl J Med. 2016;374(11):1065–1075. | ||
Pancione M, Remo A, Colantuoni V. Genetic and epigenetic events generate multiple pathways in colorectal cancer progression. Patholog Res Int. 2012;2012:509348. | ||
Ewing I, Hurley JJ, Josephides E, Millar A. The molecular genetics of colorectal cancer. Frontline Gastroenterol. 2014;5:26–30. | ||
Jawad N, Direkze N, Leedham SJ. Inflammatory bowel disease and colon cancer. Recent Results Cancer Res. 2011;185:99–115. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Andersen NN, Jess T. Has the risk of colorectal cancer in inflammatory bowel disease decreased? World J Gastroenterol. 2013;19(43):7561–7568. | ||
Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319(9):525–532. | ||
Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8(4):945–954. | ||
Bromberg J, Darnell JE Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19(21):2468–2473. | ||
Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000;19(56):6613–6626. | ||
Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19(21):2474–2488. | ||
Chiarle R, Simmons WJ, Cai H, et al. STAT3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11(6):623–629. | ||
Kortylewski M, Kujawski M, Wang T, et al. Inhibiting STAT3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11(12):1314–1321. | ||
Darnell JE. Validating STAT3 in cancer therapy. Nat Med. 2005;11(6):595–596. | ||
Xiong H, Zhang ZG, Tian XQ, et al. Inhibition of JAK1, 2/STAT3 signaling induces apoptosis, cell cycle arrest, and reduces tumor cell invasion in colorectal cancer cells. Neoplasia. 2008;10(3):287–297. | ||
Lin Q, Lai R, Chirieac LR, et al. Constitutive activation of JAK3/STAT3 in colon carcinoma tumours and cell lines: inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am J Pathol. 2005;167(4):969–980. | ||
Nicholson DW. From bench to clinic with apoptosis-based therapeutic agents. Nature. 2000;407(6805):810–816. | ||
Alafeefy AM, Ahmad R, Abdulla M, et al. Development of certain new 2-substituted-quinazolin-4-yl-aminobenzenesulfonamide as potential antitumor agents. Eur J Med Chem. 2016;109:247–253. | ||
Al-Khayal K, Alafeefy A, Vaali-Mohammed MA, et al. Novel derivative of aminobenzenesulfonamide (3c) induces apoptosis in colorectal cancer cells through ROS generation and inhibits cell migration. BMC Cancer. 2017;17:4. | ||
Zhang X, Wang X, Wu T, et al. Isoliensinine induces apoptosis in triple-negative human breast cancer through ROS generation and p38 MAPK/JNK activation. Sci Rep. 2015;5:12579. | ||
Subramani R, Gonzalez E, Arumugam A, et al. Nimbolide inhibits pancreatic cancer growth and metastasis through ROS-mediated apoptosis and inhibition of epithelial-to-mesenchymal transition. Sci Rep. 2016;6:19819. | ||
Sreevalsan S, Safe S. Reactive oxygen species and colorectal cancer. Curr Colorectal Cancer Rep. 2013;9(4):350–357. | ||
Trachootham D, Alexandr J, Huang P. Targeting cancer cells by ROS-mediated mechanism: a radical therapeutic approach. Nat Rev Drug Discov. 2009;8(7):579–591. | ||
Gomez-Lazaro M, Galindo MF, de Melero-Fernandez Mera RM, et al. Reactive oxygen species and p38 mitogen-activated protein kinase activate Bax to induce mitochondrial cytochrome c release and apoptosis in response to malonate. Mol Pharmacol. 2007;71(3):736–743. | ||
Wang S, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, Kalyanaraman B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. Intermediacy of H2O2 and p53-dependent pathway. J Biol Chem. 2004;279(24):25535–25543. | ||
Zhu S, Li T, Tan J, et al. Bax is essential for death receptor-mediated apoptosis in human colon cancer cells. Cancer Biother Radiopharm. 2012;27(9):577–581. | ||
Cherbonnel-Lasserre C, Dosanjh ML. Suppression of apoptosis by overexpression of Bcl2 or BclxL promotes survival and mutagenesis after oxidative damage. Biochimie. 1997;79(9–10):613–617. | ||
Bonnotte B, Favre N, Moutet M, et al. Bcl2-mediated inhibition of apoptosis prevents immunogenicity and restores tumorigenicity of spontaneously regressive tumors. J Immunol. 1998;161(13):1433–1438. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.