Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
Novel MYBPC3 Mutations in Indian Population with Cardiomyopathies
Authors Rani DS ![]() , Kasala A, Dhandapany PS, Muthusami U, Kunnoth S, Rathinavel A, Ayapati DR, Thangaraj K
, Kasala A, Dhandapany PS, Muthusami U, Kunnoth S, Rathinavel A, Ayapati DR, Thangaraj K ![]()
Received 17 March 2023
Accepted for publication 11 August 2023
Published 20 September 2023 Volume 2023:16 Pages 883—893
DOI https://doi.org/10.2147/PGPM.S407179
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Deepa Selvi Rani,1 Apoorva Kasala,1 Perundurai S Dhandapany,2 Uthiralingam Muthusami,3 Sreejith Kunnoth,3 Andiappan Rathinavel,4 Dharma Rakshak Ayapati,5 Kumarasamy Thangaraj1,6
1Department of Population and Medical Genomics, CSIR-Centre for Cellular and Molecular Biology, Hyderabad, Telangana, India; 2Department of Cardiovascular Biology and Medicine, Institute for Stem Cell Science and Regenerative Medicine, Bangalore, Karnataka, India; 3Department of Advanced Zoology and Biotechnology, Loyola College, Chennai, Tamil Nadu, India; 4Department of Cardiology, Government Rajaji Hospital, Madurai, Tamil Nadu, India; 5Department of Cardiology, Nizam’s Institute of Medical Sciences, Hyderabad, Telangana, India; 6DBT-Centre for DNA Fingerprinting and Diagnostics, Hyderabad, Telangana, India
Correspondence: Deepa Selvi Rani; Kumarasamy Thangaraj, CSIR-Centre for Cellular and Molecular Biology, Uppal Road, Hyderabad, 500 007, India, Tel +91-40-271926, Email [email protected]; [email protected]
Background: Mutations in Myosin Binding Protein C (MYBPC3) are one of the most frequent causes of cardiomyopathies in the world, but not much data are available in India.
Methods: We carried out targeted direct sequencing of MYBPC3 in 115 hypertrophic (HCM) and 127 dilated (DCM) cardiomyopathies against 197 ethnically matched healthy controls from India.
Results: We detected 34 single nucleotide variations in MYBPC3, of which 19 were novel. We found a splice site mutation [(IVS6+2T) T>G] and 16 missense mutations in Indian cardiomyopathies [5 in HCM; E258K, T262S, H287L, R408M, V483A: 4 in DCM; T146N, V321L, A392T, E393K and 7 in both HCM and DCM; L104M, V158M, S236G, R272C, T290A, G522E, A626V], but those were absent in 197 normal healthy controls. Interestingly, we found 7 out of 16 missense mutations (V158M, E258K, R272C, A392T, V483A, G522E, and A626V) in MYBPC3 were altering the evolutionarily conserved native amino acids, accounted for 8.7% and 6.3% in HCM and DCM, respectively. The bioinformatic tools predicted that those 7 missense mutations were pathogenic. Moreover, the co-segregation of those 7 mutations in families further confirmed their pathogenicity. Remarkably, we also identified compound mutations within the MYBPC3 gene of 6 cardiomyopathy patients (5%) with more severe disease phenotype; of which, 3 were HCM (2.6%) [(1. K244K + E258K + (IVS6+2T) T>G); (2. L104M + G522E + A626V); (3. P186P + G522E + A626V]; and 3 were DCM (2.4%) [(1. 5’UTR + A392T; 2. V158M+G522E; and 3.V158M + T262T + A626V].
Conclusion: The present comprehensive study on MYBPC3 has revealed both single and compound mutations in MYBPC3 and their association with disease in Indian Population with Cardiomyopathies. Our findings may perhaps help in initiating diagnostic strategies and eventually recognizing the targets for therapeutic interventions.
Keywords: MYBPC3, cardiomyopathy, sarcomere genes, HCM, DCM
Introduction
Cardiac Myosin Binding Protein C (MyBP-C_OMIM-600958), one of the thick filaments exhibited across the C zone of A-bands of sarcomeres, binds ß-myosin (ß-MYH7_OMIM-160710) in thick filaments and titin (TTN_OMIM-188840) in elastic filaments.1,2 It serves as a control that limits cross-bridge interactions between myosin and actin.3,4 Phosphorylation of MYBPC3 modulates contraction and is believed to play both structural and regulatory functions. A total of 3 isoforms of MyBP-C (a cardiac and two skeletal) have been reported, all 3 share a conserved region composed of 7 IgI (immunoglobulin) and 3 FnIII (fibronectin type III) domains.4 The cardiac isoform cMyBP-C contains a unique IgI domain (C0) at the N-terminus and four distinctive phosphorylation sites and an exclusive proline-rich 25 residue insertion at the C5 domain.5,6 Cardiomyopathy (CM), a heart muscle disease, is classified by its morphological features leading to subtypes called hypertrophic (HCM), dilated (DCM), left ventricular noncompaction (LVNC), restrictive (RCM), and arrhythmogenic right ventricular cardiomyopathy (ARVD/C).7,8 The former two (HCM and DCM) are the most frequent forms of cardiomyopathies, usually affecting the cardiac wall thickness, chamber size and ultimately pumping efficiency.9 Describing features of HCM include a hypertrophied/thickened left ventricle with weakened diastolic relaxation, myocyte disarray, and replacement fibrosis, with an estimated prevalence of 1 in 500.10 HCM is known as a “disease of the sarcomere”. Sarcomere consists of thick filaments of myosin and thin filaments of actin, tropomyosin, troponin complex, along with the assembly proteins cardiac myosin binding protein C and titin. To date, hundreds of mutations in sarcomere genes have been reported to cause cardiomyopathies,10–24 most of the mutations (~75%) were found in HCM and a few mutations (~10–16%) were in hereditary DCM. Mutations were predominantly reported in two sarcomere genes: ß-Myosin heavy chain (MYH7) and Myosin binding protein C (MYBPC3). Defining characteristics of DCM are a left ventricular dilatation, myocardial fibrosis, and myocyte disease, which affect systolic function with an estimated prevalence of 1 in 2500.13,17 Currently, more than 40 genes, TTN,25 LMNA,26 DES27 RBM20, etc.,28 along with sarcomere genes were reported to cause familial DCM.2,14,29–35 The essence of mutation screening in disease genetics mostly relayed on the interpretation of genotype and phenotype correlation. Sometimes the factors that govern the variable phenotypic expressions are largely due to unknown factors like epigenetics, environment, lifestyle, etc., which may also possibly implicate a significant role in disease phenotypes.9,36 Mutations in myosin binding protein C (MYBPC3) gene, one of the most frequent causes of cardiomyopathies, studied extensively in various other populations9–19,22,36 but not much studied in the Indian population with cardiomyopathies.37,38 Therefore, here, we performed a targeted screening of the MYBPC3 gene in 242 cardiomyopathy patients against 197 controls (ethnically matched healthy individuals).
Materials and Methods
Study Population and Ethical Approval
The Institutional Ethical Committees (IECs) of CSIR-Centre for Cellular and Molecular Biology (CCMB) and other two participating hospitals [(1) Government Rajaji Hospital (GH), Madurai, India, and (2) Nizam’s Institute of Medical Sciences (NIMS), Hyderabad, India] have approved the study (Table 1). We obtained informed written consent from all the participated individuals before sample collection. We then collected ~5.0 mL of blood samples from each of the 242 patients consisting of 115 HCM, and 127 DCM along with 197 ethnically matched controls (who are healthy individuals without heart problems) (Table 1). To get permission to research human subjects the required guidelines and regulations were followed according to the principles outlined in the Declaration of Helsinki, the World Medical Association.
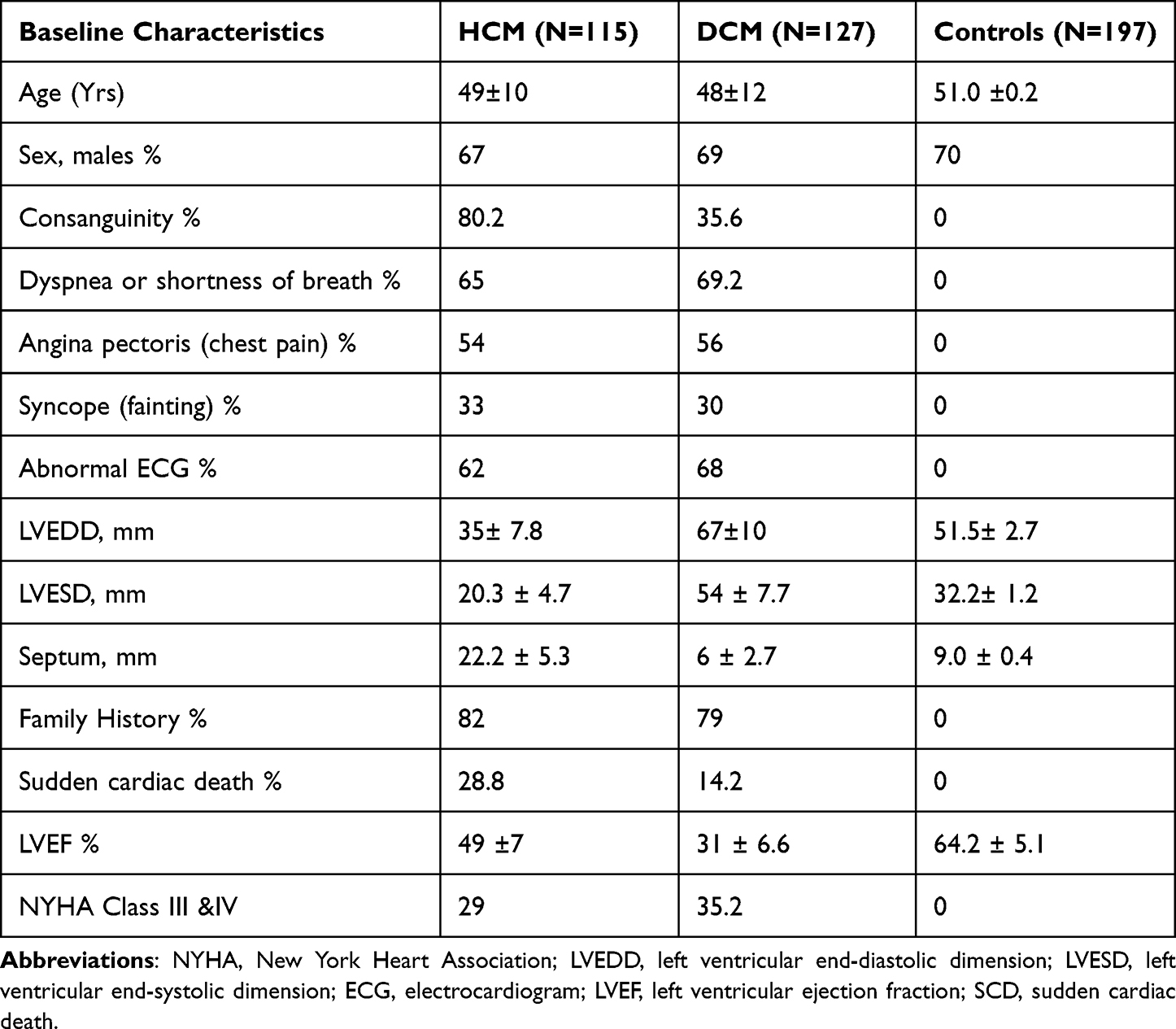 |
Table 1 Baseline Clinical Characteristics of HCM and DCM Patients along with Controls |
Genomic DNA
DNA was isolated from the peripheral blood of all cardiomyopathy patients and normal healthy controls using a standard protocol as follows. To the peripheral blood (5.0mL), we added 15mL of erythrocyte lysis buffer [containing 10mM Tris at pH 8.0, 320mM Sucrose, 5mM MgCl2, and 1% Triton X-100; Sigma Chemical Company, St. Louis, MO] for 5 minutes to lysis the erythrocytes. Leucocytes were pelleted by spinning for 5 minutes at 500g in a centrifuge. Leucocytes were lysed using 8mL of leucocyte lysis buffer (400mM Tris, 60mM EDTA, 150mM NaCl, and 1% SDS; Sigma Chemical Company). To this lysate, 2.0mL of 5M sodium perchlorate (E. Merck, Darmstadt, Germany) was added and mixed thoroughly for 2–3 min. DNA was precipitated using isopropanol, after extracting once with phenol:chloroform (1:1) and then with chloroform. DNA was washed 2 times with 70% ethanol, and the pellet was dissolved in the TE (10mM Tris at pH 8.0 and 1mM EDTA) buffer.
Polymerase Chain Reaction (PCR) and Sequence Analysis
The targeted primer sequences for PCR (Supplement Table S1) covering the exons, exon-intron boundaries of MYBPC3 were designed and synthesized using an ABI 392 oligo synthesizer (Perkin–Elmer, Foster City, CA). Using 50ng of genomic DNA as a template, Polymerase Chain Reactions (PCR) were carried out using 5pM of both forward and reverse primers (Supplementary Table S1), 200mM dNTPs, 10X PCR buffer containing 1.5mM MgCl2, and 1U of AmpliTaq Gold (Perkin–Elmer). Amplifications were carried out in a thermal cycler (MJ Research, Waltham, MA, USA) using the following cycling conditions: 94°C for 5 min, 35 cycles at 94°C for 1 min, 55–60°C for 1 min, 72°C for 1 min, followed by a final extension at 72°C for 10 min. Resulted PCR products (amplicons) were checked using 2% agarose gel electrophoresis. The PCR products were then purified using ExoSAP-IT enzyme (USB Corporation, USA) and subjected to cycle sequencing reaction in GeneAmp 9700 thermal cycler using Big Dye Terminator ready reaction cycle sequencing kit (Applied Biosystems, Foster City, USA). The cycle sequencing products were precipitated with ethanol, dried and dissolved in Hi-Di formamide, and bi-directional sequencing was performed in an ABI 3730XL automated DNA analyzer (Applied Biosystems, Foster City, USA). The MYBPC3 gene sequences obtained were noted and aligned with the Reference MYBPC3 gene sequences using Sequence Analyzer and Auto Assembler tools. We followed the American College of Medical Genetics and Genomics (ACMG) guidance for the interpretation of sequence variants.39 All the mismatched nucleotide sequences were carefully noted and compared against 197 healthy control sequences to detect their significance. We used two bioinformatics tools, Polyphen-2 (Polymorphism phenotyping 2)40 and SIFT (Sorting intolerant from tolerant),41 to predict the possible pathogenic effects of missense mutations.
Results
In the present study, we identified 34 genetic variants in MYBPC3 gene (Table 2), of which 19 were novel (https://www.ncbi.nlm.nih.gov/SNP/snp_viewTablecgi?handle=THANGARAJ_DEEPA_CCMB). We found a splice site mutation [(IVS6+2T) T>G] and 16 were missense mutations [5 in HCM (E258K, T262S, H287L, R408M, V483A), 4 in DCM (T146N, V321L, A392T, E393K) and 7 in both HCM and DCM (L104M, V158M, S236G, R272C, T290A, G522E, A626V)], but all those 16 mutations were absent in 197 controls (Figure 1A and B, Supplementary Figure S1; Table 2). We found 7 out of 16 heterozygous missense mutations in MYBPC3 [V158M, E258K, R272C, A392T, V483A, G522E, and A626V in MYBPC3 (Figure 1C and Table 3)] were altering the native evolutionarily conserved amino acids (Figure 1D). The Polyphen-2 or SIFT bioinformatics tools predicted that those 7 missense mutations were pathogenic (Table 2). The co-segregation of those 7 missense mutations in families also confirmed their pathogenicity, accounting for 8.7% and 6.3% in HCM and DCM, respectively. Interestingly, in the present study, 6 patients [3 HCM and 3 DCM (Table 4)] with more severe disease phenotypes have shown compound mutations in the MYBPC3 gene. They were as follows; one was a 51-year-old HCM patient, who carried allelic heterogeneity by possessing three heterozygous mutations in the MYBPC3; [a splice site mutation (IVS6+2T) T>G, a missense mutation E258K, and a silent mutation K244K (Figure 1C)]. Another was a 53-year-old HCM patient with three missense mutations; L104M, G522E, and A626V. The third was a 49-year-old HCM patient, who possessed two missense mutations, G522E, A626V, and a silent mutation P186P. The following are the 3 dilated cardiomyopathy patients with more than one mutation in the MYBPC3 gene. The 1st DCM patient carried a 5’UTR mutation and a missense mutation A392T. The 2nd DCM patient carried 2 missense mutations: V158M and G522E. The 3rd DCM patient possessed 3 missense mutations: V158M, T262T and A626V (Figure 1 and Table 3). In addition, we too detected 10 silent mutations exclusively in cardiomyopathy patients [3 in HCM (P186P, K24K, and A482A), 5 in DCM; A58A, T146T, P150P, G291G, D303D, 2 in both HCM and DCM; T262T and E402E], with unknown implication (Figure S1; Table 2).
 |
Table 2 Comparing the Allele Frequencies of Detected Mutations in the MYBPC3 Gene of Indian Population vs Other Populations |
 |
Table 3 Missense Mutations in Myosin Binding Protein C (MYBPC3) Gene |
 |
Table 4 Compound Mutations in Myosin Binding Protein C (MYBPC3) Gene |
 |
Figure 1 (A) Schematic representation of the MYBPC3 structure. (B) Highlighted are the observed exonic, and splice sites variations. (The 10 amino acid substitutions, and 1 splice-site mutation were indicated in red color). (C) Electropherograms (arrows) showing 10 missense mutations [V158M, E258K, R272C, H287L, V321L, E392T, R408M, V483A, and a G522E, A626V), and a splice-donor mutation (T>G[IVS6+2T]) in the MYBPC3 gene. (D) Multiple alignments of amino acid sequences in the MYBPC3gene of several species, showing that those were highly conserved across many species. |
Discussion
One of the interesting outcomes of the present study was the identification of compound variations within the MYBPC3 gene of 6 cardiomyopathy patients with more severe disease presentation. Of which, 3 were HCM [(1. K244K + E258K + (IVS6+2T) T>G), (2. L104M + G522E + A626V) and (3. P186P + G522E + A626V)]; and 3 were DCM [(1. 5’UTR + A392T, (2. V158M+G522E), and (3. V158M + T262T + A626V)] (Table 2 and Figure 1), accounting for 2.6% and 2.4% in Indian HCM and DCM patients, respectively. Further analysis of family members of all 6 patients with compound variations has demonstrated the segregation of those variations along with the disease. When we extracted clinical data from hospital records, we noticed increased susceptibility to ventricular arrhythmias in affected patients and a history of sudden cardiac deaths (SCDs) in the families. Studies suggested that the patients possessing compound variations in MYBPC3,21,42,43 have shown dosage-dependent effects,42,44–46 therefore, required constant monitoring to avoid adverse outcomes.36,47–49
We studied a few familial samples to understand the co-segregation of variations in families and their association with disease phenotypes. As, the genotype of the patient’s family members are extremely important to understand the genotype-phenotype correlation,23,33,34 and how it might modify the clinical course and prognosis of the disease along with other factors like epigenetics, lifestyle, environment, etc.50–53 We understood from our study that most of the patient’s family members possessing mutations in MYBPC3 gene did not show any symptom when they were below 15 years of age, and they all started having the symptom in their 3rd decade of life, ie, late onset of the disease symptoms. Therefore, we too strongly suggest that the mutations in MYBPC3 showed slightly lower penetrance, delayed onset, and milder forms of disease progression.48,54,55
Studies reported deletions, insertions, and splice site variations in the MYBPC3 gene.56–61 In our previous study on MYBPC3, we observed a founder 25bp del in MYBPC3 of HCM, DCM, and RCM and evaluated its distribution among the South-Asian population, and pinpointed an association with familial cardiomyopathies with an increased chance of heart failure [overall OR, 6.99; p = 4 × 10 (−11)].23 In the present study, we report 16 missense mutations in the MYBPC3 gene of Indian cardiomyopathy patients and those were absent in 197 normal healthy controls (Table 2 and Figure 1A). Remarkably, we found that 7 out of 16 heterozygous missense mutations in MYBPC3 (V158M, E258K, R272C, A392T, V483A, G522E, and A626V) (Figure 1C) were altering the native evolutionarily conserved amino acids (Figure 1D), accounting for 8.7% and 6.3% in HCM and DCM, respectively. Polyphen-2 or SIFT bioinformatics tools also predicted that those 7 missense mutations were pathogenic (Table 2). The co-segregation of those mutations in families also confirmed their pathogenicity. Except for two missense mutations, R272C and V483A, the remaining 5 missense mutations (V158M, E258K, A392T, G522E, and A626V) were also detected along with other variations in the MYBPC3 gene as compound mutations (Table 4).
In our previous studies, we reported a few genetic variations in other sarcomere genes of Indian cardiomyopathy patients: Tropomyosin (α-TMP1),45 Troponin I3 (TNNI3),24,62 Troponin T2 (TNNT2),34,63 Actin (ACTC),64 Myosin (β-MYH7),35,65,66 and MYL2 & MYL3.67 Therefore, our present and previous studies clearly illustrated the prevalence and spectrum of variations in sarcomere genes and their associations in Indian populationwith cardiomyopathies.23,24,34,35,45,62–67
Our categorization as pathogenic variations relied on the result that we could detect those missense mutations exclusively in patients and their family members. In the present study, we found 19 novel SNPs in the MYBPC3 gene, along with accumulated compound variations that were responsible for more severe disease phenotypes in Indian cardiomyopathies. Our present comprehensive genetic analysis of the MYBPC3 gene in Indian HCM and DCM patients has given important insight into risk stratification. Based on our present and previous studies on sarcomere genes in Indian population,23,24,34,35,45,62–67 we fully agree that the mutations in myosin heavy chain and myosin binding protein C are the frequent causes of cardiomyopathies; therefore, these two genes should be screened first, secondly, the thin filament regulatory genes (TNNT2, TNNI3, and TPM1), and finally, the rarely involved genes like TTN, MYL2, MYL3, and ACTC. Importantly, it is not sufficient and advisable to screen only the known reported mutations, because we could miss unique, rare, and population-specific disease-causing mutations.
Limitations of the Study
For any genetic study, it is crucial to extend the study with their family members to understand the inheritance pattern and to correlate the mutations with the disease phenotype. However, generally, it is tough to collect the samples from family member for genetic analysis, mainly because the patients are not strictly adhering to the follow-up procedures that would have allowed us to invite the family members for counselling and further genetic studies. Though we identified missense mutations in many individuals, we could not establish the genotype-to-phenotype correlation within the family in many cases. Thus, for some patients, performing genetic testing, understanding their family history, and giving counselling is not an easy task!
Conclusion
The present comprehensive study has revealed both single and compound mutations in MYBPC3 and their association with disease in Indian population with cardiomyopathies. Our findings may perhaps help in initiating diagnostic strategies and eventually recognizing the targets for therapeutic interventions.
Acknowledgments
We thank all cardiomyopathy patients both HCM and DCM and their family members and all the healthy volunteers as normal controls, who participated in this study.
Funding
Rani DS has been supported by the CSIR-CCMB, Hyderabad, Telangana, India. K Thangaraj has been supported by the JC Bose Fellowship (JCB/2019/000027) from SERB, DST, and The Government of India. However, the funders had no role in designing the study, the collection of data, the analysis of sequence data, the decision to publish, or the preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Previs MJ, Beck Previs S, Gulick J, Robbins J, Warshaw DM. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science. 2012;337(6099):1215–1218. doi:10.1126/science.1223602
2. Daehmlow S, Erdmann J, Knueppel T, et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;298(1):116–120. doi:10.1016/s0006-291x(02)02374-4
3. Lee KH, Sulbaran G, Yang S, et al. Interacting-heads motif has been conserved as a mechanism of myosin II inhibition since before the origin of animals. Proc Natl Acad Sci USA. 2018;115(9):E1991–E2000. doi:10.1073/pnas.1715247115
4. Pfuhl M, Gautel M. Structure, interactions and function of the N-terminus of cardiac myosin binding protein C (MyBP-C): who does what, with what, and to whom? J Muscle Res Cell Motil. 2012;33(1):83–94. doi:10.1007/s10974-012-9291-z
5. Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol. 2010;48(5):866–875. doi:10.1016/j.yjmcc.2009.11.014
6. Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14(9):1952–1960. doi:10.1002/j.1460-2075.1995.tb07187.x
7. Barry M, Hall M. Familial cardiomyopathy. Br Heart J. 1962;24(5):613–624. doi:10.1136/hrt.24.5.613
8. Gerull B, Klaassen S, Brodehl A. The genetic landscape of cardiomyopathies. In: Genetic Causes of Cardiac Disease. Switzerland AG: Springer Nature; 2020:45–91.
9. Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001;104(4):557–567. doi:10.1016/j.jacc.2006.09.014
10. Maron BJ, Nichols PF 3rd, Pickle LW, Wesley YE, Mulvihill JJ. Patterns of inheritance in hypertrophic cardiomyopathy: assessment by M-mode and two-dimensional echocardiography. Am J Cardiol. 1984;53(8):1087–1094. doi:10.1016/0002-9149(84)90643-x
11. Alders M, Jongbloed R, Deelen W, et al. The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur Heart J. 2003;24(20):1848–1853. doi:10.1016/s0195-668x(03)00466-4
12. Andersen PS, Havndrup O, Bundgaard H, et al. Genetic and phenotypic characterization of mutations in myosin-binding protein C (MYBPC3) in 81 families with familial hypertrophic cardiomyopathy: total or partial haploinsufficiency. Eur J Hum Genet. 2004;12(8):673–677. doi:10.1038/sj.ejhg.5201190
13. Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994;331(23):1564–1575. doi:10.1056/NEJM199412083312307
14. Ehlermann P, Weichenhan D, Zehelein J, et al. Adverse events in families with hypertrophic or dilated cardiomyopathy and mutations in the MYBPC3gene. BMC Med Genet. 2008;9(1):95. doi:10.1186/1471-2350-9-95
15. Erdmann J, Daehmlow S, Wischke S, et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet. 2003;64(4):339–349. doi:10.1034/j.1399-0004.2003.00151.x
16. Frank-Hansen R, Page SP, Syrris P, McKenna WJ, Christiansen M, Andersen PS. Micro-exons of the cardiac myosin binding protein C gene: flanking introns contain a disproportionately large number of hypertrophic cardiomyopathy mutations. Eur J Hum Genet. 2008;16(9):1062–1069. doi:10.1038/ejhg.2008.52
17. Michels VV, Moll PP, Miller FA, et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326(2):77–82. doi:10.1056/NEJM199201093260201
18. Nanni L, Pieroni M, Chimenti C, et al. Hypertrophic cardiomyopathy: two homozygous cases with “typical” hypertrophic cardiomyopathy and three new mutations in cases with progression to dilated cardiomyopathy. Biochem Biophys Res Commun. 2003;309(2):391–398. doi:10.1016/j.bbrc.2003.08.014
19. Niimura H, Patton KK, McKenna WJ, et al. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002;105(4):446–451. doi:10.1161/hc0402.102990
20. Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107(17):2227–2232. doi:10.1161/01.CIR.0000066323.15244.54
21. Van Driest SL, Vasile VC, Ommen SR, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44(9):1903–1910. doi:10.1016/j.jacc.2004.07.045
22. Watkins H, Conner D, Thierfelder L, et al. Mutations in the cardiac myosin binding protein–C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11(4):434–437. doi:10.1038/ng1295-434
23. Dhandapany PS, Sadayappan S, Xue Y, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41(2):187–191. doi:10.1038/ng.309
24. Rani DS, Nallari P, Priyamvada S, Narasimhan C, Singh L, Thangaraj K. High prevalence of Arginine to Glutamine substitution at 98, 141 and 162 positions in Troponin I (TNNI3) associated with hypertrophic cardiomyopathy among Indians. BMC Med Genet. 2012;13(1):69. doi:10.1186/1471-2350-13-69
25. Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30(2):201–204. doi:10.1038/ng815
26. Keller H, Finsterer J, Steger C, et al. Novel c.367_369del LMNA mutation manifesting as severe arrhythmias, dilated cardiomyopathy, and myopathy. Heart Lung. 2012;41(4):382–386. doi:10.1016/j.hrtlng.2011.07.007
27. Brodehl A, Dieding M, Biere N, et al. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J Mol Cell Cardiol. 2016;91:207–214. doi:10.1016/j.yjmcc.2015.12.015
28. Gaertner A, Klauke B, Felski E, et al. Cardiomyopathy-associated mutations in the RS domain affect nuclear localization of RBM20. Hum Mutat. 2020;41(11):1931–1943. doi:10.1002/humu.24096
29. Kamisago M, Sharma SD, DePalma SR, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343(23):1688–1696. doi:10.1056/NEJM200012073432304
30. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531–547. doi:10.1038/nrcardio.2013.105
31. Hershberger RE, Lindenfeld J, Mestroni L, et al. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J Card Fail. 2009;15(2):83–97. doi:10.1016/j.cardfail.2009.01.006
32. Dhandapany PS, Razzaque MA, Muthusami U, et al. RAF1 mutations in childhood-onset dilated cardiomyopathy. Nat Genet. 2014;46(6):635–639. doi:10.1038/ng.2963
33. Morita H, Larson MG, Barr SC, et al. Single-gene mutations and increased left ventricular wall thickness in the community: the Framingham Heart Study. Circulation. 2006;113(23):2697–2705. doi:10.1161/circulationaha.105.593558
34. Rani DS, Dhandapany PS, Nallari P, Narasimhan C, Thangaraj K. A novel arginine to tryptophan (R144W) mutation in troponin T (cTnT) gene in an Indian multigenerational family with dilated cardiomyopathy (FDCM). PLoS One. 2014;9(7):e101451. doi:10.1371/journal.pone.0101451
35. Rani DS, Vijaya Kumar A, Nallari P, et al. Novel mutations in beta-MYH7 gene in Indian patients with dilated cardiomyopathy. CJC Open. 2022;4(1):1–11. doi:10.1016/j.cjco.2021.07.020
36. Marian AJ. Phenotypic plasticity of sarcomeric protein mutations. J Am Coll Cardiol. 2007;49(25):2427–2429. doi:10.1016/j.jacc.2007.04.016
37. Bashyam MD, Purushotham G, Chaudhary AK, et al. A low prevalence of MYH7/MYBPC3 mutations among familial hypertrophic cardiomyopathy patients in India. Mol Cell Biochem. 2012;360(1–2):373–382. doi:10.1007/s11010-011-1077-x
38. Waldmuller S, Sakthivel S, Saadi AV, et al. Novel deletions in MYH7 and MYBPC3 identified in Indian families with familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2003;35(6):623–636. doi:10.1016/s0022-2828(03)00050-6
39. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
40. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi:10.1038/nmeth0410-248
41. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi:10.1093/nar/gkg509
42. Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005;42(10):e59. doi:10.1136/jmg.2005.033886
43. Lekanne Deprez RH, Muurling-Vlietman JJ, Hruda J, et al. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J Med Genet. 2006;43(10):829–832. doi:10.1136/jmg.2005.040329
44. Gaertner A, Bloebaum J, Brodehl A, et al. The combined human genotype of truncating TTN and RBM20 Mutations is associated with severe and early onset of dilated cardiomyopathy. Genes. 2021;12(6). doi:10.3390/genes12060883
45. Selvi Rani D, Nallari P, Dhandapany PS, et al. Coexistence of digenic mutations in both thin (TPM1) and thick (MYH7) filaments of sarcomeric genes leads to severe hypertrophic cardiomyopathy in a South Indian FHCM. DNA Cell Biol. 2015;34(5):350–359. doi:10.1089/dna.2014.2650
46. Ito K, Patel PN, Gorham JM, et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc Natl Acad Sci USA. 2017;114(29):7689–7694. doi:10.1073/pnas.1707741114
47. Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008;51(14):1369–1374. doi:10.1016/j.jacc.2007.11.071
48. Girolami F, Ho CY, Semsarian C, et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010;55(14):1444–1453. doi:10.1016/j.jacc.2009.11.062
49. Myasnikov R, Brodehl A, Meshkov A, et al. The double mutation DSG2-p. S363X and TBX20-p.D278X is associated with left ventricular non-compaction cardiomyopathy: case report. Int J Mol Sci. 2021;22(13). doi:10.3390/ijms22136775
50. Wang P, Zou Y, Fu C, Zhou X, Hui R. MYBPC3 polymorphism is a modifier for expression of cardiac hypertrophy in patients with hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2005;329(2):796–799. doi:10.1016/j.bbrc.2005.02.004
51. Liu J, Zhao S, Yu S, et al. Patterns of replacement fibrosis in hypertrophic cardiomyopathy. Radiology. 2022;302(2):298–306. doi:10.1148/radiol.2021210914
52. Jurgens SJ, Choi SH, Morrill VN, et al. Analysis of rare genetic variation underlying cardiometabolic diseases and traits among 200,000 individuals in the UK Biobank. Nat Genet. 2022;54(3):240–250. doi:10.1038/s41588-021-01011-w
53. Biddinger KJ, Jurgens SJ, Maamari D, et al. Rare and common genetic variation underlying the risk of hypertrophic cardiomyopathy in a national biobank. JAMA Cardiol. 2022;7(7):715–722. doi:10.1001/jamacardio.2022.1061
54. van Velzen HG, Schinkel AFL, Oldenburg RA, et al. Clinical characteristics and long-term outcome of hypertrophic cardiomyopathy in individuals with a MYBPC3 (Myosin-Binding Protein C) founder mutation. Circ Cardiovasc Genet. 2017;10(4). doi:10.1161/circgenetics.116.001660
55. Page SP, Kounas S, Syrris P, et al. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long-term outcome. Circ Cardiovasc Genet. 2012;5(2):156–166. doi:10.1161/circgenetics.111.960831
56. Xin B, Puffenberger E, Tumbush J, Bockoven JR, Wang H. Homozygosity for a novel splice site mutation in the cardiac myosin-binding protein C gene causes severe neonatal hypertrophic cardiomyopathy. Am J Med Genet A. 2007;143A(22):2662–2667. doi:10.1002/ajmg.a.31981
57. Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338(18):1248–1257. doi:10.1056/NEJM199804303381802
58. Bonne G, Carrier L, Bercovici J, et al. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet. 1995;11(4):438–440. doi:10.1038/ng1295-438
59. Rottbauer W, Gautel M, Zehelein J, et al. Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy. characterization of cardiac transcript and protein. J Clin Invest. 1997;100(2):475–482. doi:10.1172/JCI119555
60. Moolman JA, Reith S, Uhl K, et al. A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation. 2000;101(12):1396–1402. doi:10.1161/01.cir.101.12.1396
61. Carrier L, Bonne G, Bahrend E, et al. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res. 1997;80(3):427–434. doi:10.1161/01.res.0000435859.24609.b3
62. Ramachandran G, Kumar M, Selvi Rani D, et al. An in-silico analysis of troponin I mutations in hypertrophic cardiomyopathy of Indian origin. PLoS One. 2013;8(8):e70704. doi:10.1371/journal.pone.0070704
63. Rani DS, Nallari P, Dhandapany PS, et al. Cardiac Troponin T (TNNT2) mutations are less prevalent in Indian hypertrophic cardiomyopathy patients. DNA Cell Biol. 2012;31(4):616–624. doi:10.1089/dna.2011.1366
64. Rangaraju A, Rani DS, Satyanarayana M, Calambur N, Swapna N, Nallari P. Genetic variations of alpha-cardiac actin and cardiac muscle LIM protein in hypertrophic cardiomyopathy in South India. Exp Clin Cardiol. 2012;17(1):26–29.
65. Rani DS, Nallari P, Narasimhan C, Thangaraj K. Novel variations in β-myosin heavy-chain gene (β-MYH7) and its association in south Indian women with cardiomyopathies. Ind J Cardio Dis Women. 2019;04(02):072–078. doi:10.1055/s-0039-1694829
66. Rani DS, Veera Subhashini G, Sharadhadevi A, Cyril E, Thangaraj K. A missense mutation (R723H) in the head motor domain of β-MYH7 gene in an Indian HCM patient and phenotypic plasticity. J Clin Cardiol Cardiovasc Interv. 2021;4(17). doi:10.31579/2641-0419/219
67. Rani DS, Nallari P, Rani J, et al. A complete absence of missense mutation in myosin regulatory and essential light chain genes of South Indian hypertrophic and dilated cardiomyopathies. Cardiology. 2018;141(3):156–166. doi:10.1159/000495027
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.