Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Novel lactoferrin-conjugated amphiphilic poly(aminoethyl ethylene phosphate)/poly(L-lactide) copolymer nanobubbles for tumor-targeting ultrasonic imaging
Authors Luo B ![]() , Liang H
, Liang H ![]() , Zhang S, Qin X, Liu X, Liu W, Zeng F, Wu Y, Yang X
, Zhang S, Qin X, Liu X, Liu W, Zeng F, Wu Y, Yang X
Received 27 February 2015
Accepted for publication 30 April 2015
Published 16 September 2015 Volume 2015:10(1) Pages 5805—5817
DOI https://doi.org/10.2147/IJN.S83582
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Binhua Luo,1,2,* Huageng Liang,3,* Shengwei Zhang,3 Xiaojuan Qin,4 Xuhan Liu,1 Wei Liu,1,5 Fuqing Zeng,3 Yun Wu,6 Xiangliang Yang1,5
1College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 2College of Pharmacy, Hubei University of Science and Technology, Xianning, 3Department of Urology, Union Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 4Department of Medical Ultrasound, Union Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 5National Engineering Research Center for Nanomedicine, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 6Department of Biomedical Engineering, University at Buffalo, State University of New York, Buffalo, NY, USA
*These authors contributed equally to this work
Abstract: In the study reported here, a novel amphiphilic poly(aminoethyl ethylene phosphate)/poly(L-lactide) (PAEEP-PLLA) copolymer was synthesized by ring-opening polymerization reaction. The perfluoropentane-filled PAEEP-PLLA nanobubbles (NBs) were prepared using the O1/O2/W double-emulsion and solvent-evaporation method, with the copolymer as the shell and liquid perfluoropentane as the core of NBs. The prepared NBs were further conjugated with lactoferrin (Lf) for tumor-cell targeting. The resulting Lf-conjugated amphiphilic poly(aminoethyl ethylene phosphate)/poly(L-lactide) nanobubbles (Lf-PAEEP-PLLA NBs) were characterized by photon correlation spectroscopy, polyacrylamide gel electrophoresis, Fourier transform infrared spectroscopy, and transmission electron microscopy. The average size of the Lf-PAEEP-PLLA NBs was 328.4±5.1 nm, with polydispersity index of 0.167±0.020, and zeta potential of -12.6±0.3 mV. Transmission electron microscopy imaging showed that the Lf-PAEEP-PLLA NBs had a near-spherical structure, were quite monodisperse, and there was a clear interface between the copolymer shell and the liquid core inside the NBs. The Lf-PAEEP-PLLA NBs also exhibited good biocompatibility in cytotoxicity and hemolysis studies and good stability during storage. The high cellular uptake of Lf-PAEEP-PLLA NBs in C6 cells (low-density lipoprotein receptor-related protein 1-positive cells) at concentrations of 0–20 µg/mL indicated that the Lf provided effective targeting for brain-tumor cells. The in vitro acoustic behavior of Lf-PAEEP-PLLA NBs was evaluated using a B-mode clinical ultrasound imaging system. In vivo ultrasound imaging was performed on tumor-bearing BALB/c nude mice, and compared with SonoVue® microbubbles, a commercial ultrasonic contrast agent. Both in vitro and in vivo ultrasound imaging indicated that the Lf-PAEEP-PLLA NBs possessed strong, long-lasting, and tumor-enhanced ultrasonic contrast ability. Taken together, these results indicate that Lf-PAEEP-PLLA NBs represent a promising nano-sized ultrasonic contrast agent for tumor-targeting ultrasonic imaging.
Keywords: PAEEP-PLLA copolymer, in vitro acoustic behavior, in vivo ultrasonic imaging, SonoVue® microbubbles
Introduction
Ultrasonography is a widely used, non-invasive diagnostic technique, which can provide real-time imaging in clinical diagnosis. Recently, contrast-enhanced ultrasonic imaging has become an indispensable part of clinical ultrasonography.1,2 As effective contrast agents, gas-filled microbubbles have been widely employed in contrast-enhanced ultrasound imaging.3 Microbubbles are filled with gas and stabilized by lipids, surfactants, polymers, or other proprietary shell materials; therefore, they can be injected intravenously to produce echogenic contrast.4 In addition, microbubbles can be “explosive” under ultrasonic illumination, which promotes the destruction of bubbles and enhances cellular-membrane permeability change. Consequently, microbubbles are considered ultrasound-mediated drug/gene delivery vehicles to facilitate drug release and extravascular delivery besides diagnostic application.5–7 However, the diameters of microbubbles are usually a few microns (2–8 μm), which makes microbubbles trapped in the blood pool and poses severe limitations in tumor ultrasonic imaging and drug/gene delivery due to their large sizes.8
Recently, nanoscale bubbles (nanobubbles [NBs]) have attracted investigators in extravascular ultrasonic imaging and drug/gene delivery.9–12 NBs of less than 700 nm can pass through the larger-than-usual pores in the leaky vasculature of tumors. The endothelial cells of the blood vessels in normal tissue, however, have tighter inter-endothelial junctions, which would not allow the penetration of NBs. In contrast, tumor tissue usually retains defective vasculature with large gaps.13 As a matter of fact, it appears that the characteristic pore cutoff size ranges from 380 nm to 780 nm for a large variety of tumors.14 Consequently, NBs can effectively accumulate in the tumor interstitium due to the enhanced permeability and retention effect, which naturally enables them to serve as promising contrast agents for tumor ultrasonic imaging and treatment.11,15,16
NBs usually contain a gaseous perfluorocarbon or sulfur hexafluoride as cores, and their shells are composed of albumin, lipids, nonionic surfactants, and polymers.16–19 Polymer-based hard-shell NBs are superior to phospholipid/surfactant-based soft-shell NBs because the hard shell makes those NBs more stable and thus have longer circulation life.20,21 However, the mechanical properties, degradation time, and hydrophilic/lipophilic feature of the reported copolymers, such as polylactic acid, poly(lactic-co-glycolic acid), and polycaprolactone, are difficult to adjust to meet the requirements of clinical ultrasound imaging. These polymers lack active sites for conjugating tumor-targeting molecules. Moreover, previous studies have focused on enhancing the contrast capabilities of the NBs, and their active tumor-targeting potential has not been well explored.
Lactoferrin (Lf) is a single-chain iron-binding glycoprotein belonging to the transferrin (Tf) family.22 Tf is used as a tumor-targeting ligand because Tf receptors are overexpressed in several human carcinomas including brain, breast, and ovary cancers.23,24 In our recent work, Lf-conjugated nanoparticles were developed as the specific and multifunctional magnetic resonance imaging contrast agent for the diagnosis of brain glioma.25,26 In the study reported here, a novel amphiphilic biodegradable poly(aminoethyl ethylene phosphate)/poly(L-lactide) (PAEEP-PLLA) copolymer was synthesized by ring-opening polymerization reaction. The NBs were prepared with PAEEP-PLLA copolymer as the shell and liquid perfluoropentane (PFP) as the core. Lf was conjugated to the PAEEP-PLLA NBs for tumor targeting. The physiochemical properties (size, size distribution, zeta potential, and morphology), biocompatibility, and Lf targeting effect of the Lf-PAEEP-PLLA NBs were investigated. The ultrasound-triggered phase transition and in vitro and in vivo ultrasonic imaging behavior of the NBs were evaluated using a B-mode clinical ultrasound imaging system.
Materials and methods
Materials
L-lactide was purchased from Daigang Biotechnology Co, Ltd (Shandong, People’s Republic of China) and was recrystallized from dry ethyl acetate prior to use. Stannous octoate [Sn(Oct)2], Lf from bovine colostrum, and 2-iminothiolane hydrochloride were purchased from Sigma-Aldrich Co (St Louis, MO, USA). Poly(vinyl alcohol) (PVA-217, the degree of polymerization is 1700, and the degree of hydrolysis is 88.5%) was obtained from Kuraray Co, Ltd (Tokyo, Japan). PFP was obtained from Strem Chemicals, Inc (Newburyport, MO, USA), while 2-chloro-2-oxo-1,3,2-dioxaphospholane (COP) was purchased from Tokyo Chemical Industry Co, Ltd (Tokyo, Japan); α-malemidyl-ω-N-hydroxysuccinimidylpoly (ethylene glycol) (MW3400) was obtained from Jenkem Technology Co, Ltd (Beijing, People’s Republic of China); and ethanolamine and di-tert-butyl dicarbonate were purchased from Aladdin Industrial Corporation (Shanghai, People’s Republic of China). Triethylamine and tetrahydrofuran were purchased from Sinopharm Chemical Reagent Co, Ltd (Shanghai, People’s Republic of China) and dried and distilled prior to use.
Synthesis of amino-terminated PLLA130-OH
PLLA130-OH was synthesized with the ring-opening polymerization reaction of L-lactide. EABoc and Sn(Oct)2 were used as the initiator and catalyst, respectively. In a glove box (H2O and O2 contents less than 0.1 ppm), L-lactide (1.4771 g, 10 mmol), EABoc (0.0161 g, 0.1 mmol), and Sn(Oct)2 (0.0045 g, 0.01 mmol) were added into a freshly flamed nitrogen-purged round-bottomed flask. The resulting mixture was maintained at 120°C for 3 hours. The crude material was first dissolved in 30 mL of chloroform. Then the desired product was gradually precipitated, followed by washing with 100 mL of cold methanol twice. The precipitates were dried under vacuum until a constant weight was obtained. The average degree of polymerization of L-lactide was calculated based on its GPC results. The proton nuclear magnetic resonance (1H NMR) spectrum is shown in Figure 1A (100 MHz; CDCl3; Me4Si; ppm).
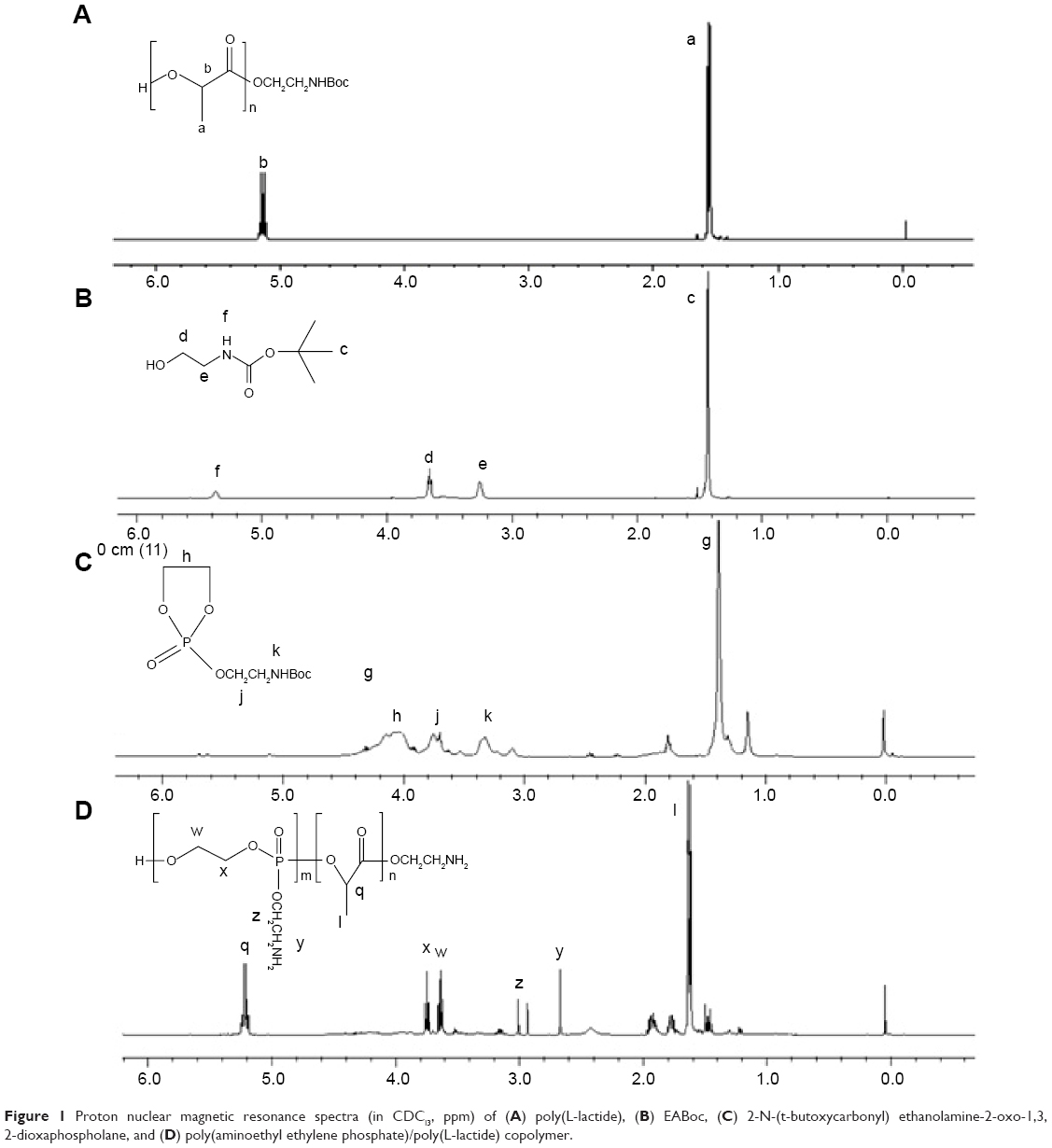 | Figure 1 Proton nuclear magnetic resonance spectra (in CDCl3, ppm) of (A) poly(L-lactide), (B) EABoc, (C) 2-N-(t-butoxycarbonyl) ethanolamine-2-oxo-1,3, 2-dioxaphospholane, and (D) poly(aminoethyl ethylene phosphate)/poly(L-lactide) copolymer. |
Synthesis of N-(t-butoxycarbonyl) ethanolamine
Sodium bicarbonate solution (0.84 g, 0.01 mol in 10 mL water) was added to the solution of ethanolamine (0.6 g, 0.01 mol) in 10 mL tetrahydrofuran at 0°C. Di-tert-butyl dicarbonate (2.18 g, 0.01 mol) was added dropwise over a period of 0.5 hours with magnetic stirring. The resulting mixture was stirred overnight at room temperature and extracted three times with diethyl ether. The organic phase was combined and dried with sodium sulfate. N-(t-butoxycarbonyl) ethanolamine (EABoc) was obtained after vacuum distillation. The 1H NMR spectrum is shown in Figure 1B (100 MHz; CDCl3; Me4Si; ppm).
Synthesis of monomer 2-N-(t-butoxycarbonyl) ethanolamine-2-oxo-1,3,2-dioxaphospholane
At −10°C, COP solution (1.425 g, 0.01 mol in 3 mL anhydrous tetrahydrofuran) was added dropwise to the solution of EABoc (1.61 g, 0.01 mol) and triethylamine (1.0120 g, 0.01 mol) in 12 mL anhydrous tetrahydrofuran over a period of 0.5 hours. The mixture was maintained at −10°C overnight under magnetic stirring. Subsequently, the solid precipitates, which should have been triethylammonium chloride, were removed by filtration using a Schlenk funnel. The filtrate was concentrated under vacuum, then re-precipitated from 40 mL of cold anhydrous diethyl ether. The resulting material was dried under vacuum at 40°C, affording the final monomer product. The 1H NMR spectrum is shown in Figure 1C (100 MHz; CDCl3; Me4Si; ppm).
Synthesis of PAEEP-PLLA block copolymers
The PAEEP-PLLA block copolymers featuring different PEEA chain lengths were synthesized by the ring-opening polymerization reaction of N-(t-butoxycarbonyl) ethanolamine-2-oxo-1,3,2-dioxaphospholane (PEEABoc). The PLLA130-OH macro-initiator with Sn(Oct)2 was employed as the catalyst. In a glove box (H2O and O2 content less than 0.1 ppm), PLLA130-OH (0.47 g, 0.025 mmol) and PEEABoc (0.557 g, 2.5 mmol) were dissolved in 5 mL of anhydrous tetrahydrofuran, then Sn(Oct)2 (0.01 g, 0.025 mmol) was added to the solution. The resulting mixture was stirred at 40°C for 2 hours, and the concentrated polymer product was precipitated twice using a mixture of diethyl ether/isopropyl alcohol (4:1, v/v) and dried under vacuum until a constant weight was obtained.
De-protection of t-butoxycarbonyl (Boc)
At 0°C, 4 mol/L hydrogen chloride solution in tetrahydrofuran was added to the copolymers under magnetic stirring over 1 hours. The product was collected by centrifugation and dried under vacuum. The reaction yield of the final product was 70.7%. The 1H NMR spectrum is shown in Figure 1D (100 MHz; CDCl3; Me4Si; ppm). A Diamond™ DSC (differential scanning calorimeter; PerkinElmer Inc, Waltham, MA, USA) was employed to detect the thermodynamic properties of the PAEEP-PLLA copolymer. The lyophilized copolymer was weighed and placed in a sealed aluminum crimp cell then heated from 50°C to 200°C at a rate of 5°C/minute.
Preparation of PAEEP-PLLA NBs
PAEEP-PLLA NBs were prepared by a double-emulsion (oil-in-oil-in-water, O1/O2/W) and solvent-evaporation method. PAEEP-PLLA (50 mg) was first dissolved in 2 mL dichloromethane. PFP (0.5 mL) was added to the solution then the solution was placed in a high-shear mixer (Fluko FA25 homogenizer; Fluko Equipment Shanghai Co, Ltd, Shanghai, People’s Republic of China) for 1 minute at 10,000 rpm in an ice bath while in the dark, ultimately forming a primary emulsion. The primary emulsion was added dropwise into 12 mL 0.3% PVA solution, with magnetic stirring in an ice bath, to produce the coarse double emulsion. The coarse double emulsion was sonicated for 10 seconds at 100 Watts, then stirred in an ice bath overnight. Organic solvents were removed by evaporation, and NBs were formed after solidification. The NB dispersion was centrifuged (10,000 rpm, 10 minutes) to obtain NB precipitates, which were dispersed in normal saline solution and then stored at 4°C for further studies.
Conjugation of Lf to PAEEP-PLLA NBs
Sulfhydryl groups were linked to Lf molecules using Traut’s reagent. A cold solution of 0.0024 g Lf in 1 mL of 0.1 M sodium phosphate buffer (pH 8.0, containing 5 mM EDTA) was mixed with 1.0 mL of 2-iminothiolane solution (0.24 mg/mL in water). The resulting mixture was allowed to react at room temperature for 1 hour, then the modified Lf was isolated by eluting through a Sephadex™ PD-10 column with phosphate-buffered saline (PBS) containing 5 mM EDTA. The product obtained was Lf sulfhydryl.
The NB suspension (1.0 mL) was incubated with 300 μL of α-Malemidyl-ω-N-hydroxysuccinimidylpoly (ethylene glycol) solution (0.0047 g). The resulting mixture was maintained at room temperature for 2 hours in the dark. Subsequently, 50 μL of glycine solution (1.0 M) was added to quench the reaction. The mixture obtained from the last-mentioned step was further incubated with 2.0 mL of Lf sulfhydryl in PBS for 24 hours at room temperature. The Lf-conjugated PAEEP-PLLA NBs (Lf-PAEEP-PLLA NBs) were purified by centrifugation.
Characterization of Lf-PAEEP-PLLA NBs
The hydrodynamic size and polydispersity index (PDI) of the Lf-PAEEP-PLLA NBs were measured by photon correlation spectroscopy (Zetasizer Nano ZS90; Malvern Instruments Ltd, Malvern, UK) at 25°C using a He-Ne laser of 633 nm. The zeta potential was measured in folded capillary cells using the Zetasizer. The conductivity of the NB solution was adjusted to 50 μS/cm by 0.1 mmol/L sodium chloride solution for zeta potential measurement. The zeta potential values were calculated using the Helmholtz–Smoluchowski equation.
The concentration of Lf-PAEEP-PLLA NBs was determined with a hemocytometer, using a calculation approach similar to the cell-counting method. In order to obtain direct visualization, the NBs were examined on an inverted fluorescence microscope (Olympus IX71; Olympus Corporation, Tokyo, Japan) with a 400× magnification in bright and fluorescent visual fields. The formation of conjugated NBs was confirmed by polyacrylamide gel electrophoresis and Fourier transform infrared (FTIR) spectroscopy (Bruker Vertex 70; Bruker Corporation, Billerica, MA, USA).
The morphology of the Lf-PAEEP-PLLA NBs was examined by transmission electron microscopy (TEM) with a 100 kV accelerating voltage. Samples were dropped on an amorphous carbon-coated copper grid. After drying naturally, the grid was negatively stained with 2% phosphotungstic acid and was observed with TEM.
Cell culture
Rat C6 glioma (C6) cells, human umbilical vein endothelial (ECV304) cells, and the human normal liver cell line (HL-7702) were purchased from the Shanghai Institute of Life Science Cell Culture Center (Shanghai, People’s Republic of China). These cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal calf serum (Gibco®; Thermo Fisher Scientific, Waltham, MA, USA), penicillin (100 IU/mL), and streptomycin (100 mg/mL), under humidified air containing 5% CO2 at 37°C. Ethical approval was not sought for the use of human cell lines.
Cytotoxicity assay
C6 cells and HL-7702 cells were used to evaluate the cytotoxicity with MTT assay. Cells were seeded in 96-well plates at a density of 8,000 cells/well in DMEM. The NBs were added to the cell medium at concentrations of 0.0%, 0.5%, 1.0%, 2.5%, 5.0%, 7.5%, 10.0%, 12.5%, 15.0%, and 20.0% (v/v) and incubated for 24 hours and 48 hours. The control was the culture medium only. The absorbance at 492 nm was recorded on a microplate reader (318C Microplate Reader; People’s Republic of China).
Hemolysis assay
Blood samples were collected from the main abdominal artery of Sprague Dawley rats, and placed in a tube containing heparin sodium (15 IU/mL). The blood samples were centrifuged at 1,500 rpm for 15 minutes to harvest erythrocytes. Erythrocytes collected were resuspended in normal saline to achieve a concentration of 2% (v/v). Lf-PAEEP-PLLA NBs were diluted in normal saline to produce suspensions with different concentrations (0.5%, 1.0%, 2.5%, 5.0%, 7.5%, 10.0%, 12.5%, 15.0%, and 20.0% [v/v]). In addition, 150 μL of blood diluents was added to 150 μL of normal saline as the negative control, and 150 μL of double-distilled water was added as the positive control. After 1 hour’s incubation in a water bath at 37°C, the samples were centrifuged at 3,000 rpm for 25 minutes. The resulting supernatant was analyzed using a spectrophotometer at 540 nm. The percentage of hemolysis was calculated with respect to the 100% hemolysis control.
Effect of Lf on cellular uptake
Both C6 cells (low-density lipoprotein receptor-related protein 1 (LRP1) positive) and ECV304 cells (LRP1 negative) were used to investigate the cellular uptake of Lf-PAEEP-PLLA NBs and non-targeted PAEEP-PLLA NBs. Cells were seeded in 12-well plates at a density of 150,000 cells/well in DMEM, and incubated for 24 hours. Then Lf-PAEEP-PLLA NBs or PAEEP-PLLA NBs were added in cell culture medium at concentrations of 5, 10, 15, 20 μg/mL. After 2 hours’ incubation, the medium was removed and cells were washed with PBS three times. To remove cell debris, samples were centrifuged at 2,000 rpm for 10 minutes, followed by resuspension in double-distilled water. The supernatant was analyzed by flow cytometry (Beckman Coulter Inc, Brea, CA, USA). The NBs were labelled with fluorescent aminoluciferin for the flow cytometric cell-uptake study.
In vitro acoustic behavior
Lf-PAEEP-PLLA NBs solution was added to the wells of a custom-made 2% (w/v) agarose mode, which was positioned in a degassed water bath. The ultrasound contrast results were obtained from a B-mode clinical ultrasound imaging system (Philips NV, Amsterdam, Netherlands). To collect ultrasonic images, the 10 MHz probe of the clinical ultrasound imaging system (L12–1) was employed and immersed in the liquid. All contrast images was recorded in the B-mode, with low persistence, a frame rate of 37 Hz, and a mechanical index of 0.07. Photoshop software (Adobe Systems, Mountain View, CA, USA) was used to perform grayscale image analysis of the samples.
In vivo ultrasonic imaging study
All animal studies were approved by the Animal Experimentation Ethics Committee of Huazhong University of Science and Technology, and carried out in compliance with guidelines approved by the Science and Technology Department of Hubei Province.
BALB/c nude mice (male, aged 4 weeks, 18–20 g) were used in this study (Animals Center at Tongji Medical College). C6 cells (2×106 cells/mouse in 100 μL PBS) were subcutaneously injected into the right lower back of the mice. When the tumor volume reached 350–400 mm3, 100 μL of NBs were injected intra-tumorally. A probe (10 MHz) was placed at the tumor region to obtain images at the time points of 0 seconds, 30 seconds, 10 minutes, and 120 minutes. SonoVue® microbubbles (Bracco Imaging SpA, Milan, Italy), a commercial ultrasonic contrast agent, were used as control.
Statistical analysis
All data are reported as mean ± standard deviation. The statistical analysis of the differences among various treatments was performed using unpaired Student’s t-test. A 0.05 level of probability was used as the level of significance. Analysis of variance was also employed when necessary.
Results and discussion
Synthesis of PAEEP-PLLA copolymer
Polyphosphoesters have attracted a great deal of interest in biomedical fields, due to their biocompatibility, biodegradability, functional possibility, and flexibility.27–29 As previously reported, polyphosphoesters featuring a linear molecular structure can be synthesized through the ring-opening polymerization reaction of EEP in tetrahydrofuran, utilizing ROH or RNH2 as the co-initiator, and Sn(Oct)2 as the catalyst.30 In this work, the PAEEP-PLLA copolymer was synthesized as illustrated in Figure 2. Specifically, EABoc reacted with ethanolamine, and the resulting product was protected with Boc, while the active center of ROH was retained as the co-initiator. At this stage, the obtained PEEABoc reacted with EABoc and COP. The amino-terminated poly(L-lactide) (Mn =18,000) was synthesized by the ring-opening polymerization reaction of L-lactide using EABoc as the initiator and Sn(Oct)2 as the catalyst. PAEEP-PLLA was synthesized by the ring-opening polymerization reaction of PEEABoc, using amino-terminated poly(L-lactide) as the initiator and Sn(Oct)2 as the catalyst. The structure of products was confirmed by 1H NMR spectroscopy (Figure 1). The polymerization degree of PAEEP was determined using the integrated peak areas at 3.7 ppm (4H) and 5.2 ppm (1H), which correspond to the protons of the PLLA block. The PAEEP-PLLA copolymer was composed of a PAEEP segment with a polymerization degree of 30, and a PLLA segment with a polymerization degree of 130. Differential scanning calorimetry was employed to investigate the thermal behavior of the crystalline PAEEP-PLLA copolymer. As shown in Figure 3, the differential scanning calorimetry diagrams showed the melting temperature range of PAEEP-PLLA was from 129.05°C to 142.47°C and the glass-transition temperature (Tg) was 135.71°C.
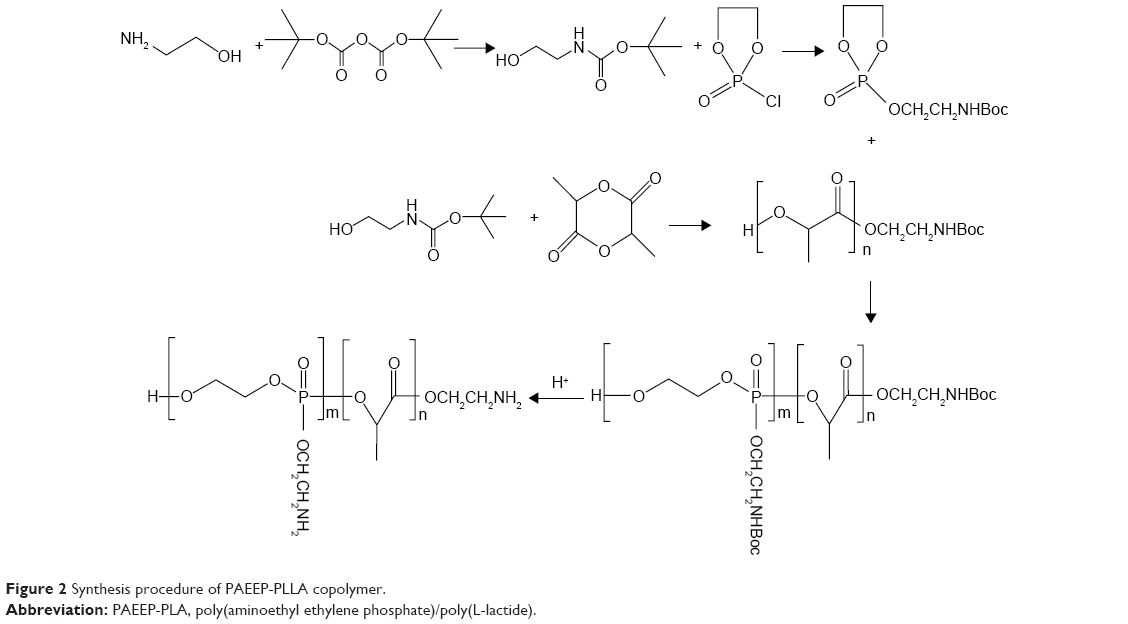 | Figure 2 Synthesis procedure of PAEEP-PLLA copolymer. |
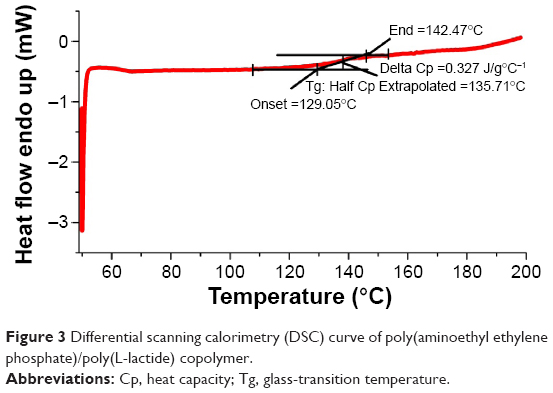 | Figure 3 Differential scanning calorimetry (DSC) curve of poly(aminoethyl ethylene phosphate)/poly(L-lactide) copolymer. |
Theoretically, the PLLA segment is hydrophobic and the PAEEP segment is highly hydrophilic. The obtained PAEEP-PLLA block copolymer was amphipathic with the combined appealing features of PLLA and PAEEP. The mechanical properties, degradation time, and hydrophilic/lipophilic feature of the copolymer can be regulated by selecting a suitable molecular weight, PAEEP/PLLA ratio, and side chain. Therefore, NBs made from PAEEP-PLLA copolymer, as an innovative ultrasonic imaging contrast agent, may have appropriate mechanical properties, good stability, appropriate hydrophilic/lipophilic properties, and moderate degradation time. Hydrophilic polyphosphate chains can effectively prevent the NBs from being cleared by the reticuloendothelial system in the blood. Moreover, the AEEP segment contains a large number of amino groups, which can be readily modified with tumor-targeting molecules.
Preparation of PAEEP-PLLA NBs
PAEEP-PLLA NBs were prepared using a O1/O2/W double-emulsion and solvent-evaporation method. PFP, a liquid with a boiling point of 29.5°C, was employed as the inner core of the NBs. PAEEP-PLLA was used as the shell material of the NBs. The liquid PFP core enhanced the storage stability and ensured the ultrasound-triggered phase-transition capability of the NBs, and can be converted to gaseous PFP at physiological temperature. The PAEEP-PLLA on the surface of the NBs can be conveniently coupled with tumor-targeting molecules.
Conjugation of Lf to PAEEP-PLLA NBs
Lf was conjugated to PAEEP-PLLA NBs for tumor targeting. The conjugation of Lf through a polyethylene glycol spacer is illustrated in Figure 4. Polyacrylamide gel electrophoresis was used to confirm the conjugation of Lf on the NBs. With the continuous buffer system, the separation by electrophoresis mainly depends on charge and volume effect. Because the isoelectric point of Lf is about 8.65, free Lf migrated easily onto the gel at pH 7.4. On the other hand, the Lf-PAEEP-PLLA NBs were negatively charged, as determined by determination of zeta potential, and had large volume. As shown in Figure 5, no migration of Lf was detected after it was conjugated to the NBs, indicating the coupling reaction had been successful.
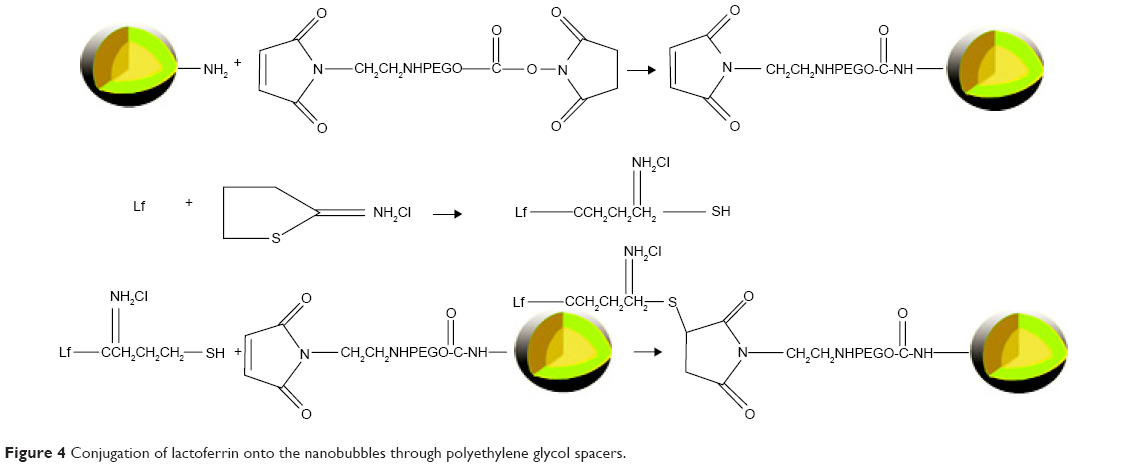 | Figure 4 Conjugation of lactoferrin onto the nanobubbles through polyethylene glycol spacers. |
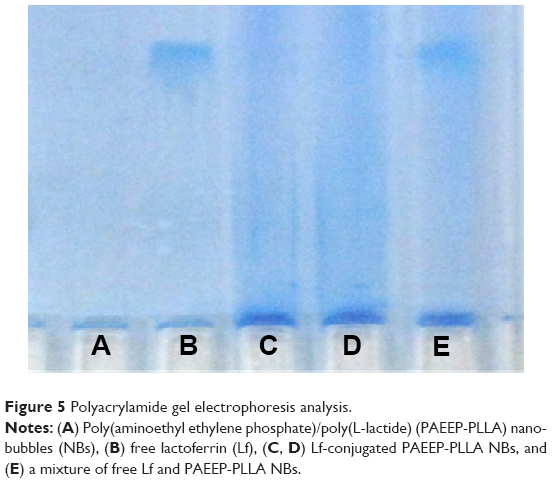 | Figure 5 Polyacrylamide gel electrophoresis analysis. |
In the FTIR spectra of Lf-conjugated NBs, two carbonyl absorption peaks at 1,600 cm−1 and 450 cm−1 were observed (Figure 6). However, these peaks were not present in the FTIR spectra of plain NBs. Both electrophoresis and FTIR analysis demonstrated the successful conjugation of Lf to NBs. The concentration of the conjugated Lf was 0.297 mg/mL, as determined by the bicinchoninic acid method.
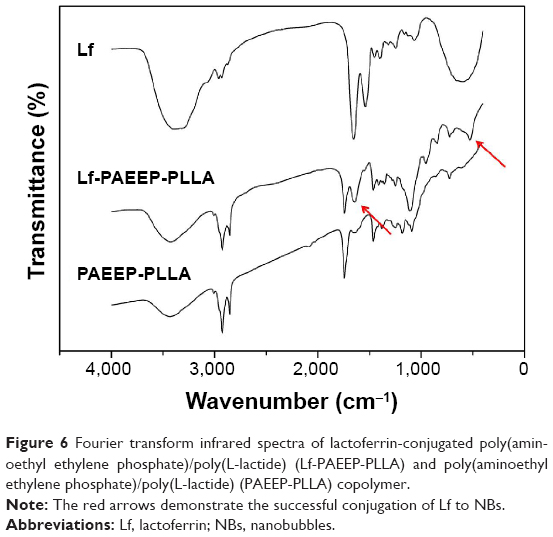 | Figure 6 Fourier transform infrared spectra of lactoferrin-conjugated poly(aminoethyl ethylene phosphate)/poly(L-lactide) (Lf-PAEEP-PLLA) and poly(aminoethyl ethylene phosphate)/poly(L-lactide) (PAEEP-PLLA) copolymer. |
Characterization of Lf-PAEEP-PLLA NBs
The appearance, size/size distribution, and TEM images of Lf-PAEEP-PLLA NBs are shown in Figure 7. The size and size distribution of Lf-PAEEP-PLLA NBs were measured by photon-correlation spectroscopy. An average size of 328.4±5.1 nm was found, with a PDI of 0.167±0.020, suggesting the NBs were relatively small and quite uniform. The zeta potential of the Lf-PAEEP-PLLA NBs was -12.6±0.3 mV, which indicates that the NBs were negatively charged when the Lf was conjugated. The number concentration of the Lf-PAEEP-PLLA NB solution was 1.5×108 bubbles/mL, which was determined using a hemocytometer and cell-counting method. TEM images revealed detailed morphological information about the the NBs. Lf-PAEEP-PLLA NBs existed as near-spherical particles and were quite monodisperse. There was a clear interface between the shell and the liquid core inside the NBs.
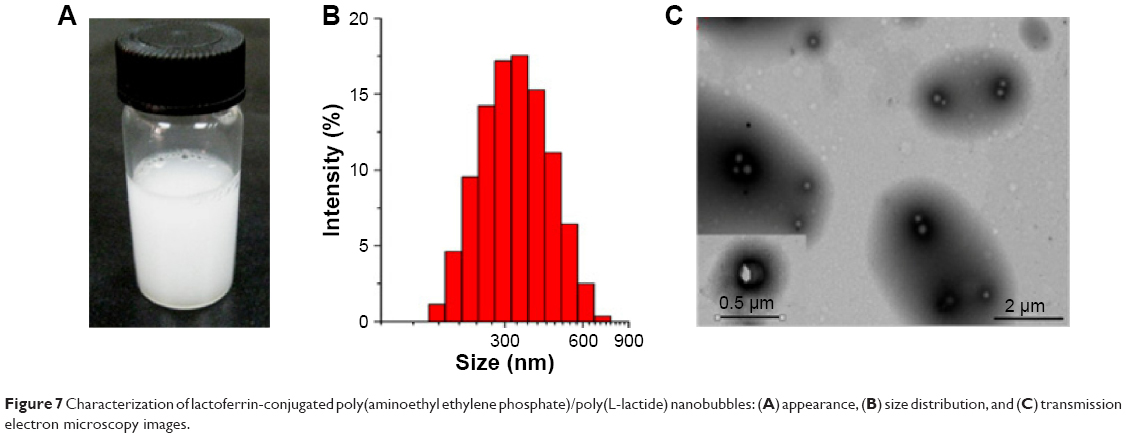 | Figure 7 Characterization of lactoferrin-conjugated poly(aminoethyl ethylene phosphate)/poly(L-lactide) nanobubbles: (A) appearance, (B) size distribution, and (C) transmission electron microscopy images. |
To evaluate the stability of the NBs, Lf-PAEEP-PLLA NB aqueous dispersion was stored at 4°C for 3 months. Little change was observed in particle size, PDI, and zeta potential during the storage period (0.0, 0.5, 1.0, and 3 months), suggesting the Lf-PAEEP-PLLA NBs had good storage stability (data not shown).
Biocompatibility evaluation of Lf-PAEEP-PLLA NBs
The biocompatibility of the Lf-PAEEP-PLLA NBs was evaluated by cytotoxicity assay and hemolysis assay. The cytotoxicity of Lf-PAEEP-PLLA NBs was tested using MTT assay for C6 cells and HL7702 cells. The results showed that the NBs did not induce cytotoxicity in C6 cells and HL7702 cells within the concentration range of 0.5%~20.0% (v/v). Less than 5% of hemolysis was observed at all evaluated concentrations of the NBs compared to double-distilled water (the positive control) with 100% of hemolysis under the experimental conditions. Thus, the cytotoxicity and hemolysis studies strongly suggest that the NBs had good biocompatibility for biomedical applications.
Effect of Lf on cellular uptake
C6 cells are rat glioma cells, which highly express LRP1. ECV304 cells are normal human umbilical vein endothelial cells with no LRP1 expression. Although LRP1 is expressed in some normal tissues, the level of LRP1 in tumor tissues is much higher.31 Previous studies have shown that Lf is a LRP1 ligand, which is a promising targeting ligand for cancer cells.26,32 As shown in Figure 8, for C6 cells, the uptake of Lf-PAEEP-PLLA NBs showed a significant increase in comparison with PAEEP-PLLA NBs. However, no obvious change in cellular uptake was observed in the ECV304 cells. The results clearly indicate that the Lf-PAEEP-PLLA NBs exhibited an adequate active tumor-cell targeting ability due to the introduction of the targeting ligand Lf.
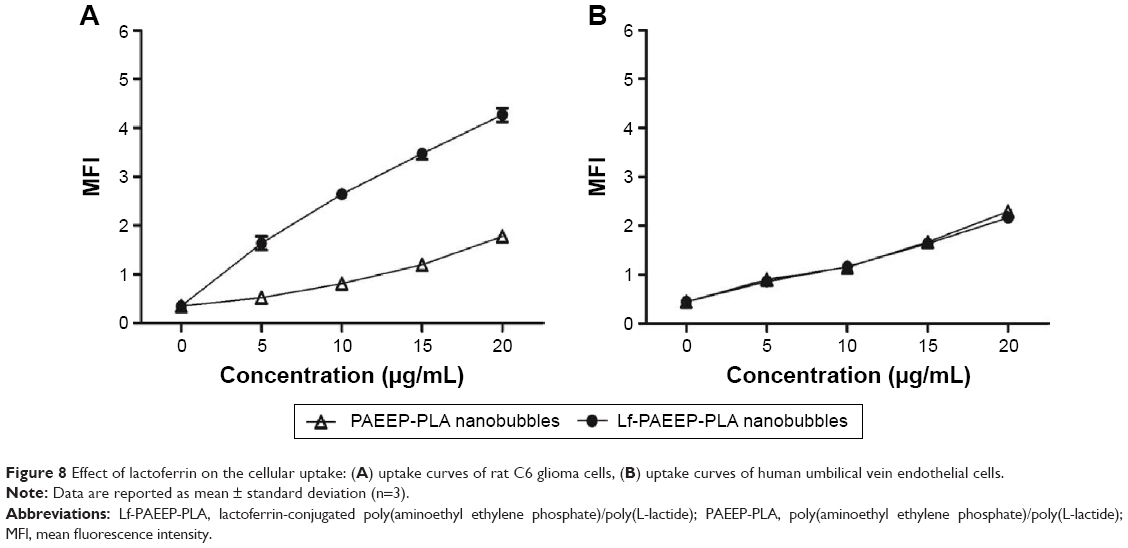 | Figure 8 Effect of lactoferrin on the cellular uptake: (A) uptake curves of rat C6 glioma cells, (B) uptake curves of human umbilical vein endothelial cells. |
Ultrasound-triggered phase transition
At room temperature, PFP maintained liquid state, and the size of the Lf-PAEEP-PLLA NBs was 328 nm. At physiological temperature (37°C), although the temperature is above the boiling point of PFP (29.5°C), the size of the NBs was only changed to about 340 nm; so no significant phase transition of PFP occurred. When the temperature reached 50°C, the size of the NBs increased to about 690 nm, suggesting that phase transition had occurred.
PFP is stabilized by elastic copolymer shells, and its boiling temperature is substantially increased by Laplace pressure. Laplace pressure is generally produced by the pressure difference between the inside and outside of a bubble, due to the surface tension at the interface between the bulk liquid and the bubble liquid.33–35
Laplace pressure is given as: 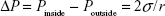 in which Pinside is the pressure inside the bubble, Poutside is the pressure outside the bubble, σ is the surface tension, and r is the bubble radius. Excessive pressure inside the bubble would result in an increase in the boiling temperature of PFP. According to the equation, Laplace pressure is reversely proportional to the radius of the bubble. Therefore, smaller bubbles should have a higher Pinside, leading to a higher boiling temperature of PFP.
in which Pinside is the pressure inside the bubble, Poutside is the pressure outside the bubble, σ is the surface tension, and r is the bubble radius. Excessive pressure inside the bubble would result in an increase in the boiling temperature of PFP. According to the equation, Laplace pressure is reversely proportional to the radius of the bubble. Therefore, smaller bubbles should have a higher Pinside, leading to a higher boiling temperature of PFP.
To evaluate the ultrasonic-triggered phase transition in the NBs, in vitro ultrasonic imaging experiments were performed. During the ultrasonic imaging, the NBs in the agarose model were placed in a water bath, and the temperature was increased from 20°C to 38°C gradually. As shown in Figure 9, the ultrasonic signals increased with the rise of temperature, and the signals strength reached its maximum at 29°C. But the signal strength started to decrease when the temperature was gradually increased to 38°C. Theoretically, the phase transition of PFP is related to two factors, heating and sonication.32 In the experiment, the phase transition started at 20°C under ultrasonic conditions, indicating that sonication reduced the PFP phase-transition temperature. Sonication appeared to be the most effective factor for PFP phase transition in the NBs.
 | Figure 9 In vitro ultrasonic images of lactoferrin-conjugated poly(aminoethyl ethylene phosphate)/poly(L-lactide) nanobubbles in agarose mold. |
In vitro ultrasonic imaging
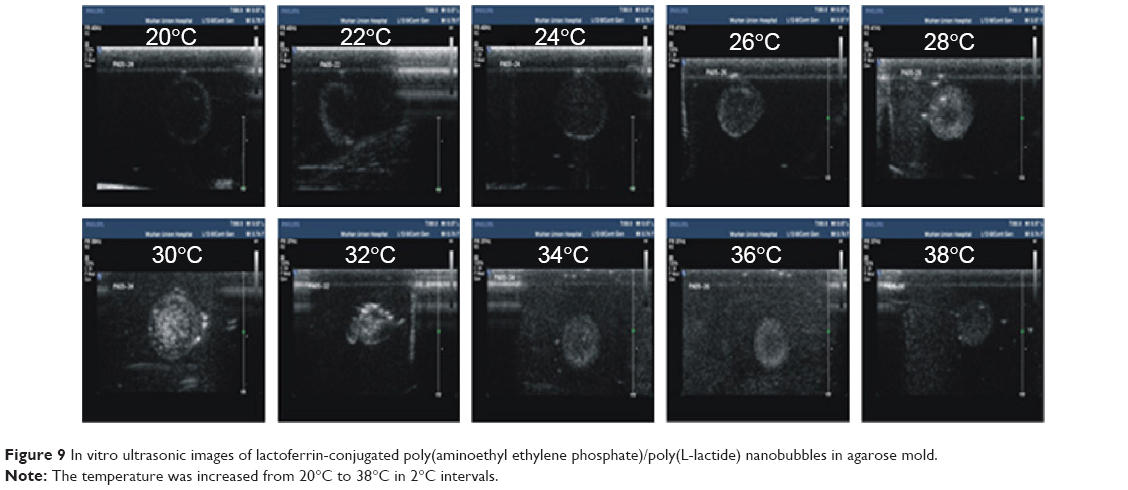
The PAEEP-PLLA copolymer used as the shell material for the NBs had good mechanical properties, which stretched under ultrasound irradiation. The NBs were turned into larger NBs or microbubbles when the PFP core in the copolymer shell transformed from liquid to gas. It appeared that the NBs in the agarose model oscillated in response to the ultrasonic wave. There was no signal observed when saline was placed in the agarose model. As shown in Figure 10A, the acoustic behavior of the NBs was presented as bright spots, which reflected the echogenic movement signals of the NBs. Naturally, this observation confirms that the NBs possessed acoustic activity.
 | Figure 10 Representative in vitro ultrasonic images. |
Ultrasound-targeted microbubble destruction is attractive as a safe, effective, non-invasive, and time- and region-controlled drug-release technology.36,37 However, microbubbles prepared from different materials behave differently. For instance, polymer-based shell materials, such as polylactic acid and poly(lactic-co-glycolic acid), have a high destruction threshold under ultrasonic irradiation. Consequently, it is extremely difficult to break up the shell and release its encapsulated cargo. Meanwhile, lipid-based shell materials generally possess a low destruction threshold under ultrasonic irradiation,20,38 thus they can be readily destroyed. The shell made from PAEEP-PLLA copolymer developed in the work reported here was effectively destroyed upon ultrasonication. As shown in Figure 10B and C, the signal intensity dramatically decreased compared with that originating from the NBs without flash treatment. Therefore, in addition to a contrast agent for ultrasound, Lf-PAEEP-PLLA NBs may serve as a controlled drug-release system.
In vivo ultrasonic imaging
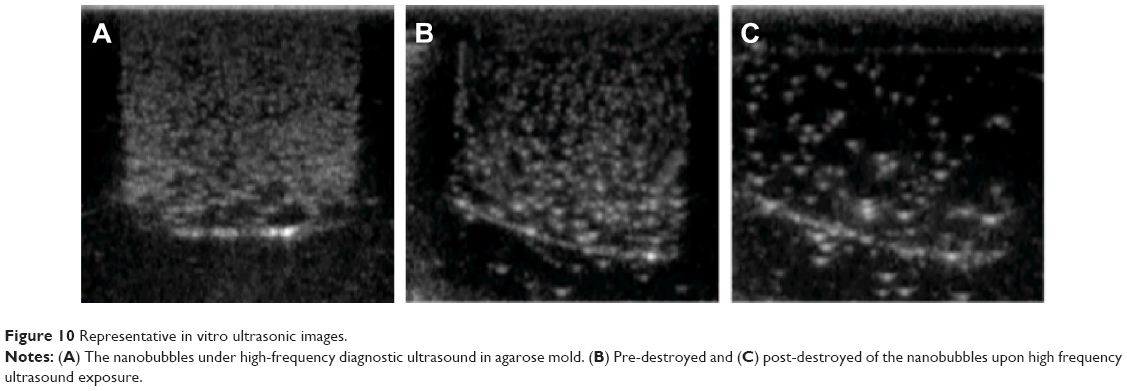
Ultrasonic imaging of subcutaneous tumors in BALB/c nude mice after intra-tumoral injection of 100 μL Lf-PAEEP-PLLA NBs was performed using a 10 MHz transducer. Images were recorded at 0 second, 30 seconds, 10 minutes, and 120 minutes after injection (Figure 11). A dark hypoechoic area was found in the tumors before the NBs injection (Figure 11A). Thirty seconds after the injection, some hyperechoic bright spots were found to have been generated in the tumor (Figure 11B). Subsequently, more and more bright spots appeared and a large number of bright spots was observed in the tumor 10 minutes after the injection (Figure 11C). The number of bright spots was not found to have decreased significantly in the tumor at 120 minutes after injection (Figure 11D). The prepared NBs were formed by PFP and stabilized by an outer wall composed of the biodegradable block copolymer. As a result, the PFP stabilized by the outer layer could not diffuse away from the NBs, so the NBs would have long-lasting ultrasonic contrast ability. These results suggest that such NBs can be used as a stable and long-lasting contrast agent in clinical ultrasonography.
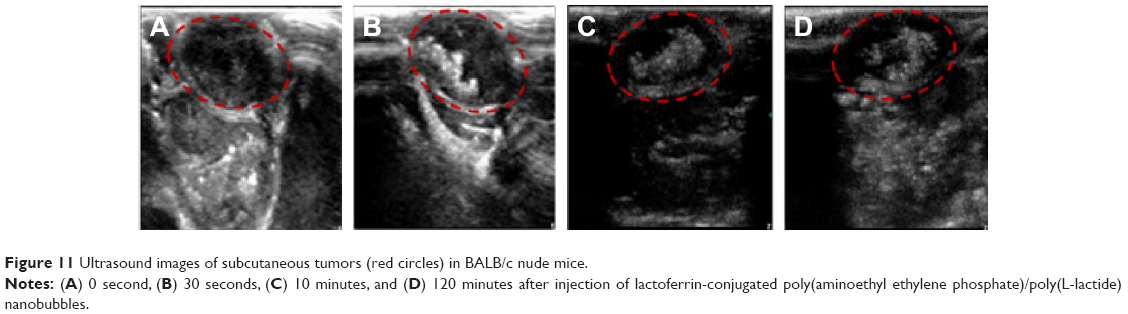 | Figure 11 Ultrasound images of subcutaneous tumors (red circles) in BALB/c nude mice. |
The performance of the Lf-PAEEP-PLLA NBs was also compared with that of SonoVue, a commercial ultrasonic contrast agent. As shown in Figure 12, the Lf-PAEEP-PLLA NBs produced a remarkable and complete, powerful Doppler enhancement throughout the tumors, while SonoVue only generated signals in a small part of the tumor. SonoVue is a kind of microbubble, with a larger diameter (2–8 μm) compared to our Lf-PAEEP-PLLA NBs, so it has been limited to use in diseases of the cardiovascular system. When the SonoVue microbubbles were injected into the tumor, they could not distribute throughout the tumor due to their larger diameter, so only induced contrast enhancement in a small portion of the tumor. However, as the Lf-PAEEP-PLLA NBs were of nanoscale size, they were able to distribute inside the whole tumor, and significant contrast enhancement was observed throughout the tumor. These results successfully demonstrate the great potential of Lf-PAEEP-PLLA NBs as a tumor-targeting imaging reagent.
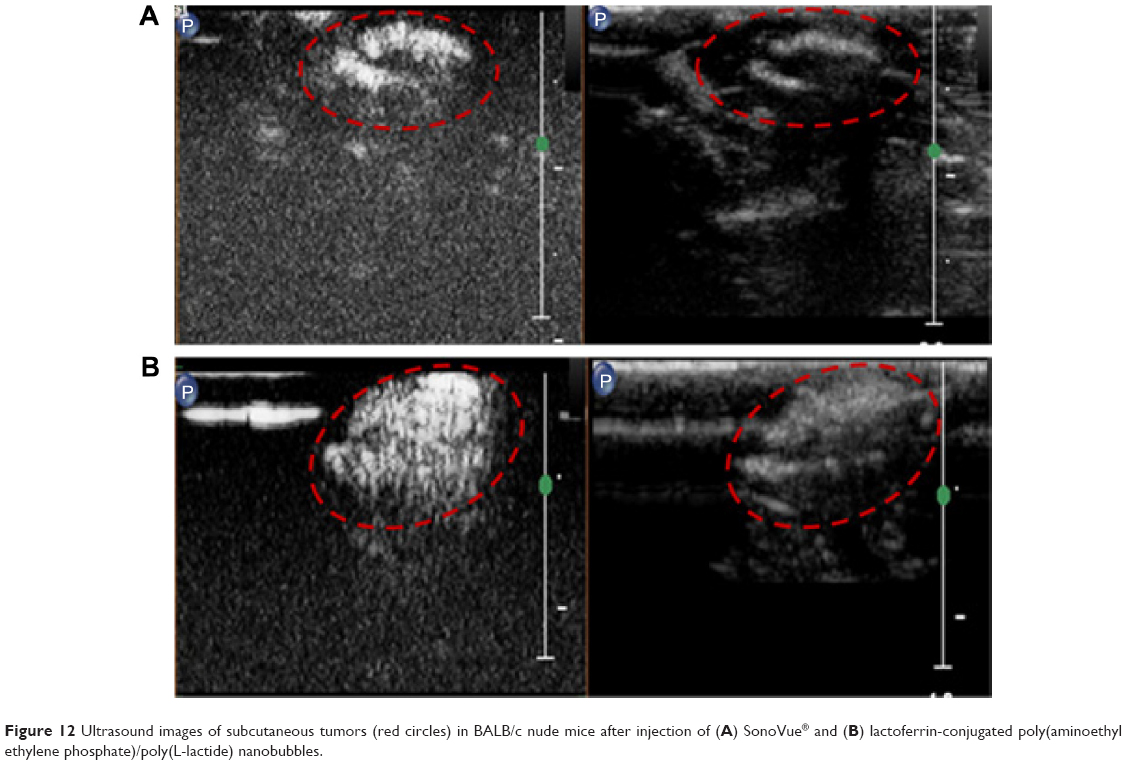 | Figure 12 Ultrasound images of subcutaneous tumors (red circles) in BALB/c nude mice after injection of (A) SonoVue® and (B) lactoferrin-conjugated poly(aminoethyl ethylene phosphate)/poly(L-lactide) nanobubbles. |
Conclusion
In this study, amphiphilic PAEEP-PLLA copolymer was successfully synthesized by ring-opening polymerization reaction. A novel ultrasound contrast agent based on PFP-filled PAEEP-PLLA NBs were prepared using O1/O2/W double-emulsion and solvent-evaporation method. Lf, an effective targeting ligand for cancer cells, was conjugated to the PAEEP-PLLA NBs. The experimental results show that the Lf-PAEEP-PLLA NBs were highly biocompatible and exhibited remarkable tumor-targeting capability. The in vitro and in vivo ultrasound imaging indicate that the Lf-PAEEP-PLLA NBs possessed strong, long-lasting, tumor-targeting, and contrast-enhanced ultrasonic ability. Furthermore, the Lf-PAEEP-PLLA NBs were effectively destroyed upon ultrasonication, indicating they may serve as a controlled drug-release system. In short, the novel Lf-PAEEP-PLLA NBs hold great potential for broad biomedical applications, from ultrasound imaging to the targeted delivery and controlled release of drugs to tumors.
Acknowledgments
This work was financially supported by the National Science Foundation of China (numbers 31170960, 31470968, and 81271582), the National Basic Research Program of China (973 Program, 2012CB932500), and the National High Technology Research and Development Program of China (863 Program, 2012AA022704).
Disclosure
The authors report no conflicts of interest in this work.
References
Dănilă M, Popescu A, Sirli R, Sporea I, Martie A, Sendroiu M. Contrast enhanced ultrasound (CEUS) in the evaluation of liver metastases. Med Ultrason. 2010;12(3):233–237. | ||
Kiessling F, Fokong S, Koczera P, Lederle W, Lammers T. Ultrasound microbubbles for molecular diagnosis, therapy, and theranostics. J Nucl Med. 2012;53(3):345–348. | ||
Wilson SR, Burns PN. Microbubble-enhanced US in body imaging: what role? Radiology. 2010;257(1):24–39. | ||
Geers B, Lentacker I, Sanders NN, Demeester J, Meairs S, De Smedt SC. Self-assembled liposome-loaded microbubbles: The missing link for safe and efficient ultrasound triggered drug-delivery. J Control Release. 2011;152(2):249–256. | ||
Hernot S, Klibanov AL. Microbubbles in ultrasound-triggered drug and gene delivery. Adv Drug Deliv Rev. 2008;60(10):1153–1166. | ||
Lentacker I, De Smedt SC, Sanders NN. Drug loaded microbubble design for ultrasound triggered delivery. Soft Matter. 2009;5(11):2161–2170. | ||
Melino S, Zhou M, Tortora M, Paci M, Cavalieri F, Ashokkumar M. Molecular properties of lysozyme-microbubbles: towards the protein and nucleic acid delivery. Amino Acids. 2012;43(2):885–896. | ||
Gessner R, Dayton PA. Advances in molecular imaging with ultrasound. Mol Imaging. 2010;9(3):117–127. | ||
Yin T, Wang P, Li J, et al. Ultrasound-sensitive siRNA-loaded nanobubbles formed by hetero-assembly of polymeric micelles and liposomes and their therapeutic effect in gliomas. Biomaterials. 2013;34(18):4532–4543. | ||
Suzuki R, Oda Y, Utoguchi N, Maruyama K. Progress in the development of ultrasound-mediated gene delivery systems utilizing nano- and microbubbles. J Control Release. 2011;149(1):36–41. | ||
Yang H, Cai W, Xu L, et al. Nanobubble-Affibody: Novel ultrasound contrast agents for targeted molecular ultrasound imaging of tumor. Biomaterials. 2014;37:279–288. | ||
Xu JS, Huang J, Qin R, et al. Synthesizing and binding dual-mode poly (lactic-co-glycolic acid) (PLGA) nanobubbles for cancer targeting and imaging. Biomaterials. 2010;31(7):1716–1722. | ||
Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–135. | ||
Rapoport NY, Kennedy AM, Shea JE, Scaife CL, Nam KH. Controlled and targeted tumor chemotherapy by ultrasound-activated nanoemulsions/microbubbles. J Control Release. 2009;138(3):268–276. | ||
Yin T, Wang P, Zheng R, et al. Nanobubbles for enhanced ultrasound imaging of tumors. Int J Nanomedicine. 2012;7:895–904. | ||
Zhang X, Zheng Y, Wang Z, et al. Methotrexate-loaded PLGA nanobubbles for ultrasound imaging and Synergistic Targeted therapy of residual tumor during HIFU ablation. Biomaterials. 2014;35(19):5148–5161. | ||
Hernot S, Unnikrishnan S, Du Z, et al. Nanobody-coupled microbubbles as novel molecular tracer. J Control Release. 2012;158(2):346–353. | ||
Zhang X, Uddin MH, Yang H, et al. Effects of surfactants on the formation and the stability of interfacial nanobubbles. Langmuir. 2012;28(28):10471–10477. | ||
Yin T, Wang P, Li J, et al. Tumor-penetrating codelivery of siRNA and paclitaxel with ultrasound-responsive nanobubbles hetero-assembled from polymeric micelles and liposomes. Biomaterials. 2014;35(22):5932–5943. | ||
Böhmer MR, Chlon CH, Raju BI, Chin CT, Shevchenko T, Klibanov AL. Focused ultrasound and microbubbles for enhanced extravasation. J Control Release. 2010;148(1):18–24. | ||
Cheng X, LI H, Chen YC, et al. Ultrasound-triggered phase transition sensitive magnetic fluorescent nanodroplets as a multimodal imaging contrast agent in rat and mouse model. PLoS One. 2013; 8(12):e85003. | ||
Kim SE, Yun YP, Shim KS, Park K, Choi SW, Suh DH. Effect of lactoferrin-impregnated porous poly(lactide-co-glycolide) (PLGA) microspheres on osteogenic differentiation of rabbit adipose-derived stem cells (rADSCs). Colloid Surfaces B: Biointerfaces. 2014;122:457–464. | ||
Li Y, He H, Jia X, Lu WL, Lou J, Wei Y. A dual-targeting nanocarrier based on poly(amidoamine) dendrimers conjugated with transferrin and tamoxifen for treating brain gliomas. Biomaterials. 2012;33(15):3899–3908. | ||
Ha NH, Nair VS, Reddy DN, et al. Lactoferrin-endothelin-1 axis contributes to the development and invasiveness of triple-negative breast cancer phenotypes. Cancer Res. 2011;71(23):7259–7269. | ||
Xie H, Zhu Y, Jiang W, et al. Lactoferrin-conjugated superparamagnetic iron oxide nanoparticles as a specific MRI contrast agent for detection of brain glioma in vivo. Biomaterials. 2011;32(2):495–502. | ||
Jiang L, Zhou Q, Mu K, et al. pH/temperature sensitive magnetic nanogels conjugated with Cy5.5-labled lactoferrin for MR and fluorescence imaging of glioma in rats. Biomaterials. 2013;34(30):7418–7428. | ||
Wang YC, Yuan YY, Du JZ, Yang XZ, Wang J. Recent progress in polyphosphoesters: from controlled synthesis to biomedical applications. Macromol Biosci. 2009;9(12):1154–1164. | ||
Zhang P, Hu L, Wang Y, Wang J, Feng L, Li Y. Poly(ε-caprolactone)-block-poly(ethyl ethylene phosphate) micelles for brain-targeting drug delivery: in vitro and in vivo valuation. Pharm Res. 2010;27(12):2657–2669. | ||
Sun TM, Du JZ, Yan LF, Mao HQ, Wang J. Self-assembled biodegradable micellar nanoparticles of amphiphilic and cationic block copolymer for siRNA delivery. Biomaterials. 2008;29(32):4348–4355. | ||
Xiao CS, Wang YC, Du JZ, Chen XS, Wang J. Kinetics and mechanism of 2-ethoxy-2-oxo-1,3,2-dioxaphospholane polymerization initiated by stannous octoate. Macromolecules. 2006;39:6825–6831. | ||
Demeule M, Currie JC, Bertrand Y, et al. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J Neurochem. 2008;106(4):1534–1544. | ||
Huang R, Ke W, Liu Y, Jiang C, Pei Y. The use of lactoferrin as a ligand for targeting the polyamidoamine-based gene delivery system to the brain. Biomaterials. 2008;29(2):238–246. | ||
Cavalli R, Bisazza A, Trotta M, et al. New chitosan nanobubbles for ultrasound-mediated gene delivery: preparation and in vitro characterization. Int J Nanomedicine. 2012;7:3309–3318. | ||
Rapoport N, Gao Z, Kennedy A. Multifunctional nanoparticles for combining ultrasonic tumor imaging and targeted chemotherapy. J Natl Cancer Inst. 2007;99(14):1095–1106. | ||
Rapoport NY, Efros AL, Christensen DA, Kennedy AM, Nam KH. Microbubble Generation in Phase-Shift Nanoemulsions used as Anticancer Drug Carriers. Bubble Sci Eng Technol. 2009;1(1–2):31–39. | ||
Zhou S, Li S, Liu Z, et al. Ultrasound-targeted microbubble destruction mediated herpes simplex virus-thymidine kinase gene treats hepatoma in mice. J Exp Clin Canc Res. 2010;29:170. | ||
Luo J, Zhou X, Diao L, Wang Z. Experimental research on wild-type p53 plasmid transfected into retinoblastoma cells and tissues using an ultrasound microbubble intensifier. J Int Med Res. 2010;38(3):1005–1015. | ||
Bloch SH, Wan M, Dayton PA, Ferrara KW. Optical observation of lipid- and polymer-shelled ultrasound microbubble contrast agents. Appl Phys Lett. 2004;84(4):631–633. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.