Back to Journals » The Application of Clinical Genetics » Volume 14
Novel Genetic Causes of Gastrointestinal Polyposis Syndromes
Authors Jelsig AM ![]() , Byrjalsen A
, Byrjalsen A ![]() , Busk Madsen M
, Busk Madsen M ![]() , Kuhlmann TP, van Overeem Hansen T, Wadt KAW
, Kuhlmann TP, van Overeem Hansen T, Wadt KAW ![]() , Karstensen JG
, Karstensen JG
Received 2 September 2021
Accepted for publication 10 November 2021
Published 27 November 2021 Volume 2021:14 Pages 455—466
DOI https://doi.org/10.2147/TACG.S295157
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Anne Marie Jelsig,1 Anna Byrjalsen,1 Majbritt Busk Madsen,2 Tine Plato Kuhlmann,3 Thomas van Overeem Hansen,1 Karin AW Wadt,1,4 John Gásdal Karstensen4,5
1Department of Clinical Genetics, University Hospital of Copenhagen, Copenhagen, Denmark; 2Center for Genomic Medicine, University Hospital of Copenhagen, Copenhagen, Denmark; 3Department of Pathology, University Hospital of Copenhagen, Herlev Hospital, Herlev, Denmark; 4Department of Clinical Medicine, University of Copenhagen, Copenhagen, Denmark; 5Danish Polyposis Registry, Gastro Unit, Copenhagen University Hospital – Amager and Hvidovre, Copenhagen, Denmark
Correspondence: Anne Marie Jelsig
Department of Clinical Genetics, University Hospital of Copenhagen, Rigshospitalet, Blegdamsvej 9, Copenhagen, 2100, Denmark
Tel +45 20 36 18 85
Email [email protected]
Abstract: Hereditary polyposis syndromes are characterized by a large number and/or histopathologically specific polyps in the gastrointestinal tract and a high risk of both colorectal cancer and extracolonic cancer at an early age. While the genes responsible for some of the syndromes, eg, APC in familial adenomatous polyposis and STK11 in Peutz-Jeghers syndrome, have been known for decades, novel genetic causes have recently been detected that have shed light on the broader clinical spectrum of syndromes. Genetic diagnoses are important because they can facilitate a personalized surveillance program. Furthermore, at-risk members of the patient’s family can be tested and enrolled in surveillance as needed. In some cases, prenatal diagnostics should be offered. In this paper, we describe the development in germline genetics of the hereditary polyposis syndromes over the last 10– 12 years, their clinical characteristics, as well as how to implement genetic analyses in the diagnostic pipeline.
Keywords: polyposis, hereditary, familial adenomatous polyposis, cancer, management
Introduction
Hereditary polyposis syndromes (HPS) are the cause of severe morbidity and increased mortality in a significant number of patients worldwide. HPS patients often have an increased risk of extraintestinal cancer and extraintestinal manifestations that require tailored surveillance, depending on the specific genetic cause. The syndromes can be present in childhood, adolescence, and adulthood with a broad spectrum of symptoms ranging from very few (asymptomatic) polyps to severe polyposis and cancer. As the syndromes are genetic in origin, patients’ family members may also be affected.
In 1991, APC was the first known gene to be causative of a polyposis syndrome: familial adenomatous polyposis (FAP).1–3 Since then, several other genes have been discovered that are associated with gastrointestinal (GI) polyposis and for which causal pathogenic variants (PVs) can be detected (Figures 1 and 2). Next-generation sequencing (NGS) methods in genetic research and diagnostics have further accelerated the search for novel genes and have lowered the threshold for performing genetic analyses in suspected HPS patients as analyses become faster and cheaper.
Most of the novel HPS are rare and have only been reported in a small number of patients and their families. Thus, we are only now beginning to understand the full phenotypic spectrum of the syndromes and their medical implications. In this paper, we focus on describing the discoveries made of HPS’ clinical characteristics during the last 10–12 years, and we discuss implementing genetic analyses in the diagnostic pipeline. Detailed descriptions of FAP, juvenile polyposis syndrome (JPS), Peutz-Jeghers syndrome (PJS), PTEN hamartoma tumor syndrome, AXIN2-related polyposis, and MUTYH-associated polyposis (MAP) are beyond the remit of this review.
Phenotypic Spectrum and Histopathology
Traditionally, HPS have been classified according to the dominant histopathology of the polyposis, eg, in hamartomatous, adenomatous polyposis, etc. (Figures 2 and 3), and their mode of inheritance. This approach is still useful, but for many of the syndromes a patient presents with a more mixed picture of polyps and hence establishing a clinical diagnosis can prove more difficult. The histologic distinction between different polyps might not always be obvious, due to overlapping morphology and the difficulty in recognizing features such as subtle dysplasia. As a pathologist, one should of course always pay special attention to a patient’s former pathology reports and the number, size, histology, and location of polyps. Suspected polyposis and hereditary cases should be clearly stated in such reports and, ideally, discussed at multidisciplinary team meetings.
Genetic analysis can make possible a precise diagnosis in many patients with GI polyposis, and as more patients are offered genetic testing, knowledge of the phenotypic spectrum grows. Not every patient with a genetic diagnosis and pathogenic variants in a polyposis-related gene presents with the classic multiple polyps; some patients present with only a small number of polyps. Although many of the HPS have full penetrance, most have considerable inter- and intrafamilial expressivity, making it impossible to predict the course of disease and development of cancer in an individual carrying PV. For some syndromes, we are only now beginning to understand how some variants lead to a milder or more severe phenotype (ie, the genotype–phenotype correlation).
Both novel causes of autosomal dominant and autosomal recessive HPS have been discovered. Here, we will focus on both. The AD syndromes comprise the polymerase proofreading-associated polyposis (POLD1- and POLE-associated polyposis), GREM1-associated polyposis, gastric adenocarcinoma and proximal polyposis syndrome (GAPPS) and RNF43-associated serrated polyposis, while the autosomal recessive syndromes include NTHL1-, MLH3-, MSH3- and MBD4-associated polyposis (Table 1).
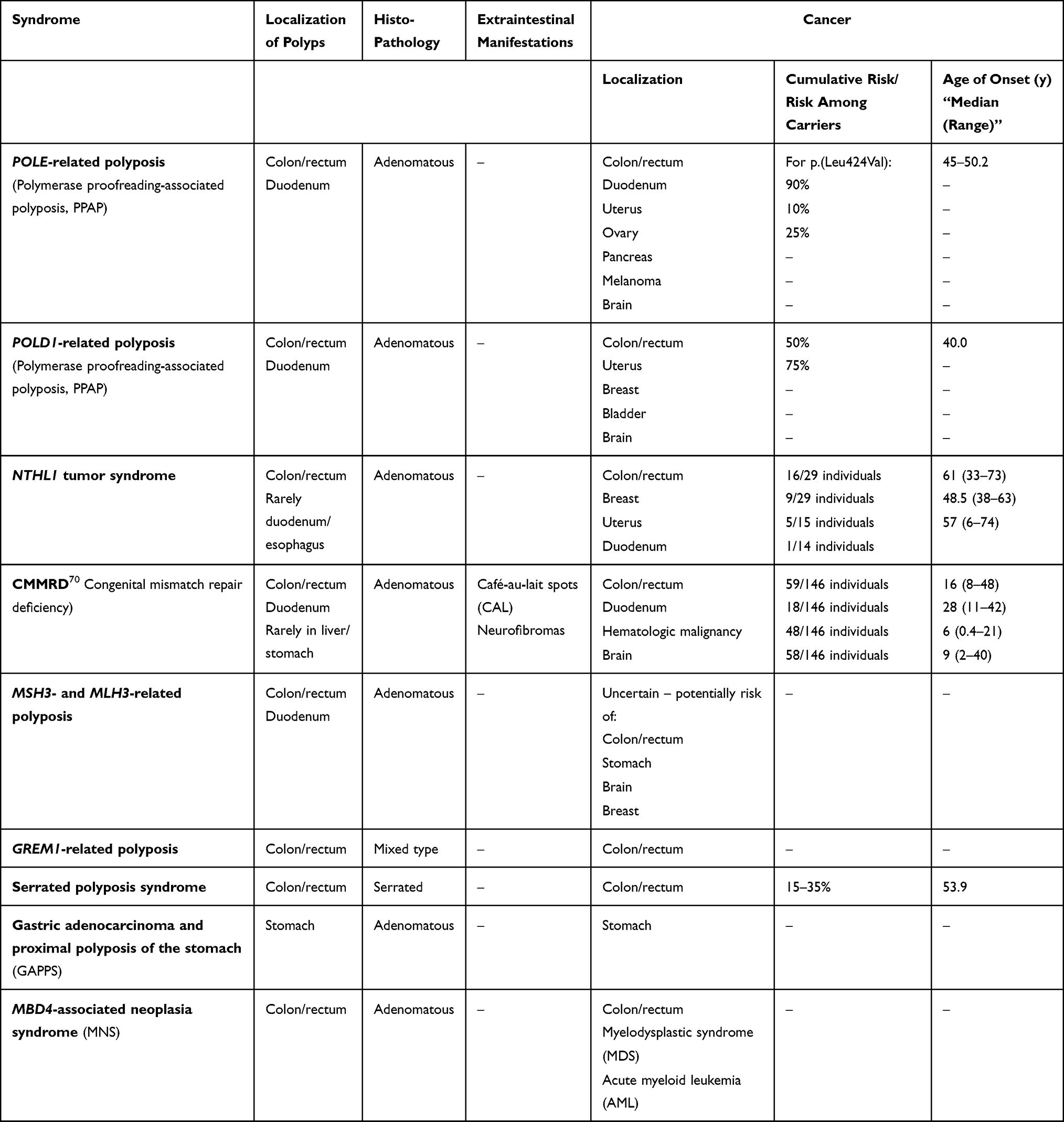 |
Table 1 Novel Polyposis Syndromes |
Adenomatous Polyposis Syndromes
Polymerase Proofreading-associated Polyposis (POLD1- and POLE-associated Polyposis)
In 2013, Palles et al demonstrated that pathogenic variants in the exonuclease domain of DNA polymerase ε (POLE) and DNA polymerase δ (POLD1) are associated with hereditary adenomatous polyposis and an increased risk of CRC, now named “proofreading-associated polyposis” (PPAP, OMIM 615083 and 612591).4 Since then, more than 100 patients with PPAP have been described in the literature.5,6 POLE and POLD1 synthesize each of the two new DNA strands during DNA replication. Both proteins have proofreading exonuclease abilities, which corrects synthesized DNA for replication errors.7
PPAP is characterized by adenomatous colonic polyps/polyposis that develops in adulthood. The median age for detection of polyps and CRC is typically in the third and fourth decades, but can develop in adults of any age.6,8 The polyp burden is variable and ranges from none to around 100. Upper GI-polyposis has been reported in a smaller proportion of patients and tends to be diagnosed later than colorectal polyps. The lifetime risk of CRC is estimated to be 50–90% and highest for POLE-carriers5,6 and 25–75% for endometrial cancer with POLD1 carriers to be at the highest risk. Duodenal (10/105 POLE patients) ovarian (5/43 POLE patients) and breast cancer (10/60) have been observed at a higher frequency than in the background population.6 Brain cancer has been detected in both POLE and POLD1 carriers and comprises astrocytomas, glioblastomas and oligodendrogliomas.6,8 PPAP is inherited in an autosomal dominant manner, with most cases inherited from an affected parent rather than being de novo. Variants in POLD1 and POLE correlated with polyposis and CRC/endometrial cancer that are missense variants found in the exonuclease domain of the proteins. There is no evidence that truncating variants are associated with PPAP. However, homozygote or compound heterozygote pathogenic loss-of-function (LoF) variants in POLE have been detected in IMAGE syndrome (OMIM 614732), which is characterized by intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita and genital anomalies,9 and in FILS syndromes (OMIM 615139), characterized by dysmorphic features including malar hypoplasia, livedo, short stature and immunodeficiency.10,11 Moreover, pathogenic de novo LoF variants in POLD1 have been associated with subcutaneous lipodystrophy, deafness, mandibular hypoplasia and hypogonadism in males (MPD syndrome),12,13 as well as with Werner syndrome.14 Most PPAP patients with a PV in POLE carry the p.(Leu424Val) variant and, although other variants have been reported, most data derive from people carrying this variant. Patients with other missense variants in the exonuclease domain of POLD1 and POLE are rare and although only based on a few cases, patients might have a phenotype mimicking constitutional MMR deficiency syndrome (CMMRD) with a childhood onset of tumors and polyposis15,16 if a variant causes a more severe proofreading defect. In light of these facts, the interpretation of missense variants in the exonuclease domain of POLE and POLD1 should be carefully evaluated.
Gastric Adenocarcinoma and Proximal Polyposis Syndrome
For the last couple of decades, APC has been associated with FAP, but we now know that specific PVs in the promotor 1B of APC cause gastric adenocarcinoma and proximal polyposis syndrome (GAPPS)17 (OMIN 619182). The syndrome is considered to be a rare autosomal dominant condition and was first recognized by Worthley et al in 2012.18 The genetic cause was published 4 years later, with the detection of PVs in the promotor 1B by Li et al.17 The promotor activates transcription of APC in gastric mucosa and the presence of variants in the 1B promotor impairs the binding of the transcription factor YY1, resulting in reduced expression of the APC gene. The variants reported so far include c.-190G>A, c.-191T>C, c.-192A>G, c.-192A>T and c.-195A>C substitutions.
Until now, around 100 patients from 25 families have been described in the literature.19 The penetrance is not complete, and the age of debut of symptoms varies. These patients typically have fundic gland polyposis in the gastric body and are at increased risk of gastric cancer, but lack the colorectal manifestations seen in FAP. The earliest onset of polyps was reported in a 10 year-old,18 but if symptomatic GAPPS primarily manifests in adulthood. The number of polyps can range from a few to a “carpetlike appearance.” Patients are at increased risk of gastric cancer (adenocarcinoma). Some carriers stay asymptomatic for life. Currently, prophylactic gastrectomy is recommended in the presence of dysplastic changes in the stomach. However, and contrary to FAP, there seems to be a mild colorectal phenotype.
NTHL1-associated Tumor Syndrome
NTHL1-associated polyposis (or NTHL1 tumor syndrome) was described for the first time by Weren et al in patients with adenomatous polyposis in the lower GI tract.20 The syndrome is rare, and until now, only around 40 cases have been described. The syndrome is estimated to account for approximately 2% of unexplained polyposis.21 Most patients are homozygous to the recurrent pathogenic variant p.(Gln90Ter), but a few other nonsense and splice variants have been reported.22 Endonuclease III-like 1 (NTHL1) encodes a DNA glycosylase gene involved in the base excision repair (BER) pathway. NTHL1 removes damaged nucleotides, specifically oxidized pyrimidines, from the DNA strand. The resulting gap is filled further down the pathway.23
The polyp burden is variable, from fewer than 10 polyps to 50–100, with CRC typically diagnosed between the ages of 40 and 65 years24 and polyposis developing in adulthood. Adenomatous polyposis has also been observed in the upper GI tract, but only a few cases of duodenal cancer have been reported. Due to the rarity of the syndrome, the full phenotypic spectrum is not known, but based on the largest cohort of patients so far (n=29) homozygous, or compound heterozygote, carriers of PVs are also at risk of non-GI- malignancies25 Breast cancer (including bilateral) have been reported in a large proportion of female carriers (with a median age of 49 years), as well as endometrial cancer (median age of 57).25,26 It is a matter of debate whether monoallelic carriers are at increased risk of cancer (as with monoallelic MUTYH carriers), but so far, the evidence for this is not convincing, with only a few reported cases of heterozygous carriers with cancer and loss of heterozygosity.27,28
MSH3-associated Polyposis
MSH3 associated polyposis is considered a rare polyposis syndrome with an autosomal recessive inheritance pattern. MutS homolog 3 (MSH3) encodes a protein, which in a heterodimer formation with MSH2 plays a role in the mismatch repair system. Biallelic PVs were reported in four patients (two pairs of siblings) in 2016.29 These patients presented with several benign and malignant tumors, indicating a new subtype of a polyposis syndrome with a broad tumor spectrum. All four patients had colonic adenomatous polyposis, which was diagnosed when they were in their 30s, 40s, or 50s, with one female patient presenting with CRC at age 55, followed by a gastric carcinoma at age 59. The other patients (two females, one male) suffered from several other tumors, including astrocytoma (one patient), duodenal adenomas (two patients), and thyroid adenomas (two patients). Breast papilloma (two patients) and uterine benign tumors (two patients) were also reported. Currently, these are the only reported cases of MSH3-associated polyposis and subsequent studies indicate that the syndrome is very rare.30
MLH3-associated Polyposis
MLH3-associated polyposis was reported for the first time by Olkinuora et al in 2018 in four patients who were found to be homozygous for p.(Ser1188Ter), an apparent Finnish founder mutation.31 MutL homolog 3 (MLH3) encodes a protein that forms a heterodimer with MLH1 and is involved in the mismatch repair system. Olkinuora et al’s patients had a variable number of adenomas (1–200) diagnosed in their 40s and 50s. One patient had CRC at the time polyps were detected (48 years old), and two patients in their 50s had breast cancer. However, evidence of MLH3 as the cause of a high penetrant polyposis syndrome is scarce, and homozygous truncating variants have also been found in patients with infertility and no reported polyposis.32,33
Constitutional MMR Deficiency Syndrome (CMMRD)
Biallelic PVs in the Lynch genes – MLH1, MSH2, PMS2 and MSH6 (mainly PMS2) – can cause another polyposis syndrome: constitutional MMR deficiency syndrome (CMMRD, OMIM 276300). It is a highly penetrant syndrome in which patients are at high risk of GI polyposis and can suffer from a spectrum of malignancies, including brain tumors, lymphoma and leukemia, with onset during childhood and adolescence. Approximately 80% develop their first malignancy before age 18, and many have multiple primary malignancies.34 In addition, patients often present with dermatological features, such as multiple café-au-lait spots typically with irregular margins. Both neurofibromatosis type 1 and Li Fraumeni syndrome should be considered as differential diagnoses for CMMRD.
MBD4-associated Polyposis (MBD4-associated Neoplasia Syndrome)
MBD4 encodes a BER glycosylase that repairs G:T mismatches resulting from the deamination of 5ʹ-methylcytosine.44 The gene is frequently mutated in MMR-defective colorectal tumors.35
In 2018, Sanders et al described three patients with biallelic germline PVs in MBD4. The patients had early onset (before 40 years-old) acute myeloid leukemia (AML), and two of the patients also had a history of colonic polyps.36 Palles et al recently coined the term “MBD4-associated neoplasia syndrome” (MANS) and linked MBD4 to polyposis based on the report of a homozygous 4bp deletion in MPD4 in a patient with approximately 60 colonic and rectal adenomas at age 36.37 No upper gastrointestinal polyps were reported. A few months after the detection of polyposis and a subsequent proctocolectomy, the patient was diagnosed with myelodysplastic syndrome, which progressed to AML. The authors retrospectively evaluated the patients of Sanders et al and of the two patients with colonic polyps, one developed CRC at age 40 and accumulated 17 adenomas. The other patient had a hemicolectomy due to multiple polyps.
Although still mindful of the limited evidence, these few cases indicate that MBD4 is associated with an autosomal recessive polyposis syndrome that entails a high risk of polyposis and myeloid malignancies; however, these risk estimates, and those for other malignancies, have yet to be determined with any precision. The risk of cancer in monoallelic carriers of PVS in MBD4 is unknown. Tankaya et al reported a woman with over 30 colorectal adenomas and a heterozygous MBD4 PV and expression of the MBD4 protein with immunohistochemical staining was absent.38 Palles et al did not find that monoallelic carriers had an increased risk of polyposis and CRC.37
Other Polyposis Syndromes
RNF43-associated Serrated Polyposis Syndrome
Serrated polyposis syndrome (SPS, OMIM 617108), formerly known as hyperplastic polyposis, is now defined according to the updated World Health Organization (WHO) criteria as 1) serrated polyps (SPs) proximal to the rectum, all 5 mm or larger in size, with at least two 10 mm or larger in size (criterion I), and 2) a more distal phenotype that presents with more than 20 SPs of any size throughout the large bowel, with five being proximal to the rectum (criterion II). There are three histologic subtypes of SPs: hyperplastic polyps (HP), sessile serrated lesions (SSL), and traditional serrated adenomas (TSA). The number of polyps is cumulative and can be found over the course of several colonoscopies, sometimes making the diagnosis difficult. The number of cumulative polyps is often between 30 and 40 but may vary from just a few to several hundred. SPS is a more common syndrome than previously thought but is still rare: in colonoscopy screenings of patients with a positive fecal occult blood test the prevalence of SPS ranged from 0.31% to 0.66%.39 However, these polyps seldom bleed, and traditional screening may not be the best way to detect them. The number of cases is probably going to rise anyway as a result of the aforementioned revised diagnostic criteria.
SPS is not, in general, considered to be a monogenic syndrome, but rather a sporadic syndrome with multifactorial etiology including both environmental and genetic factors.40 Yet, a small proportion of patients may have SPS as an autosomal dominant polyposis syndrome due to a monoallelic PV in RNF43. In 2014, Gala et al were the first to describe two unrelated patients with PVs in RNF43 with SPS.41 This has been followed by several other similar reports.42,43
Patients with SPS have an increased risk of CRC, with an overall risk of approximately 20%, highest at the time of diagnosis.44 Several studies point towards an increased risk of CRC among first-degree relatives of those with the syndrome.45,46 Some studies suggest that cancer can develop rapidly, especially in SLLs with dysplasia,47 and most recommendations for surveillance suggest a colonoscopy every 1–2 years,48,49 often preceded by a “polyp clearance colonoscopy,” during which all visible polyps/lesions are removed.
The spectrum of other extraintestinal cancers is unknown, although not found in all reports, some studies have reported a high incidence of other types of malignancies.50–52
GREM1-associated Mixed Polyposis
Hereditary mixed polyposis syndrome (OMIM: 601228) was first described by Whitelaw et al in 199753 in an Ashkenazi Jewish family with mixed GI polyposis and CRC. The genetic cause was described in 2012 by Jaeger et al, who detected a 40kb duplication upstream of Gremlin 1 (GREM1).54 Subsequently, there have been several cases of the 40kb duplication, but also a 16kb duplication and 24kb duplication of the regulatory region of GREM1, as well as a duplication of the entire gene.55–57 The 40kp duplication seems to be a founder mutation in the Ashkenazi Jewish population. GREM1 encodes a bone morphogenetic protein (BMP) antagonist and dysregulation of the BMP pathway can promote tumorigenesis. The colorectal polyposis in these families showed a mixed histopathology, including adenomatous, hyperplastic and hamartomatous polyps, all of which developed in adulthood. While there appears to be an increased risk of CRC, a precise risk estimate has not been determined.
Surveillance and Treatment
Recommendations for several of the well-established hereditary syndromes have been published, including FAP, MAP, PJS and JPS.58–60 However, for most of the rare syndromes, it is difficult to provide general recommendations for surveillance, both for affected patients as well as for asymptomatic carriers of a PV. Surveillance with endoscopy of gastrointestinal manifestations will be sufficient for the majority of syndromes. Exceptions include FAP, where colectomy is recommended, and GAPPS, where gastrectomy will often be indicated. As for the preferred endoscopic modality, the existing knowledge is too scarce to draw firm conclusions, and a surveillance program must be personalized for each patient. As several of the syndromes have a broader tumor spectrum (Table 1), surveillance of other organs should be included, but surveillance should always be guided by family history, knowledge of phenotype-genotype correlations, and the most recent literature. Preferably, patients should be allocated to expert centers, and management should be multidisciplinary.
Diagnostic Approach
In clinical settings, the diagnosis of an HPS is sometimes straightforward, eg, if a patient has polyps with a specific histopathology and characteristic extraintestinal manifestations. However, in a large proportion of patients – especially those with mixed or dominantly adenomatous polyposis – it is impossible to make a precise diagnosis. In addition, we know that patients with HPS may not have a classical phenotype and present with only a few polyps. Considering this, the threshold for performing genetic testing should be low.
Due to the phenotypic overlap and the many possible causative genes, a NGS panel of relevant polyposis/CRC-associated genes should be investigated from a blood sample. Exceptions are a convincing clinical phenotype pointing towards a specific syndrome like PJS. The genes can be analyzed with either a physical panel or a virtual panel based on whole-exome sequencing (WES) or whole-genome sequencing (WGS). WGS allows one to detect regulatory variants, deep intronic variants and structural abnormalities. NGS with high read depth is also better at detecting mosaicism, which should be considered in patients where a PV is not initially detected but who have a convincing clinical presentation. Somatic mosaicism has been found in patients with both PJS and FAP,61,62 and sometimes, it is necessary to perform genetic testing on biological samples other than blood, eg, from a polyp. The diagnostic approach is laid out in Figure 4.
Genetic Counseling and Prenatal Diagnostics
Genetic counseling is an essential part of managing patients with (or who are suspected of) an HPS. Genetic testing will identify if germline alterations are present in the known HPS-associated genes, and thereby help to identify the risk of additional tumors and tailor the surveillance offered. In addition, when a genetic pathogenic variant is identified, patients can be provided with detailed knowledge of the risk of recurrence and informed about family planning. In some countries, patients with an HPS and a high recurrence risk are offered prenatal diagnostics. These options, however, depend on the national rules, regulations and comprise ethical considerations as well. Prenatal diagnosis can be performed by invasive testing of a chorion villus biopsy at around week 11 of pregnancy. Another option is preimplantation genetic testing for monogenic disease (PGT-M). This technique comprises hormone stimulation followed by oocyte retrieval. Oocytes are then fertilized through intracytoplasmic sperm injection. The fertilized oocyte is cultivated at the blastocyst stage, and the trophectodermal cells are sampled and tested for the specific genetic condition. Unaffected blastocysts can then be transferred to the uterus. Prenatal diagnosis raises ethical and technical challenges, but several guidelines for polyposis emphasize that patients should be made aware of their reproductive options and the preimplantation and prenatal genetic testing available to them.60
Polyposis of Unknown Etiology
In a large proportion of patients with polyposis, it is impossible to detect a genetic cause, but the detection rate of pathogenic variants depends on the phenotype, eg the detection rate is high in PJS (80–90%) but lower in patients with adenomatous polyposis. The estimation of “unsolved” cases varies from 10% to 80% of patients.31 Diagnostic approaches are primarily based on genetic analysis of blood and/or polyps, and over time NGS has replaced Sanger sequencing as the method of choice. This has resulted in a number of candidate genes, including DSC2, PIEZO1, ZSWIM7, MCM963 and SMAD9,64,65 but a significant number of cases remain unsolved. WGS will increasingly be used and is expected to reveal PVs in already known genes, as well as novel genes and other genetic causes.
For patients and families with polyposis of unknown etiology, decisions about surveillance are difficult to make. A few studies have addressed this question with varying results, likely influenced by selection bias and variation in the genetic analyses performed. The occurrence of cancer has been reported in relatives, both in the colon and in the upper GI tract.66–68 The National Comprehensive Cancer Network (NCCN) suggests that surveillance and management be guided by the phenotype of the patient and by the family history.69 Finally, some patients with multiple polyps (eg, more than 100) are treated as FAP patients.
Conclusion
Currently, 19 genes associated with GI polyposis are known, including the four Lynch genes where biallelic carriers have CMMRD. The genes and associated syndromes comprise both autosomal recessive and autosomal dominant syndromes. Sequencing of these genes should be implemented in the diagnostic pipeline for patients suspected of a polyposis syndrome, and patients should be offered genetic counseling. Patients and at-risk members of their families should be offered surveillance as several of the syndromes are associated with an increased risk of both CRC and extraintestinal cancers. For several of the syndromes, the phenotypic spectrum is not fully understood, and the rarity of the conditions makes evidence-based, long-term follow-up studies difficult to conduct. As technologies improve, we will hopefully be able to identify new genetic causes and expand upon our knowledge of these syndromes.
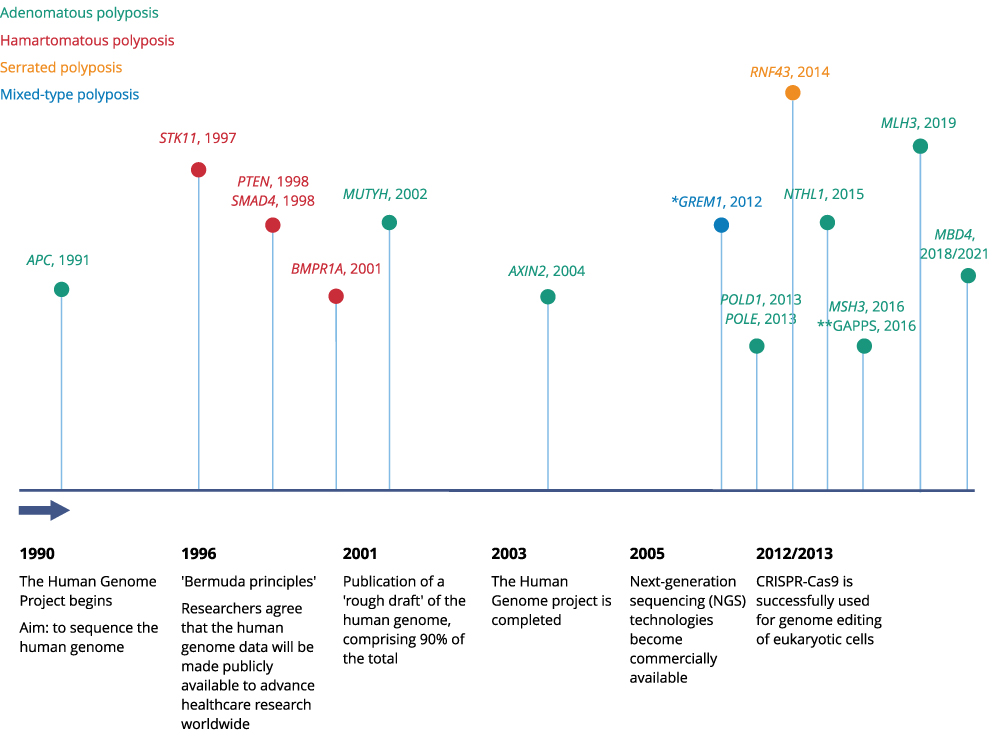 |
Figure 1 Timeline polyposis syndromes. *Upstream 40 kb duplication. **Gastric adenocarcinoma and proximal polyposis of the stomach syndrome (GAPPS) caused by variants in the APC promoter 1B. |
 |
Figure 2 Polyposis syndromes distributed according to histology, inheritance pattern and pathway. *Pathogenic variants in the APC promoter 1B predisposes to gastric adenocarcinoma and proximal polyposis of the stomach syndrome (GAPPS). **Caused by a duplication 40kb upstream of GREM1. |
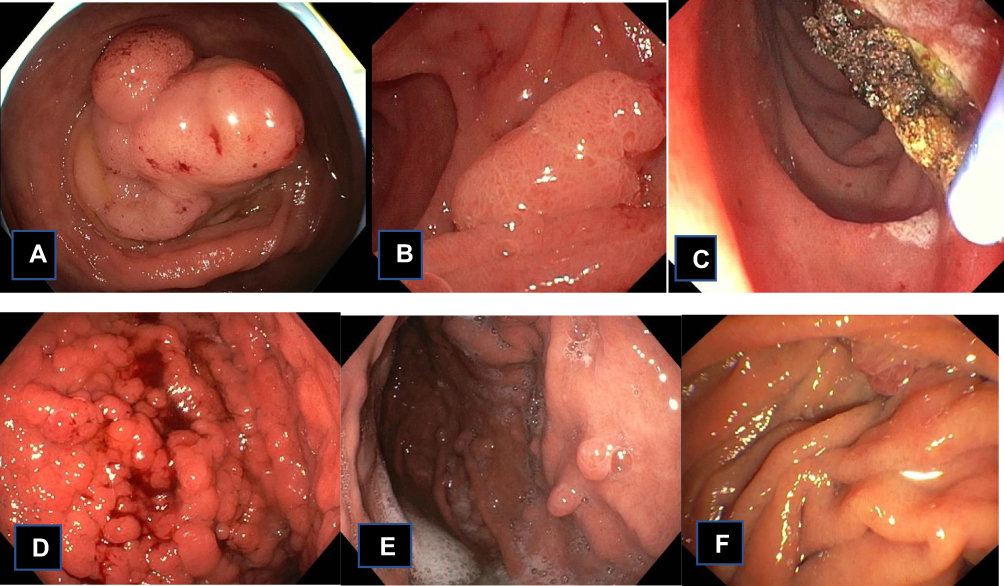 |
Figure 3 Clinical pictures of patients with polyposis Adenoma in the right colon (A) from a patient with MUTYH-associated Polyposis. (B) A 15 mm adenoma in the duodenum from the same patient. It was removed by argon plasma coagulation (C). (D) Diffuse fundic gland polyposis in a patient with GAPSS and a pathogenic variant in the promotor 1B of APC: c.-191T>C. The lesions were random biopsied, and the histopathology showed low-grade dysplasia. (E and F) is from a 60y old patient that had a colectomy as a teenager. At regular GI surveillance few small rectal adenomas were detected, while the duodenum and the major papilla were without polyps. Following genetic counseling a pathogenic variant in POLE, p.(Leu424Val) was detected, and gynecological surveillance was added to her surveillance program. |
 |
Figure 4 Genetic work up of patients suspected of a underlying polyposis syndrome. *The list of relevant genes is dynamic, but at present (2021) the following genes should be included: APC, POLE, POLD1, MBD4, MUTYH, NTHL1, MSH2, MLH1, MSH3, MSH6, STK11, SMAD4, BMPR1A, PTEN, RNF43, GREM1, MLH3, PMS2, AXIN2. |
Abbreviations
CRC, colorectal cancer; FAP, familial adenomatous polyposis; GI, gastrointestinal; HPS, hereditary polyposis syndromes; JPS, juvenile polyposis syndrome; MAP, MUTYH-associated polyposis; NGS, next-generation sequencing; PJS, Peutz-Jeghers syndrome; PV, pathogenic variants; WES, whole exome sequencing; WGS, whole-genome sequencing.
Disclosure
Dr Karin AW Wadt reports personal fees from advisory board meetings with MSD and AstraZeneca, outside the submitted work. The authors report no conflicts of interest in this work.
References
1. Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66(3):589–600. doi:10.1016/0092-8674(81)90021-0
2. Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science (New York, NY). 1991;253(5020):661–665. doi:10.1126/science.1651562
3. Joslyn G, Carlson M, Thliveris A, et al. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. 1991;66(3):601–613. doi:10.1016/0092-8674(81)90022-2
4. Palles C, Cazier J-B, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–144. doi:10.1038/ng.2503
5. Buchanan DD, Stewart JR, Clendenning M, et al. Risk of colorectal cancer for carriers of a germ-line mutation in POLE or POLD1. Genetics Med. 2018;20:890–895. doi:10.1038/gim.2017.185
6. Palles C, Martin L, Domingo E, et al. The clinical features of polymerase proof-reading associated polyposis (PPAP) and recommendations for patient management. Fam Cancer. 2021. doi:10.1007/s10689-021-00256-y
7. Magrin L, Fanale D, Brando C, et al. POLE, POLD1, and NTHL1: the last but not the least hereditary cancer-predisposing genes. Oncogene. 2021;40(40):5893–5901. doi:10.1038/s41388-021-01984-2
8. Spier I, Holzapfel S, Altmüller J, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer. 2015;137(2):320–331. doi:10.1002/ijc.29396
9. Logan CV, Murray JE, Parry DA, et al. DNA polymerase epsilon deficiency causes IMAGe syndrome with variable immunodeficiency. Am J Hum Genet. 2018;103(6):1038–1044.
10. Pachlopnik Schmid J, Lemoine R, Nehme N, et al. Polymerase ε1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (“FILS syndrome”). J Exp Med. 2012;209(13):2323–2330. doi:10.1084/jem.20121303
11. Eason C, Aleisa A, Jones JR, Prijoles EJ, Wine Lee L. Filling in the gaps on FILS syndrome: a case report and literature review. Pediatr Dermatol. 2020;37(5):915–917. doi:10.1111/pde.14274
12. Weedon MN, Ellard S, Prindle MJ, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45(8):947–950. doi:10.1038/ng.2670
13. Elouej S, Beleza-Meireles A, Caswell R, et al. Exome sequencing reveals a de novo POLD1 mutation causing phenotypic variability in mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome (MDPL). Metabolism. 2017;71:213–225. doi:10.1016/j.metabol.2017.03.011
14. Lessel D, Hisama FM, Szakszon K, et al. POLD1 germline mutations in patients initially diagnosed with Werner syndrome. Hum Mutat. 2015;36(11):1070–1079. doi:10.1002/humu.22833
15. Wimmer K, Beilken A, Nustede R, et al. A novel germline POLE mutation causes an early onset cancer prone syndrome mimicking constitutional mismatch repair deficiency. Fam Cancer. 2017;16:67–71. doi:10.1007/s10689-016-9925-1
16. Lindsay H, Scollon S, Reuther J, et al. Germline POLE mutation in a child with hypermutated medulloblastoma and features of constitutional mismatch repair deficiency. Cold Spring Harbor Mol Case Studies. 2019;5:a004499.
17. Li J, Woods SL, Healey S, et al. Point mutations in Exon 1B of APC reveal gastric adenocarcinoma and proximal polyposis of the stomach as a familial adenomatous polyposis variant. Am J Hum Genet. 2016;98(5):830–842. doi:10.1016/j.ajhg.2016.03.001
18. Worthley DL, Phillips KD, Wayte N, et al. Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS): a new autosomal dominant syndrome. Gut. 2012;61(5):774–779. doi:10.1136/gutjnl-2011-300348
19. Tacheci I, Repak R, Podhola M, et al. Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) - A Helicobacter-opposite point. Best Pract Res Clin Gastroenterol. 2021;50-51:101728. doi:10.1016/j.bpg.2021.101728
20. Weren RD, Ligtenberg MJ, Kets CM, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet. 2015;47:668–671. doi:10.1038/ng.3287
21. Terradas M, Munoz-Torres PM, Belhadj S, et al. Contribution to colonic polyposis of recently proposed predisposing genes and assessment of the prevalence of NTHL1- and MSH3-associated polyposes. Hum Mutat. 2019;40(11):1910–1923. doi:10.1002/humu.23853
22. Rivera B, Castellsague E, Bah I, van Kempen LC, Foulkes WD. Biallelic NTHL1 Mutations in a Woman with Multiple Primary Tumors. N Engl J Med. 2015(373):1985–1986.
23. Das L, Quintana VG, Sweasy JB. NTHL1 in genomic integrity, aging and cancer. DNA Repair (Amst). 2020;93:102920. doi:10.1016/j.dnarep.2020.102920
24. Weren RD, Ligtenberg MJ, Geurts van kessel A, De voer RM, Hoogerbrugge N, Kuiper RP. NTHL1 and MUTYH polyposis syndromes: two sides of the same coin? J Pathol. 2018;244(2):135–142. doi:10.1002/path.5002
25. Grolleman JE, de Voer RM, Elsayed FA, et al. Mutational signature analysis reveals NTHL1 deficiency to cause a multi-tumor phenotype. Cancer Cell. 2019;35:256–266e255.
26. Belhadj S, Quintana I, Mur P, et al. NTHL1 biallelic mutations seldom cause colorectal cancer, serrated polyposis or a multi-tumor phenotype, in absence of colorectal adenomas. Sci Rep. 2019;9:9020. doi:10.1038/s41598-019-45281-1
27. Elsayed FA, Grolleman JE, Ragunathan A, Buchanan DD, van Wezel T, de Voer RM. Monoallelic NTHL1 loss-of-function variants and risk of polyposis and colorectal cancer. Gastroenterology. 2020;159(6):2241–2243.e2246. doi:10.1053/j.gastro.2020.08.042
28. Salo-Mullen EE, Maio A, Mukherjee S, et al. Prevalence and characterization of biallelic and monoallelic NTHL1 and MSH3 variant carriers from a pan-cancer patient population. JCO Precis Oncol. 2021;5:455.
29. Adam R, Spier I, Zhao B, et al. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am J Hum Genet. 2016;99:337–351. doi:10.1016/j.ajhg.2016.06.015
30. Terradas M, Munoz-Torres PM, Belhadj S, et al. Contribution to colonic polyposis of recently proposed predisposing genes and assessment of the prevalence of NTHL1- and MSH3-associated polyposes. Hum Mutat. 2019(40):1910–1923.
31. Olkinuora A, Nieminen TT, Martensson E, et al. Biallelic germline nonsense variant of MLH3 underlies polyposis predisposition. Genetics Med. 2019(21):1868–1873.
32. Nawaz S, Ullah MI, Hamid BS, et al. A loss-of-function variant in DNA mismatch repair gene MLH3 underlies severe oligozoospermia. J Hum Genet. 2021;66(7):725–730. doi:10.1038/s10038-021-00907-z
33. Chen S, Wang G, Zheng X, et al. Whole-exome sequencing of a large Chinese azoospermia and severe oligospermia cohort identifies novel infertility causative variants and genes. Hum Mol Genet. 2020;29(14):2451–2459. doi:10.1093/hmg/ddaa101
34. Valle L, de Voer RM, Goldberg Y, et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol Aspects Med. 2019;69:10–26. doi:10.1016/j.mam.2019.03.001
35. Tricarico R, Cortellino S, Riccio A, et al. Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis. Oncotarget. 2015;6(40):42892–42904. doi:10.18632/oncotarget.5740
36. Sanders MA, Chew E, Flensburg C, et al. MBD4 guards against methylation damage and germ line deficiency predisposes to clonal hematopoiesis and early-onset AML. Blood. 2018;132(14):1526–1534. doi:10.1182/blood-2018-05-852566
37. Palles C, Chew E, Grolleman JE, et al. Germline loss-of-function variants in the base-excision repair gene MBD4 cause a Mendelian recessive syndrome of adenomatous colorectal polyposis and acute myeloid leukaemia. bioRxiv. 2021. doi:10.1101/2021.04.27.441137
38. Tanakaya K, Kumamoto K, Tada Y, et al. A germline MBD4 mutation was identified in a patient with colorectal oligopolyposis and early‑onset cancer: a case report. Oncol Rep. 2019;42(3):1133–1140.
39. Fousekis FS, Mitselos IV, Christodoulou DK. Diagnosis, epidemiology and management of serrated polyposis syndrome: a comprehensive review of the literature. Am J Transl Res. 2021;13(6):5786–5795.
40. Buchanan DD, Clendenning M, Zhuoer L, et al. Lack of evidence for germline RNF43 mutations in patients with serrated polyposis syndrome from a large multinational study. Gut. 2017;66:1170–1172. doi:10.1136/gutjnl-2016-312773
41. Gala MK, Mizukami Y, Le LP, et al. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology. 2014;146:520–529. doi:10.1053/j.gastro.2013.10.045
42. Taupin D, Lam W, Rangiah D, et al. A deleterious RNF43 germline mutation in a severely affected serrated polyposis kindred. Human Genome Variation. 2015;2:15013. doi:10.1038/hgv.2015.13
43. Yan HHN, Lai JCW, Ho SL, et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut. 2017;66:1645–1656. doi:10.1136/gutjnl-2016-311849
44. Muller C, Yamada A, Ikegami S, et al. Risk of Colorectal cancer in serrated polyposis syndrome: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2021. doi:10.1016/j.cgh.2021.05.057
45. Win AK, Walters RJ, Buchanan DD, et al. Cancer risks for relatives of patients with serrated polyposis. Am J Gastroenterol. 2012;107:770–778. doi:10.1038/ajg.2012.52
46. Boparai KS, Reitsma JB, Lemmens V, et al. Increased colorectal cancer risk in first-degree relatives of patients with hyperplastic polyposis syndrome. Gut. 2010;59(9):1222–1225. doi:10.1136/gut.2009.200741
47. Edelstein DL, Axilbund JE, Hylind LM, et al. Serrated polyposis: rapid and relentless development of colorectal neoplasia. Gut. 2013;62(3):404–408. doi:10.1136/gutjnl-2011-300514
48. Carballal S, Rodríguez-Alcalde D, Moreira L, et al. Colorectal cancer risk factors in patients with serrated polyposis syndrome: a large multicentre study. Gut. 2016;65(11):1829–1837. doi:10.1136/gutjnl-2015-309647
49. Bleijenberg AG, Je IJ, van Herwaarden YJ, et al. Personalised surveillance for serrated polyposis syndrome: results from a prospective 5-year international cohort study. Gut. 2020;69:112–121. doi:10.1136/gutjnl-2018-318134
50. Hazewinkel Y, Reitsma JB, Nagengast FM, et al. Extracolonic cancer risk in patients with serrated polyposis syndrome and their first-degree relatives. Fam Cancer. 2013;12(4):669–673. doi:10.1007/s10689-013-9643-x
51. Edelstein DL, Cruz-Correa M, Soto-Salgado M, et al. Risk of colorectal and other cancers in patients with serrated polyposis. Clin Gastroenterol Hepatol. 2015;13(9):1697–1699. doi:10.1016/j.cgh.2015.02.003
52. Jasperson KW, Kanth P, Kirchhoff AC, et al. Serrated polyposis: colonic phenotype, extracolonic features, and familial risk in a large cohort. Dis Colon Rectum. 2013;56(11):1211–1216. doi:10.1097/DCR.0b013e3182a11cca
53. Whitelaw SC, Murday VA, Tomlinson IP, et al. Clinical and molecular features of the hereditary mixed polyposis syndrome. Gastroenterology. 1997;112:327–334. doi:10.1053/gast.1997.v112.pm9024286
54. Jaeger E, Leedham S, Lewis A, et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet. 2012;44:699–703. doi:10.1038/ng.2263
55. McKenna DB, Van Den Akker J, Zhou AY, et al. Identification of a novel GREM1 duplication in a patient with multiple colon polyps. Fam Cancer. 2019;18:63–66. doi:10.1007/s10689-018-0090-6
56. Rohlin A, Rambech E, Kvist A, et al. Expanding the genotype-phenotype spectrum in hereditary colorectal cancer by gene panel testing. Fam Cancer. 2017;16:195–203. doi:10.1007/s10689-016-9934-0
57. Lieberman S, Walsh T, Schechter M, et al. Features of patients with hereditary mixed polyposis syndrome caused by duplication of GREM1 and implications for screening and surveillance. Gastroenterology. 2017;152(1876–1880):e1871. doi:10.1053/j.gastro.2017.02.014
58. Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110:
59. Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020;69:411–444. doi:10.1136/gutjnl-2019-319915
60. Wagner A, Aretz S, Auranen A, et al. The management of Peutz-Jeghers syndrome: European Hereditary Tumour Group (EHTG) Guideline. J Clin Med. 2021;10(3):473. doi:10.3390/jcm10030473
61. Jelsig AM, Bertelsen B, Forss I, Karstensen JG. Two cases of somatic STK11 mosaicism in Danish patients with Peutz-Jeghers syndrome. Fam Cancer. 2020;20:55.
62. Spier I, Drichel D, Kerick M, et al. Low-level APC mutational mosaicism is the underlying cause in a substantial fraction of unexplained colorectal adenomatous polyposis cases. J Med Genet. 2016;53:172–179. doi:10.1136/jmedgenet-2015-103468
63. Goldberg Y, Halpern N, Hubert A, et al. Mutated MCM9 is associated with predisposition to hereditary mixed polyposis and colorectal cancer in addition to primary ovarian failure. Cancer Genet. 2015;208(12):621–624. doi:10.1016/j.cancergen.2015.10.001
64. Spier I, Kerick M, Drichel D, et al. Exome sequencing identifies potential novel candidate genes in patients with unexplained colorectal adenomatous polyposis. Fam Cancer. 2016;15(2):281–288. doi:10.1007/s10689-016-9870-z
65. Sweet K, Willis J, Zhou XP, et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA. 2005;294:2465–2473. doi:10.1001/jama.294.19.2465
66. Tieu AH, Edelstein D, Axilbund J, et al. Clinical characteristics of multiple colorectal adenoma patients without germline APC or MYH mutations. J Clin Gastroenterol. 2016;50:584–588. doi:10.1097/MCG.0000000000000416
67. Kallenberg FGJ, Latchford A, Lips NC, et al. Duodenal adenomas in patients with multiple colorectal adenomas without germline APC or MUTYH mutations. Dis Colon Rectum. 2018;61:58–66. doi:10.1097/DCR.0000000000000868
68. Giarola M, Stagi L, Presciuttini S, et al. Screening for mutations of the APC gene in 66 Italian familial adenomatous polyposis patients: evidence for phenotypic differences in cases with and without identified mutation. Hum Mutat. 1999;13:116–123. doi:10.1002/(SICI)1098-1004(1999)13:2<116::AID-HUMU3>3.0.CO;2-2
69. NCCN.Available from: https://www.nccn.org.
70. Wimmer K, Kratz CP, Vasen HF, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet. 2014;51(6):355–365.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.