")
Back to Journals » OncoTargets and Therapy » Volume 13
Novel mTOR Inhibitor Enhances the Sensitivity of Hepatocellular Carcinoma Cells to Molecular Targeting Agents
Authors Feng YQ, Li BA, Feng F , Chen YS, Ren YX, Zhang H, Cao S
Received 4 January 2020
Accepted for publication 2 July 2020
Published 27 July 2020 Volume 2020:13 Pages 7165—7176
DOI https://doi.org/10.2147/OTT.S244474
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
Ying-Qi Feng,1,* Bo-An Li,2,* Fan Feng,2,* Yong-Shou Chen,1 Yi-Xin Ren,1 Heng Zhang,1 Shuang Cao1
1Key Laboratory for Green Chemical Process of Ministry of Education, School of Chemical Engineering and Pharmacy, Wuhan Institute of Technology, Wuhan 430072, People’s Republic of China; 2Center for Clinical Laboratory, The Fifth Medical Center, General Hospital of Chinese PLA, Beijing 100039, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shuang Cao; Heng Zhang
Key Laboratory for Green Chemical Process of Ministry of Education, School of Chemical Engineering and Pharmacy, Wuhan Institute of Technology, Wuhan 430072, People’s Republic of China
Tel/ Fax +86-18701418117
; +86-15994288097
Email [email protected]; [email protected]
Background: Although molecular-targeted agents are still the first choice for advanced hepatocellular carcinoma (HCC) treatment, the therapeutic efficacy of these agents is not satisfactory. Recently, the mammalian target of rapamycin (mTOR) is considered to be a promising molecular target that can enhance the sensitivity of HCC cells to antitumor therapy. However, the reported mTOR inhibitors have some shortcomings, and novel mTOR inhibitors need to be developed to enhance the antitumor effect of molecularly targeted agents on advanced HCC.
Methods: In this study, five small-molecular compounds that could serve as potential mTOR-specific inhibitors were identified by virtual screening. The activity of tert-butyl (4-(9-(2-(1,3-dioxolan-2-yl)ethyl)-6-morpholino-9H-purin-2-yl)phenyl)carbamate (compound 4) was measured by enzyme test and Western blot, and its antitumor effect on HCC was examined in nude mice subcutaneous tumor model.
Results: The results showed that 4 is the most effective one in inhibiting the activation of mTOR kinase (mTOR IC50 = 17.52± 3.67 nmol/L) among the five lead compounds. Further research in this study indicated that treatment with 4 enhanced the sensitivity of HCC cells to the molecular-targeted agents, such as sorafenib, regorafenib, lenvatinib, anlotinib, and apatinib. In addition, this research indicated that mTOR was correlated with the poor prognosis in patients with advanced HCC who received sorafenib.
Conclusion: Our study identified a new type of small-molecular inhibitors of mTOR and confirmed their ability to enhance the antitumor effect of molecular-targeted agents on advanced HCC.
Keywords: hepatocellular carcinoma, mTOR, molecular-targeted agents, virtual screening
Introduction
Due to the high infection rates of hepatitis viruses (HBV [hepatitis B virus] or HCV [hepatitis C virus]) in China, HCC is now one of the most fatal diseases.1–3 Moreover, most diagnosed HCC patients are often at the advanced stage (the Barcelona Clinic Liver Cancer [BCLC] stage C of disease) and unsuitable for receiving surgeries or liver transplant; chemotherapy and radiotherapy are the only few treatments for them.4–6 Until now, given the multi-drug resistance (MDR) in HCC chemotherapy, molecular-targeted agents are still the first-line or second-line treatment for HCC patients.7–9 However, molecular-targeted therapies for advanced HCC are also facing many challenges: (1) there are individual differences in the sensitivity of patients to molecular-targeted agents and only a few patients are sensitive to them;10 (2) patients are prone to developing a resistance to molecular-targeted agents during the treatment;10 (3) molecular-targeted therapies require large doses of drugs (for example, a dose of 800 mg sorafenib per day), which not only imposes a huge economic burden on patients but also seriously affects their health condition.11 Therefore, it is very urgent to study and improve the sensitivity of HCC to molecular-targeted therapies in order to reduce the drug doses while retaining their antitumor efficacy.
As it is known, mTOR is a key regulator in cancer cells to regulate important physical processes in response to the changes of microenvironment, such as nutrient alterations, cell stress and hormone.3,12 Abnormally activated mTOR is involved in the development of human cancer,13–15 and inhibiting mTOR may have antitumor effect.16,17 In recent years, a number of ATP-competitive mTOR inhibitors have been identified and developed.18–20 However, nearly all researches using selective mTOR inhibitors alone for antitumor treatment are only in clinical stage I or II until now.21–23 More and more studies have shown that use of specific mTOR inhibitors alone has a limited inhibitory effect on tumors because of the compensatory effect of certain pathways, especially in the treatment of HCC.24–29 Although the dual PI3K/mTOR inhibitors have more significant antitumor effect than mTOR-specific inhibitors, they also lead to greater side effects due to affecting mTOR and PI3K signaling pathways at the same time.30,31
Recently, multiple studies have shown that mTOR expression is related to poor prognosis in patients with advanced cancer, and mTOR inhibitors can alleviate drug resistance in human cancer cells, which indicates that application of mTOR inhibitors to sensitize cancer cells to other antitumor treatment may be a promising strategy.32,33 In other words, mTOR inhibitors would be better used as tumor sensitizers rather than independent medicines for cancers.
In addition, classical mTOR inhibitors, such as rapamycin and everolimus, have poor performance in pharmacokinetics owning to the large molecular weight (molecular weights > 950) and poor chemical properties (logP > 6). However, ATP-competitive mTOR inhibitors can overcome these defects and are very suitable for treating HCC patients with liver damage.34 In this study, we used structure-based virtual screening and biological evaluation to find new compounds that could serve as selective and ATP-competitive mTOR inhibitors. The inhibitory activity and selectivity of these compounds towards mTOR were analyzed separately.35 The effect of mTOR inhibitors on the antitumor activity of molecular-targeted agents was examined in vitro and in vivo. By revealing that the novel mTOR inhibitors could enhance the antitumor effect of molecular-targeted agents, we not only extended our knowledge about mTOR inhibitors but also provided a new promising therapeutic strategy for more effective HCC treatment.
Materials and Methods
Molecular Docking
Molecular docking was implemented using the surflex-docking package of Sybyl-X 2.1. A cocrystal structure of mTOR with ADP (4JSV) was obtained from the Protein Data Bank. Before docking, 4JSV was prepared by removing water and magnesium ions and extracting the ligand. Addition of hydrogen and charges and treatment of the terminal residues were also performed on 4JSV. Then, the “protomol” was generated using the ligand-based mode, and an appropriate binding pocket was formed. The reliability of the surflex-docking was validated by redocking the original ligand (ADP) into the binding pocket. Next, all of the candidate compounds were docked into the binding pocket, and 20 possible docked conformations were obtained with different scores.36,37 Molecular modeling figure was drawn using PyMOL software (http://www.pymol.org).38
mTOR Enzyme Assay
LANCE® ultra time-resolved fluorescence resonance energy transfer (TR-FRET) assay (Invitrogen, Carlsbad, CA, USA) was used to determine the mTOR kinase activities of all the compounds following the manufacturer’s instructions, with compound GSK2126458 (Selleck, China) as positive control.39 Briefly, mTOR enzyme (0.1 μg/mL, Invitrogen, Carlsbad, CA, USA), ATP (3 μmol/L (μM)), GFP-4EBP1 peptide (0.4 μM), and test compounds were diluted in kinase buffer (50 mM HEPES pH 7.5, 1 mM EGTA, 3 mmol/L (mM) MnCl2, 10 mM MgCl2, 2 mM DTT, and 0.01% Tween-20). The reaction was performed in black 384-well proxiplates (Corning, New York, NY, USA) at room temperature for 1 h, then stopped by adding EDTA to 10 mM. Tb-antiphospho-4EBP1 (Thr37/46) antibody (PerkinElmer, Fremont, CA, USA) was added to each well, and the mixture was incubated at room temperature for 30 min. Test compound concentrations were 10,000, 2500, 625, 156.25, 39.06, 9.77, 2.44, 0.61, 0.15, 0.04 and 0.01 nM. The final DMSO concentration was 1%. A Spectramax 190 reader (Molecular Devices, Valley, CA, USA) was used to measure the intensity of the light in TR-FRET mode (excitation 320 nm, emission 665 nm). All compounds were tested twice, and the results were expressed as the average IC50 (inhibitory concentration 50%) of the two experiments.
Patients and Antitumor Agents
The collection and usage of clinical specimens of HCC were approved by the ethics committee of No. 302nd hospital, Chinese People’s Liberation Army (now named as the fifth medical center of the General Hospital, Chinese People’s Liberation Army). All the related researches were performed in compliance with the Helsinki Declaration. In our previous work (Table S1), a total of 52 HCC cases were included and described. The cDNA samples extracted from the clinical specimens were conserved in our lab at −80°C condition.40,41 As described in our previous work, patients whose tissues were used in this research provided written informed consent.40,42 The clinical specimens obtained from the twice liver puncture (at initial or recurrence) or the autopsy were persevered in our lab and described in our previous work.43 Antitumor agents such as sorafenib, regorafenib, lenvatinib, anlotinib, or apatinib and the small-molecular inhibitors such as mTOR inhibitor (Rapamycin), PI3K/AKT inhibitor (LY294002), MAPK inhibitor (PD98059) or Jak/STAT inhibitor (CP690550) were purchased from Selleck Corporation (Houston, TX, USA). Agents were dissolved by using Dimethyl sulfoxide (DMSO, Sigma Aldrich Corporation, St Louis, MO, USA) in the cell-based experiments and then diluted by using the Dulbecco’s Modified Eagle Medium (DMEM, Hyclone, Thermo Scientific Corporation, Waltham, MA, USA) without Fetal Bovine Serum (FBS, Invitrogen, Thermo Scientific Corporation, Waltham, MA, USA).44,45 According to the methods described by Xie et al and Wang et al, we prepared the formulation of agents (the oral liquids) with PEG (polyethylene glycol) and Tween 80 in animal experiments.46,47
Quantitative Polymerase Chain Reaction
With the cDNA samples from the clinical specimens as temples, the expression level of mTOR in clinical specimens was detected following the qPCR methods. According to the protocol provided by the manufacturer’s instruction (Applied Biosystems, Thermo Fisher Scientific, Corporation, Waltham, MA, USA) and the methods described by Ji et al and Liang et al, the qPCR experiments were performed.48 The β-actin was used in loading control. In the present work, the primers chosen are listed as: (1) mTOR, Forward Sequence 5ʹ-AGCATCGGA TGCTTAGGAGTGG-3ʹ; Reverse Sequence 5ʹ-CAGCCAGTCATCTTTGGAGACC-3ʹ; β-Actin (gene symbol ACTB): Forward Sequence 5ʹ-CACCATTGGCAATGAGCGGTTC-3ʹ; Reverse Sequence 5-AGGTCTTTGCGGATGTCCACGT-3ʹ.
Cell Line and Cell-Survival Examination
The present work did not include any materials obtained directly from human participants and only used MHCC97-H cells purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The usage of the cell lines was permitted by the ethics committee of the Fifth Medical Center, General Hospital of the Chinese PLA (previously named the Beijing 302 Hospital). All experiments were performed according to the Declaration of Helsinki (World Health Organization).45
For proliferation analysis, cells were seeded in 96-well plates (5 × 103 cells per well) (Corning, NY, USA). Cells were cultured in DMEM (complete Dulbecco’s modified Eagle’s medium, Invitrogen, USA) with 10% FBS (fetal bovine serum, Invitrogen, USA) at 37°C with 5% CO2 for 24 h.41 Treat the MHCC97-H cells with indicated concentration (10.0 μmol/L, 3.0 μmol/L, 1.0 μmol/L, 0.3 μmol/L, 0.1 μmol/L, 0.03 μmol/L or 0.01 μmol/L) of molecular targeting agents for 48 h in MTT experiments.49,50 The relative survival-cell number was reflected by OD 490 nm and the inhibitory rates of molecular targeting agents on MHCC97-H cells were calculated as (control group’s OD 490 nm – administration group’s OD 490 nm)/(control group’s OD 490 nm) × 100%. The IC50 values of molecular targeting agents on MHCC97-H cells were calculated by inhibitory rates.51,52
Enzyme-Activation Examination and the Western Blot Experiments
MHCC97-H cells (5x106 cells per 75T flask) were incubated for 24 h following pretreatment with solvent control, compound 4, LY294002, GSK2118436, CP690550 and rapamycin for 12 h. The dosing concentrations of solvent control, compound 4, LY294002, GSK2118436, CP690550 and rapamycin all were set to 1 μmol/L. At the end of the exposure period, cells were lysed in RIPA (Radio Immunoprecipitation Assay) buffer (Cell Signaling Technology, Danvers, MA, USA). Protein concentration was determined using the Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Inc.) according to the manufacturer’s instructions. Protein samples (40 μg) were loaded into each well of a 10–12% polyacrylamide gel and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (Millipore, Billerica, USA). The membrane was blocked with 5% skim milk in Tris-buffered saline with 0.1% Tween-20 and incubated with antibodies (1:1000) at 4°C. β-actin was employed as the loading control. Rabbit antibodies against human phospho-p70S6K (Thr389), p70S6K, phospho-AKT (Ser473), phospho-AKT (Thr308), AKT, phospho-ERK, ERK, phospho-rpS6 (Ser235/6), rpS6, β-actin and secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).53–55
In vivo Bioactivity Assessment of the Lead Compounds
The nude mice model was used to test the bioactivity of candidate compounds in vivo. The animal experiments were approved by the Animal Ethics Committee of the Fifth Medical Center, Chinese PLA, and carried out in accordance with the UK Animals (Scientific Procedures) Act of 1986 and its associated guidelines. In order to produce the nude mice subcutaneous tumor model, MHCC97-H cells were cultured, prepared and subcutaneously injected into the 4–5 week-old nude mice.42,56 Four to 5 days after injection, the assigned concentrations of agents were orally administrated into the mice every 2 days. Mice were cultivated in cages and their tumor tissues were collected after 30 days of oral administration (15 times). The tumor volume (V) was calculated using the formula V = (tumor length) × (tumor width) × (tumor width)/2 and the tumor weight was measured by using a precision balance. The tumor weight and volume reflected the inhibitory effect of agents on the subcutaneous growth of MHCC97-H cells.57,58 In addition, we acknowledge that the nude mouse model is absent host immunity so its generalizability for larger animal or human use is limited.
Statistical Analysis
In this study, by using a SPSS Statistics software (IBM Corporation, Armonk, NY, USA), the Bonferroni correction with two-way analysis of variance was used to carry out the statistical analysis. Origin software (Version No 6.1, OriginLab Corporation, Northampton, MA, USA) was used to calculate the IC50 values of molecular targeting agents on MHCC97-H cells. A P-value that less than 0.05 (P<0.05) was considered statistically significant between groups.
Results and Discussion
Virtual Screening
In this study, we established a virtual docking model based on the crystal structure of mTOR (PDB: 4JSV) with complete substrate-binding pocket and ligand. Then, approximately 1200 compounds in our own compound library were screened by virtual docking and ranked according to various molecular characteristics, including hydrophobicity, polarity, entropy, etc. The 50 top-ranked compounds were selected, of which, 22 compounds were retained after manual selection based on visual inspection. The selected compounds were clustered into five types according to their structural characteristics.
In order to further investigate the accuracy of the docking, five representative mTOR inhibitors (OSI-027, GDC-0349, CC-223, AZD-2014, AZD-8055) were selected, which all had been used in Phase II clinical trial, and docked into the binding pocket of mTOR.59–63 By comparing docking sites of the five compounds, we found that all these inhibitors formed hydrogen bonds with LYS2187, ASP2357 and VAL2240, indicating the significant role of these three residues. Finally, the docking sites of the selected 22 compounds were examined, revealing that five compounds (compounds 1-5) had hydrogen–bond interaction with the three key residues. Therefore, these compounds (Figure S1) with purine structure were selected out as lead compounds for further study. The structural information of them was reported in the Supplementary Materials ("Structural identification of compounds 1-5" and "1H-NMR, 13C-NMR and MS spectra of compounds 1-5").
Correlation Test
The relationship between the expression of mTOR and the poor prognosis of advanced HCC in patients having received sorafenib treatment was examined. The mRNA level of mTOR in the HCC patients was examined by qPCR and then these patients were divided into two groups (the high-mTOR group and low-mTOR group) according to the median mRNA level of mTOR; the prognosis, the OS (overall survival) and the TTP (time to progress) of patients were compared between the two groups. As shown in Figure 1 and Table 1, the patients in the high-mTOR group had a poorer prognosis compared with those in the low-mTOR group; TTP (median value of 8.0 [M, month] of 95% confidence intervals (CI): 5.3–10.7 [M]) or OS (median value of 10.0 [M, month] of 95% CI: 8.1–11.5 [M]) of patients in the high-mTOR group was significantly shorter than TTP (median value of 12.0 [M, month] of 95% CI: 10.5–15.1 [M]; log-rank P=0.014) or OS (median value of 16.0 [M, month] of 95% CI: 11.6–20.4 [M]; log-rank P=0.011) of patients in the low-mTOR group.
 |
Table 1 mTOR Expression and Clinical Outcome of Sorafenib Treatment |
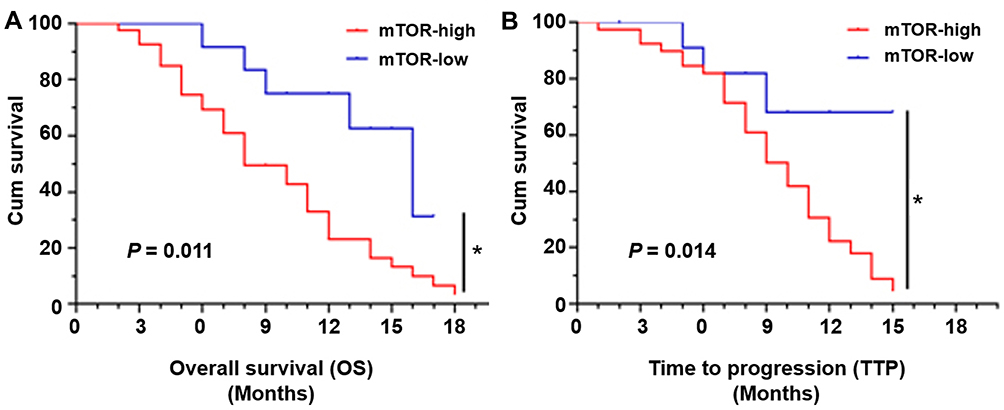 |
Figure 1 High level of endogenous mTOR is associated with the poor prognosis of advanced HCC patients received sorafenib treatment. The expression of mTOR in clinical specimens from advanced HCC patients received sorafenib treatment was examined by qPCR. Patients were divided into two groups (the mTOR-high group and the mTOR-low group) based on the median level of mTOR’s expression. The OS (overall survival) or TTP (time to progress) was examined to reveal the prognosis of patients. *P<0.05. |
To further examine the correlation between mTOR activation and advanced HCC, the twice percutaneous liver puncture (at the initial diagnosis or the recurrence time point) specimens and an autopsy liver puncture specimen which were obtained from same advanced HCC patient were used. The results indicated that the phospho-p70S6K1 (Thr389) and the phospho-rpS6 (Ser235/6) level but not p70S6K1 or rpS6 was increased with sorafenib treatment (Figure S2). Moreover, similar results were obtained from 5 advanced HCC patients with twice percutaneous liver puncture samples (Figure S3). The above results indicated that mTOR was not only correlated with the poor prognosis of advanced HCC patients having received sorafenib treatment but also activated as the cellular-injury response in HCC specimens. The mTOR pathway could serve as a promising target to attenuate drug resistance and enhance the activity of molecular-targeted agents in treating HCC.
Enzyme Inhibitory Activity and Selectivity
Firstly, the activity of five lead compounds was examined. 1 had an IC50 value against mTOR of 153.20±21.77 nmol/L; 2 had an IC50 value of 394.20±45.17 nmol/L; 3 had a IC50 values of 53.77±10.22 nmol/L; 4 had an IC50 value of 17.52±3.67 nmol/L and 5 had an IC50 value of 104.80±20.05 nmol/L. Among the five lead compounds, 4 had the most significant inhibitory activity on mTOR enzyme.
After identifying 4 as the best compound for enzyme inhibition activity, Western blot was used to investigate its selectivity to mTOR. In order to confirm that 4 acted selectively on mTOR, it was first necessary to exclude its effects on the upstream PI3K/AKT signaling pathway (Figure 2 and Figure S4). The PI3K inhibitor LY294002 was selected as the control agent. The Western blotting results showed that LY294002 significantly inhibited the phosphorylation of AKT and P70S6K1 simultaneously, whereas 4 only inhibited the phosphorylation of P70S6K1. This indicated that 4 did not affect PI3K or AKT (upstream of mTOR). Second, whether 4 had an inhibitory effect on the MAPK pathway was also investigated, as the MAPK signaling pathway could also regulate mTOR activation owing to cross-talk between signaling pathways. The MAPK inhibitor GSK2118436 was used as the control reagent, which significantly inhibited ERK phosphorylation. As expected, 4 did not show a significant inhibitory effect on ERK phosphorylation. This suggested that 4 did not inhibit mTOR by inhibiting the MAPK pathway. Finally, the JAK/STAT pathway inhibitor CP690550 (which does not affect the phosphorylation of AKT, ERK, or P70S6K1) was used as the negative control in this study. The JAK/STAT inhibitors had no significant effect on mTOR. None of the agents above affected the protein level of AKT, ERK, or P70S6K1. The above results allowed us to rule out the possibility that 4 was an upstream inhibitor of the mTOR signaling pathway, and confirmed that 4 could act directly and selectively on mTOR.
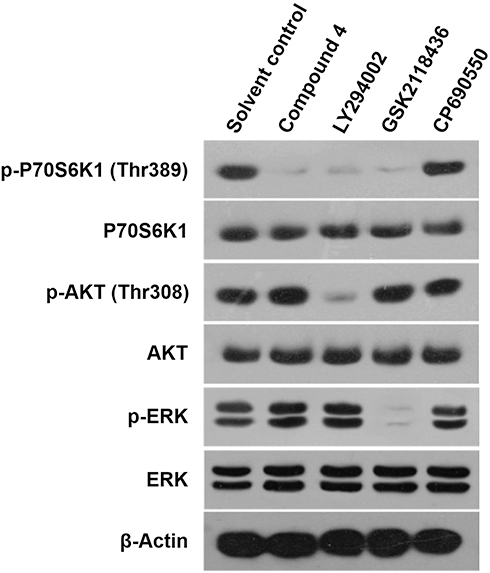 |
Figure 2 The specificity of 4 on activation of mTOR. MHCC97-H cells were treated by 4, LY294002, GSK2118436 and CP690550. Then, cells were harvested for Western blot and the expression level or the phosphorylation level of P70S6K1, AKT or ERK was examined by their antibodies. β-actin was chosen as the loading control. |
In addition, the inhibitory activity of 4 on mTORC1 and mTORC2 was examined by Western blot analysis. Phospho-p70S6K1 and phospho-AKT (SER473) are downstream factors of mTORC1 and mTORC2, respectively (Figure S5). The results of the analysis showed that 4 had a definite inhibitory effect on both mTORC1 and mTORC2, and the inhibitory effect on mTORC2 was weaker than that on mTORC1.
Structure and Activity Relationship
Molecular docking model was used to explain the significant activity and selectivity of 4 towards mTOR. In this model (Figure 3), 4 formed four hydrogen bonds with the amino acid residues SER2165, LYS2187, VAL2240 and ASP2244, respectively. By analyzing the docking model of other reported mTOR selective inhibitors, we observed that the residues SER2165, LYS2187 and VAL2240 played an important role, which proved that this docking mode was valid. VAL2240 was a vital residue in mTOR for forming a stable hydrogen bond with the 1, 3-dioxolane group in 4. Thus, 4 has high selectivity to mTOR owing to this key hydrogen bond. At the same time, purine base ring was anchored by hydrogen bond with LYS2187. Hydrogen bond between SER2165 and morpholine could be found in many mTOR inhibitors. Furthermore, ASP2244 formed two additional hydrogen bonds with the side-chain group of 4, which was rarely mentioned in previous studies and made 4 more potent than 1 or 2 (Figure S1). The n-butyl structure at the end of side chain of 4 just matched the hydrophobic cavity of mTOR in size, making 4 easier to bind to mTOR than 3 or 5.
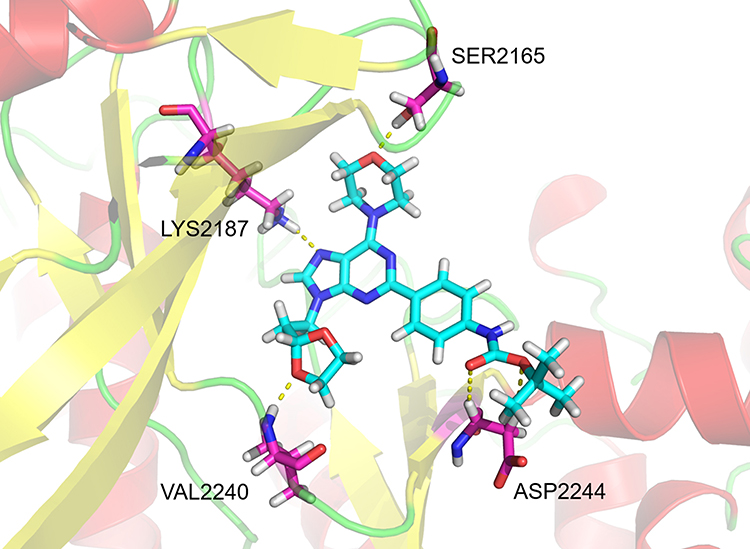 |
Figure 3 Schematic of the binding mode of 4 with mTOR. |
Sensitization Activity Test
In vitro Experiment
To investigate the sensitization effect of 4 on MHCC97-H cell in vitro, appropriate dose concentration should be determined firstly; and at this concentration, 4 should not directly inhibit the growth of MHCC97-H cells, while significantly inhibit the activation of mTOR kinase (phosphorylation of P70S6K1) in cells. The data (Table S2) showed that 4 significantly inhibited the activation of mTOR at a concentration of 0.03 μmol/L (30 nmol/L), but had no obvious cytotoxicity on MHC97H cells. Therefore, the concentration of 30 nmol/L was determined for subsequent experiments. As shown in Table 2, treatment with 4 at 30 nmol/L significantly enhanced the antitumor effect of molecular-targeted drugs on MHCC97-H cells, thereby reducing the IC50 value of the five molecular-targeted agents (Sorafenib, Regorafenib, Lenvatinib, Apatinib and Anlotinib).
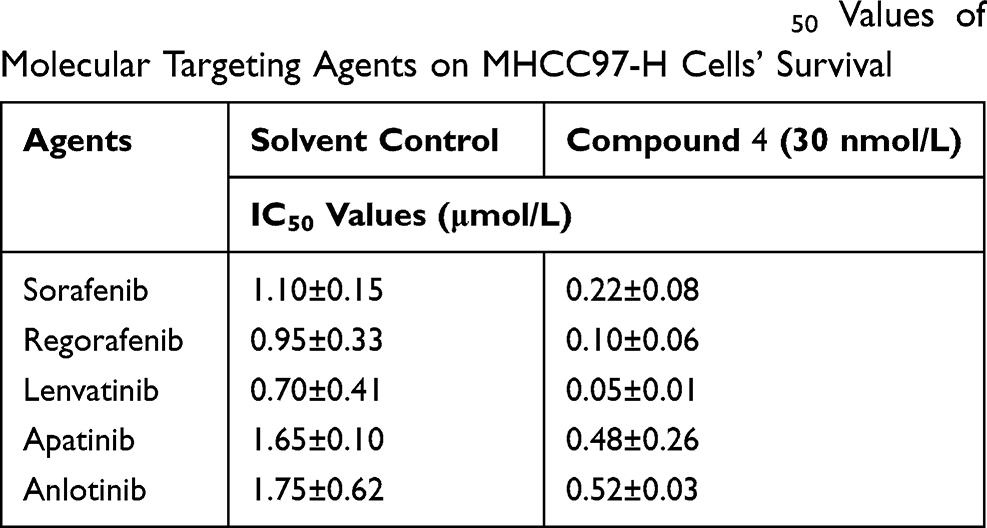 |
Table 2 The Effect of Compound 4 on the IC50 Values of Molecular Targeting Agents on MHCC97-H Cells’ Survival |
In vivo Experiment
In addition, the effect of 4 on the antitumor activity of molecular-targeted agents was also examined in vivo. An appropriate concentration was determined firstly. As shown in Figure 4 and Table S3, 4 at 1 mg/kg significantly inhibited the activation of mTOR without significantly affecting MHCC97-H cells’ subcutaneous growth. Therefore, the concentration of 4 was set at 1 mg/kg in vivo.
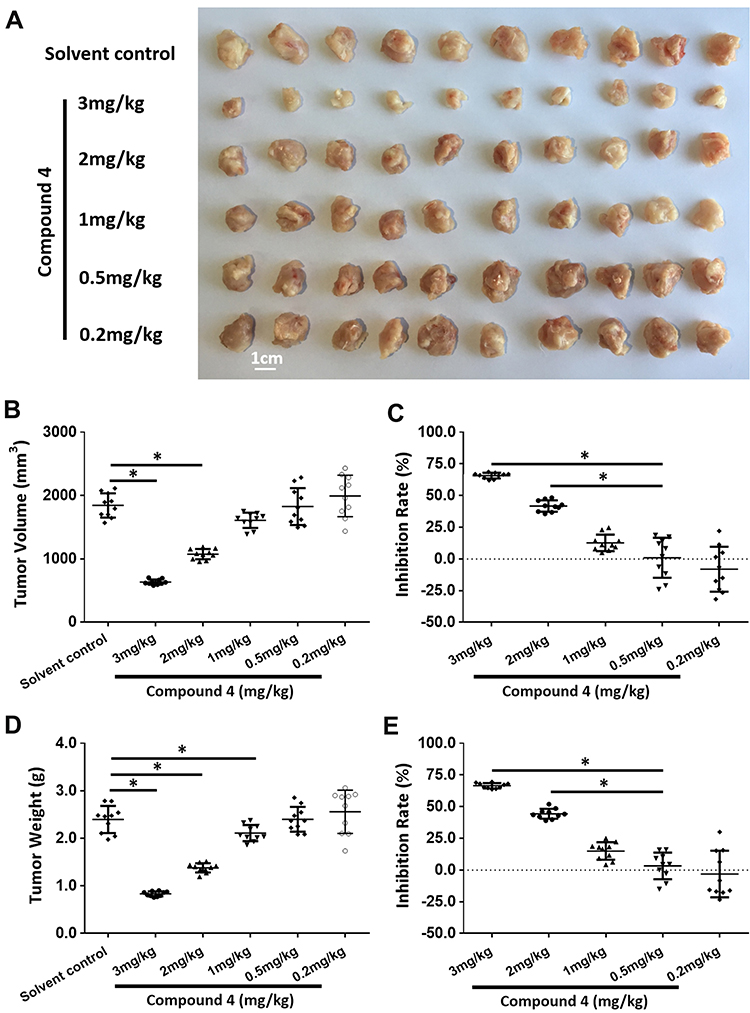 |
Figure 4 The antitumor effect of compound 4 on the subcutaneous growth of MHCC97-H cells in nude mice. MHCC97-H cells were injected into the subcutaneous position of nude mice to form subcutaneous tumor. The mice were received 4 via oral administration. Results were shown as the images of tumors (A), tumor volumes (B), inhibitory rates calculated by tumor volumes (C), tumor weights (D) and inhibitory rates calculated by tumor weights (E). *P<0.05. |
In the following experiment in vivo, as shown in Table 3, 4 at a dose of 1 mg/kg was used in combination with five molecular-targeted agents, which obviously inhibited the growth of subcutaneous tumor. And this experiment showed that combining 4 with molecular-targeted agents was more effective than using molecular-targeted agents alone. Take sorafenib as example (Figure 5), at different concentrations, combination of 4 and sorafenib showed more significant antitumor effect than treatment with sorafenib alone. Moreover, combination of 0.5 mg/kg sorafenib with 4 could achieve the equal antitumor effect as treatment with 2 mg/kg sorafenib alone. The result showed that mTOR inhibitors significantly enhanced the efficacy of molecular-targeted drugs against HCC in the nude mice subcutaneous tumor model, and indicated that using mTOR inhibitors as chemotherapeutic sensitizers could be a promising treatment strategy to hepatoma carcinoma.
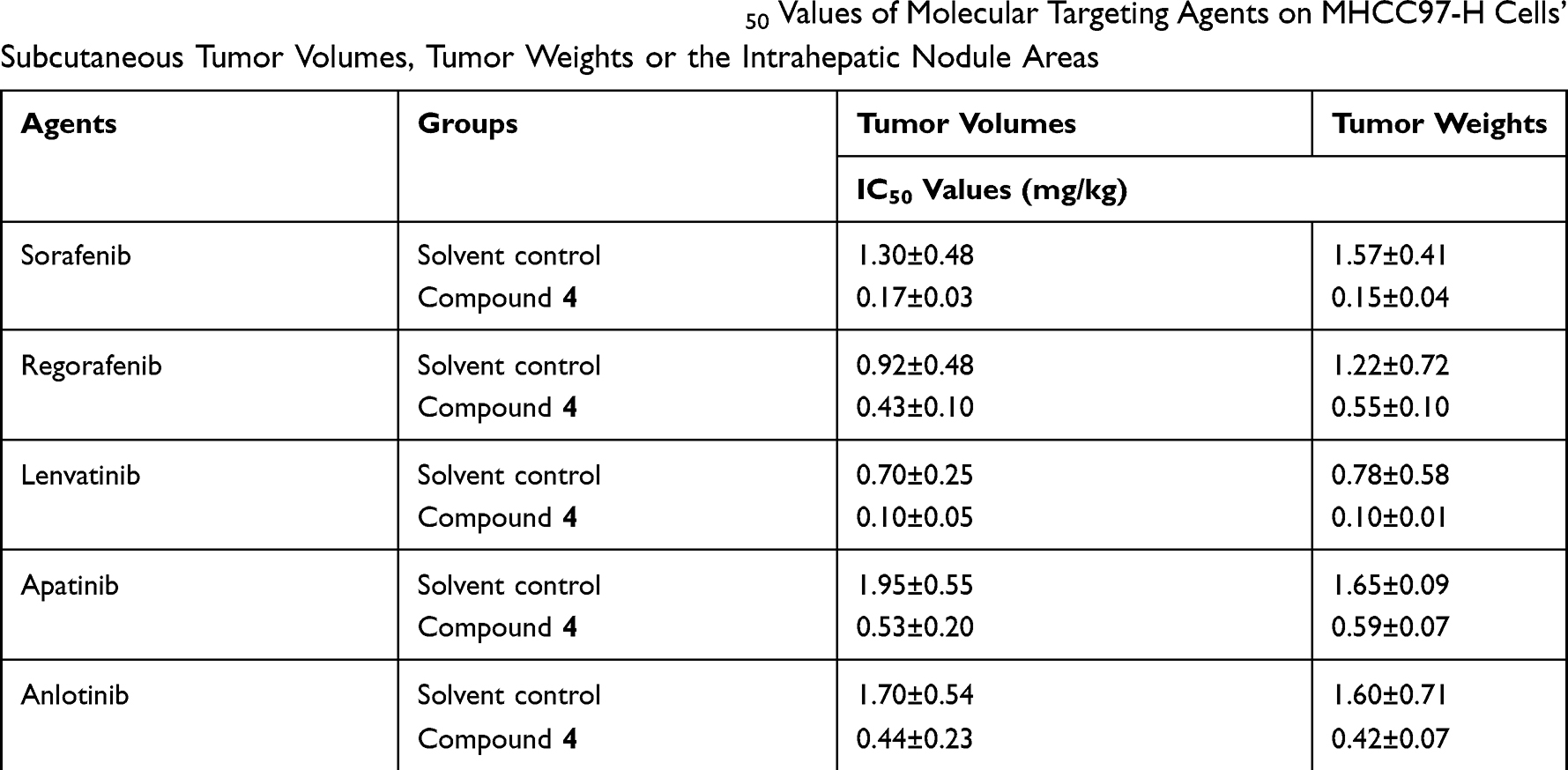 |
Table 3 The Effect of Compound 4 (1 mg/kg) on the IC50 Values of Molecular Targeting Agents on MHCC97-H Cells’ Subcutaneous Tumor Volumes, Tumor Weights or the Intrahepatic Nodule Areas |
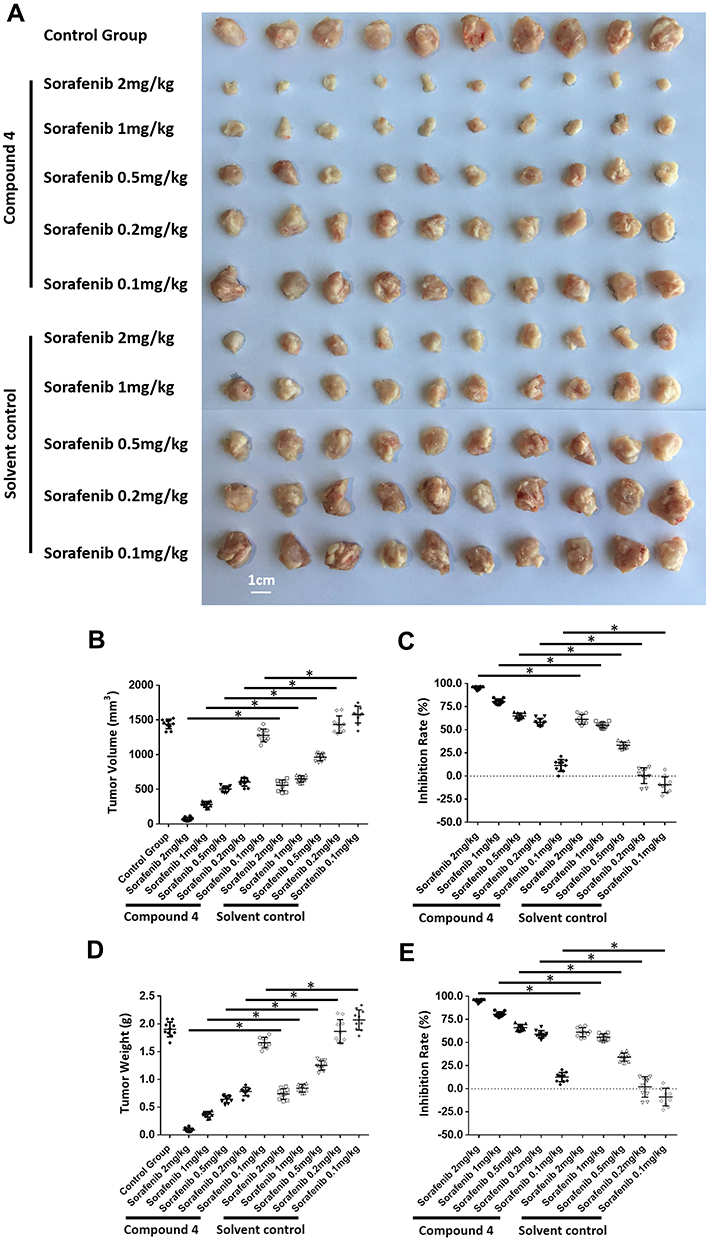 |
Figure 5 Compound 4 enhanced the sensitivity of MHCC97-H cells to sorafenib via subcutaneous tumor model. MHCC97-H cells were injected into the subcutaneous position of nude mice to form subcutaneous tumor. The mice were received sorafenib or sorafenib + 4 via oral administration. Results were shown as the images of tumors (A); (B) tumor volumes; (C) inhibitory rates calculated by tumor volumes; (D) tumor weights; (E) inhibitory rates calculated by tumor weights. *P<0.05. |
Conclusions
In recent years, molecular-targeted agents are still the main choice for treating advanced liver cancer.64–66 However, the insensitivity and drug resistance of hepatoma carcinoma cells to molecular-targeted therapy seriously affect the application of these drugs. In this study, the experimental results on HCC patients have proved that mTOR was correlated with the poor prognosis of advanced HCC patients having received sorafenib treatment, and mTOR pathway could serve as a promising target to attenuate drug resistance and enhance the activity of molecular-targeted agents in treating HCC.
A series of novel mTOR inhibitors were developed as chemosensitizer to enhance the activity of molecular-targeted agents in treating HCC. Based on the reported mTOR inhibitors, we screened out 5 lead compounds with the purine structure from our own compound library by virtual docking technology. Among the five lead compounds, 4 has the best inhibitory activity on mTOR (mTOR IC50 = 17.52±3.67 nmol/L), and its selectivity to mTOR was confirmed by Western blot. The In vitro MHCC97-H cell lines, nude mice subcutaneous tumor model was used to verify the sensitizing effect of 4 on the molecular-targeted therapies. The results showed that combined use of 4 and molecular-targeted agents was more effective than treatment with molecular-targeted agents alone. This suggests that mTOR inhibitors could be potential tumor sensitizers. The virtual docking technology was used to elucidate the significant activity of 4 and the result showed that four amino acid residues in mTOR, including SER2165, LYS2187, VAL2240 and ASP2244, contributed to binding of 4 to mTOR. VAL2240, a vital amino acid residue in mTOR, forms a hydrogen bond with the 1, 3-dioxolane group in 4, leading to the selectivity of 4 for mTOR. This study confirmed that mTOR was correlated with the poor prognosis of patients with advanced HCC receiving sorafenib, and developed a number of potential mTOR inhibitors. Research on mTOR inhibitors is ongoing, and further efforts are in process to find effective candidates for potential tumor sensitizers.
Acknowledgments
We gratefully acknowledge financial support from the program for the National Natural Science Foundation of China-Youth Foundation (No. 81502906), Research Fund of Wuhan Institute of Technology (No. K201756) and Graduate Innovative Fund of Wuhan Institute of Technology (No. CX2018001).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Shuang Cao reports grants from Wuhan Institute of Technology, during the conduct of the study. In addition, Shuang Cao has a patent under writing pending. The authors report no other possible conflicts of interest in this work.
References
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
3. Wang FS, Fan JG, Zhang Z, et al. The global burden of liver disease: the major impact of China. Hepatology. 2014;60(6):2099–2108. doi:10.1002/hep.27406
4. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. doi:10.1016/S0140-6736(18)30010-2
5. Xie H, Yu H, Tian S, et al. What is the best combination treatment with transarterial chemoembolization of unresectable hepatocellular carcinoma a systematic review and network meta-analysis. Oncotarget. 2017;8(59):100508–100523. doi:10.18632/oncotarget.20119
6. Feng F, Jiang Q, Jia H, et al. Which is the best combination of TACE and sorafenib for advanced hepatocellular carcinoma treatment A systematic review and network meta-analysis. Pharmacol Res. 2018;135:89–101. doi:10.1016/j.phrs.2018.06.021
7. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised Phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–1173. doi:10.1016/S0140-6736(18)30207-1
8. Meyer T. Treatment of advanced hepatocellular carcinoma: beyond sorafenib. Lancet Gastroenterol Hepatol. 2018;3(4):218–220. doi:10.1016/S2468-1253(17)30255-8
9. Kim DW, Talati C, Kim R. Hepatocellular carcinoma (HCC): beyond sorafenib-chemotherapy. J Gastrointest Oncol. 2017;8(2):256–265. doi:10.21037/jgo.2016.09.07
10. Zhu YJ, Zheng B, Wang HY, et al. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38(5):614–622. doi:10.1038/aps.2017.5
11. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi:10.1056/NEJMoa0708857
12. Zeng JD, Wu WKK, Wang HY, et al. Serine and one-carbon metabolism, a bridge that links mTOR signaling and DNA methylation in cancer. Pharmacol Res. 2019;149:104352–104380. doi:10.1016/j.phrs.2019.104352
13. Margaria JP, Campa CC, De Santis MC, et al. The PI3K/Akt/mTOR pathway in polycystic kidney disease: a complex interaction with polycystins and primary cilium. Cell Signal. 2019;66:109468–109511. doi:10.1016/j.cellsig.2019.109468
14. Dong X, Qin J, Ma J, et al. BAFF inhibits autophagy promoting cell proliferation and survival by activating Ca(2+)-CaMKII-dependent Akt/mTOR signaling pathway in normal and neoplastic B-lymphoid cells. Cell Signal. 2019;53:68–79. doi:10.1016/j.cellsig.2018.09.012
15. Luan W, Pang Y, Li R, et al. Akt/mTOR-mediated autophagy confers resistance to BET inhibitor JQ1 in ovarian cancer. Onco Targets Ther. 2019;12:8063–8074. doi:10.2147/OTT.S220267
16. Mortensen DS, Fultz KE, Xu S, et al. CC-223, a potent and selective inhibitor of mTOR kinase: in vitro and in vivo characterization. Mol Cancer Ther. 2015;14(6):1295–1305. doi:10.1158/1535-7163.MCT-14-1052
17. Zhang Q, Shi C, Han L, et al. Inhibition of mTORC1/C2 signaling improves anti-leukemia efficacy of JAK/STAT blockade in CRLF2 rearranged and/or JAK driven Philadelphia chromosome-like acute B-cell lymphoblastic leukemia. Oncotarget. 2018;9(8):8027–8041. doi:10.18632/oncotarget.24261
18. Cao S, Cao R, Liu X, et al. Design, synthesis and biological evaluation of novel benzothiazole derivatives as selective PI3Kbeta inhibitors. Molecules. 2016;21(7):876–890. doi:10.3390/molecules21070876
19. Cao S, Zhou X, Yang Y, et al. Selective substitution of 31/42-OH in rapamycin guided by an in situ IR technique. Molecules. 2014;19(6):7770–7784. doi:10.3390/molecules19067770
20. Badawi M, Kim J, Dauki A, et al. CD44 positive and sorafenib insensitive hepatocellular carcinomas respond to the ATP-competitive mTOR inhibitor INK128. Oncotarget. 2018;9(40):26032–26045. doi:10.18632/oncotarget.25430
21. Schmid P, Zaiss M, Harper-Wynne C, et al. Fulvestrant plus vistusertib vs fulvestrant plus everolimus vs fulvestrant alone for women with hormone receptor-positive metastatic breast cancer: the MANTA phase 2 randomized clinical trial. JAMA Oncol. 2019;5(11):1556. doi:10.1001/jamaoncol.2019.2526
22. Lapointe S, Mason W, MacNeil M, et al. A Phase I study of vistusertib (dual mTORC1/2 inhibitor) in patients with previously treated glioblastoma multiforme: a CCTG study. Invest New Drugs. 2019:1–8.
23. Behbakht K, Sill MW, Darcy KM, et al. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: a gynecologic oncology group study. Gynecol Oncol. 2011;123(1):19–26. doi:10.1016/j.ygyno.2011.06.022
24. Yu K, Shi C, Toral-Barza L, et al. Beyond rapalog therapy: preclinical pharmacology and antitumor activity of WYE-125132, an ATP-competitive and specific inhibitor of mTORC1 and mTORC2. Cancer Res. 2010;70(2):621–631. doi:10.1158/0008-5472.CAN-09-2340
25. Atkin J, Halova L, Ferguson J, et al. Torin1-mediated TOR kinase inhibition reduces Wee1 levels and advances mitotic commitment in fission yeast and HeLa cells. J Cell Sci. 2014;127(Pt 6):1346–1356. doi:10.1242/jcs.146373
26. Yu K, Toral-Barza L, Shi C, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69(15):6232–6240. doi:10.1158/0008-5472.CAN-09-0299
27. Li S, Liang Y, Wu M, et al. The novel mTOR inhibitor CCI-779 (temsirolimus) induces antiproliferative effects through inhibition of mTOR in Bel-7402 liver cancer cells. Cancer Cell Int. 2013;13(1):30. doi:10.1186/1475-2867-13-30
28. Llerena S, Garcia-Diaz N, Curiel-Olmo S, et al. Applied diagnostics in liver cancer. Efficient combinations of sorafenib with targeted inhibitors blocking AKT/mTOR. Oncotarget. 2018;9(56):30869–30882. doi:10.18632/oncotarget.25766
29. de Melo AC, Paulino E, Garces AH. A review of mTOR pathway inhibitors in gynecologic cancer. Oxid Med Cell Longev. 2017;2017:4809751. doi:10.1155/2017/4809751
30. Suzuki Y, Enokido Y, Yamada K, et al. The effect of rapamycin, NVP-BEZ235, aspirin, and metformin on PI3K/AKT/mTOR signaling pathway of PIK3CA-related overgrowth spectrum (PROS). Oncotarget. 2017;8(28):45470–45483. doi:10.18632/oncotarget.17566
31. Djuzenova CS, Fiedler V, Katzer A, et al. Dual PI3K- and mTOR-inhibitor PI-103 can either enhance or reduce the radiosensitizing effect of the Hsp90 inhibitor NVP-AUY922 in tumor cells: the role of drug-irradiation schedule. Oncotarget. 2016;7(25):38191–38209. doi:10.18632/oncotarget.9501
32. Rodrik-Outmezguine VS, Okaniwa M, Yao Z, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534(7606):272–276. doi:10.1038/nature17963
33. Li A, Zhang R, Zhang Y, et al. BEZ235 increases sorafenib inhibition of hepatocellular carcinoma cells by suppressing the PI3K/AKT/mTOR pathway. Am J Transl Res. 2019;11(9):5573–5585.
34. Shapiro GI, Bell-McGuinn KM, Molina JR, et al. First-in-human study of PF-05212384 (PKI-587), a small-molecule, intravenous, dual inhibitor of PI3K and mTOR in patients with advanced cancer. Clin Cancer Res. 2015;21(8):1888–1895. doi:10.1158/1078-0432.CCR-14-1306
35. Meng Q, Xia C, Fang J, et al. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal. 2006;18(12):2262–2271. doi:10.1016/j.cellsig.2006.05.019
36. Liu G, Wang W, Wan Y, et al. Application of 3D-QSAR, pharmacophore, and molecular docking in the molecular design of diarylpyrimidine derivatives as HIV-1 nonnucleoside reverse transcriptase inhibitors. Int J Mol Sci. 2018;19(5):1436–1452. doi:10.3390/ijms19051436
37. Caballero J. 3D-QSAR (CoMFA and CoMSIA) and pharmacophore (GALAHAD) studies on the differential inhibition of aldose reductase by flavonoid compounds. J Mol Graph Model. 2010;29(3):363–371. doi:10.1016/j.jmgm.2010.08.005
38. Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8.. New York, NY: Schrödinger; 2015.
39. Sun N, Li B, Shao J, et al. A general and facile one-pot process of isothiocyanates from amines under aqueous conditions. Beilstein J Org Chem. 2012;8:61–70. doi:10.3762/bjoc.8.6
40. Shao Z, Li Y, Dai W, et al. ETS-1 induces Sorafenib-resistance in hepatocellular carcinoma cells via regulating transcription factor activity of PXR. Pharmacol Res. 2018;135:188–200. doi:10.1016/j.phrs.2018.08.003
41. Zhang Y, Li D, Jiang Q, et al. Novel ADAM-17 inhibitor ZLDI-8 enhances the in vitro and in vivo chemotherapeutic effects of Sorafenib on hepatocellular carcinoma cells. Cell Death Dis. 2018;9(7):743–756. doi:10.1038/s41419-018-0804-6
42. Yang B, Wang C, Xie H, et al. MicroRNA-3163 targets ADAM-17 and enhances the sensitivity of hepatocellular carcinoma cells to molecular targeted agents. Cell Death Dis. 2019;10(10):784–799. doi:10.1038/s41419-019-2023-1
43. Feng F, Jiang Q, Cao S, et al. Pregnane X receptor mediates sorafenib resistance in advanced hepatocellular carcinoma. Biochim Biophys Acta. 2018;1862(4):1017–1030. doi:10.1016/j.bbagen.2018.01.011
44. Feng F, Lu YY, Zhang F, et al. Long interspersed nuclear element ORF-1 protein promotes proliferation and resistance to chemotherapy in hepatocellular carcinoma. World J Gastroenterol. 2013;19(7):1068–1078. doi:10.3748/wjg.v19.i7.1068
45. Chen Y, Feng F, Gao X, et al. MiRNA153 reduces effects of chemotherapeutic agents or small molecular kinase inhibitor in HCC cells. Curr Cancer Drug Targets. 2015;15(3):176–187. doi:10.2174/1568009615666150225122635
46. Xie H, Tian S, Yu H, et al. A new apatinib microcrystal formulation enhances the effect of radiofrequency ablation treatment on hepatocellular carcinoma. Onco Targets Ther. 2018;11:3257–3265. doi:10.2147/OTT.S165000
47. Wang Y, Tang Z. A novel long-sustaining system of apatinib for long-term inhibition of the proliferation of hepatocellular carcinoma cells. Onco Targets Ther. 2018;11:8529–8541. doi:10.2147/OTT.S188209
48. Ji Q, Xu X, Li L, et al. miR-216a inhibits osteosarcoma cell proliferation, invasion and metastasis by targeting CDK14. Cell Death Dis. 2017;8(10):e3103. doi:10.1038/cddis.2017.499
49. Hou J, Hong Z, Feng F, et al. A novel chemotherapeutic sensitivity-testing system based on collagen gel droplet embedded 3D-culture methods for hepatocellular carcinoma. BMC Cancer. 2017;17(1):729–737. doi:10.1186/s12885-017-3706-6
50. Zhao J, Bai Z, Feng F, et al. Cross-talk between EPAS-1/HIF-2alpha and PXR signaling pathway regulates multi-drug resistance of stomach cancer cell. Int J Biochem Cell Biol. 2016;72:73–88. doi:10.1016/j.biocel.2016.01.006
51. Li F, Wei A, Bu L, et al. Procaspase-3-activating compound 1 stabilizes hypoxia-inducible factor 1alpha and induces DNA damage by sequestering ferrous iron. Cell Death Dis. 2018;9(10):1025–1040. doi:10.1038/s41419-018-1038-3
52. Guan F, Ding R, Zhang Q, et al. WX-132-18B, a novel microtubule inhibitor, exhibits promising anti-tumor effects. Oncotarget. 2017;8(42):71782–71796. doi:10.18632/oncotarget.17710
53. Lu CC, Chiang JH, Tsai FJ, et al. Metformin triggers the intrinsic apoptotic response in human AGS gastric adenocarcinoma cells by activating AMPK and suppressing mTOR/AKT signaling. Int J Oncol. 2019;54(4):1271–1281. doi:10.3892/ijo.2019.4704
54. Hu Y, Tao Y, Hu J. Cannabinoid receptor 2 deletion deteriorates myocardial infarction through the down-regulation of AMPK-mTOR-p70S6K signaling-mediated autophagy. Biosci Rep. 2019;39(4):BSR20180650. doi:10.1042/BSR20180650
55. Su CC, Hsieh KL, Liu PL, et al. AICAR induces apoptosis and inhibits migration and invasion in prostate cancer cells through an AMPK/mTOR-dependent pathway. Int J Mol Sci. 2019;20(7):1647–1660. doi:10.3390/ijms20071647
56. Feng F, Li X, Li R, et al. The multiple-kinase inhibitor lenvatinib inhibits the proliferation of acute myeloid leukemia cells. Animal Model Exp Med. 2019;2(3):178–184. doi:10.1002/ame2.12076
57. Jia H, Yang Q, Wang T, et al. Rhamnetin induces sensitization of hepatocellular carcinoma cells to a small molecular kinase inhibitor or chemotherapeutic agents. Biochim Biophys Acta. 2016;1860(7):1417–1430. doi:10.1016/j.bbagen.2016.04.007
58. Yin F, Feng F, Wang L, et al. SREBP-1 inhibitor betulin enhances the antitumor effect of sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis. 2019;10(9):672–684. doi:10.1038/s41419-019-1884-7
59. Bhagwat SV, Gokhale PC, Crew AP, et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol Cancer Ther. 2011;10(8):1394–1406. doi:10.1158/1535-7163.MCT-10-1099
60. Zhou Y, Peng Y, Tang H, et al. Autophagy induction contributes to GDC-0349 resistance in head and neck squamous cell carcinoma (HNSCC) cells. Biochem Biophys Res Commun. 2016;477(2):174–180. doi:10.1016/j.bbrc.2016.06.039
61. Wolin E, Mita A, Mahipal A, et al. A phase 2 study of an oral mTORC1/mTORC2 kinase inhibitor (CC-223) for non-pancreatic neuroendocrine tumors with or without carcinoid symptoms. PLoS One. 2019;14(9):e0221994. doi:10.1371/journal.pone.0221994
62. Zheng B, Mao JH, Qian L, et al. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 2015;357(2):468–475. doi:10.1016/j.canlet.2014.11.012
63. Liu M, Gu P, Guo W, et al. C6 ceramide sensitizes the anti-hepatocellular carcinoma (HCC) activity by AZD-8055, a novel mTORC1/2 dual inhibitor. Tumour Biol. 2016;37(8):11039–11048. doi:10.1007/s13277-015-4598-1
64. Roskoski R
65. Roskoski R
66. Roskoski R
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.