Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
Novel aspects of pathogenesis and regeneration mechanisms in COPD
Authors Bagdonas E, Raudoniute J, Bruzauskaite I, Aldonyte R
Received 9 February 2015
Accepted for publication 19 March 2015
Published 2 June 2015 Volume 2015:10(1) Pages 995—1013
DOI https://doi.org/10.2147/COPD.S82518
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Edvardas Bagdonas, Jovile Raudoniute, Ieva Bruzauskaite, Ruta Aldonyte
State Research Institute Center for Innovative Medicine, Vilnius, Lithuania
Abstract: Chronic obstructive pulmonary disease (COPD), a major cause of death and morbidity worldwide, is characterized by expiratory airflow limitation that is not fully reversible, deregulated chronic inflammation, and emphysematous destruction of the lungs. Despite the fact that COPD is a steadily growing global healthcare problem, the conventional therapies remain palliative, and regenerative approaches for disease management are not available yet. We aim to provide an overview of key reviews, experimental, and clinical studies addressing lung emphysema development and repair mechanisms published in the past decade. Novel aspects discussed herein include integral revision of the literature focused on lung microflora changes in COPD, autoimmune component of the disease, and environmental risk factors other than cigarette smoke. The time span of studies on COPD, including emphysema, chronic bronchitis, and asthmatic bronchitis, covers almost 200 years, and several crucial mechanisms of COPD pathogenesis are described and studied. However, we still lack the holistic understanding of COPD development and the exact picture of the time-course and interplay of the events during stable, exacerbated, corticosteroid-treated COPD states, and transitions in-between. Several generally recognized mechanisms will be discussed shortly herein, ie, unregulated inflammation, proteolysis/antiproteolysis imbalance, and destroyed repair mechanisms, while novel topics such as deviated microbiota, air pollutants-related damage, and autoimmune process within the lung tissue will be discussed more extensively. Considerable influx of new data from the clinic, in vivo and in vitro studies stimulate to search for novel concise explanation and holistic understanding of COPD nowadays.
Keywords: dysbiosis in COPD, autoimmune COPD, occupational COPD, chronic obstructive pulmonary disease
Pathogenesis mechanisms in COPD
Several clinical features comprise COPDs such as chronic bronchitis, destruction of small airways, and enlargement/disorganization of alveoli, ie, loss of alveolar tissue. Our review will focus on the changes taking place in alveolar space, ie, emphysema component of COPD. We have illustrated emphysematous vs healthy alveoli with several well-known pathogenesis processes in Figure 1. Some novel aspects are also added therein, eg, microbiota-produced inflammatory influences. The schematic overview of the complex interplay among structural, inflammatory, and progenitor cells; mediators’ milieu; and simplified structural shifts within the lung alveoli and its surroundings represent the challenges, both inner and exogenous, that lung tissue components are exposed to.
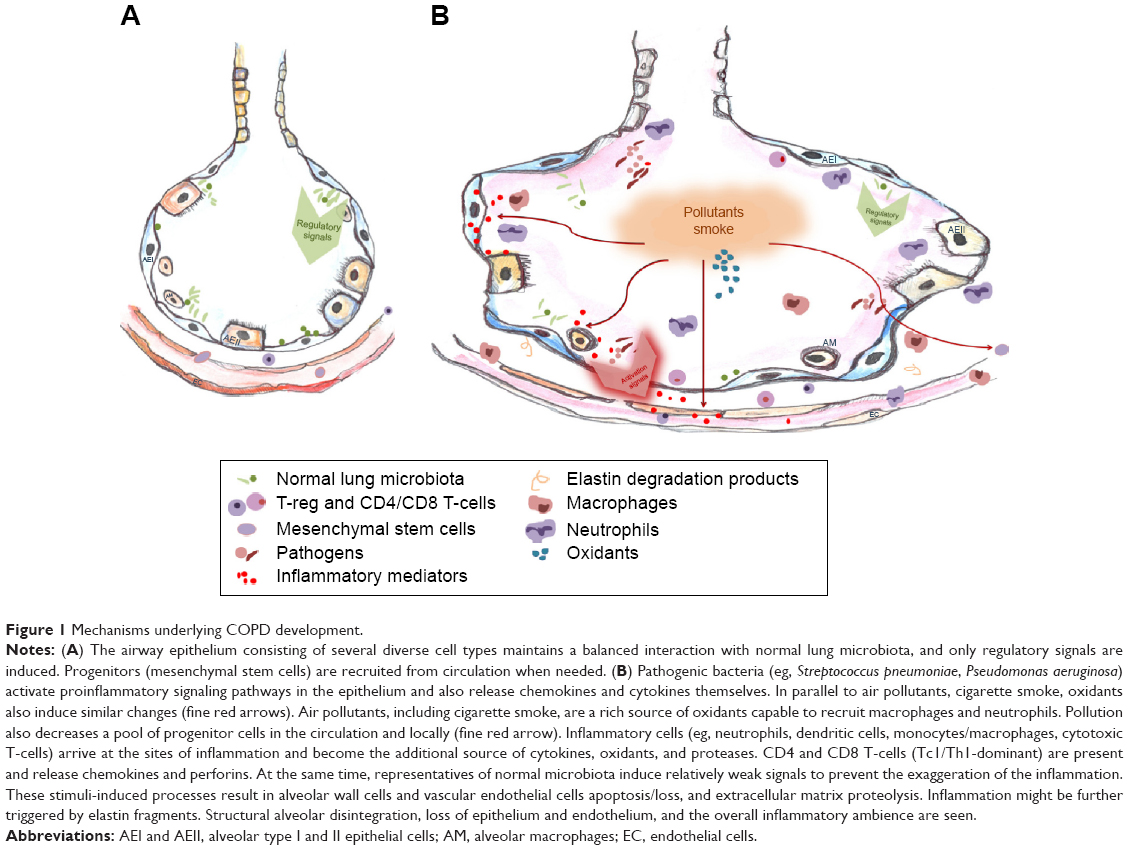 | Figure 1 Mechanisms underlying COPD development. |
In general, COPD remains unappreciated, and COPD prevalence surveys are remarkably lower in numbers in comparison with prevalence surveys available in asthma, cancer, cardiovascular, or other major diseases. This may be among the reasons why COPD was accepted as exceptionally smoking-related disease for a long time. However, this is not acceptable any longer, since extensive search reveals more new risk factors. As elegantly reviewed by Salvi and Barnes, data from population-based studies show that cigarette smoking as a risk factor may only be attributed to half of COPD cases, and some unpublished data included in the review present nearly 70% nonsmokers among COPD cases collected at the city slum in India.1 Some of the reports originating from South Africa, the People’s Republic of China, and Korea state distinct proportions of nonsmoking-COPD among men and women (where nonsmoking-COPD morbidity among women exceeds 50%) suggesting that exposure to household fumes might be implicated.2–4 Tuberculosis infection, occupational exposures, and frequent childhood infections are also discussed as major risks. Thus, other than smoking factor-associated COPD, morbidity is equally huge and unrecognized. The alternative environmental risk factors implicated in the nonsmoking COPD development include indoor and outdoor air pollution, ie, biomass fuel, dust, and fumes5,6 and the use of agricultural pesticides (comprising herbicides and insecticides). In the recent systematic review, hundreds of studies that were published after 1990 (final number of relevant papers included in the calculations was 23) were revised, and statement on the correlation between COPD and the use of pesticides was made.7 Pesticides are regarded as a very important source of reactive oxygen species (ROS) and other insults, and a direct association between the common pesticide use and COPD development has been reported a long time ago.8 Indeed, farming is recognized as a risk factor for COPD, and a population-based study reveals that risk for nonreversible airways obstruction attributable to farming is 7.7%.9 Moreover, recent study has extended the existing knowledge by following a general population cohort for 25 years and assessing the impact of pesticides on lung function. They have found that exposure to pesticides is associated with accelerated decline in lung function, as much as loss of 6.9 mL/year of forced expiratory volume in 1 second (FEV1).10 This is also corroborated by the data from other cohorts and short term, specific pesticide-related studies.11,12
Another theory relates COPD to the fetal stage (fetal origin hypothesis), ie, findings from large retrospective studies suggest that COPD might be programmed early in life. Indeed, Barker et al have found that low birth-weight and respiratory infections in infancy lead to reduced lung function in adults.13 Other “reprogramming” factors include use of anti-inflammatory medicines in pregnancy, vitamin D deficiency, certain diet, and vitamin C intake during pregnancy and inadequate duration of breastfeeding.14–17 Another important group of factors related to birth, ie, limited exposure to microbial world prior to, during, and after the delivery due to maternal use of antibiotics; increased rates of C-section; and sterile environment around the infant, will be discussed later in our paper.
Autoimmunity also plays a role in COPD as it becomes increasingly accepted. Autoimmune mechanisms can explain the unregulated inflammatory process perpetuation that continues after smoking cessation, since the initial inflammation and environmental insults in lungs expose certain epitopes for the autoimmune attack. Naming COPD an autoimmune disease opens new horizons for the treatment strategies and for the research lines to develop. Identifying, blocking, or masking auto-epitopes; preserving and enriching lung microbiota; cleaning the breathable air; and staying physically active may become major COPD management directions as opposed to conventionally used anti-inflammatory treatment, corticosteroids, and antibiotics.
In addition to factors mentioned earlier, accelerated cell senescence has been implicated in the pathogenesis of COPD. It is clearly shown that senescence affects lung structural and inflammatory cells, fibroblasts, and progenitors, rendering repair and regeneration insufficient.18–22 In corroboration, autophagy was found significantly deregulated in the cells from patients with COPD.23,24 Insufficient autophagy contributes to the senescence by increasing accumulation of damaged cellular content. While in normal lung, autophagy mechanism maintains tissue homeostasis, ie, balance among production, degradation, and recycling of organelles and proteins, and helps tissue in adaptation, in COPD lung, chronic deficiency in autophagy leads to increased tissue senescence and insufficient repair.
COPD pathogenesis evolves with the disease exacerbations that are main drivers of health deterioration in patients with COPD. Exacerbations of COPD are defined as acute episodes of symptom worsening beyond day-to-day variations requiring change in regular medication. There have been an improvement in uniformity of the definition for the exacerbations during recent years, although current treatment guidelines for the exacerbations often remain unrelated to clinical data and contribute to inappropriate use of corticosteroids and antibiotics.25 A distinct patients’ group, “frequent exacerbation type”, is recognized and reflects a heterogeneity and multiplicity of exacerbations-related pathogenesis’ factors and mechanisms. Susceptibility to exacerbations is defined by background inflammation in the lung tissue, microbiome shifts and patterns, status of the immune system, comorbidities, and medications’ background. Major mechanisms described in COPD are further upregulated during exacerbations, including inflammatory process exaggeration26 and obstruction in the airways with a subsequent hyperinflation.27 Infectious pathogens, ie, viruses and bacteria, are responsible for driving the exacerbation process in more than half of all cases as reviewed by Miravitlles and Anzueto.28 Markers of infectious process are well documented,29,30 and controversial continuous prophylactic macrolide therapy for COPD is proposed.31 Authors claim that continuous therapy reduces COPD exacerbation frequency, although their conclusion is based on only three clinical trials, and the target group remains unclear. Among other triggers for COPD exacerbations, the indoor and outdoor air pollution5,32,33 and second-hand smoke34 should be mentioned.
Inflammatory process: participating cells, proteolysis, and oxidative injury
It is well proven that lung tissue in every smoker is inflamed. Proofs come from the studies on biopsy material,35 sputum studies,36 and experimental in vitro data37 that show inflammatory cell infiltration and presence of various proinflammatory molecules at the increased levels in smoker’s lungs. At certain smoking/exposure level and other predisposing situations, yet enigmatic to us, the destructive process overcomes the local protective mechanisms and lung tissue damage and loss manifests. In COPD, inflammatory tissue damage is endless, ie, inflammation perpetuates further on long after patients have quit smoking.38 Inflammation-related contributors to lung tissue injury include cells and mediators of both initiative and adaptive immunity, ROS, imbalance of local proteolysis/antiproteolysis state, and other insults.
Participating cells: neutrophils
Circulating and lung-resident inflammatory cells, including neutrophils, for a long time are recognized participants of smoking-induced tissue injury.39 In corroboration with many previous reports, a recent study finds that neutrophils are increased in sputum of patients with COPD along with increased interleukin (IL)-6 signaling.36 The same tendency was detected in bronchoalveolar lavage fluid (BALF) long time ago.40,41 Neutrophils are known as the major destructors of the elastic matrix of the alveoli. They act via proteases and small cationic peptides and are able to attack invading bacteria, viruses, pollutants, and, under certain autoimmune circumstances, own tissue, like in Crohn’s disease and psoriasis.42,43 Under the influence of environmental insults, including cigarette smoke, neutrophil-released enzymes and peptides are able to cleave collagen into fragments that may activate inflammatory cells and drive chronic inflammation further.44 Moreover, study shows that neutrophils from patients with COPD are loaded with intracellular proteolytic enzymes by 25 folds more than the cells from healthy donors. Authors also report that immunohistologically proteolytic enzymes are distributed along with neutrophils, macrophages, and epithelial cells.
Signals that drive neutrophils to infiltrate the damaged lung parenchyma are partially identified. It is known that neutrophils rapidly emerge at the sites of inflammation in response to interleukin (IL)-8, a chemoattractant produced by multiple cell types, mainly, exposed and damaged epithelium and endothelium.45,46 A variety of other attractants have been shown to induce neutrophil migration, including chemokine CXC motif ligands 2 (CXCL2), leukotriene B4 (LTB4), and formyl-met-leu-phe (fMLP).47–51 The latter is a host–microorganism interaction-associated chemoattractant, while others are generated by organism own immune cells and injured tissues at the site of disease. A modified alpha-1-antitrypsin (AAT) needs to be mentioned as a candidate attractant for neutrophils.52–54 Various modified, ie, oxidized, polymerized, cleaved, nytrosylated, and citrullinated, AAT forms are especially relevant on the inflammatory lung destruction stage, where proteolysis and ROS attack are predominant processes and AAT is a major antiprotease.55 There is also a comparative data presented on chemotaxis induction where fMLP is shown as a most powerful attractant in vitro.56 Overall, it is accepted that cellular migration behavior (orientation and motility) is defined by integrate multiple signals, both dominant and secondary. Interestingly, stem cells also home via similar vectors, including our own data on AAT/mesenchymal stem cells (MSC) interactions in vitro,57 neural stem cells and LTB4 partnership reported,58 bone progenitors and CXC motif ligands interplay,59 and so on.
Earlier findings that neutrophils and neutrophil-produced proteinases are major contributors to tissue breakdown in COPD lungs were challenged by the observation that macrophage-released metalloproteinases (MMP) might be critical mediators in COPD.60 However, neutrophils are still regarded as initiating cells and also producers of attractants and epitopes for the further perpetuation of the inflammation. It is proven that neutrophils appear in the airways immediately following the insult in animal models and remain abundantly distributed.61,62 Moreover, the numbers of submucosal neutrophils correlate significantly with the intensity of the cigarette smoking.62 Some studies, however, reveal that neutrophilic infiltration in the smokers airways does not reflect the disease state.63 The scenario of neutrophils participation in the lung tissue breakdown is still being under evaluation, while new data emerge and certain issues are being revised, eg, the distinct features of neutrophils from smokers with COPD vs smokers without COPD in a case of infection.64
Participating cells: macrophages
When parenchyma infiltrating inflammatory cells, ie, T and B cells, macrophages and neutrophils were counted in situ (per cubic millimeter of lung tissue), the numbers of neutrophils were found to inversely correlate with the degree of emphysema.65 This surprising correlation was accompanied by the finding that the count of T lymphocytes and macrophages closely correlated with the degree of lung destruction. And furthermore, the numbers of T-cells correlated with the numbers of macrophages leading to the conclusions that these cells may act in a team.
Macrophages play a crucial role in COPD and are regarded as major producers of MMPs in the COPD lung.66 They also participate in the propagation of the inflammation by releasing chemoattractant molecules. Moreover, we and others have shown that circulating progenitors of lung macrophages, ie, peripheral blood-derived monocytes, differ if compared between patients with COPD and healthy controls.67 Such early, systemic proinflammatory commitment of monocytes may explain why COPD affects the whole organism. Lung macrophages colocalize with emphysematous zones and are found in increased numbers in patients’ airways, parenchyma, BALF, and sputum.65,68 Moreover, macrophages’ numbers in the airways correspond to the severity of COPD.69 In COPD, macrophages are recruited to the lungs and enhances the release of tumor necrosis factor (TNF)-alpha, IL-8, other CXC chemokines, monocyte chemotactic peptide (MCP)-1, LTB4, and so on as reviewed by Barnes et al.70 Alveolar macrophages also secrete elastolytic enzymes, including MMP-2, MMP-9, MMP-12, cathepsins K, L, and S.71,72 The increased recruitment of monocytes from the circulation in response to monocyte-selective chemokines that are increased in COPD airways73 and local proliferation of lung macrophages may contribute to increase in lung macrophages’ numbers. Macrophage actions might be conducted by nuclear factor (NF)- kappaB, which is activated in alveolar macrophages of patients with COPD, particularly during exacerbations.35,74,75 However, other pathways are also described, ie, activator protein-1 and tyrosine kinase c-Src.76,77
Tissue resident macrophages are now recognized as a multifunctional cell, central for tissue maintenance. It is proven that macrophages contribute to tissue regeneration, compensatory lung growth and may recruit progenitor cells.78,79 They are equally important in fighting the infection and cutting/resolving inflammatory signaling, removing apoptotic cells, and inducing certain regenerative processes.
Participating cells: T lymphocytes
It is becoming increasingly evident that T-cells reside at the epithelial surfaces throughout the human organism, including lungs, and mediate the host defense. Their contribution in so-called T-cell-mediated diseases is being revisited now. A recent study demonstrates abundant resident T-cells in human lung (more than 10 billion).80 Detected T-cells are of effectors’ memory phenotype; some of them are central memory T-cells and some are T-regulatory cells. T-helper cells (CD4) can be further subdivided into Th1 and Th2 cytokine types. Th1-type cytokines participate in perpetuating autoimmune responses with interferon gamma as the main cytokine and resulting excessive proinflammatory responses that can lead to uncontrolled tissue damage. Lung emphysema is generally accepted as a type Th1 disease.81 In support for this, an increased level of Th1 response-associated molecules was found in peripheral blood from subjects with COPD.82
T-cell roles in COPD and relevant T-cell fractions are only partially elucidated. As described by Cosio et al, all T-cell phenotypes are increased in smokers with COPD.83 Despite the fact that neutrophils are the predominant cell in the lung parenchyma of non-COPD smokers, with the signs of emphysema comes also an increase in T-cells (CD3 and CD8).84 Moreover, T-cell numbers correlate with the smoking history. Study suggests that the development of emphysema might be mediated by T lymphocytes, mainly CD8 cytotoxic T-cells, and that apoptosis might be one of the mechanisms of lung destruction leading to the development of emphysema. Cytotoxic (CD8) cells are a subgroup of cells that kill infected or damaged cells, while T-helper (CD4) cells upon activation release cytokines and orchestrate the activity of other inflammatory and related cells. In emphysema, CD8 T-cell numbers correlate with the severity of the tissue destruction, and their accumulation continues even after smoking cessation.85 This correlation is not linear according to other authors. As disease progresses, T-cytotoxic cell numbers may even decline.69 These cells require antigen to accumulate and produce their damaging effects. However, the subtle interrelation between regulatory T-cells and CD8 cells might become disrupted in autoimmune process, and regulatory T-cell-released factors might become inadequate to keep cytotoxic T-cells silent.83 Interestingly, a subset of T-cells, ie, CD56 lymphocytes, can kill own lung structural cells spontaneously, as it was shown in the recent study. This natural cytotoxicity was increased in subjects with severe COPD in support of their potential role in autoimmune process-driven emphysema progression.86 All this complex data map suggests that differential interrelationship among T-cell subtypes in COPD may be important for the progression of inflammation.87
The importance of T-cells in emphysema development was recently proven in mice model with the demonstration that T-cells from cigarette smoke-exposed mice are able to transfer the diseased process, ie, tissue destruction, to unexposed mice. Both CD8 and CD4 T-cells were required for that, and the process was antigen recognition dependent.88 These results further support and prove cigarette smoke-induced T-cell-driven autoimmune mechanism in COPD development.
Inflammatory mediators, ROS, and environmental factors
Tens of cytokines and chemokines have been found associated with COPD but overall interplay and leading role players remain unclear. Locally produced proinflammatory molecules come not only from the host cells but also from microorganisms, ie, bacterial lipopolysaccharide (LPS), fMLP, and other mediators contribute to the inflammation at the alveolar setting. Pulmonary epithelium when exposed to noxious environmental substances releases TNF-alpha, IL-1beta, granulocyte-macrophage colony-stimulating factor (GM-CSF), transforming growth factor (TGF)-beta1, MCP-1, LTB4, IL-8.89–91 Macrophages also release chemokines, such as CXCL9, CXCL10, and CXCL11, which are chemotactic.90 In addition, proteolytic enzymes, eg, MMP-2, MMP-9, MMP-12, and cathepsins, are also secreted in response to ROS-rich environment.92 Transcription factor NF-kappaB orchestrates these responses in the cells of patients with COPD.93 What cytokines and at what times are the major players on the constantly evolving stage of COPD remains unclear. There was an attempt to target TNF-alpha and specifically to silence its signaling in patients with COPD. Three studies on chimeric monoclonal antibody against TNF-alpha were performed in patients with mild-to-severe COPD. However, none of the clinical parameters assessed, ie, exacerbation rate, dyspnea, or FEV1, were improved.94–96 Even opposite, functional blockage of TNF-alpha has led to an increased rate of infectious complications and malignancy.96 Therefore, it is clear that cytokines in COPD may have several important and opposing roles and eliminating one may lead to even deeper deregulation of the inflammatory process.
ROS are oxygen-rich unstable molecules that can become donors or recipients of free electrons. The subsequent products of their reactivity are even more unstable. Intracellular ROS can induce functional and structural changes, such as membrane lipid peroxidation, DNA strand breaks, changes in enzymatic activity, and modulate intracellular signaling cascades.97 The effect of ROS depends on their concentration, ie, structural changes are associated with high ROS concentrations,98 while lower levels of ROS decrease cell proliferation capacity, induce apoptosis, and necrosis.99 Effects of petrol exhaust particles on cell viability, oxidative stress generation, DNA damage and inflammation in human lung epithelial cells, and murine macrophages are described.100 Increasing awareness on air pollution-related lung destruction process stimulates to study ROS-induced processes closer. Recently, several studies were published in this field, including report on ozone and diesel exhaust particles-modulated transcription factor NF-kappaB activity101 and LPS-induced production of inflammatory cytokines that is redox sensitive.102,103
Intracellular redox state is defined by the oxidant load in the breathable air and a pooled capacity of lung-protective mechanisms to absorb the oxidative agents. Alveolar lining fluid, epithelial cells in the alveoli, local macrophages, and pulmonary fibroblasts are all primary cellular targets for ROS. In turn, they may also become a secondary source of the ROS. According to data available, ROS production might be detected or induced in almost any cell, and all lung cell types might be implicated in redox state shifts in COPD.104 Generation of novel epitopes for autoimmune process might also be ROS exposure associated. Indeed, some findings suggest that ROS can modify the conformation of structural proteins leaving them exposed and immunogenic.105 Sophisticated detection methods that address lung tissue architecture and assess cells in situ may reveal some complex redox state fluctuations in COPD lung.
An indoor air should also be regarded as a source of environmental ROS. It is sometimes chemically very close to outdoor air with combustion gases and exhaust particles, while sometimes it is much cleaner. However, in some cases, indoor air can become an independent pool of hazardous particles and gases. This is especially true for office spaces in high-rise buildings with parking garages or loading docks underneath. Ventilation system may become a carrier of carbon monoxide, second-hand cigarette smoke (if smokers stay at the intake openings), and other pollutants. Printers and other machines emit ozone; many types of organic chemicals are detected in the workplace as well.106,107 A recent study suggests106 that laser printers significantly increase indoor air pollution with ozone and volatile organic compounds (VOCs), and that appropriate filters may reduce this but not the formaldehyde emissions. Authors state that there is no (and would not be) clear distinction between “dangerous” and “harmless” concentrations. The crucial preventive action should be to use low-VOC-containing materials if possible. Proper ventilation and aeration are also essential.
Moreover, office buildings are layered with pesticide-sprayed carpets; aggressive cleaning products are used creating a pool of chemicals and particles that are inhalable. In addition, VOCs and other toxic substances may be delivered by aerosol spray products, air fresheners, chlorine bleach, detergents, dry cleaning chemicals, furniture, and floor polish.108–111
Building a healthier house/office also involves certain construction decisions, such as avoiding revolving door that may suck in outdoor pollutants, refusing too tight insulation, installing proper ventilation and aeration possibilities, and so-called sick buildings expose inhabitants and office workers to disease-inducing conditions that last long. Sick building-associated diseases are COPD, asthma, easy-spreading viral infections, and cancer. Understanding of “sick building” problem has started with the first outbreak of legionellosis when 182 cases of pneumonia were diagnosed to American Legion members staying in one building. Since then, understanding and policies have improved a lot, although increasing indoor work intensities, duration of time-spent indoors, outsourcing office jobs to countries with lower regulatory standards for indoor air quality, novel unsafe office supplies and materials, and several other factors contribute to increasingly poor situation with indoor air quality and morbidity caused by it. As declared by the American Lung Association in its bulletin State of Lung Disease in Diverse Communities, COPD should now be recognized as a common occupational disease.112
Unbalanced proteolysis
Unbalanced proteolysis is a long-lived and simplified explanatory mechanism to explain processes within emphysematous lung. It is becoming increasingly clear that this theory explains COPD development only partially. Proteolytic enzymes in the healthy lung are counterbalanced by antiproteases, but the balance shifts toward proteolysis when cigarette smoke exposure recruits neutrophils and macrophages able to produce large quantities of proteases and destroy the tissue and, particularly, elastin. Indeed, lung specimens from patients with emphysema have significantly decreased elastin content.83 The levels of elastin degradation products may therefore serve as a good index of lung damage. In corroboration, an increased plasma and urine levels of elastin-derived peptides have been reported in patients with COPD compared with those in nonsmokers.113,114 However, it is also reported that elastin breakdown products are also elevated in smokers without COPD and varies independently of smoking or COPD state.115,116
The role of proteolysis is proven and recognized from the very beginning, when emphysema was first described as an antiprotease deficiency-associated disease.117 As reported, cigarette smoke or polluted air-derived irritants attracts inflammatory cells and create an overwhelming proteolytic environment mainly comprised of neutrophil-derived protease 3 and elastase, macrophage-derived MMP-12 or so-called macrophage metallo-elastase,118,119 cathepsins L and S120,121 and collagenases MMP-2 and MMP-9.122 The antiproteolytic shield is in response created by AAT, secretory leukocyte protease inhibitor, tissue inhibitors of MMPs (TIMPs), and other inhibitors as reviewed by Abboud and Vimalanathan.123 In addition to host inflammatory cells-derived proteolytic and inhibiting substances, bacterial enzymes and inhibitors should be considered. Accumulating data suggest that bacterial proteases contribute to inflammatory process within the lung tissue, eg, Pseudomonas aeruginosa-delivered elastase activates epidermal growth factor receptor (EGFR)/extracellular signal-regulated kinase (ERK) signaling pathway and enhances IL-8 production by upregulating NF-kappaB in lung fibroblasts.124
Unbalanced proteolysis results in extracellular matrix (ECM) proteolytic degradation and fragments that may act as chemokines and promote inflammation locally.125,126 Specifically, laminin and fibronectin fragments are chemotactic to human neutrophils and monocytes.125,127 In that way, proteolysis of ECM generates fragments that may perpetuate inflammation even after smoking cessation.
Epigenetic changes in COPD development
Imbalanced proteolysis theory is also backed by data from genomic studies such as genome-wide association studies (GWAS) and gene expression studies. Recent COPD GWAS studies have identified following loci at genome-wide significance: FAM13A on 4q22, at the upstream enhancer of HHIP on 4q31, IREB2 and nicotinic acetylcholine receptors (CHRNA3 and CHRNA5) on 15q25, the 19q13 locus with genes RAB4B, EGLN2 and CYP2A6, RIN3 on 14q32, MMP12 on 11q22, and TGFB2 on 1q41.128,129 Yu et al have also performed a meta-analysis and indicated that MMP12 as well as COX2 genetic polymorphisms may be strongly implicated in the development of COPD.130 The meta-analysis has revealed an association of six loci at disintegrin and MMP33 (ADAM33) gene with the risk of COPD.131 Population-scale genetic analysis revealed that the IL1β (-511), (-31), and IL1RN (VNTR) polymorphisms are associated with COPD risk in East Asians132, IREB2 (rs13180) is associated with COPD in Hainan population (People’s Republic of China),133 TT genotype of MDR-1 gene is significantly more frequent in patients with COPD from the Aegean part of Turkey.134 The second-generation analytical approach is a combined analysis of GWAS together with gene expression meta-analysis data. Recently, Lamontagne et al combined genome-wide genotyping and gene expression in 1,111 human lung specimens to map expression quantitative trait loci (eQTL).135 They found that upregulated cystatin C (CST3) and CD22 were associated with worse lung function in COPD. Gene expression microarray studies allow genome-wide assessment of transcriptional activity as a means of exploring the biologic pathways involved in COPD pathogenesis. Hobbs and Hersh have reviewed gene expression profiling studies performed in respiratory and nonrespiratory tissues, including lung tissue (nine studies), airway epithelium (seven studies), and peripheral blood (four studies).128 Depending on the severity of emphysema, different pathways were found affected. Among differentially expressed pathways in the lung tissue inflammation, ECM-, and TGF-beta-related signaling were stated in two and/or more studies. In the airway epithelium studies, there was no matching in the pathways affected except oxidation (in two studies). Inflammation pathway was differentially expressed in blood samples in three of four COPD studies. Zeskind et al have compared several lung tissue transcriptome studies using computational approaches and have found some fundamental similarities and identified common biological processes underlying COPD, despite each study having identified largely nonoverlapping lists of differentially expressed genes.136 Computational approaches to identify gene regulatory networks are also likely to provide insights into the primary causes and secondary consequences of the complex biology that underlies COPD. Regardless of the complexity of multifactorial analysis, a promising tool would be the implication of the epigenomics into already complexed COPD data networks of genetics and transcriptomics. Examples of epigenetic changes are post-translational modifications of histones, DNA methylation, and the expression of noncoding RNAs (microRNAs [miRNAs]), which modulate gene expression without altering the sequence of the target genes. Ezzie et al screened lung samples from smokers with and without COPD for miRNA and messenger RNA (mRNA) profiles.137 They found 70 miRNAs and 2667 mRNAs differentially expressed and proposed several miRNAs, including members of the miR15/107 family, which deserve further investigation in the regulation of TGF-beta signaling in COPD. DNA methylation is an important regulator of gene transcription, which is strongly modulated by environmental factors. In the large-scale gene-specific investigation of DNA methylation marks that associate with COPD and limited lung function, Qiu et al have identified genes both studied in COPD in the past (such as SERPINA1) as well as new candidate genes (such as FUT7).138 Vucic et al have performed DNA methylation and gene expression arrays on small airway epithelium from COPD subjects and controls.139 They found 1,120 differentially methylated genes, mostly hypermethylated, which showed enrichment for three pathways: G-protein-coupled receptor signaling, aryl hydrocarbon receptor signaling, and cAMP-mediated signaling. Methylation state in 144 of these genes was negatively correlated with gene expression in three pathways: phosphatase and tensin homolog (PTEN) signaling, nuclear factor erythroid-derived 2-related factor 2 (also known as Nrf2) oxidative stress response, and IL-17F effects in allergic inflammatory diseases. A total of 15 of these 141 genes (11%) have been previously associated with COPD, at either the DNA or mRNA level, but none has been previously associated with differential DNA methylation in COPD.
Future directions from large-scale epigenetic assessments like mentioned above focus on the genes and pathways to interrogate for functional impact on COPD susceptibility and severity. The emerging role of epigenetics in COPD development stimulates new approaches. Knowledge in this field would enable reprogramming, minimizing risks, explaining etiology, and creating novel treatments for COPD.
Lung microbiome variations in COPD
Human microbiome concept pursued by National Institutes of Health-sponsored Human Microbiome Project have contributed to our knowledge about the human microbiome, and its methodologies have opened new horizons in our understanding on many human/microbial interaction aspects, including lung microflora and its physiologic and pathologic fluctuations.140,141 Suddenly scientists, clinicians, and educators have realized that dogma about lungs being a sterile space below the larynx is untrue and that we have denied the holistic picture of the life in the respiratory space for ages and only the pathogens were identified and studied at the certain conditions.
Today scientists are able to test and identify microbial content of human lung without getting the microorganisms out and culturing them in the laboratory. With powerful PCR technology and specific primers, the entire microorganism pool in a certain location can now be amplified. Technologies enable not only the global identification of microbiome but also potential functional abilities of the microbes identified. Together with such metagenomics, one can also expect metatranscriptomics and metaproteomics to emerge, where RNA and protein content would be selectively identified for each area of interest as suggested by Chambers et al.142 However, what works perfectly for skin or gut may fail in the lung. Lung tissue presents challenges due to its anatomy (not easy to reach), high risk of contamination from upper parts of respiratory system or mouth, and low availability of tissue explants from healthy donors. Thus, data on healthy lung microbiome composition are still under construction.143 It is clear that multiple factors, such as anatomical architecture, varying epithelial defense system abilities, nonuniform influx of immune cells, subject’s age, and sex, determine nonhomogenous microbiome within the human lung. Microbiome evolution dynamics, uniqueness, and seeding routes, ie, aspiration, inhaling, and hematogenous route, are intensively investigated by scientists worldwide. It is known that microbiota in the lower airways do not come entirely via mouth.144 It is also accepted now that lung microbiota are acquired and constantly build up perinatally, ie, within the womb (under influence of placenta microbiome), during and after the birth (affected by delivery route, feeding, etc.). And the more diverse microbiota become, the better it is for the health of the lungs later in life.145,146 The baseline of the healthy lung microlife forms was set, and the upper and lower airways microbiota were declared indistinguishable recently.147 Authors also stated that airways are inhabited by rather homogenous microbiota and that biomass decreases from upper to the lower tract. In healthy smokers and healthy nonsmokers, only small part of the upper airways, ie, mouth, is inhabited differently.144 This statement comes from lung microbiome group where the largest collection of the lower respiratory tract microbiome in healthy individuals was studied and the lung microbiome was compared with the mouth for the first time. Study results show that the lung microbiome is originally separate from the mouth, that mouth microbiome differs in nonsmokers and smokers, while lung communities are not significantly altered by smoking.
Our improving understanding of lung microorganisms’ ecosystem brings new explanations for microbe-related lung diseases. Apparently, we need to review the pathologic process not as a consequence of overwhelming bacterial intervention but rather as an increasing failure of normal lung ecosystem.148 Lung ecosystem’s capacity to adapt is based on heterogenous complex entities with complex interrelationships and joint actions. Moreover, in the same study, lung biogeography was explained by “island model”, and the environmental gradients were described together with previously unexplored positive feedback loops in pneumonia development. It is now accepted that exacerbations of COPD should be regarded as an increasingly pronounced local dysbiosis with the pathogenic bacteria focally disrupting the airway ecosystem. Host immune responses at the dysbiosis site might also become divergent and perpetuate in the so-called vicious cycle.
Attributing COPD and its exacerbations to dysbacteriosis-type diseases opens new treatment possibilities, somewhat similar to those used in other organ systems where preserving and restoring microbiome could protect against disease developing and/or exacerbating. Still too few studies are available today, and data reported need proofs and expansion. To date, several groups have started to evaluate lung microbiota content in COPD.141,149,150 Their main findings suggest that patients with COPD have distinct microbiome in comparison with healthy individuals with the higher prevalence of Afipia, Brevundimonas, Curvibacter, Moraxella, Neisseria, Undibacterium, Corynebacterium, Capnocytophaga, and Leptolyngbya genera. Significant decrease of the bacterial diversity associated with P. aeruginosa presence was detected in some cases.151,152
In light of the above-mentioned findings, it is not surprising to see reports on corticosteroid use-associated infectious complications in COPD.153–156 One can also speculate that the use of antibiotics might be associated with the changes in microbiome content in the lungs and also lung health overall, although data on this are lacking. It was reported that treatment with antibiotics decreases the abundance of Proteobacteria and suppression of other microbiota follows.157 Data from other organ systems and overall understanding also suggest and antibiotic-treatment contribute as a major factor to the decreasing diversity of lung microbiome. Indirectly, this notion is also supported by the very recent data on decreasing diversity of the bronchial microbiome with the increasing severity of COPD and increasing antibiotic exposure of the patients.158 A loss and replacement of microbiota by pathogenic species are also reported in the study. Similarly, in patients with cystic fibrosis, there was an increase in the total bacterial count, while microbial diversity was significantly decreased. Authors speculate that such shift might be caused by the repeated antibiotic exposure.159 Data from other human organ systems, eg, gut, suggest that treatment with antibiotics significantly modulates microbial ecosystem and its interactions with host metabolism.160 However, proofs of antibiotic-related lung microbiota changes would require specifically designed trials, sensitive assessment techniques, and frequent sampling. Assuming that antibiotics are most often prescribed and targeted toward various airway infections, the impact of these treatments on lung microbial ecosystem might be devastating. In addition, reports show the importance of gut microbiome on lung health, ie, antibiotic-exposed intestinal flora modulates immunity in the respiratory mucosa via the activation of inflammasomes.161 Moreover, airway hyper-responsiveness was reported in antibiotic-treated mice in comparison with untreated,162 specifically, when antibiotics were given perinatally, gut flora was found affected, and, subsequently, specific predisposition to hyper-responsive lung diseases was also detected. In corroboration, similar effects were proven in humans by large-scale retrospective and prospective epidemiological studies and meta-analyses, ie, children exposed to antibiotics early in life were more prone to develop bronchial asthma and transient wheezing.163–165 However, some confounding familial factors and confounding respiratory infections might have been involved, and the association might have been biased as a recent study that involves siblings has reported.166 To further prove a link between lung and gut health and healthy microflora, several studies report COPD and inflammatory bowel disease correlation in patients and, possibly, common pathways.167,168 COPD-associated comorbidities, such as inflammatory bowel disease, atherosclerosis, cardiac failure, diabetes, osteoporosis, cachexia, gastroesophageal reflux disease, and depression, affect the whole human organism. Some of them are consequential (eg, following steroid use), some etiological and, probably, dysbiosis related. The latter might be illustrated by the results from the study where smoking was shown as a modulating factor for the human gut microbiota, and potential pathogenetic link between smoking and inflammation in the gut was discovered.169 It illustrates how COPD affects person’s multiple body systems rendering them inflamed, dysbiotic, and metabolically disturbed.
Authors of the recent intelligent review also underscore the meaning of microbiome and virome.170 The fungal composition of the respiratory tract biomass appears to be more environment- and dietary habits-related than bacterial is. In addition, fungal participants are important for the post-antibiotic treatment rehabilitation when colonization may take place due to diminished colonization resistance followed by inflammatory reactions, alteration of microbial metabolic networks, and disturbed quorum sensing.171 Another entity of microbiome, virome, is distinct and considerably stable between patients, including upper respiratory tract,172 and it is partially bacteriome integrated since a large number of viruses are bacteriophages.173 Since recently, it is accepted that viruses can also be found beyond lungs and gut, ie, in the circulation of healthy individuals.174 The importance of virome in disease development, expanding an auto-epitope repertoire, influencing microbial counterparts and host cells is being recognized now. Retamales et al have found that lung destruction correlates with the number of alveolar epithelial cells expressing adenoviral proteins,175 which suggests that latent expression of the adenoviral protein may have a role in exaggerating inflammation. Moreover, persistent viral infection in animal model is shown to amplify lung destruction and increases cigarette smoke exposure-induced effects.176
Mechanisms of microbiome–host interactions that are important in COPD remain unclear. One of the mechanisms might be dependent on the potential inhibition of macrophage immunological functions induced by cigarette smoke.177 In addition; microbial colonization-related COPD exacerbations might be realized via toll-like receptor (TLR) signaling. Indeed, TLR expression on CD8 T-cells from subjects with COPD was found to be increased compared with subjects with normal pulmonary function.178 Furthermore, authors show how upon stimulation with TLR ligands, COPD patients-derived T-cells increase production of cytokines, cytotoxic molecules, and chemokines compared with controls.
Major inflammatory lung diseases appear to have distinct microbiome shifts, ie, healthy airway microbiota are diverse and balanced, asthma might be characterized by dysbiosis with an outgrowth of Proteobacteria, and a shift in the proportion of streptococci in the Firmicutes phylum, while COPD microbiome manifests with an increase in staphylococci and streptococci, in addition to an outgrowth of Proteobacteria phylum.170 Knowing the microbial ecology of human lung will help us understand the pathogenesis of COPD exacerbations and disease progression in general. We still need to know what normal and abnormal lung microbiota are and if it is possible to repopulate diseased airways with beneficial microbes the same way that we manage gut dysbiosis. Hygiene hypothesis, which focuses on the deteriorating effects of nonbiodynamic diet, improved sanitary conditions, and increased use of antibiotics and anti-inflammatory agents on adequate immune maturation and balanced T-cell system leading to immune diseases, merits some reevaluation and recognition. Indeed, data from two studies (PARSIFAL – Prevention of Allergy Risk Factors for Sensitization in Children Related to Farming and Anthroposophic Lifestyle) and GABRIEL (Multidisciplinary Study to Identify the Genetic and Environmental Causes of Asthma in the European Community) reveal that children from families living in farms and adherent to anthroposophic lifestyle (low vaccination rates, low use of antibiotics and antipyretics, biodynamic food, etc.) have lower prevalence of asthma and atopy and are exposed to a greater variety of environmental microorganisms than city children.179
Autoimmune COPD component
Autoimmunity is described as an excessive immune response prompted by antigens of the host organism (autoantigens). Vast variety of mechanisms are known today that expose or modify host molecules rendering them antigenic, including oxidized, nitrosylated, polymerized, citrullinated, or otherwise chemically and structurally affected proteins and peptides, mutated under the influence of environmental insults proteins, aberrantly exposed molecules (eg, in the event of abundant apoptosis) and viral or bacterial invasion-affected cells. All of those might be important in emphysema. In the review by Cosio et al, cigarette smoking is represented as a major promoter of parenchymal inflammation, and possible multiple mechanisms for the expression of novel epitopes are discussed.83 Consequences of such self-attack induce lung tissue damage and further lead to structural alterations and further creation of neoantigens.180 The same study reports that adoptive transfer of CD8 cytotoxic T lymphocytes specific for an alveolar neoantigen (influenza hemagglutinin) leads to progressive lethal injury and lung destruction in transgenic mice. Similarly, proofs that T-cells mediation is essential for the lung tissue injury were reported long ago, when experiments were carried out in influenza model in T-cell-deficient mice. Experimental influenza was significantly milder and histologically less pronounced than in wild-type animals.181
Recognition of the autoimmune component importance in the COPD development calls for epitope(s) identification.182 Whether it is unusually exposed protein, eg, neutrophil degranulation products,183 viral epitopes on host cell surface, apoptotic cell content exposed, or a mix of several remains to be elucidated. There is an old observation that patients with COPD blood sera reacts with bronchial secretions and the so-called mucus antibody, and this is followed by mucus accumulation and disease progression.184 More recently, several reports have demonstrated that alveolar epithelium or small vascular endothelium might be the target, eg, global antiepithelial antibodies (against type II respiratory epithelium) were found in all patients with COPD in comparison with minority of controls.185 Some of those antibodies were associated with decreased body mass index. And furthermore, cytotoxicity of pulmonary epithelial cells by allogeneic mononuclear cells also increased after activation by patients’ with COPD blood plasma, compared with controls. Such results prove that vast array of pulmonary epithelium-directed autoantibodies are prevalent in COPD and participate in the disease development. Next to epithelium, endothelium of the lung vasculature is also exposed to autoimmune attacks. Indeed, in the unique animal model, Taraseviciene-Stewart et al have shown that xenogeneic endothelial cells injected induce autoantibody production that subsequently leads to lung emphysema in rats, involving an accumulation of CD4 T-cells in the host lungs.186 Moreover, adoptive transfer of pathogenic, spleen-derived CD4 cells into naive immune-competent animal also results in emphysema. The study further proves that tissue destruction in emphysema depends (in part) on immune system and that CD4 cell-dependent mechanisms can induce emphysema development without other insults. Corroboratively, another study demonstrates that patients with COPD have significantly higher serum antiendothelial cell antibodies than subjects in the reference population.187 Recently, antineutrophil antibodies directed against granule content – lactoferrin were detected in the circulation of AAT deficiency carriers, a subset of patients with COPD.188 Supplementation therapy did reduce the levels of autoantibodies and the overall proinflammatory content in the patients’ plasma and neutrophils. Other studies report more candidate epitopes and autoimmune activity, including antihuman cytokeratin 18 protein antibodies that were detected in patients with COPD sera in correlation with lung functional parameters.189 Antielastin antibodies have been proposed to drive COPD progression, but the data accumulated today are conflicting.190,191
Environmental insults, particularly oxidative stress inducers, induce structural protein carbonylation in exposed lungs. It was demonstrated that carbonylated proteins serve as auto-epitopes in patients with COPD and in an animal model of chronic exposure to ozone.192 Cigarette smoke is also clearly associated with citrullination of proteins and contributes to the developmental mechanisms of rheumatoid arthritis. In COPD, levels of anticitrullinated peptide antibodies are also increased.193 This response is even more exaggerated in wood smoke-induced-COPD compared with tobacco smoke-induced COPD. The important role in the disease process should be attributed to the other autoimmune disease-related autoantibodies, eg, rheumatoid factor. Recent study suggests that in COPD, rheumatoid factor antibodies and autoantibodies to heat shock protein (HSP) 70 are detectable and elevated in comparison with controls. Moreover, they remain elevated after smoking cessation.194 In a complex disease as COPD, systemic autoantigens are also involved. Patients with COPD were found to have high titers of circulating antinuclear and antitissue antibodies in Spanish Phenotype and Course of Chronic Obstructive Pulmonary Disease study.195 The relationship between antibody titers and lung function was also demonstrated. There is a category of nonspecific autoantibodies, which are present in everyone’s circulation and may play a role in disease development as suggested by recent study.196 Moreover, the amount of IgG antibodies recognizing lung tissue components was significantly lower in the circulation of COPD compared with nonsmoking control subjects, and the target for some of those antibodies was identified as collagen-1. Thus, authors speculate that naturally occurring antibodies or rather lack of them is caused by lower availability due to their increased reactivity or abundance of their epitopes. Whether their role is destructive or beneficial remains to be elucidated.
All these data stimulate discussion if autoimmune process in COPD is a causative process or just a consequence or even a coincidence. Lymphoid organ neogenesis and persistence in smoke-exposed mice lungs suggest that autoimmune process is causative rather than simple coincidence.197 This notion is further supported by the fact that the subset of lymphocytes, particularly CD56 lymphocytes, is able to rapidly kill autologous lung cells. It was a spontaneous process, and it was upregulated in subjects with severe COPD.86 There is an individual variability and, probably, individual COPD-related epitope array in every case. In the future, revealing specific autoantibodies for each patient or patients’ group might initiate possibilities to diagnose and treat etiologically distinct COPD entities individually.
At the crossroads of microbiota and autoimmunity theories, virus- and other microorganism-associated epitopes emerge. Viral epitopes, ie, viral peptides presented on the cell surface by major histocompatibility complex molecules, signal for the destruction of the infected cells. Chemical modifications of viral peptides and proteins may be crucial for their inadequate proceeding by host cells, generation of new epitopes, and increase in antigenic insult. Human organism as a habitat for numerous micro-roommates goes into disease as a whole system, and two main hypotheses are proposed to explain the interrelationships and predisposition to the disease. The hygiene hypothesis suggests that extensive microbial exposure in early childhood is essential for proper immune-regulatory mechanisms to engage and control autoimmune reactions and/or allergy. And in parallel, the triggering hypothesis suggests that specific microbes, particularly, viruses, may trigger autoimmunity.198 Similarly, enteroviral infections cause autoimmune diabetes in animals and are associated with an increased risk of autoimmune diabetes in epidemiological and other studies.199 And opposite, the same review discusses protective effects of microbes that have been studied in animal models and in epidemiological studies, ie, hepatitis A virus and Helicobacter pylori persistence is associated with lower risk of autoimmune diabetes.
Inefficient repair, current therapeutic approaches, and study models
It is increasingly clear that slowing down the progression of the disease might be very complicated due to all ongoing chronic inflammatory, proteolytic, autoimmune, and dysbiotic processes reviewed earlier. In addition, it is generally accepted that repair mechanisms in emphysematous lung are deficient. The exact reasons for that are not known. Some compensatory lung tissue regrowth is possible, and several factors, such as, thyroid transcription factor 1, vascular endothelial growth factor (VEGF), hypoxia-inducible factor 1, and keratinocyte growth factor, are known to be responsible for it.200–203 Central process for the repair according to some authors is epithelial–mesenchymal transition (EMT).204 Particularly, epithelial cells participate in EMT and restores integrity directly.205 In the elegant study by Barkauskas et al, a reparative behavior of type II epithelial cells in the alveoli was studied.206 They show that type II epithelial cells can self-renew in culture and differentiate into alveolar-like structures “alveolospheres” containing type II and type I epithelial cells. In alveolar tissue context, MSC also has been shown to contribute to regeneration in numerous experimental settings, ie, home to sites of asbestos-induced lung injury, contribute to tissue remodeling in a rat monocrotaline model of pulmonary hypertension, decrease chronic airway inflammation in ovalbumin model of asthma, restore alveolar and lung fluid balance after endotoxin-induced acute lung injury, and modulate the fibrotic response to radiation-induced injury.207–214 A new study shows that transplanted MSC is capable to integrate into damaged (pulmonary fibrosis model) mice lungs and differentiate into type II lung cells expressing surfactant proteins.215 Moreover, in the cigarette smoke induced rat emphysema model, amniotic fluid-derived MSC has generated type II alveolar epithelial cells, integrated at the sites of destruction and induced local regeneration of the lung alveolar epithelium.216 Promising potential of MSC for the repair of the lung tissue might be harnessed in several ways: via endogenous activation and recruitment, delivery from outside via circulation, or via airways. The latter possibility was explored in the new study, and authors have demonstrated that intratracheal MSC engrafts at the hyperoxia-induced dysplasia sites in the rat lungs and expresses surfactant proteins.217 However, authors claim that therapeutic benefit was achieved due to paracrine mechanisms, since engraftment was very low. Moreover, this and other studies show that MSC secretion products reduce structural lung cells apoptosis, accelerate healing, and enhance endothelial functions.217,218 Reports show that bacteria-induced tissue damage promotes the airway engraftment of heterologous bone marrow–derived stem cells and their epithelial transformation.219 The ability of MSC of various origins to transform and express phenotypic markers of airway epithelium and to participate in airway remodeling in vivo is documented.219,220
Since cures and regeneration induction strategies are still in infancy, the best treatment is prevention. In this context, healthy living, clean air, proper use of antibiotics, and adequate exposure to microorganisms become increasingly important. Physical activity was also reported as a factor associated with lower rates of hospital readmission in patients with COPD.221 Also, it should be clear that current therapeutic approaches needs constant revisions and reapproval. It is particularly important for oral corticoids, antibiotics, and anti-inflammatory agents. If we picture COPD as dysbiosis, the quality and diversity of microbiome in the human airways become highly valuable. From the birth and with the every breath after birth, microbiome deserves protection since it comprises an essential part of our own health. In addition to balanced microbial ecosystem, there is also discussion on dietary interventions, ie, increasing antioxidant content.222 Interestingly, living in altitude might also be a therapeutic approach depending on the lung disease.223
To date, many COPD (both emphysema and bronchitis component) models are in use and different approaches are employed, ie, protease challenge,224,225 toxic gases,226 and cigarette smoke.227 In addition, numerous species have been used to study of COPD, eg, rats, mice, dogs, guinea pigs, monkeys, and sheep as reviewed by Mahadeva and Shapiro.228 Cigarette smoke-exposed mouse remains the most often used in vivo model. Relatively low costs of the animals and the molecular biology tools available make this model attractive. However, detailed local cellular involvement (migration of neutrophils, macrophages, T-cells, release of proteases, and various proinflammatory molecules), loss of epithelium and endothelium, transformation of the mesenchyme, and ongoing repair processes are easier to study and visualize in larger models in vivo or, also, in vitro. Primary cells obtained ex vivo play a crucial role in COPD research by helping to assess the differential behavior of cells from diseased and healthy persons. In addition, a number of immortalized cell lines derived from carcinomas or transformed from primary tissue are in use, eg, A549 (type II pneumocyte), several bronchial epithelial cell lines.229 These cells exhibit phenotypic characteristics of their original cell type and are easy to use and well documented. In addition, they enable to easily reproduce results. However, all ex vivo tissue cultures have their shortcomings, eg, lack of the native environment, contacts with the ECM, the inflammatory ambience, and interactions. Lung-on-chip technologies enable high throughout and convenient cytotoxicity and genotoxicology assays. However, they lack epithelium–ECM interactions and discrete spatial architecture that defines parenchymal structure and function within the lungs. There are few attempts reported to use native lung matrix, ie, decellularized and reseeded lung tissue matrix. Such modeling provides with relevant structural and biochemical scaffold for the tissue cultures and greatly resembles in vivo situation. However, native organs require specific processing and are complicated to handle. With all the possibilities available, evaluation and choice of the most relevant study model depends on the individual project’s aim, capabilities, and complex questions that are going to be addressed.
Conclusion
COPD as a lifestyle and/or occupational disease should be regarded as a complex problem related not only on what we breathe in but also on what we eat, how we move, what are we treated with, and so on. With all these factors in mind, we should direct our efforts toward prevention and regeneration, since symptomatic treatment of the disease symptoms has led us nowhere. Balancing immune response in close relation to maintaining a diverse microbiome (ie, low antibiotic exposure), elimination of air pollutants and smoke (safe smart homes and offices, air pollution management policies, etc.), full resolution of inflammatory processes (limited anti-inflammatory medication use), healthy diet with good antioxidant load, and physical activity are among the measures that need to be taken into consideration when managing COPD. Similar research lines should follow.
Acknowledgments
This work was supported by European Social Fund (ESF) under the Human Resources Development Action Programme by implementation of a project “Execution of functions assigned to Lithuanian Research Council for implementation of the Global Grant measure”.
Disclosure
The authors report no conflicts of interest in this work.
References
Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non-smokers. Lancet. 2009;374(9691):733–743. doi:10.1016/S0140-6736(09)61303-9. | ||
Kim DS, Kim YS, Jung K-S, et al. Prevalence of chronic obstructive pulmonary disease in Korea: a population-based spirometry survey. Am J Respir Crit Care Med. 2005;172(7):842–847. doi:10.1164/rccm.200502-259OC. | ||
Zhou Y, Wang C, Yao W, et al. COPD in Chinese nonsmokers. Eur Respir J. 2009;33(3):509–518. doi:10.1183/09031936.00084408. | ||
Ehrlich RI, White N, Norman R, et al. Predictors of chronic bronchitis in South African adults. Int J Tuberc Lung Dis. 2004;8(3):369–376. | ||
Forbes LJL, Kapetanakis V, Rudnicka AR, et al. Chronic exposure to outdoor air pollution and lung function in adults. Thorax. 2009;64(8):657–663. doi:10.1136/thx.2008.109389. | ||
Hopkinson NS, Polkey MI. Chronic obstructive pulmonary disease in non-smokers. Lancet. 2015;374(9706):1964. doi:10.1016/S0140-6736(09)62115-2. | ||
Doust E, Ayres JG, Devereux G, et al. Is pesticide exposure a cause of obstructive airways disease? Eur Respir Rev. 2014;23(132):180–192. doi:10.1183/09059180.00005113. | ||
Arroyave ME. Pulmonary obstructive disease in a population using paraquat in Colombia. 1993:1–7. | ||
Lamprecht B, Schirnhofer L, Kaiser B, Studnicka M, Buist AS. Farming and the prevalence of non-reversible airways obstruction – results from a population-based study. Am J Ind Med. 2007;50(6):421–426. doi:10.1002/ajim.20470. | ||
Jong K De, Boezen HM, Kromhout H, Vermeulen R, Postma DS, Vonk JM. Original contribution association of occupational pesticide exposure with accelerated longitudinal decline in lung function. Am J Epidemiol. 2014;179(11):1323–1330. doi:10.1093/aje/kwu053. | ||
Callahan C, Al-Batanony M, Ismail A, et al. Chlorpyrifos exposure and respiratory health among adolescent agricultural workers. Int J Environ Res Public Health. 2014;11:13117–13129. doi:10.3390/ijerph111213117. | ||
De Jong K, Boezen H, Kromhout H, Vermeulen R, Postma D, Vonk J. Pesticides and other occupational exposures are associated with airway obstruction: the LifeLines cohort study. Occup Env Med. 2014;71:88–96. doi:10.1136/oemed-2013-101639. | ||
Barker DJ, Godfrey KM, Fall C, Osmond C, Winter PD, Shaheen SO. Relation of birth weight and childhood respiratory infection to adult lung function and death from chronic obstructive airways disease. BMJ. 1991;303(6804):671–675. | ||
Zosky GR, Berry LJ, Elliot JG, James AL, Gorman S, Hart PH. Vitamin D deficiency causes deficits in lung function and alters lung structure. Am J Respir Crit Care Med. 2011;183(10):1336–1343. doi:10.1164/rccm.201010-1596OC. | ||
Shaheen SO, Newson RB, Smith GD, Henderson AJ. Prenatal paracetamol exposure and asthma: further evidence against confounding. Int J Epidemiol. 2010;39(3):790–794. doi:10.1093/ije/dyq049. | ||
Martindale S, McNeill G, Devereux G, Campbell D, Russell G, Seaton A. Antioxidant intake in pregnancy in relation to wheeze and eczema in the first two years of life. Am J Respir Crit Care Med. 2005;171(2):121–128. doi:10.1164/rccm.200402-220OC. | ||
Guilbert TW, Stern DA, Morgan WJ, Martinez FD, Wright AL. Effect of breastfeeding on lung function in childhood and modulation by maternal asthma and atopy. Am J Respir Crit Care Med. 2007;176(9):843–848. doi:10.1164/rccm.200610-1507OC. | ||
Mui TSY, Man JM, McElhaney JE, et al. Telomere length and chronic obstructive pulmonary disease: evidence of accelerated aging. J Am Geriatr Soc. 2009;57(12):2372–2374. doi:10.1111/j.1532-5415.2009.02589.x. | ||
Savale L, Chaouat A, Bastuji-Garin S, et al. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;179(7):566–571. doi:10.1164/rccm.200809-1398OC. | ||
Walters MS, De BP, Salit J, et al. Smoking accelerates aging of the small airway epithelium. Respir Res. 2014;15(1):94. doi:10.1186/s12931-014-0094-1. | ||
Houben JMJ, Mercken EM, Ketelslegers HB, et al. Telomere shortening in chronic obstructive pulmonary disease. Respir Med. 2009;103(2):230–236. doi:10.1016/j.rmed.2008.09.003. | ||
Noordhoek JA, Postma DS, Chong LL, et al. Different proliferative capacity of lung fibroblasts obtained from control subjects and patients with emphysema. Exp Lung Res. 2003;29(5):291–302. doi:10.1080/01902140303789. | ||
Fujii S, Hara H, Araya J, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012;1(5):630–641. doi:10.4161/onci.20297. | ||
Ryter SW, Lee S-J, Choi AMK. Autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. Expert Rev Respir Med. 2010;4(5):573–584. doi:10.1586/ers.10.61. | ||
Laue J, Reierth E, Melbye H. When should acute exacerbations of COPD be treated with systemic corticosteroids and antibiotics in primary care: a systematic review of current COPD guidelines. NPJ Prim Care Respir Med. 2015;25:15002. doi:10.1038/npjpcrm.2015.2. | ||
Seemungal T, Harper-Owen R, Bhowmik A, et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164(9):1618–1623. doi:10.1164/ajrccm.164.9.2105011. | ||
O’Donnell DE, Laveneziana P. Physiology and consequences of lung hyperinflation in COPD. Eur Respir Rev. 2006;15:61–67. doi:10.1183/09059180.00010002. | ||
Miravitlles M, Anzueto A. Role of infection in exacerbations of chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2015. doi:10.1097/MCP.0000000000000154. | ||
Bathoorn E, Liesker JJW, Postma DS, et al. Change in inflammation in out-patient COPD patients from stable phase to a subsequent exacerbation. Int J Chron Obstruct Pulmon Dis. 2009;4:101–109. | ||
Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173(10):1114–1121. doi:10.1164/rccm.200506-859OC. | ||
Herath SC, Poole P. Prophylactic antibiotic therapy for chronic obstructive pulmonary disease (COPD). Cochrane Database Syst Rev. 2013;11:CD009764. doi:10.1002/14651858.CD009764.pub2. | ||
Arbex MA, de Souza Conceicao GM, Cendon SP, et al. Urban air pollution and chronic obstructive pulmonary disease-related emergency department visits. J Epidemiol Community Health. 2009;63(10):777–783. doi:10.1136/jech.2008.078360. | ||
Peacock JL, Anderson HR, Bremner SA, et al. Outdoor air pollution and respiratory health in patients with COPD. Thorax. 2011;66(7):591–596. doi:10.1136/thx.2010.155358. | ||
Eisner MD, Iribarren C, Yelin EH, et al. The impact of SHS exposure on health status and exacerbations among patients with COPD. Int J Chron Obstruct Pulmon Dis. 2009;4:169–176. | ||
Di Stefano A, Caramori G, Oates T, et al. Increased expression of nuclear factor-κB in bronchial biopsies from smokers and patients with COPD. Eur Respir J. 2002;20(3):556–563. doi:10.1183/09031936.02.00272002. | ||
Ravi AK, Khurana S, Lemon J, et al. Increased levels of soluble interleukin-6 receptor and CCL3 in COPD sputum. Respir Res. 2014;15(1):103. doi:10.1186/s12931-014-0103-4. | ||
Shao MXG, Nakanaga T, Nadel JA. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-α-converting enzyme in human airway epithelial (NCI-H292) cells. Am J Physiol Lung Cell Mol Physiol. 2004;287(2):L420–L427. | ||
Shapiro SD. End-stage chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164(3):339–340. doi:10.1164/ajrccm.164.3.2105072c. | ||
Hunninghake GW, Crystal RG. Cigarette smoking and lung destruction. Accumulation of neutrophils in the lungs of cigarette smokers. Am Rev Respir Dis. 1983;128(5):833–838. | ||
Martin TR, Raghu G, Maunder RJ, Springmeyer SC. The effects of chronic bronchitis and chronic air-flow obstruction on lung cell populations recovered by bronchoalveolar lavage. Am Rev Respir Dis. 1985;132(2):254–260. | ||
Hunninghake GW, Gadek JE, Kawanami O, Ferrans VJ, Crystal RG. Inflammatory and immune processes in the human lung in health and disease: evaluation by bronchoalveolar lavage. Am J Pathol. 1979;97(1): 149–206. | ||
Paone G, Conti V, Vestri A, et al. Analysis of sputum markers in the evaluation of lung inflammation and functional impairment in symptomatic smokers and COPD patients. Dis Markers. 2011;31(2):91–100. doi:10.3233/DMA-2011-0807. | ||
Frasca L, Lande R. Role of defensins and cathelicidin LL37 in auto-immune and auto-inflammatory diseases. Curr Pharm Biotechnol. 2012;13(10):1882–1897. | ||
Overbeek SA, Braber S, Koelink PJ, et al. Cigarette smoke-induced collagen destruction; key to chronic neutrophilic airway inflammation? PLoS One. 2013;8(1):e55612. doi:10.1371/journal.pone.0055612. | ||
Kobayashi SD, DeLeo FR. Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdiscip Rev Syst Biol Med. 2009;1(3):309–333. doi:10.1002/wsbm.32. | ||
Kobayashi SD, Voyich JM, Burlak C, DeLeo FR. Neutrophils in the innate immune response. Arch Immunol Ther Exp (Warsz). 2005;53(6):505–517. | ||
Barnett ML, Lamb KA, Costello KM, Pike MC. Characterization of interleukin-8 receptors in human neutrophil membranes: regulation by guanine nucleotides. Biochim Biophys Acta. 1993;1177(3):275–282. | ||
Reilly IA, Knapp HR, Fitzgerald GA. Leukotriene B4 synthesis and neutrophil chemotaxis in chronic granulocytic leukaemia. J Clin Pathol. 1988;41(11):1163–1167. | ||
Mathis SP, Jala VR, Lee DM, Haribabu B. Nonredundant roles for leukotriene B4 receptors BLT1 and BLT2 in inflammatory arthritis. J Immunol. 2010;185(5):3049–3056. doi:10.4049/jimmunol.1001031. | ||
Kreisle RA, Parker CW. Specific binding of leukotriene B4 to a receptor on human polymorphonuclear leukocytes. J Exp Med. 1983;157(2):628–641. | ||
Cavicchioni G, Fraulini A, Falzarano S, Spisani S. Oligomeric formylpeptide activity on human neutrophils. Eur J Med Chem. 2009;44(12):4926–4930. doi:10.1016/j.ejmech.2009.08.010. | ||
Al-Omari M, Korenbaum E, Ballmaier M, et al. Acute-phase protein alpha1-antitrypsin inhibits neutrophil calpain I and induces random migration. Mol Med. 2011;17(9–10):865–874. doi:10.2119/molmed.2011.00089. | ||
Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol. 2002;26(6):723–730. doi:10.1165/ajrcmb.26.6.4739. | ||
Mulgrew AT, Taggart CC, Lawless MW, et al. Z alpha1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractant. Chest. 2004;125(5):1952–1957. | ||
Janciauskiene S. Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological roles. Biochim Biophys Acta. 2001;1535(3):221–235. | ||
Kim D, Haynes CL. Neutrophil chemotaxis within a competing gradient of chemoattractants. Anal Chem. 2012;84:6070–6078. doi:10.1021/ac3009548. | ||
Aldonyte R, Tunaitis V, Surovas A, et al. Effects of major human antiprotease alpha-1-antitrypsin on the motility and proliferation of stromal cells from human exfoliated deciduous teeth. Regen Med. 2010;5(4):633–643. | ||
Wada K, Arita M, Nakajima A, et al. Leukotriene B4 and lipoxin A4 are regulatory signals for neural stem cell proliferation and differentiation. FASEB J. 2006;20(11):1785–1792. doi:10.1096/fj.06-5809com. | ||
Stich S, Loch A, Leinhase I, et al. Human periosteum-derived progenitor cells express distinct chemokine receptors and migrate upon stimulation with CCL2, CCL25, CXCL8, CXCL12, and CXCL13. Eur J Cell Biol. 2008;87(6):365–376. doi:10.1016/j.ejcb.2008.03.009. | ||
Hunninghake GW, Crystal RG. Cigarette smoking and lung destruction. Accumulation of neutrophils in the lungs of cigarette smokers. Am Rev Respir Dis. 1983;128(5):833–838. | ||
Dhami R, Gilks B, Xie C, Zay K, Wright JL, Churg A. Acute cigarette smoke-induced connective tissue breakdown is mediated by neutrophils and prevented by alpha1-antitrypsin. Am J Respir Cell Mol Biol. 2000;22(2):244–252. doi:10.1165/ajrcmb.22.2.3809. | ||
Lams BE, Sousa AR, Rees PJ, Lee TH. Immunopathology of the small-airway submucosa in smokers with and without chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;158(5 Pt 1):1518–1523. doi:10.1164/ajrccm.158.5.9802121. | ||
Bosken CH, Hards J, Gatter K, Hogg JC. Characterization of the inflammatory reaction in the peripheral airways of cigarette smokers using immunocytochemistry. Am Rev Respir Dis. 1992;145(4 Pt 1):911–917. doi:10.1164/ajrccm/145.4_Pt_1.911. | ||
Mallia P, Footitt J, Sotero R, et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(11):1117–1124. doi:10.1164/rccm.201205-0806OC. | ||
Finkelstein R, Fraser RS, Ghezzo H, Cosio MG. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med. 1995;152(5 Pt 1):1666–1672. doi:10.1164/ajrccm.152.5.7582312. | ||
Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev. 2007;87(1):69–98. doi:10.1152/physrev.00022.2006. | ||
Aldonyte R, Jansson L, Piitulainen E, Janciauskiene S. Circulating monocytes from healthy individuals and COPD patients. Respir Res. 2003;4:11. | ||
Shapiro SD. The macrophage in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(5 Pt 2):S29–S32. doi:10.1164/ajrccm.160.supplement_1.9. | ||
Di Stefano A, Capelli A, Lusuardi M, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med. 1998;158(4):1277–1285. doi:10.1164/ajrccm.158.4.9802078. | ||
Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22:672–688. doi:10.1183/09031936.03.00040703. | ||
Punturieri A, Filippov S, Allen E, et al. Regulation of elastinolytic cysteine proteinase activity in normal and cathepsin K-deficient human macrophages. J Exp Med. 2000;192(6):789–799. | ||
Russell REK, Thorley A, Culpitt SV, et al. Alveolar macrophage-mediated elastolysis: roles of matrix metalloproteinases, cysteine, and serine proteases. Am J Physiol Lung Cell Mol Physiol. 2002;283(4):L867–L873. doi:10.1152/ajplung.00020.2002. | ||
Saetta M, Mariani M, Panina-Bordignon P, et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;165(10):1404–1409. | ||
Caramori G, Romagnoli M, Casolari P, et al. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax. 2003;58(4):348–351. | ||
Anto RJ, Mukhopadhyay A, Shishodia S, Gairola CG, Aggarwal BB. Cigarette smoke condensate activates nuclear transcription factor-kappaB through phosphorylation and degradation of IkappaB(alpha): correlation with induction of cyclooxygenase-2. Carcinogenesis. 2002;23(9):1511–1518. | ||
Walters MJ, Paul-Clark MJ, McMaster SK, Ito K, Adcock IM, Mitchell JA. Cigarette smoke activates human monocytes by an oxidant-AP-1 signaling pathway: implications for steroid resistance. Mol Pharmacol. 2005;68(5):1343–1353. doi:10.1124/mol.105.012591. | ||
Geraghty P, Hardigan A, Foronjy RF. Cigarette smoke activates the proto-oncogene c-src to promote airway inflammation and lung tissue destruction. Am J Respir Cell Mol Biol. 2014;50(3):559–570. doi:10.1165/rcmb.2013-0258OC. | ||
Chamoto K, Gibney BC, Ackermann M, et al. Alveolar macrophage dynamics in murine lung regeneration. J Cell Physiol. 2012;227:3208–3215. doi:10.1002/jcp.24009. | ||
Lolmede K, Campana L, Vezzoli M, et al. Inflammatory and alternatively activated human macrophages attract vessel-associated stem cells, relying on separate HMGB1- and MMP-9-dependent pathways. J Leukoc Biol. 2009;85:779–787. doi:10.1189/jlb.0908579. | ||
Purwar R, Campbell J, Murphy G, Richards WG, Clark Ra, Kupper TS. Resident memory T cells (TRM) are abundant in human lung: diversity, function, and antigen specificity. PLoS One. 2011;6(1):e16245. doi:10.1371/journal.pone.0016245. | ||
Shirai T, Suda T, Inui N, Chida K. Correlation between peripheral blood T-cell profiles and clinical and inflammatory parameters in stable COPD. Allergol Int. 2010;59:75–82. doi:10.2332/allergolint.09-OA-0126. | ||
Majori M, Corradi M, Caminati A, Cacciani G, Bertacco S, Pesci A. Predominant TH1 cytokine pattern in peripheral blood from subjects with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 1999;103(3 Pt 1):458–462. | ||
Cosio MG, Majo J, Cosio MG. Inflammation of the airways and lung parenchyma in COPD: role of T cells. Chest. 2002:121(5 Suppl):160S–165S. doi:10.1378/chest.121.5. | ||
Majo J, Ghezzo H, Cosio MG. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J. 2001;17(5):946–953. | ||
Wang J, Urbanowicz RA, Tighe PJ, Todd I, Corne JM, Fairclough LC. Differential activation of killer cells in the circulation and the lung: a study of current smoking status and chronic obstructive pulmonary disease (COPD). PLoS One. 2013;8(3):e58556. doi:10.1371/journal.pone.0058556. | ||
Freeman CM, Stolberg VR, Crudgington S, et al. Human CD56+ cytotoxic lung lymphocytes kill autologous lung cells in chronic obstructive pulmonary disease. PLoS One. 2014;9(7):1–12. doi:10.1371/journal.pone.0103840. | ||
Mikko M, Forsslund H, Cui L, et al. Increased intraepithelial (CD103+) CD8+ T cells in the airways of smokers with and without chronic obstructive pulmonary disease. Immunobiology. 2013;218(2):225–231. doi:10.1016/j.imbio.2012.04.012. | ||
Eppert BL, Wortham BW, Flury JL, Borchers MT. Functional characterization of T cell populations in a mouse model of chronic obstructive pulmonary disease. J Immunol. 2013;190(3):1331–1340. doi:10.4049/jimmunol.1202442. | ||
Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31(6):1334–1356. doi:10.1183/09031936.00018908. | ||
Caramori G, Di Stefano A, Casolari P, et al. Chemokines and chemokine receptors blockers as new drugs for the treatment of chronic obstructive pulmonary disease. Curr Med Chem. 2013;20(35):4317–4349. | ||
Barnes PJ. The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2009;41(6):631–638. doi:10.1165/rcmb.2009-0220TR. | ||
Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 2004;56(4):515–548. doi:10.1124/pr.56.4.2. | ||
Caramori G, Casolari P, Adcock I. Role of transcription factors in the pathogenesis of asthma and COPD. Cell Commun Adhes. 2013;20(1–2):21–40. doi:10.3109/15419061.2013.775257. | ||
Rennard SI, Fogarty C, Kelsen S, et al. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(9):926–934. doi:10.1164/rccm.200607-995OC. | ||
Van der Vaart H, Koeter GH, Postma DS, Kauffman HF, ten Hacken NHT. First study of infliximab treatment in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(4):465–469. doi:10.1164/rccm.200501-147OC. | ||
Dentener MA, Creutzberg EC, Pennings H-J, Rijkers GT, Mercken E, Wouters EFM. Effect of infliximab on local and systemic inflammation in chronic obstructive pulmonary disease: a pilot study. Respiration. 2008;76(3):275–282. doi:10.1159/000117386. | ||
Ray PD, Huang B-W, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–990. doi:10.1016/j.cellsig.2012.01.008. | ||
Schraufstätter I, Hyslop PA, Jackson JH, Cochrane CG. Oxidant-induced DNA damage of target cells. J Clin Invest. 1988;82(3):1040–1050. | ||
Durga M, Nathiya S, Rajasekar A, Devasena T. Effects of ultrafine petrol exhaust particles on cytotoxicity, oxidative stress generation, DNA damage and inflammation in human A549 lung cells and murine RAW 264.7 macrophages. Environ Toxicol Pharmacol. 2014;38(2):518–530. doi:10.1016/j.etap.2014.08.003. | ||
Moller P, Danielsen PH, Karottki DG, et al. Oxidative stress and inflammation generated DNA damage by exposure to air pollution particles. Mutat Res Rev Mutat Res. 2014;762C:133–166. doi:10.1016/j.mrrev.2014.09.001. | ||
Kafoury RM, Kelley J. Ozone enhances diesel exhaust particles (DEP)-induced interleukin-8 (IL-8) gene expression in human airway epithelial cells through activation of nuclear factors-kappaB (NF-kappaB) and IL-6 (NF-IL6). Int J Environ Res Public Health. 2005;2(3–4):403–410. | ||
Haddad JJ, Land SC. Redox signaling-mediated regulation of lipopolysaccharide-induced proinflammatory cytokine biosynthesis in alveolar epithelial cells. Antioxid Redox Signal. 2002;4(1):179–193. doi:10.1089/152308602753625942. | ||
Tzeng H-P, Yang R Sen, Ueng T-H, Liu S-H. Upregulation of cyclooxygenase-2 by motorcycle exhaust particulate-induced reactive oxygen species enhances rat vascular smooth muscle cell proliferation. Chem Res Toxicol. 2007;20(8):1170–1176. doi:10.1021/tx700084z. | ||
Tkaczyk J, Vízek M. Oxidative stress in the lung tissue – sources of reactive oxygen species and antioxidant defence. Prague Med Rep. 2007;108(2):105–114. | ||
Kalluri R, Cantley LG, Kerjaschki D, Neilson EG. Reactive oxygen species expose cryptic epitopes associated with autoimmune goodpasture syndrome. J Biol Chem. 2000;275(26):20027–20032. doi:10.1074/jbc.M904549199. | ||
Barrese E, Gioffrè A, Scarpelli M, Turbante D, Trovato R, Iavicoli S. Indoor pollution in work office: VOCs, formaldehyde and ozone by printer. Occup Dis Environ Med. 2014;2:49–55. | ||
Lee C-W, Dai Y-T, Chien C-H, Hsu D-J. Characteristics and health impacts of volatile organic compounds in photocopy centers. Environ Res. 2006;100(2):139–149. doi:10.1016/j.envres.2005.05.003. | ||
Institute of Medicine (US) Committee on the Assessment of Asthma and Indoor Air. Clearing the Air: Asthma and Indoor Air Exposures. Washington, DC: National Academies Press (US); 2000. | ||
Nazaroff WW, Weschler CJ. Cleaning products and air fresheners: exposure to primary and secondary air pollutants. Atmos Environ. 2004;38(18):2841–2865. doi:10.1016/j.atmosenv.2004.02.040. | ||
Steinemann AC, MacGregor IC, Gordon SM, et al. Fragranced consumer products: chemicals emitted, ingredients unlisted. Environ Impact Assess Rev. 2011;31:328–333. doi:10.1016/j.eiar.2010.08.002. | ||
Sarwar G, Olson DA, Corsi RL, Weschler CJ. Indoor fine particles: the role of terpene emissions from consumer products. J Air Waste Manag Assoc. 2004;54(3):367–377. | ||
Reid Pa, Reid PT. Occupational lung disease. J R Coll Physicians Edinb. 2013;43:44–48. doi:10.4997/JRCPE.2013.111. | ||
Schriver EE, Davidson JM, Sutcliffe MC, Swindell BB, Bernard GR. Comparison of elastin peptide concentrations in body fluids from healthy volunteers, smokers, and patients with chronic obstructive pulmonary disease. Am Rev Respir Dis. 1992;145(4 Pt 1):762–766. doi:10.1164/ajrccm/145.4_Pt_1.762. | ||
Harel S, Janoff A, Yu SY, Hurewitz A, Bergofsky EH. Desmosine radioimmunoassay for measuring elastin degradation in vivo. Am Rev Respir Dis. 1980;122(5):769–773. | ||
Davies SF, Offord KP, Brown MG, Campe H, Niewoehner D. Urine desmosine is unrelated to cigarette smoking or to spirometric function. Am Rev Respir Dis. 1983;128(3):473–475. | ||
Betsuyaku T, Nishimura M, Yoshioka A, Takeyabu K, Miyamoto K, Kawakami Y. Elastin-derived peptides and neutrophil elastase in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1996;154(3 Pt 1):720–724. doi:10.1164/ajrccm.154.3.8810611. | ||
Laurell CB, Eriksson S. Hypo-alpha-1-antitrypsinemia. Verh Dtsch Ges Inn Med. 1964;70:537–539. | ||
Molet S, Belleguic C, Lena H, et al. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res. 2005;54(1):31–36. doi:10.1007/s00011-004-1319-4. | ||
Churg A, Wang RD, Tai H, et al. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am J Respir Crit Care Med. 2003;167(8):1083–1089. doi:10.1164/rccm.200212-1396OC. | ||
Reilly JJJ, Chen P, Sailor LZ, Wilcox D, Mason RW, Chapman HAJ. Cigarette smoking induces an elastolytic cysteine proteinase in macrophages distinct from cathepsin L. Am J Physiol. 1991;261(2 Pt 1):L41–L48. | ||
Shi GP, Munger JS, Meara JP, Rich DH, Chapman HA. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J Biol Chem. 1992;267(11):7258–7262. | ||
Atkinson JJ, Lutey BA, Suzuki Y, et al. The role of matrix metalloproteinase-9 in cigarette smoke-induced emphysema. Am J Respir Crit Care Med. 2011;183(7):876–884. doi:10.1164/rccm.201005-0718OC. | ||
Abboud RT, Vimalanathan S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int J Tuberc Lung Dis. 2008;12(4):361–367. | ||
Azghani AO, Neal K, Idell S, et al. Mechanism of fibroblast inflammatory responses to Pseudomonas aeruginosa elastase. Microbiology. 2014;160(Pt 3):547–555. doi:10.1099/mic.0.075325-0. | ||
Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–2004. | ||
Hunninghake GW, Davidson JM, Rennard S, Szapiel S, Gadek JE, Crystal RG. Elastin fragments attract macrophage precursors to diseased sites in pulmonary emphysema. Science. 1981;212(4497):925–927. | ||
Adair-Kirk TL, Atkinson JJ, Broekelmann TJ, et al. A site on laminin alpha 5, AQARSAASKVKVSMKF, induces inflammatory cell production of matrix metalloproteinase-9 and chemotaxis. J Immunol. 2003;171(1):398–406. | ||
Hobbs BD, Hersh CP. Integrative genomics of chronic obstructive pulmonary disease. Biochem Biophys Res Commun. 2014;452(2):276–286. doi:10.1016/j.bbrc.2014.07.086. | ||
Castaldi PJ, Cho MH, Zhou X, et al. Genetic control of gene expression at novel and established chronic obstructive pulmonary disease loci. Hum Mol Genet. 2015;24(4):1200–1210. doi:10.1093/hmg/ddu525. | ||
Yu XL, Zhang J, Zhao F, Pan XM. Relationships of COX2 and MMP12 genetic polymorphisms with chronic obstructive pulmonary disease risk: a meta-analysis. Mol Biol Rep. 2014;41(12):8149–8162. doi:10.1007/s11033-014-3715-3. | ||
Zhou DC, Zhou CF, Toloo S, Shen T, Tong SL, Zhu QX. Association of a disintegrin and metalloprotease 33 (ADAM33) gene polymorphisms with the risk of COPD: an updated meta-analysis of 2,644 cases and 4,804 controls. Mol Biol Rep. 2015;42(2):409–422. doi:10.1007/s11033-014-3782-5. | ||
Xie Z, Huang Q, Huang J, Xie Z. Association between the IL1B, IL1RN polymorphisms and COPD risk: a meta-analysis. Sci Rep. 2014;4:6202. doi:10.1038/srep06202. | ||
Guo Y, Gong Y, Pan C, et al. Association of genetic polymorphisms with chronic obstructive pulmonary disease in the Chinese Han population: a case-control study. BMC Med Genomics. 2012;5:64. doi:10.1186/1755-8794-5-64. | ||
Toru Ü, Ayada C, Genç O, Turgut S, Turgut G, Bulut I. MDR-1 gene C/T polymorphism in COPD: data from Aegean part of Turkey. Int J Clin Exp Med. 2014;7(10):3573–3577. | ||
Lamontagne M, Timens W, Hao K, et al. Genetic regulation of gene expression in the lung identifies CST3 and CD22 as potential causal genes for airflow obstruction. Thorax. 2014;69(11):997–1004. doi:10.1136/thoraxjnl-2014-205630. | ||
Zeskind JE, Lenburg ME, Spira A. Translating the COPD transcriptome: insights into pathogenesis and tools for clinical management. Proc Am Thorac Soc. 2008;5(15):834–841. doi:10.1513/pats.200807-074TH. | ||
Ezzie ME, Crawford M, Cho J-H, et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2012;67:122–131. doi:10.1136/thoraxjnl-2011-200089. | ||
Qiu W, Baccarelli A, Carey VJ, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med. 2012;185:373–381. doi:10.1164/rccm.201108-1382OC. | ||
Vucic EA, Chari R, Thu KL, et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am J Respir Cell Mol Biol. 2014;50(5):912–922. doi:10.1165/rcmb.2013-0304OC. | ||
Segal LN, Blaser MJ. A brave new world: the lung microbiota in an era of change. Ann Am Thorac Soc. 2014;11(Suppl 1):S21–S27. doi:10.1513/AnnalsATS.201306-189MG. | ||
Sze MA, Dimitriu PA, Hayashi S, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(19):1073–1080. doi:10.1164/rccm.201111-2075OC. | ||
Chambers DC, Gellatly SL, Hugenholtz P, Hansbro PM. JTD special edition “Hot topics in COPD” – the microbiome in COPD. J Thorac Dis. 2014;6(11):1525–1531. doi:10.3978/j.issn.2072-1439.2014.11.08. | ||
Lemon KP, Klepac-Ceraj V, Schiffer HK, Brodie EL, Lynch SV, Kolter R. Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. MBio 2010;1(3):e00129-10. doi:10.1128/mBio.00129-10. | ||
Morris A, Beck JM, Schloss PD, et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187(10):1067–1075. doi:10.1164/rccm.201210-1913OC. | ||
Ege MJ, Mayer M, Normand A-C, et al. Exposure to environmental microorganisms and childhood asthma. N Engl J Med. 2011;364(8):701–709. doi:10.1056/NEJMoa1007302. | ||
Abrahamsson TR, Jakobsson HE, Andersson AF, Björkstén B, Engstrand L, Jenmalm MC. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin Exp Allergy. 2014;44(6):842–850. doi:10.1111/cea.12253. | ||
Charlson ES, Bittinger K, Haas AR, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. doi:10.1164/rccm.201104-0655OC. | ||
Dickson RP, Erb-Downward JR, Huffnagle GB. Towards an ecology of the lung: new conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir Med. 2014;2(3):238–246. doi:10.1016/S2213-2600(14)70028-1. | ||
Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One. 2012;7(10):e47305. | ||
Zakharkina T, Heinzel E, Koczulla RA, et al. Analysis of the airway microbiota of healthy individuals and patients with chronic obstructive pulmonary disease by t-rflp and clone sequencing. PLoS One. 2013;8(7). doi:10.1371/journal.pone.0068302. | ||
Millares L, Ferrari R, Gallego M, et al. Bronchial microbiome of severe COPD patients colonised by Pseudomonas aeruginosa. Eur J Clin Microbiol Infect Dis. 2014;33:1101–1111. doi:10.1007/s10096-013-2044-0. | ||
Huang YJ, Kim E, Cox MJ, et al. A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS. 2010;14(1):9–59. doi:10.1089/omi.2009.0100. | ||
Yawn BP, Li Y, Tian H, Zhang J, Arcona S, Kahler KH. Inhaled corticosteroid use in patients with chronic obstructive pulmonary disease and the risk of pneumonia: a retrospective claims data analysis. Int J Chron Obstruct Pulmon Dis. 2013;8:295–304. doi:10.2147/COPD.S42366. | ||
Welte T. Inhaled corticosteroids in COPD and the risk of pneumonia. Lancet. 2009;374:668–670. doi:10.1016/S0140-6736(09)61540-3. | ||
Calverley PMA, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356(8):775–789. doi:10.1056/NEJMoa063070. | ||
Thornton Snider J, Luna Y, Wong KS, et al. Inhaled corticosteroids and the risk of pneumonia in medicare patients with COPD. Curr Med Res Opin. 2012;28(12):1959–1967. doi:10.1185/03007995.2012.743459. | ||
Huang YJ, Sethi S, Murphy T, Nariya S, Boushey HA, Lynch SV. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52(8):2813–2823. doi:10.1128/JCM.00035-14. | ||
Garcia-Nunez M, Millares L, Pomares X, et al. Severity-related changes of bronchial microbiome in chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52(12):4217–4223. doi:10.1128/JCM.01967-14. | ||
Stokell JR, Gharaibeh RZ, Hamp TJ, Zapata MJ, Fodor AA, Steck R. Analysis of changes in diversity and abundance of the microbial community in a cystic fibrosis patient over a multiyear period. 2015;53(1):237–247. doi:10.1128/JCM.02555-14. | ||
Perez-Cobas AE, Gosalbes MJ, Friedrichs A, et al. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut. 2013;62(11):1591–1601. doi:10.1136/gutjnl-2012-303184. | ||
Wu S, Jiang ZY, Sun YF, et al. Microbiota regulates the TLR7 signaling pathway against respiratory tract influenza a virus infection. Curr Microbiol. 2013;67:414–422. doi:10.1007/s00284-013-0380-z. | ||
Russell SL, Gold MJ, Hartmann M, et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012;13(5):440–447. doi:10.1038/embor.2012.32. | ||
Flöistrup H, Swartz J, Bergström A, et al. Allergic disease and sensitization in Steiner school children. J Allergy Clin Immunol. 2006;117:59–66. doi:10.1016/j.jaci.2005.09.039. | ||
Marra F, Lynd L, Coombes M, et al. Does antibiotic exposure during infancy lead to development of asthma? A systematic review and metaanalysis. Chest. 2006;129:610–618. doi:10.1378/chest.129.3.610. | ||
Ong M-S, Umetsu DT, Mandl KD. Consequences of antibiotics and infections in infancy: bugs, drugs, and wheezing. Ann Allergy Asthma Immunol. 2014;112(5):441–445.e1. doi:10.1016/j.anai.2014.01.022. | ||
Örtqvist AK, Lundholm C, Kieler H, et al. Antibiotics in fetal and early life and subsequent childhood asthma: nationwide population based study with sibling analysis. BMJ. 2014;349:g6979. doi:10.1136/bmj.g6979. | ||
Ekbom A, Brandt L, Granath F, Löfdahl C-G, Egesten A. Increased risk of both ulcerative colitis and Crohn’s disease in a population suffering from COPD. Lung. 2008;186:167–172. doi:10.1007/s00408-008-9080-z. | ||
Keely S, Talley NJ, Hansbro PM. Pulmonary-intestinal cross-talk in mucosal inflammatory disease. Mucosal Immunol. 2012;5(1):7–18. doi:10.1038/mi.2011.55. | ||
Biedermann L, Zeitz J, Mwinyi J, et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS One. 2013;8(3). doi:10.1371/journal.pone.0059260. | ||
Marsland BJ, Gollwitzer ES. Host – microorganism interactions in lung diseases. Nat Publ Gr. 2014;14(12):827–835. doi:10.1038/nri3769. | ||
Kennedy MJ, Volz PA. Ecology of Candida albicans gut colonization: inhibition of Candida adhesion, colonization, and dissemination from the gastrointestinal tract by bacterial antagonism. Infect Immun. 1985;49(3):654–663. | ||
Abeles SR, Robles-Sikisaka R, Ly M, et al. Human oral viruses are personal, persistent and gender-consistent. ISME J. 2014;8(9): 1753–1767. | ||
Wylie KM, Weinstock GM, Storch GA. Emerging view of the human virome. Transl Res. 2012;160(4):283–290. doi:10.1016/j.trsl.2012.03.006. | ||
Breitbart M, Rohwer F. Method for discovering novel DNA viruses in blood using viral particle selection and shotgun sequencing. Biotechniques. 2005;39(5):729–736. | ||
Retamales I, Elliott WM, Meshi B, et al. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med. 2001;164(3):469–473. doi:10.1164/ajrccm.164.3.2007149. | ||
Meshi B, Vitalis TZ, Ionescu D, et al. Emphysematous lung destruction by cigarette smoke the effects of latent adenoviral infection on the lung inflammatory response. Am J Respir Cell Mol Biol. 2002;26:52–57. doi:10.1165/ajrcmb.26.1.4253. | ||
Ishida T, Hirono Y, Yoshikawa K, et al. Inhibition of immunological function mediated DNA damage of alveolar macrophages caused by cigarette smoke in mice. Inhal Toxicol. 2009;21(14):1229–1235. doi:10.3109/08958370903176727. | ||
Ojo O, Lagan AL, Rajendran V, et al. Pathological changes in the COPD lung mesenchyme – novel lessons learned from in vitro and in vivo studies. Pulm Pharmacol Ther. 2014;29(2):121–128. doi:10.1016/j.pupt.2014.04.004. | ||
Ege MJ, Mayer M, Normand AC, et al. Exposure to environmental microorganisms and childhood asthma. N Engl J Med. 2011;364(8):701–709. | ||
Enelow RI, Mohammed AZ, Stoler MH, et al. Structural and functional consequences of alveolar cell recognition by CD8(+) T lymphocytes in experimental lung disease. J Clin Invest. 1998;102:1653–1661. doi:10.1172/JCI4174. | ||
Wells MA, Albrecht P, Ennis FA. Recovery from a viral respiratory infection. I. Influenza pneumonia in normal and T-deficient mice. J Immunol. 1981;126(3):1036–1041. | ||
Packard TA, Li QZ, Cosgrove GP, Bowler RP, Cambier JC. COPD is associated with production of autoantibodies to a broad spectrum of self-antigens, correlative with disease phenotype. Immunol Res. 2013;55(1–3):48–57. doi:10.1007/s12026-012-8347-x. | ||
Bergin DA, Reeves EP, Hurley K, et al. The circulating proteinase inhibitor alpha-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci Transl Med. 2014;6(217):217ra1. doi:10.1126/scitranslmed.3007116. | ||
Masala C, Amendolea M, Bonini S. Mucus antibodies in pulmonary tuberculosis and chronic obstructive lung disease. Lancet. 1976;308(7990):821–824. doi:10.1016/S0140-6736(76)91208-3. | ||
Feghali-Bostwick CA, Gadgil AS, Otterbein LE, et al. Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:156–163. doi:10.1164/rccm.200701-014OC. | ||
Taraseviciene-Stewart L, Scerbavicius R, Choe KH, et al. An animal model of autoimmune emphysema. Am J Respir Crit Care Med. 2005;171:734–742. doi:10.1164/rccm.200409-1275OC. | ||
Karayama M, Inui N, Suda T, Nakamura Y, Nakamura H, Chida K. Antiendothelial cell antibodies in patients with COPD. Chest. 2010;138:1303–1308. doi:10.1378/chest.10-0863. | ||
Bergin DA, Hurley K, Reeves EP, McElvaney NG. Novel neutrophil derived autoantibodies in alpha-1 antitrypsin deficiency. In: B39. Neutrophils: new insights into their activation and contribution to lung injury. American Thoracic Society International Conference Abstracts. American Thoracic Society; 2013:A2740. doi:10.1164/ajrccm-conference.2013.187.1_MeetingAbstracts.A2740. | ||
Kuo Y-B, Chang CA, Wu Y-K, et al. Identification and clinical association of anti-cytokeratin 18 autoantibody in COPD. Immunol Lett. 2010;128(2):131–136. doi:10.1016/j.imlet.2009.12.017. | ||
Lee S-H, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13(5):567–569. doi:10.1038/nm1583. | ||
Rinaldi M, Lehouck A, Heulens N, et al. Antielastin B-cell and T-cell immunity in patients with chronic obstructive pulmonary disease. Thorax. 2012;67(8):694–700. doi:10.1136/thoraxjnl-2011-200690. | ||
Kirkham Pa, Caramori G, Casolari P, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184:796–802. doi:10.1164/rccm.201010-1605OC. | ||
Sigari N, Moghimi N, Shahraki FS, Mohammadi S, Roshani D. Anti-cyclic citrullinated peptide (CCP) antibody in patients with wood-smoke-induced chronic obstructive pulmonary disease (COPD) without rheumatoid arthritis. Rheumatol Int. 2015;35(1):85–91. doi:10.1007/s00296-014-3083-2. | ||
Newkirk MM, Mitchell S, Procino M, et al. Chronic smoke exposure induces rheumatoid factor and anti-heat shock protein 70 autoantibodies in susceptible mice and humans with lung disease. Eur J Immunol. 2012;42:1051–1061. doi:10.1002/eji.201141856. | ||
Nunez B, Sauleda J, Anto JM, et al. Anti-tissue antibodies are related to lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(8):1025–1031. doi:10.1164/rccm.201001-0029OC. | ||
Daffa NI, Tighe PJ, Corne JM, Fairclough LC, Todd I. Natural and disease-specific autoantibodies in chronic obstructive pulmonary disease. Clin Exp Immunol. 2014. doi:10.1111/cei.12565. | ||
Litsiou E, Semitekolou M, Galani IE, et al. CXCL13 production in B cells via Toll-like receptor/lymphotoxin receptor signaling is involved in lymphoid neogenesis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187(11):1194–1202. doi:10.1164/rccm.201208-1543OC. | ||
Getts DR, Chastain EML, Terry RL, Miller SD. Virus infection, antiviral immunity, and autoimmunity. Immunol Rev. 2013;255(1):197–209. doi:10.1111/imr.12091. | ||
Coppieters KT, Boettler T, von Herrath M. Virus infections in type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2(1):a007682. doi:10.1101/cshperspect.a007682. | ||
Kaza AK, Kron IL, Leuwerke SM, Tribble CG, Laubach VE. Keratinocyte growth factor enhances post-pneumonectomy lung growth by alveolar proliferation. Circulation. 2002;106(12 Suppl 1):I120–I124. | ||
Zhang Q, Bellotto DJ, Ravikumar P, et al. Postpneumonectomy lung expansion elicits hypoxia-inducible factor-1alpha signaling. Am J Physiol Lung Cell Mol Physiol. 2007;293(2):L497–L504. doi:10.1152/ajplung.00393.2006. | ||
Sakurai MK, Lee S, Arsenault DA, et al. Vascular endothelial growth factor accelerates compensatory lung growth after unilateral pneumonectomy. Am J Physiol Lung Cell Mol Physiol. 2007;292(3):L742–L747. doi:10.1152/ajplung.00064.2006. | ||
Takahashi Y, Izumi Y, Kohno M, et al. Thyroid transcription factor-1 influences the early phase of compensatory lung growth in adult mice. Am J Respir Crit Care Med. 2010;181(12):1397–1406. doi:10.1164/rccm.200908-1265OC. | ||
Nowrin K, Sohal SS, Peterson G, Patel R, Walters EH. Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: fibrosis, remodeling and cancer. Expert Rev Respir Med. 2014;8(5):547–559. doi:10.1586/17476348.2014.948853. | ||
Sohal SS, Reid D, Soltani A, et al. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology. 2010;15(6):930–938. doi:10.1111/j.1440-1843.2010.01808.x. | ||
Barkauskas CE, Cronce MJ, Rackley CR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123(7):3025–3036. doi:10.1172/JCI68782DS1. | ||
Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. 2007;179(3):1855–1863. | ||
Lee JW, Fang X, Gupta N, Serikov V, Matthay MA. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci U S A. 2009;106(38):16357–16362. doi:10.1073/pnas.0907996106. | ||
Bonfield TL, Koloze M, Lennon DP, Zuchowski B, Yang SE, Caplan AI. Human mesenchymal stem cells suppress chronic airway inflammation in the murine ovalbumin asthma model. Am J Physiol Lung Cell Mol Physiol. 2010;299(6):L760–L770. doi:10.1152/ajplung.00182.2009. | ||
Sueblinvong V, Loi R, Eisenhauer PL, et al. Derivation of lung epithelium from human cord blood-derived mesenchymal stem cells. Am J Respir Crit Care Med. 2008;177(7):701–711. doi:10.1164/rccm.200706-859OC. | ||
Spees JL, Pociask DA, Sullivan DE, et al. Engraftment of bone marrow progenitor cells in a rat model of asbestos-induced pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(4):385–394. doi:10.1164/rccm.200607-1004OC. | ||
Ishizawa K, Kubo H, Yamada M, et al. Bone marrow-derived cells contribute to lung regeneration after elastase-induced pulmonary emphysema. FEBS Lett. 2004;556(1–3):249–252. | ||
Westbury CB, Yarnold JR. Radiation fibrosis – current clinical and therapeutic perspectives. Clin Oncol. (R Coll Radiol). 2012;24(10):657–672. doi:10.1016/j.clon.2012.04.001. | ||
Toonkel RL, Hare JM, Matthay MA, Glassberg MK. Mesenchymal stem cells and idiopathic pulmonary fibrosis potential for clinical testing. Am J Respir Crit Care Med. 2013;188(8):133–140. doi:10.1164/rccm.201207-1204PP. | ||
Choi M, Ban T, Rhim T. Therapeutic use of stem cell transplantation for cell replacement or cytoprotective effect of microvesicle released from mesenchymal stem cell. Mol Cells. 2014;37(2):133–139. doi:10.14348/molcells.2014.2317. | ||
Li Y, Gu C, Xu W, et al. Therapeutic effects of amniotic fluid-derived mesenchymal stromal cells on lung injury in rats with emphysema. Respir Res. 2014;15:120. doi:10.1186/s12931-014-0120-3. | ||
Van Haaften T, Byrne R, Bonnet S, et al. Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am J Respir Crit Care Med. 2009;180(11):1131–1142. doi:10.1164/rccm.200902-0179OC. | ||
Zhang H, Fang J, Wu Y, Mai Y, Lai W, Su H. Mesenchymal stem cells protect against neonatal rat hyperoxic lung injury. Expert Opin Biol Ther. 2013;13(6):817–829. doi:10.1517/14712598.2013.778969. | ||
Rejman J, Colombo C, Conese M. Engraftment of bone marrow-derived stem cells to the lung in a model of acute respiratory infection by Pseudomonas aeruginosa. Mol Ther J Am Soc Gene Ther. 2009;17(7):1257–1265. doi:10.1038/mt.2009.96. | ||
Chang YS, Oh W, Choi SJ, et al. Human umbilical cord blood-derived mesenchymal stem cells attenuate hyperoxia-induced lung injury in neonatal rats. Cell Transplant. 2009;18(8):869–886. doi:10.3727/096368909X471189. | ||
Kaiser Permanente. Physical activity associated with lower rates of hospital readmission in patients with COPD. ScienceDaily. 2014. Available from: www.sciencedaily.com/releases/2014/04/140409204425.htm. Accessed January 1, 2015. | ||
Shaheen SO. Antioxidants and respiratory disease: the uric acid paradox. Thorax. 2014;69 (11):978–979. doi:10.1136/thoraxjnl-2014-205751. | ||
Ezzati M, Horwitz MEM, Thomas DSK, et al. Altitude, life expectancy and mortality from ischaemic heart disease, stroke, COPD and cancers: national population-based analysis of US counties. J Epidemiol Community Health. 2012;66(7):e17. doi:10.1136/jech.2010.112938. | ||
Gross P, Pfitzer EA, Tolker E, Babyak MA, Kashak M. Experimental emphysema: its production with papain in normal and silicotic rats. Arch Environ Health. 1965;11:50–58. | ||
Vidal D, Fortunato G, Klein W, et al. Alterations in pulmonary structure by elastase administration in a model of emphysema in mice is associated with functional disturbances. Rev Port Pneumol. 2012;18(3):128–136. doi:10.1016/j.rppneu.2011.12.007. | ||
Holroyd KJ, Eleff SM, Zhang LY, Jakab GJ, Kleeberger SR. Genetic modeling of susceptibility to nitrogen dioxide-induced lung injury in mice. Am J Physiol. 1997;273(3 Pt 1):L595–602. | ||
Wright JL, Postma DS, Kerstjens HAM, Timens W, Whittaker P, Churg A. Airway remodeling in the smoke exposed guinea pig model. Inhal Toxicol. 2007;19(11):915–923. doi:10.1080/08958370701515563. | ||
Mahadeva R, Shapiro SD. Chronic obstructive pulmonary disease * 3: experimental animal models of pulmonary emphysema. Thorax. 2002;57(10):908–914. | ||
Sakagami M. In vivo, in vitro and ex vivo models to assess pulmonary absorption and disposition of inhaled therapeutics for systemic delivery. Adv Drug Deliv Rev. 2006;58(9–10):1030–1060. doi:10.1016/j.addr.2006.07.012. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.