")
Back to Journals » International Journal of General Medicine » Volume 16
Novel Intronic Mutations of the SLC12A3 Gene in Patients with Gitelman Syndrome
Authors Xun Z, Gao P, Du Y, Yan X, Yang J, Wang Z
Received 3 March 2023
Accepted for publication 1 May 2023
Published 11 May 2023 Volume 2023:16 Pages 1797—1806
DOI https://doi.org/10.2147/IJGM.S408631
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Zeli Xun,1,* Pengfei Gao,2,3,* Yanan Du,1 Xue Yan,3 Jingmin Yang,2– 4 Zhihua Wang1
1Department of Endocrinology, Xi’an Children’s Hospital, Shanxi, People’s Republic of China; 2State Key Laboratory of Genetic Engineering, School of Life Sciences, Fudan University, Shanghai, People’s Republic of China; 3Shanghai WeHealth Biomedical Technology Co, Ltd, Shanghai, People’s Republic of China; 4Key Laboratory of Birth Defects and Reproductive Health of National Health and Family Planning Commission (Chongqing Key Laboratory of Birth Defects and Reproductive Health, Chongqing Population and Family Planning, Science and Technology Research Institute), Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhihua Wang, Xi’an Children’s Hospital, Shanxi, 710002, People’s Republic of China, Email [email protected]
Aim: Mutations in the SLC12A3 gene have been reported to cause Gitelman syndrome (GS), characterized by hypokalemic metabolic alkalosis. The aim of this research is to investigate the genetic mutations and clinical features of patients with clinical suspicion of GS.
Methods: Six families were enrolled. The symptoms, clinical examination, laboratory results, genotypes, and effect of mutations on mRNA splicing were analyzed. Genomic DNA was screened for gene variations using whole exome sequence and Sanger sequencing. DNA sequences were compared with reference sequences.
Results: Genetic analysis revealed nine genetic variants of SLC12A3, including three novel heterozygous mutations (c.1096– 2A>G, c.1862A>G, and c.2747+4del) and six previously characterized mutations (c.965– 1_976delinsACCGAAAATTTT, c.506– 1G>A, c.602– 16G>A, c.533C >T, c.1456 G>A, and c.1108 G>C). Probands presented with the clinical syndrome of hypokalemia, increased plasma renin, hypocalciuria and hypokalemic alkalosis.
Conclusion: These clinical manifestations and genotypes were consistent with the diagnostic criteria of GS. The study described the phenotypes and genotypes of six pedigrees involving GS patients, demonstrating the importance of SLC12A3 gene screening for GS. This study expands the mutation spectrum of SLC12A3 gene in GS.
Keywords: SLC12A3, Gitelman syndrome, gene mutation, mRNA splicing, clinical characteristics
Introduction
Gitelman syndrome (GS) is an autosomal recessive disease, characterized by hypokalemic alkalosis, accompanied by hypomagnesaemia, hypocalciuria, low blood pressure, and hypocalcemia, first described by Gitelman in 1966.1 It is caused by mutations in the SLC12A3 gene, which is located on the long arm of chromosome 16(16q13) and encodes the renal thiazide-sensitive Na/Cl co-transporter (NCCT) in the distal convoluted tubules.2
In Western countries, the incidence of GS is about 1–10 per 40,000 people and the prevalence for the heterozygote is roughly 1%.3 However, the prevalence of GS has grown remarkably to 10.3 per 10,000 population and mutation rate may be as high as 3% in Asia.4 Currently, a number of pathogenic mutations have been found to cause GS, including nonsense mutations, frameshift mutations, deletions, insertions, splicing site mutations and missense mutations. Intronic variants can not only activate noncanonical splice sites and alter splicing regulatory elements,5 but also interrupt regulatory motifs and the transcription of noncoding RNA genes.6 The incidence of GS has been increasing, and the most common missense mutation was p. Thr60Met in Chinese GS patients.7 However, the hot spot mutation site is not yet known.
Although the long-term prognosis is considered to be favorable, hypokalemia is difficult to cure. The diagnosis of GS is made through clinical assessments, biochemical screening and genetic research analyses which has become important in diagnosing GS.8 In this study, we described five compound heterozygous and one homozygous variant of SLC12A3 gene in six Chinese pediatric GS patients. Whole exome sequencing (WES) identified nine variants, three of which were novel. We verified that two variants can affect the RNA splicing of SLC12A3 gene by in vitro experiments.
Method
Subjects
Children who were clinically suspected or clinically diagnosed with GS combined with genetic diagnosis at the Department of Endocrinology of Xi’an Children’s Hospital from December 2020 to December 2021 were enrolled in this cohort. Patients’ clinical manifestation and biochemical parameters were consistent with the diagnostic criteria for kidney disease guidelines,9 which included manifestations of hypokalemia, hypomagnesemia, hypocalcemia, muscle weakness, and normal or low blood pressure, and so on. The exclusion criteria were as follows: 1) Patients with a history of diuretics, laxatives, chronic vomiting, or diarrhea; 2) Patients with other conditions that could lead to hypokalemia (renal tubular acidosis or thyrotoxic periodic paralysis, Cushing’s syndrome, primary aldosteronism, etc.). Six pedigrees were selected finally (Figure 1). Clinical data, medical history and general information were retrospectively collected. Probands and their parents underwent genetic analysis, including WES and Sanger sequencing. Genomic DNA (gDNA) was isolated from ethylenediaminetetraacetic acid-treated peripheral blood. Genetic sequencing revealed that each patient had rare/novel intronic or exonic mutation with unknown significance or pathogenic. This study protocol was approved by the ethics committees of Xi’an Children’s Hospital and carried out according to the 2013 Helsinki Declaration. Written informed consent was obtained from each patient’s parents or guardian.
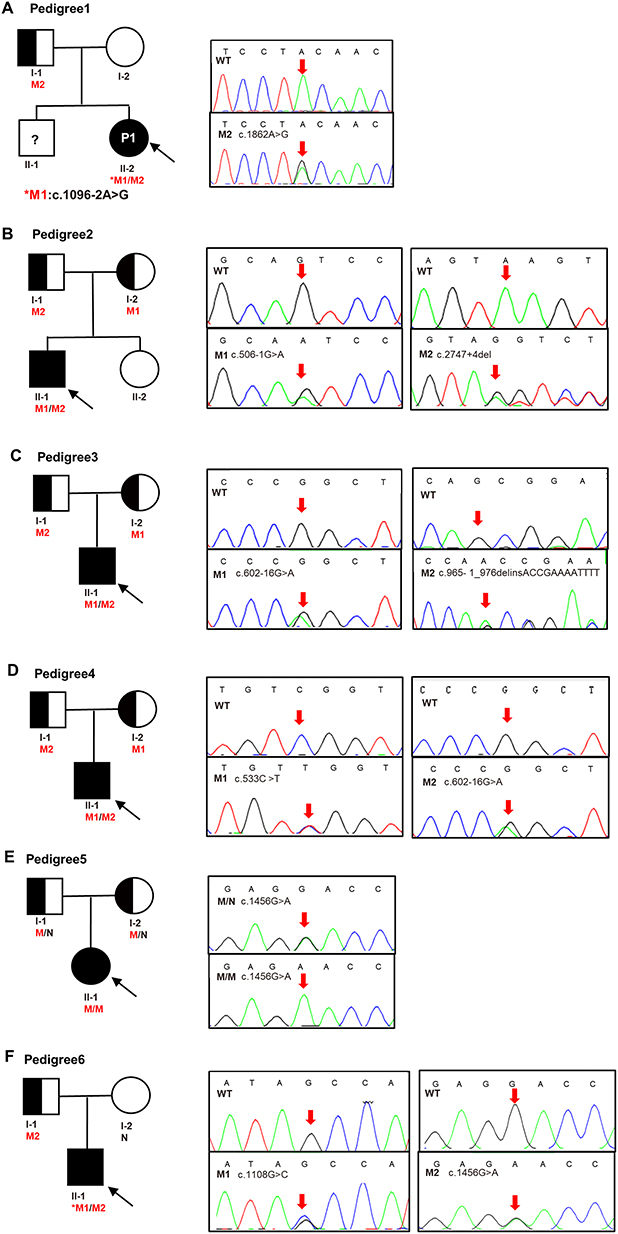 |
Figure 1 Pedigrees of the six family and Sanger sequencing. (A) Pedigree of No.1 patient, M1 (c.1096–2A>G) was de novo, M2 (c.1862A>G) was inherited from father. (B) Pedigree of No.2 patient, M1 (c.506–1G>A) and M2 (c.2747+4del) were inherited from the mother and father, respectively. (C) Pedigree of No.3 patient, M1 (c.602–16G>A) and M2 (c.965–1_976delinsACCGAAAATTTT) were inherited from the mother and father, respectively. (D) Pedigree of No.4 patient, M1 (c.533C>T) and M2 (c.602–16G>A) were inherited from the mother and father, respectively. (E) Pedigree of No.5 patient, mutation (c.1456G>A) was inherited from her mother and father. (F) Pedigree of No.6 patient, M1 (c.1108G>C) was de novo, M2 (c.1456G>A) was inherited from father. Males and females are indicated by squares and circles. An affected individual is indicated by filled black and carrier filled in half black. The black arrow indicates the proband and red arrow indicates the location of the Sanger sequencing of the mutations. An asterisk refers to a de novo mutation. N stands for normal sequencing and WT stands for wild type. M1 and M2 represent mutation 1 and mutation 2. |
Genetic Analysis
Peripheral blood of proband and parents were collected by EDTA anticoagulant tube and outsourced to Shanghai We-Health Biomedical Technology Co, Ltd. The genomic DNA extraction was undertaken with the Blood Genomic DNA Extraction Kit (TIANGEN, China). Library preparation and Clinical Exome capture were performed with Hieff NGS®OnePot DNA Library Prep Kit for Illumina® (YEASEN, China) and twist custom panel (Twist Bioscience Corporation, USA). Captured libraries were sequenced on DNBSEQ-T7 (MGI, China) in Pair-End 100 mode.
A bioinformatic filtering pipeline was modified from a previously described approach.10 The quality of raw data was checked by FASTQC. The clean reads were mapped to the reference genome (GRCh37/hg19) with BWA software. After removing duplicates and base quality recalibration, SNP and Indel variants were called using the GATK pipeline. Detected variants were annotated using ANNOVAR and filtered with minor allele frequencies (MAFs) of <0.5% for dbSNP, 1000G, ExAC, and gnomAD databases. Additionally, copy number variation analysis was performed of probe coverage regions using XHMM11 and CLAMMS12 tools. The list of genes related to patient phenotype used in variation analysis is presented in Supplementary Table 1.
SLC12A3 was sequenced by Sanger sequencing. The polymerase chain reaction (PCR) conditions were as follows: a pre-denaturation step (95°C for 5 min), followed by 35 cycles of denaturation (95°C for 30s), annealing (60°C for 30s), elongation (72°C for 30s), with a final extension (72°C for 5 min). Amplicon was sequenced on the ABI 3730xl Genetic Analyzer (Applied Biosystems, Inc.). Sequences were compared using Codon Code Aligner to reference sequences.
Online mutation prediction tools like PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Taster (http://www.mutationtaster.org/), and SIFT (http://sift.jcvi.org/) were used to predict the pathogenicity of missense mutations. Potential splice effects of variants were predicted using the following mutation prediction tools: Varseak (https://varseak.bio), SpliceAI (https://spliceailookup.broadinstitute.org/), and dbscSNV.
In vitro Splicing Analysis
In order to verify the effect of variation on mRNA splicing, total RNA was isolated using the PAXgene Blood RNA Kit (Qiagen #762174). Reverse transcription was performed using Hifair®II 1st Strand cDNA Synthesis Kit (YEASEN Biotech Co., Ltd, China). Primer sequences were designed to amplify the different transcripts of SLC12A3 (Table 1). The 20ul PCR reaction system included 10ul 2 × Taq Plus Master Mix (Nanjing Vazyme Biotech Co., Ltd.), 0.5ul each primer, 5ul cDNA template and 4ul H2O. PCR cycling conditions were as follows: an initial denaturation of 95°C for 3 min, 35 cycles of denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec, and extension at 72°C for 45 sec and then an extra 5 min at 72°C. Successful amplification of the PCR products was confirmed by 2% agarose gel electrophoresis. PCR products of similar lengths were isolated by TA cloning. The PCR product fragments were purified by Sanprep Column DNA Gel Extraction Kit (Sangon Biotech, China) and then cloned into the pESI-T vector using Hieff CloneTM Zero TOPO-TA Cloning Kit (YEASEN Biotech Co., Ltd, China). The recombinant DNAJB6 plasmids were transformed into E. coli competent cells of the DH5α strain. Recombinant plasmids were extracted using Sanprep Column Plasmid DNA Microextraction Kit (Sangon Biotech Co., Ltd, China), and sequencing directly was performed by ABI 3730xl DNA Analyzer.
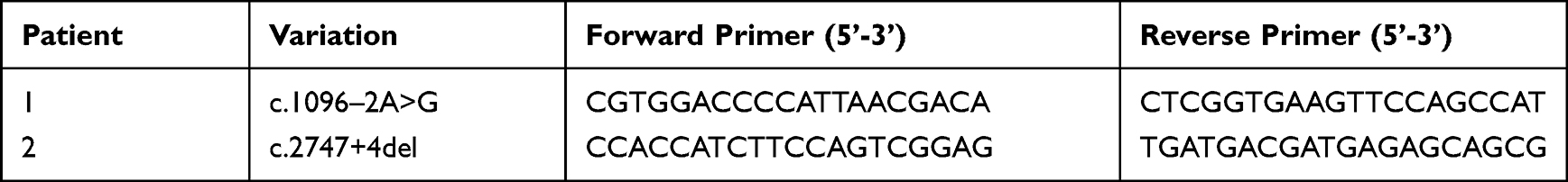 |
Table 1 RT-PCR Primer Information |
Result
Clinical Characteristics of the Patients
Six patients, including four boys and two girls, were enrolled in the study. They came from six non-consanguineous families (Figure 1). Clinical characteristics and laboratory data of 6 patients are listed in Table 2, as well as the reference values of biochemical parameters. As shown in Table 2, the typical onset of the disease occurs in young adults, with a higher incidence in males. All patients presented with hypokalemia (1.83–2.56 mmol/L), increased plasma renin (142.69–4947.54 pg/mL), hypocalciuria (0.11–0.46 mmol/24h) and hypokalemic alkalosis (pH 7.46–7.58), which are typical symptoms of GS. Milder hyponatremia was found in patient No.1, and hypochloremia were found in patient No.1, No.3, No.4 and No.6, respectively. Patient No.1, No.2, No.4 and No.5 had a typical muscle weakness phenotype, which recovered quickly after intravenous potassium infusion or ingestion of oral potassium chloride. In addition, three probands had normal–low blood pressure (90/60–104/63mmHg). The grandfather of No.1 patient had a family history of hypertension, and the grandmother and grandfather of No.3 patient had diabetes. The remaining members denied a family history of hypokalemia and other diseases. Combining the clinical features and laboratory findings of the six preceding patients, it was possible to find hypokalemia, hypocalciuria, hyperreninemia, and hypotension in almost all of them. The common causes of hypokalemia (primary aldosteronism, Cushing’s syndrome, hyperthyroidism, etc.) were also excluded. Therefore, the clinical diagnosis of GS was suggested, but further genetic testing is needed to confirm the diagnosis.
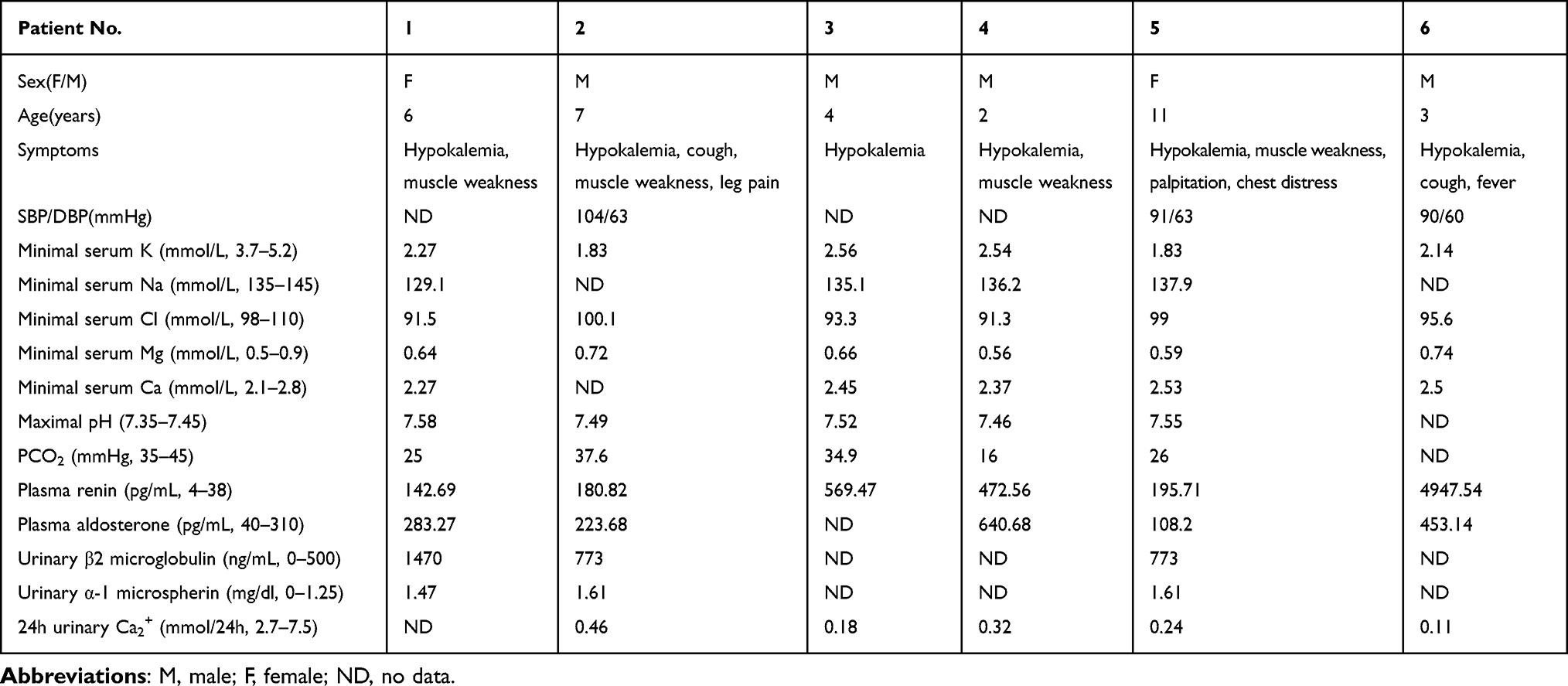 |
Table 2 Clinical Characteristics of Six Patients with GS |
Genomic Studies Identify Variant in SLC12A3
Analysis of the SLC12A3 gene revealed nine genetic variants, including three novel heterozygous mutations, c.1096–2A>G, c.1862A>G, and c.2747+4del, and six previously characterized heterozygous mutations, c.965–1_976delinsACCGAAAATTTT, c.506–1G>A, c.602–16G>A, c.533C>T, c.1456G>A, and c.1108G>C (Table 3 and Figure 2A). As shown in Table 3, there are nine variants, including 4 missense mutations, 4 intronic mutations and 1 insertion deletion mutation. According to the American College of Medical Genetics (ACMG) guidelines,13 there were six distinct pathogenic or likely pathogenic variants and three uncertain significance variants (VUS). All the parental studies of the six patients had done, and the lineage diagrams and Sanger sequencing results of the six families are shown in Figure 1. For five patients, one mutant allele was inherited from one carrier parent, and the other mutant allele was inherited from the other carrier carried or was de novo variant. Hence, five patients were compound heterozygous and 1 patient was homozygous. All the missense variants were predicted to be deleterious or probably damaging by Polyphen-2, Mutation Taster, and SIFT and were categorized into likely pathogenic or pathogenic variant according to ACMG, except c.1862A>G. There was an adenine to guanine transition (c.1862A>G), resulting in a tyrosine to cysteine substitution at position 621 (p.Y621C) in exon 15. Although c.1862A>G was predicted to be probably damaging deleterious by software and was absent in gnomAD_EAS database, no published references could be matched to illustrate the pathogenicity of this variant. Therefore, we presume that this variant may be uncertain significance variant (VUS). Another heterozygous variant harbored by No.1 patient (c.1096–2A>G) and a heterozygous variant detected in No.2 patient (c.2747+4del) were predicted to have splicing effect by several software. Both variants were no published reference reported and absent in gnomAD_EAS database. Thus, they were categorized as VUS variants and further functional studies are necessary to confirm the pathogenicity.
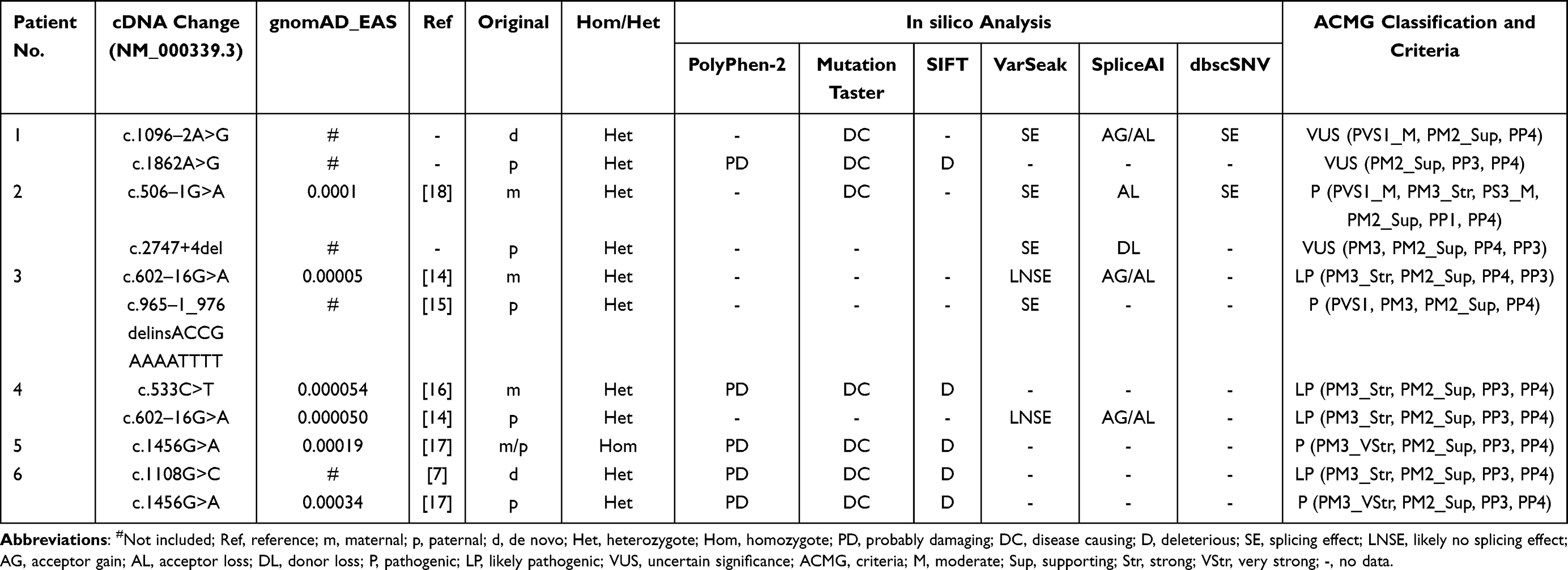 |
Table 3 Genetic Analysis of Six Patients with GS |
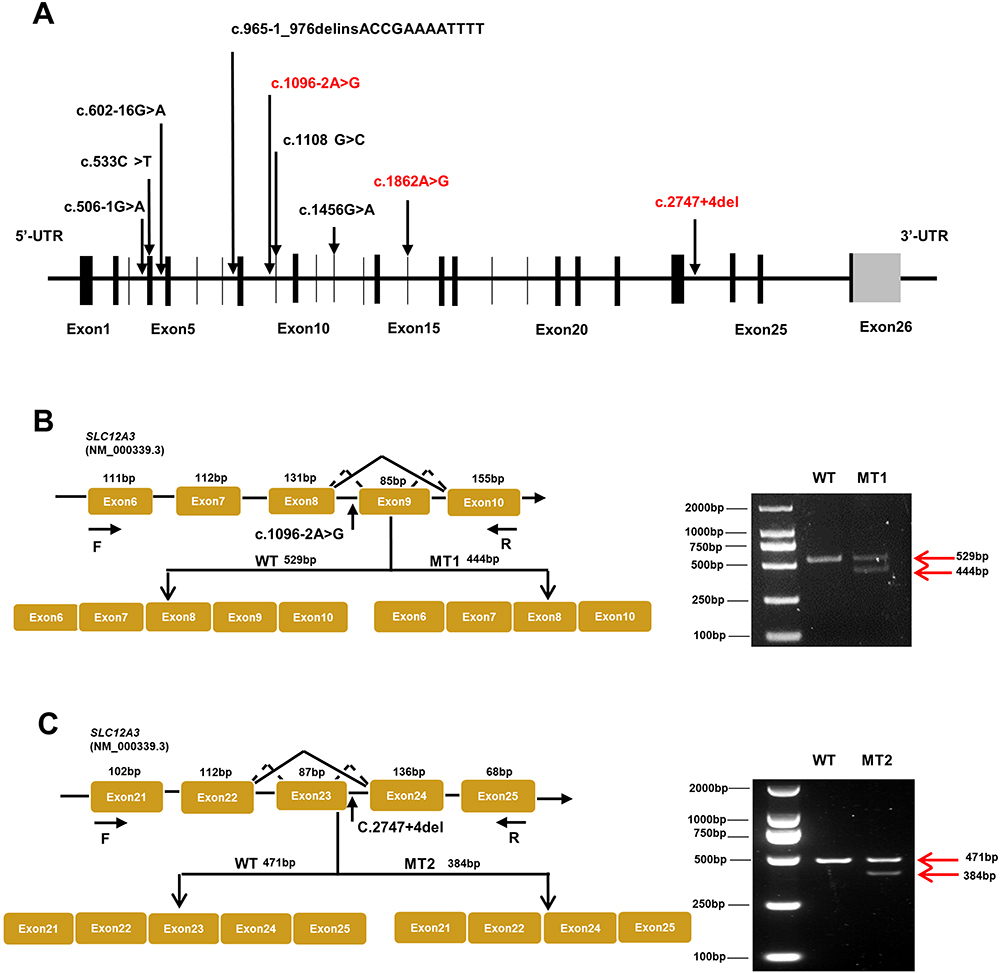 |
Figure 2 Schematic overview of SLC12A3 gene and representation of intronic variants on pre-RNA splicing in patients with GS. (A) The structure of SLC12A3 and its 26 exons which are indicated by black vertical lines. The black arrows in the diagram indicate the position of nine variations. Mutations that have been reported are shown in black, and those novel mutations in the manuscript are indicated by red. (B) Schematic overview of the region harboring c.1096–2A>G mutation in SLC12A3 and representation of the abnormal splicing. Representation of the alternative splicing of exon 9 in SLC12A3. Electrophoresis for amplification of SLC12A3 cDNA from the WT and No.1 patient (MT1). (C) Schematic overview of the region harboring c.2747+4del mutation in SLC12A3 and representation of the abnormal splicing. Representation of the alternative splicing of exon 23 in SLC12A3. Electrophoresis for amplification of SLC12A3 cDNA from the WT and No.2 patient (MT2). Red arrows indicate the position of MT and WT in agarose gel. WT represents wild-type and MT represents mutant type. |
The SLC12A3 Variants Induce Aberrant mRNA Splicing and Transcript Deletion
Due to its proximity effect to the exon splice donor site or acceptor site, we wondered whether SLC12A3 c.1096–2A>G, and c.2747+4del could induce mRNA splicing defects. To investigate this possibility, we isolated the total RNA from whole blood of No.1 and No.2 patients then reverse transcribed into cDNA. Since blood leukocytes express NCCT mRNA,18 it is feasible to detect intronic mutations in blood leukocytes from GS patients using reverse transcriptase (RT)-PCR. cDNA sequencing was performed to confirm the splicing predictions, along with normal splicing, aberrant splicing products were observed (shown in Figure 2B and C).
The presence of the splicing mutation c.1096-2A>G C in No.1 patient which was a de novo variant caused the occurrence of an abnormal transcript that lacked the entire exon 9 in addition to the normal-length transcript (Figure 2B). Electrophoresis for amplification of SLC12A3 cDNA from No.1 patient showed the large-sized fragment (529bp) corresponded to the correctly spliced mRNA and the small-sized fragment (444 bp) corresponded to the skipping of exon 9. The small-sized fragment was more apparently observed for the patient. Analysis of the No.2 patients who carried the mutation c.2747+4del revealed an aberrantly spliced transcript that lacked exon 23. The large-sized fragment (471bp) corresponded to the correctly spliced mRNA and the small-sized fragment (384 bp) corresponded to the skipping of exons 23. The sequencing chromatogram of SLC12A3 normal cDNA and two splicing variants are shown in Figure 3. These data suggest that these mutations may disrupt exon splicing enhancers (ESE), which are sequences of ESE following exon skipping. The results point to the potential pathogenicity of these mutations, which should be recognized as candidates for variant reclassification in the future.
 |
Figure 3 Sequencing chromatogram of SLC12A3 wild-type cDNA and splice variants. (A) The normal splicing fragment between exon 8 and 10; aberrant transcript of c.1096–2A>G skipping of exon 9. (B) The normal splicing fragment between exon 22 and 24; aberrant transcript of c.2747+4del skipping of exon 23. |
Discussion
In this study, we explored the novel mutations of our GS patients as well as some previously reported variants and the clinical characteristics. Notably, three novel mutations, c.1096–2A>G, c.1862A>G, and c.2747+4del, were identified. Genealogical analysis revealed a pattern consistent with autosomal recessive inheritance.
The six patients in the present study presented with clinical symptoms of hypokalemia, normal serum magnesium, increased plasma renin, hypocalciuria, hypokalemic alkalosis, milder hyponatremia, hypochloremia, muscle weakness, and normal–low blood pressure. Although some patients have symptoms of hypokalemia, they usually hospitalized due to none related diseases such as fever and cough, occasionally with mild symptoms of GS. Most clinical symptoms of GS are linked to acid-base and electrolyte disturbances.9,19 Normomagnesemia is not uncommon in GS, and the severity of the clinical manifestations of GS is negatively correlated with the level of serum magnesium.20 It has been reported that 8% to 22% of patients with GS have normal blood magnesium,21 and the pathogenesis of hypomagnesemia in GS is still unclear. Blood magnesium levels below 0.5 mmol/L result in significant clinical manifestations including neuromuscular hyperexcitability, tremors, convulsions, and coma.22 GS with normal blood magnesium showed a low frequency of wrist spasms or hand and foot twitches, as well as mild electrolyte disturbances, metabolic alkalosis, and activation of the renin angiotensin-aldosterone system (RAAS). In this study, the patient had no relevant conscious clinical symptoms, which may relate to normal magnesium levels. Three probands showed elevated micro-urinary protein, suggesting renal tubular dysfunction. The causes of proteinuria and renal impairment in patients with Gitelman syndrome are mainly related to long-term hypokalemia, which can lead to tubulointerstitial damage and cyst formation, and proteinuria is mainly tubular source;23 Secondly, due to the long-term activation of RAAS, it may directly or indirectly lead to glomerular sclerosis of the renal pool, and in rare cases, there are other glomerular diseases, such as IgA nephropathy, C1q nephropathy,24 etc., but its correlation with Gitelman syndrome is not clear.9 It is considered that proteinuria is due to angiotensin II or renin-induced damage to the glomerular basement membrane caused by podocyte injury.25 Renal potassium loss is a common clinical cause of hypokalemia. Common diseases that can cause renal potassium loss include primary aldosteronism, Cushing’s syndrome, renal tubular acidosis, Bartter syndrome and GS. Among them, most patients with primary aldosteronism have blood pressure higher than 160/100 mmHg and blood potassium is usually mildly reduced, while most patients in this study had aldosterone levels less than 300 pg/mL and normal to low blood pressure, so primary aldosteronism was not considered. In addition, none of the patients in this study had clinical manifestations such as full moon face, buffalo back, or acne, so Cushing’s syndrome was also not considered. Patients with Bartter syndrome have similar symptoms to GS and are more difficult to differentiate, but patients with Bartter syndrome generally have normal blood magnesium and urinary calcium levels, so the patients in this study are more consistent with the symptoms of GS, but still need to be differentiated by further genetic testing.
SLC12A3 gene has a total of 26 exons. The distribution of 9 mutations detected in this paper is shown in Figure 2A. SLC12A3 gene encodes the NaCl cotransport protein (NCC), which is a 12-transmembrane protein.26 Depending on the relationship between the NCC protein and the cell membrane, the mutation sites can be intracellular, extracellular and transmembrane. GS is caused by SLC12A3 gene mutation, and missense mutation is the most common. According to the genetic analysis, Thr60Met and Asp486Asn are the most common amino acid mutations in Chinese patients and possibly specific to Asian populations,27,28 while IVS9+1G>T and c.1196_1202dup7bp were reported to be the most common mutation in European populations.29,30 In this study, we found a novel missense mutation in exon 15(c.1862A>G), which have not been reported previously. The missense variant change locates at the carboxy end of the protein intracellular, where amino acids at this region are conserved among different species and human ethnicity.31
In the nucleus, mature mRNA contains only coding sequences, and intronic sequences are removed from transcripts during splicing. Splicing mutations may occur in introns and exons and disrupt existing splice sites or splicing regulatory sequences. Splicing mutations can lead not only to improper exon and intron recognition but also to incorrect intron removal, which can lead to alterations in the open reading frame and ultimately may result in the formation of abnormal transcripts of mutant genes. Therefore, precise pre-mRNA splicing is essential for protein translation.32 For the c.1096–2A>G and c.2747+4del mutations, reclassification of variants of uncertain significance is critical and additional evidence is needed to support pathogenicity. The results of RNA splicing analysis point to the potential pathogenicity of these variants, and we strongly recommend additional RNA splicing analysis, especially for variants predicted to have potential splicing alterations. Notably, No.1 patient carries a de novo mutation and a missense variant of paternal origin, whose parents are both healthy adults, and the Sanger verification results suggest that his brother has the same mutation. It is hypothesized that the source of the SLC12A3 variants in this pedigree may be due to germline mosaicism, since parental germ cells are difficult to obtain and whole exome sequencing cannot exclude the possibility of a very low percentage of germline mosaicism in the tested samples. According to the follow-up, the younger brother of the proband is in good health and has no GS related clinical characteristics. Follow-up observation of the brother is recommended, and germ cell chimerism may be considered as a source of germline mosaicism if the parents have a request to have rebirth.
Our results identified two novel mutation sites (c.1096–2A>G, c.2747+4del) in vitro experiments, which could contribute to the enrichment of database resources on GS if its pathogenicity was determined. Moreover, the establishment of a diagnostic process for GS has important implications for confirmed cases. We are fully aware that this would have been very important to define the significance of the study.
There are two major limitations in this study that could be addressed in future research. First, more functional experiments are needed to reveal the underlying molecular mechanism of the two novel splice mutations in GS. Second, we expand the sample size to further explore the correlation between genotype and phenotype to better understand GS.
Conclusions
Overall, this study identified three novel mutations of SLC12A3 gene in Chinese patients. These clinical manifestations and genotypes were consistent with the diagnostic criteria of GS. The study described the phenotypes and genotypes of six pedigrees involving GS patients, demonstrating the importance of SLC12A3 gene screening for GS and expanding the mutation spectrum of SLC12A3 gene in GS.
Ethics Approval and Consent to Participate
This protocol of this study was approved by the ethics committees of Xi’an Children’s Hospital and carried out according to the 2013 Helsinki Declaration. Written informed consent was obtained from each patient’s parents or guardian.
Funding
We thank all the patients and families for their cooperation throughout the study. The authors report no targeted funding.
Disclosure
Zeli Xun and Pengfei Gao share first authorship. This study did not receive any financial support from any individual or organization.
References
1. Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–235.
2. Mastroianni N, Bettinelli A, Bianchetti M, et al. Novel molecular variants of the Na-Cl cotransporter gene are responsible for Gitelman syndrome. Am J Hum Genet. 1996;59(5):1019–1026.
3. Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis. 2008;3:22. doi:10.1186/1750-1172-3-22
4. Tago N, Kokubo Y, Inamoto N, Naraba H, Tomoike H, Iwai N. A high prevalence of Gitelman’s syndrome mutations in Japanese. Hypertens Res. 2004;27(5):327–331. doi:10.1291/hypres.27.327
5. Rossanti R, Horinouchi T, Sakakibara N, et al. Detecting pathogenic deep intronic variants in Gitelman syndrome. Am J Med Genet A. 2022;188(9):2576–2583. doi:10.1002/ajmg.a.62885
6. Lo YF, Nozu K, Iijima K, et al. Recurrent deep intronic mutations in the SLC12A3 gene responsible for Gitelman’s syndrome. Clin J Am Soc Nephrol. 2011;6(3):630–639. doi:10.2215/cjn.06730810
7. Zeng Y, Li P, Fang S, et al. Genetic Analysis of SLC12A3 Gene in Chinese Patients with Gitelman Syndrome. Med Sci Monit. 2019;25:5942–5952. doi:10.12659/msm.916069
8. Fujimura J, Nozu K, Yamamura T, et al. Clinical and Genetic Characteristics in Patients With Gitelman Syndrome. Kidney Int Rep. 2019;4(1):119–125. doi:10.1016/j.ekir.2018.09.015
9. Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017;91(1):24–33. doi:10.1016/j.kint.2016.09.046
10. Kohda M, Tokuzawa Y, Kishita Y, et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016;12(1):e1005679. doi:10.1371/journal.pgen.1005679
11. Miyatake S, Koshimizu E, Fujita A, et al. Detecting copy-number variations in whole-exome sequencing data using the eXome Hidden Markov Model: an ‘exome-first’ approach. J Hum Genet. 2015;60(4):175–182. doi:10.1038/jhg.2014.124
12. Packer JS, Maxwell EK, O’Dushlaine C, et al. CLAMMS: a scalable algorithm for calling common and rare copy number variants from exome sequencing data. Bioinformatics. 2016;32(1):133–135. doi:10.1093/bioinformatics/btv547
13. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
14. Glaudemans B, Yntema HG, San-Cristobal P, et al. Novel NCC mutants and functional analysis in a new cohort of patients with Gitelman syndrome. Eur J Hum Genet. 2012;20(3):263–270. doi:10.1038/ejhg.2011.189
15. Shao L, Liu L, Miao Z, et al. A novel SLC12A3 splicing mutation skipping of two exons and preliminary screening for alternative splice variants in human kidney. Am J Nephrol. 2008;28(6):900–907. doi:10.1159/000141932
16. Zahed H, Sparks TN, Li B, Alsadah A, Shieh JTC. Potential Role of Genomic Sequencing in the Early Diagnosis of Treatable Genetic Conditions. J Pediatr. 2017;189:222–226.e1. doi:10.1016/j.jpeds.2017.06.040
17. Bao M, Cai J, Yang X, Ma W. Genetic screening for Bartter syndrome and Gitelman syndrome pathogenic genes among individuals with hypertension and hypokalemia. Clin Exp Hypertens. 2019;41(4):381–388. doi:10.1080/10641963.2018.1489547
18. Abuladze N, Yanagawa N, Lee I, et al. Peripheral blood mononuclear cells express mutated NCCT mRNA in Gitelman’s syndrome: evidence for abnormal thiazide-sensitive NaCl cotransport. J Am Soc Nephrol. 1998;9(5):819–826. doi:10.1681/asn.V95819
19. Filippatos TD, Rizos CV, Tzavella E, Elisaf MS. Gitelman syndrome: an analysis of the underlying pathophysiologic mechanisms of acid-base and electrolyte abnormalities. Int Urol Nephrol. 2018;50(1):91–96. doi:10.1007/s11255-017-1653-4
20. Jiang L, Chen C, Yuan T, et al. Clinical severity of Gitelman syndrome determined by serum magnesium. Am J Nephrol. 2014;39(4):357–366. doi:10.1159/000360773
21. Liu T, Wang C, Lu J, Zhao X, Lang Y, Shao L. Genotype/Phenotype Analysis in 67 Chinese Patients with Gitelman’s Syndrome. Am J Nephrol. 2016;44(2):159–168. doi:10.1159/000448694
22. Viering D, de Baaij JHF, Walsh SB, Kleta R, Bockenhauer D. Genetic causes of hypomagnesemia, a clinical overview. Pediatr Nephrol. 2017;32(7):1123–1135. doi:10.1007/s00467-016-3416-3
23. Ceri M, Unverdi S, Altay M, et al. Focal segmental glomerulosclerosis in association with Gitelman syndrome. Int Urol Nephrol. 2011;43(3):905–907. doi:10.1007/s11255-010-9802-z
24. Hanevold C, Mian A, Dalton R. C1q nephropathy in association with Gitelman syndrome: a case report. Pediatr Nephrol. 2006;21(12):1904–1908. doi:10.1007/s00467-006-0261-9
25. Chen Q, Wu Y, Zhao J, Jia Y, Wang W. A case of hypokalemia and proteinuria with a new mutation in the SLC12A3 Gene. BMC Nephrol. 2018;19(1):275. doi:10.1186/s12882-018-1083-2
26. Ellison DH. The thiazide-sensitive na-cl cotransporter and human disease: reemergence of an old player. J Am Soc Nephrol. 2003;14(2):538–540. doi:10.1681/asn.V142538
27. Ma J, Ren H, Lin L, et al. Genetic Features of Chinese Patients with Gitelman Syndrome: sixteen Novel SLC12A3 Mutations Identified in a New Cohort. Am J Nephrol. 2016;44(2):113–121. doi:10.1159/000447366
28. Wang F, Shi C, Cui Y, Li C, Tong A. Mutation profile and treatment of Gitelman syndrome in Chinese patients. Clin Exp Nephrol. 2017;21(2):293–299. doi:10.1007/s10157-016-1284-6
29. Bouwer ST, Coto E, Santos F, Angelicheva D, Chandler D, Kalaydjieva L. The Gitelman syndrome mutation, IVS9+1G>T, is common across Europe. Kidney Int. 2007;72(7):898. doi:10.1038/sj.ki.5002504
30. Gu X, Su Z, Chen M, Xu Y, Wang Y. Acquired Gitelman syndrome in a primary Sjögren syndrome patient with a SLC12A3 heterozygous mutation: a case report and literature review. Nephrology. 2017;22(8):652–655. doi:10.1111/nep.13045
31. Gil-Peña H, Coto E, Santos F, et al. A new SLC12A3 founder mutation (p.Val647Met) in Gitelman’s syndrome patients of Roma ancestry. Nefrologia. 2017;37(4):423–428. doi:10.1016/j.nefro.2017.01.007
32. Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. 2018;59(3):253–268. doi:10.1007/s13353-018-0444-7
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.