Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
Novel Anti-Inflammatory Approaches to COPD
Authors Cazzola M ![]() , Hanania NA, Page CP, Matera MG
, Hanania NA, Page CP, Matera MG
Received 27 April 2023
Accepted for publication 20 June 2023
Published 29 June 2023 Volume 2023:18 Pages 1333—1352
DOI https://doi.org/10.2147/COPD.S419056
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jill Ohar
Mario Cazzola,1 Nicola A Hanania,2 Clive P Page,3 Maria Gabriella Matera4
1Department of Experimental Medicine, University of Rome “Tor Vergata”, Rome, Italy; 2Section of Pulmonary and Critical Care Medicine, Baylor College of Medicine, Houston, TX, USA; 3Sackler Institute of Pulmonary Pharmacology, Institute of Pharmaceutical Science, King’s College London, London, UK; 4Department of Experimental Medicine, University of Campania “Luigi Vanvitelli”, Naples, Italy
Correspondence: Mario Cazzola, Department of Experimental Medicine, University of Rome “Tor Vergata”, Rome, Italy, Email [email protected]
Abstract: Airway inflammation, driven by different types of inflammatory cells and mediators, plays a fundamental role in COPD and its progression. Neutrophils, eosinophils, macrophages, and CD4+ and CD8+ T lymphocytes are key players in this process, although the extent of their participation varies according to the patient’s endotype. Anti-inflammatory medications may modify the natural history and progression of COPD. However, since airway inflammation in COPD is relatively resistant to corticosteroid therapy, innovative pharmacological anti-inflammatory approaches are required. The heterogeneity of inflammatory cells and mediators in annethe different COPD endo-phenotypes requires the development of specific pharmacologic agents. Indeed, over the past two decades, several mechanisms that influence the influx and/or activity of inflammatory cells in the airways and lung parenchyma have been identified. Several of these molecules have been tested in vitro models and in vivo in laboratory animals, but only a few have been studied in humans. Although early studies have not been encouraging, useful information emerged suggesting that some of these agents may need to be further tested in specific subgroups of patients, hopefully leading to a more personalized approach to treating COPD.
Keywords: anti-inflammatory drugs, COPD, inflammation, treatment
The Unmet Need for New Anti-Inflammatory Therapies for COPD
An aberrant inflammatory response that involves both innate and adaptive immunity characterizes COPD.1 However, determining the precise role of specific cell types in the evolution of COPD is challenging.2 While chronic inflammation is basically characterised by neutrophils and macrophages,3 20–40% of patients exhibit increased eosinophil numbers in blood and lung tissue.4 Furthermore, neutrophilic inflammation is variably associated with concomitant eosinophilic inflammation in many patients.5 T cells, B cells, dendritic cells (DCs) and epithelial cells are also involved in COPD.5
Corticosteroids (CSs) are the most often utilized anti-inflammatory medications in the treatment of COPD.6 ICSs are indicated in COPD patients when there is an overlap with asthma or frequent exacerbations associated with high blood eosinophil counts (BEC).7 Furthermore, CSs may play a cardioprotective role, reduce the risk of lung cancer, and possibly improve survival.7 However, ICSs have little to no impact on the underlying inflammation in most COPD patients,8 particularly in those who continue to smoke.9
The transcription of some anti-inflammatory genes is controlled by histone deacetylase (HDAC)2, which has been found to be downregulated by oxidative stress generated by cigarette smoke and activated neutrophils.8,9 Another factor is the reduced glucocorticoid receptor (GR) expression in airway neutrophils, but not blood neutrophils, which has been found to be associated with the poor clinical response to ICS therapy in COPD patients.10 Moreover, neutrophils from patients with COPD also showed an increase in GRβ levels,11 which is thought to be a factor in the development of corticosteroid resistance because it is capable of inhibiting HDAC2 promoter activity.12 Furthermore, several studies have revealed a connection between long-term ICS treatment and the incidence of pneumonia in COPD patients, mainly in those with lower BECs.13
Since CSs are less effective in neutrophil-driven pulmonary inflammation, the demand for therapies that act on this type of inflammation is increasing.6,14
Over the past two decades, research has focused on discovering new targets capable of inhibiting the recruitment or activation of inflammatory cells involved in COPD and the development of drugs capable of blocking the inflammatory mediators released by these cells.6 A complex network of inflammatory mediators, including chemokines, growth factors and lipid mediators, produced by the structural and inflammatory cells in the lung, are implicated in COPD and play a potential role in its pathogenesis.15,16 Therefore, their inhibition is an important strategy to tamper the ongoing inflammatory process. Unfortunately, the results have often fallen short of expectations.
This narrative review aims to examine ongoing research to identify and develop possible new anti-inflammatory therapies for use in patients with COPD.
Search Strategy
A review of the literature published up to May 2023 was performed through the PubMed and Scopus databases to identify studies related to our stated objective of reviewing novel drugs under development for treating COPD. Thereafter, evaluation of the references of the selected articles identified other relevant publications. All authors participated in evaluation of the literature.
Inhibition of Recruitment and Activation of the Inflammatory Cells
Several small molecule inhibitors and biologic therapies are able, at least in experimental models, to prevent the recruitment and activation of the cellular components of inflammation.6 Some of these have now been tested in COPD (Table 1). The development of other classes of drugs acting on targets that were considered potentially useful17 was later abandoned because when tested on humans they proved to be poorly effective and/or induced major adverse effects. This is the case with the adenosine A2a receptor agonists (regadenoson and UK432.097) due to unsatisfactory results and unwanted effects on the cardiovascular system,18,19 anti-CXCL8 (ABX-CXCL) mAbs, which only slightly improved dyspnoea in COPD patients20 probably because such drugs only block free CXCL8 and not the bound one, TNF-α inhibitors (infliximab and etanercept) that had no beneficial effects in patients with mild, moderate, or severe COPD21,22 or for the treatment of acute exacerbations of COPD (AECOPDs),23 likely because other proinflammatory cytokines drive the inflammatory process24 and, furthermore, COPD patients had a significantly increased incidence of airway tumours and lung infections caused by TNF-α antibodies,22 and anti-IL-1β mAbs (canakinumab and MEDI8986) because a lack of efficacy at least with regard to impact on the risk of AECOPDs, lung function, and HRQoL.25
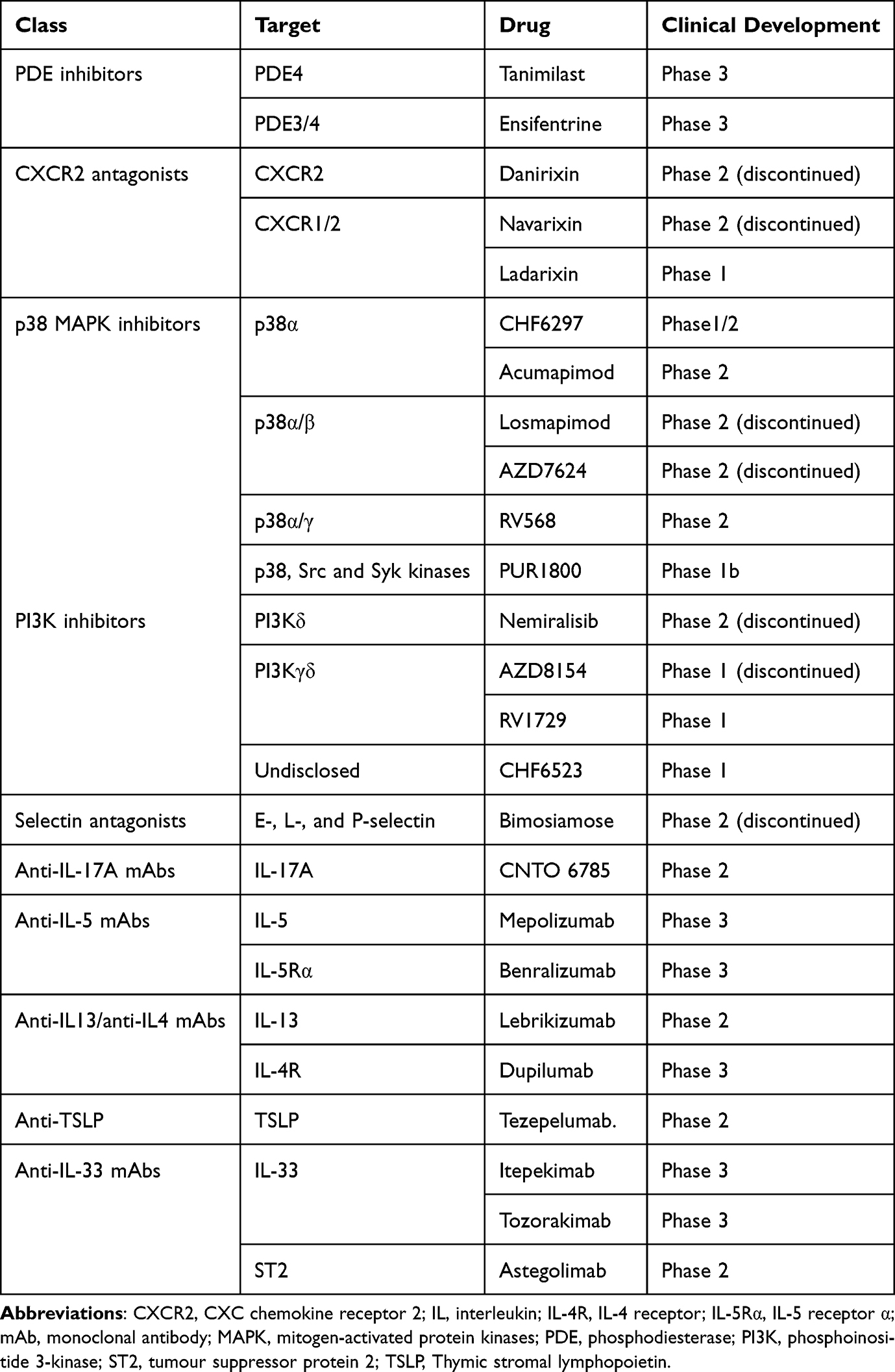 |
Table 1 Therapies That Inhibit Recruitment and Activation of the Cellular Components of Inflammation in COPD |
Small Molecule Inhibitors
The main classes of small molecule inhibitors investigated for COPD include PDE inhibitors, CXCR2 antagonists, p38 MAPK inhibitors, PI3K inhibitors, and selectin antagonists.
Phosphodiesterase Inhibitors
There are 21 PDE genes in the human genome, which are organized into PDE families (PDE1 to PDE11), with many subtypes in each family.26 PDEs catalyse the hydrolysis of cAMP and cGMP, thus controlling the intracellular levels of these cyclic nucleotides, their signalling pathways, and, ultimately, their biological responses.26 Almost all cell types implicated in the pathophysiology of COPD are modulated by cAMP, which also controls the tone of the airway smooth muscle via the β2-adrenoceptor (β2-AR)-soluble adenylyl cyclase (sAC)-cAMP signalling pathway.27 cGMP promotes vascular smooth muscle relaxation via the NO-soluble guanylyl cyclase-cGMP pathway28 and is also implicated in eliciting bronchodilation in human small airways via the β2-AR-sAC-cAMP pathway.29 PDE4, PDE7, and PDE8 are cAMP-specific PDEs, while PDE5, PDE6, and PDE9 are cGMP-specific PDEs, and PDE1, PDE2, PDE3, PDE10, and PDE11 hydrolyse both cAMP and cGMP.30
PDE4 inhibitors are likely the most studied because PDE4 is the predominant PDE expressed in T-cells, eosinophils, neutrophils, monocytes, and macrophages, and therefore its inhibition may also have inhibitory effects upon both inflammatory and immune cells.30 Nevertheless, only one drug in this class, roflumilast N-oxide, has been approved for treating severe COPD.31 However, roflumilast is not widely used because of a narrow therapeutic window with a range of dose-limiting side effects in the gastrointestinal tract and the central nervous system.32 The addition of roflumilast is only recommended for patients who continue to exacerbate despite triple therapy with LABA + LAMA + ICS therapy or for those with a BEC of <100 cells/μL, especially if they have chronic bronchitis and a FEV1 <50%.33
Therefore, other PDE4 inhibitors have been developed to be administered by the inhaled route to potentially reduce these adverse effects.34 Unfortunately, the development of most of the inhaled PDE4 inhibitors has been discontinued due to modest effects in patients with COPD.35 This lack of beneficial effects may be because inhaled drugs are generally designed to be retained in the lung and have no effect on systemic inflammation.36 However, adding the inhaled PDE4 inhibitor, tanimilast, to inhaled triple therapy reduced AECOPDs.37 Tanimilast exerts specific immunomodulatory effects linked to a T2 endotype and CD141 overexpression in DCs which suggests that it may have complementary anti-inflammatory effects to ICSs.38 Tanimilast is currently being evaluated in two Phase 3 studies that are investigating its efficacy and safety as an add-on to triple maintenance therapy in subjects with COPD and chronic bronchitis (NCT04636801 and NCT04636814).
PDE4 is divided into four subfamilies: PDE4A, PDE4B, PDE4C, and PDE4D. The genes in each subfamily can express between 3 and 11 proteins, resulting in at least 25 distinct PDE4 protein isoforms.39 PDE4 isoforms are essential spatiotemporal regulators of cAMP signalling due to their distinct intracellular distribution patterns, dynamic activity regulation, and isoform-specific regulation.40 PDE4 isoform selective inhibitors do not yet exist. Therefore, it will be important to create novel isoform specific PDE4 inhibitors also because this approach may help to reduce side effects.39 However, due to the structural similarities and highly conserved sequences of the isoforms, designing PDE4 isoform selective inhibitors is difficult.30
Agents capable of simultaneously inhibiting PDE3 and PDE4 are also under development.41 This approach induces additional or synergistic anti-inflammatory and bronchodilator benefits compared to PDE3 or PDE4 inhibition alone42 (Figure 1). Furthermore, dual PDE3/4 inhibitors have been shown to improve mucociliary clearance.43 Ensifentrine, a first-in-class inhaled “bifunctional” dual PDE3/4 inhibitor, exhibits both bronchodilator and anti-inflammatory activities.43 The two Phase 3 ENHANCE (Ensifentrine as a Novel inHAled Nebulized COPD thErapy)-1 and −2 clinical trials, not yet published in full, demonstrated substantial improvements in lung function, a clinically significant reduction of approximately 40% in AECOPD rate following 24 weeks of treatment with favourable safety results, although the improvement in symptoms and quality of life reached statistical significance only in ENHANCE-1.44,45 The reduction in exacerbations was associated with a numerical reduction in circulating inflammatory biomarkers (IL-6, IL-8 and CRP).46 The fact that ensifentrine was able to reduce the frequency and risk of exacerbations in patients with eosinophils >150 cells/μL44 is fascinating because it suggests that in many patients, there would be no need to use ICSs. In ENHANCE-1, a higher percentage of patients were also under regular treatment for COPD (about 69% of subjects received LAMA or LABA and about 20% received ICS with concomitant LAMA or LABA), while in ENHANCE-2, this percentage was lower (about 55% of subjects received LAMA or LABA and about 15% received ICS with concomitant LAMA or LABA). Therefore, because few patients in these studies were on dual bronchodilation or triple therapy, it remains to be established what additional clinical benefit this drug brings to current standard of care.
 |
Figure 1 Effect of PDE inhibition in COPD. Combined inhibition of PDE3 and PDE4 provides additive and synergistic anti-inflammatory and bronchodilator effects when compared to PDE3 or PDE4 inhibition alone. It also improves mucociliary clearance. The primary PDE implicated in the activity of the given cell is shown in red. |
Dual acting PDE4/PDE5 inhibitors, PDE4/PDE7 inhibitors, and PDE4/PDE1 inhibitors are also being investigated. The creation of pan-PDE inhibitors that can block a variety of PDE isoforms is another novel pharmacological approach being pursued.41 An intriguing alternative is to create hybrid compounds with two or more pharmacophores, such as dual PDE4 inhibitors/muscarinic antagonists and dual PDE4 inhibitors/β2-agonists.41
CXCR2 Antagonists
Due to their role in leukocyte chemotaxis, chemokines have long been thought to be involved in the onset and amplification of inflammatory reactions. One of the chemokine receptors, CXCR2, is expressed on a range of cell types and organs, which raises the possibility that these receptors have a wide functional involvement in the pathophysiology of COPD.47 CXCR2 antagonists decreased neutrophil infiltration and tissue damage in the airways in pre-clinical models of cigarette smoke-induced acute neutrophilic inflammation in the lungs.48 Some neutrophil CXCR2 antagonists have been investigated, but this drug class has only shown modest effects in patients with COPD, possibly due to redundancy in the chemokine network.6 Navarixin, a dual CXCR1 and CXCR2 inhibitor, reduced inflammation and delayed the onset of the first exacerbation but also decreased the absolute neutrophil count, leading to the withdrawal of 18% of patients.49 Danirixin, a selective CXCR2 antagonist, improved symptoms but its long-term administration caused a high incidence of AECOPDs and pneumonia, suggesting an adverse effect on host responses to infection.50 Furthermore, in a small pilot study, there was no discernible difference between danirixin and placebo in terms of the % change from baseline in NETs, as determined by the histone-elastase immunoassay.51 Ladarixin is another dual CXCR1 and CXCR2 antagonist that has been tested in a Phase 1 clinical trial (NCT04854642). It improved lung function and reduced neutrophilic airway inflammation in a corticosteroid-resistant model of cigarette smoke-induced influenza-A infection exacerbation.52
p38 Mitogen-Activated Protein Kinase Inhibitors
MAPKs significantly influence chronic inflammation. The p38 MAPK subgroup consists of four isoforms (α, β, γ, δ),53 with p38α MAPK being suggested to have a significant impact on COPD.26 It is increased in bronchial epithelial cells, macrophages and CD20+ and CD8+ lymphocytes in COPD lungs.54 Several extracellular triggers cause the p38 MAPK pathway to become activated, leading to inflammatory gene transcription with increased cytokine and chemokine production, particularly interleukin 1β (IL-1β), CXCL8 (IL-8), and TNF-α, which are associated with the neutrophilic endotype of COPD26 (Figure 2). Therefore, inhibiting p38 MAPK may be an effective treatment for patients with COPD.6,53
 |
Figure 2 The role of p38 MAPK in the pathobiology of COPD. At the level of alveolar macrophages and other inflammatory cells, airborne pollutants, cigarette smoke, and microbial pathogens activate p38 MAPK. The p38 signaling pathway leads to increased cytokine and chemokine production, particularly interleukin (IL)-1β, IL-8, and tumor necrosis factor-α (TNF-α), which are associated with the neutrophilic endotype of COPD. Therefore, inhibiting p38 MAPK may be an effective treatment for patients with COPD. |
Some p38 MAPK inhibitors have been evaluated in patients with COPD. Losmapimod, an oral dual p38α/β inhibitor was not effective in reducing the moderate/severe COPD exacerbation rate in patients with blood eosinophils ≤2% and frequent exacerbations.54 AZD7624, another p38α/β inhibitor, reduced the production of inflammatory mediators by primary human bronchial epithelial cells stimulated with polyinosinic: polycytidylic acid and lipopolysaccharide (LPS)-stimulated bronchoalveolar lavage fluid (BALF) macrophages to a greater extent than budesonide.55 It was also more effective than budesonide in reducing IL-6 expression in bronchial epithelial cells, but without impacting AECOPD.56 The inhaled narrow spectrum (p38α/γ) inhibitor RV568 improved lung function in a small 14-day clinical trial in COPD patients.57 Acumapimod, a p38α inhibitor, improved FEV1 compared to placebo in severe AECOPD.58 It decreased the number of hospital readmissions due to COPD exacerbations.59 CHF6297, a selective p38α inhibitor, has been evaluated for safety, tolerability, and pharmacokinetics in healthy participants and for its anti-inflammatory effects in patients with COPD (NCT02815488). However, the results of these studies have not yet been made public. A systematic review with meta-analysis that included 10 RCTs in patients with COPD concluded that p38 MAPK inhibition was safe when compared with placebo, however this approach at best only caused a post-bronchodilator improvement in forced vital capacity.59
Oral administration of this class of drugs at safe doses is likely insufficient to induce significant clinical benefit.60 However, inhaled p38 MAPK inhibitors may lead to a higher local drug concentrations in the lung. An alternative approach could be the development of selective inhibitors of the α-δ subgroups. However, it is now recognized that the transforming growth factor-activated kinase-1 and mixed-lineage kinase are hyperactivated due to p38 MAPK inhibitors’ blocking of the upstream MAPK kinases, which in turn causes the c-Jun N-terminal kinase to become overactive.61 These observations suggest a need to develop drugs that target downstream substrates in this signalling cascade.
Use of narrow spectrum kinase inhibitors, a new class of pharmaceutical agents that simultaneously targets key kinases involved in both innate and adaptive immune cell signalling, such as p38 MAPK, spleen tyrosine kinase (Syk), and Src family kinases (SFK), which include Src and lymphocyte-specific protein tyrosine kinase, is an additional option.62 PUR1800 is a novel dry powder iSPERSE formulation of RV1162, a narrow spectrum kinase inhibitor, targeting p38 MAPK, Src and Syk kinases. In contrast to fluticasone propionate, it reduced TNF-α-induced cytokine release from primary bronchial epithelial cells of healthy volunteers and COPD patients.63 The preliminary pharmacokinetic data indicate that PUR1800 results in low and consistent systemic exposure in COPD patients when administered via oral inhalation.64
Phosphoinositide 3-Kinase Inhibitors
PI3K is an enzyme that catalyses the production of phosphatidylinositol-3,4,5-triphosphate and is involved in the activation of macrophages and neutrophils, as well as the hyperphosphorylation and ubiquitination of HDAC2, thus influencing sensitivity of inflammatory cells to corticosteroids.65 The function of this enzyme increases in patients with COPD.65 Increased migratory speed and decreased directionality toward the cytokine gradient were seen with neutrophils from patients with COPD, and these changes were reversed by PI3Kδ inhibition.66 Furthermore, PI3Kδ inhibition reduces pro-inflammatory cytokine secretion and improves survival rates following infection.67
In a proof-of-concept trial, adding nemiralisib, an inhaled PI3Kδ inhibitor, to usual care was associated with a more effective recovery from AECOPDs and improved lung function but also resulted in cough.68 However, in another trial, nemiralisib had no positive impact on lung function or relapses after an AECOPD.69 Furthermore, this drug altered neutrophil migratory behaviour in stable patients with COPD, but not in those with AECOPD, despite improving lung function in the latter group.70
CHF6523 is another inhaled PI3K inhibitor, currently in clinical development and has been investigated for safety and tolerability in patients with COPD.6 AZD8154, a PI3Kγδ inhibitor, has been tested in healthy volunteers,71 while RV1729, another PI3Kγδ inhibitor, is being investigated in patients with COPD (NCT02140346). GSK045, a selective PI3Kδ inhibitor, and ZSTK474, a pan PI3K inhibitor, are other potential drugs for inhibiting PI3K in COPD.6
Selectin Antagonists
The selectin family consists of three members (E-, L-, and P-selectin). While L-selectin is constitutively expressed on circulating leukocytes, E- and P-selectin are expressed on the endothelium.17 Selectins regulate inflammatory cell migration from the bloodstream into the lungs.17 E-selectin, which is up-regulated on endothelial cells in the airways of COPD patients,72 is required for the first attachment of neutrophilic granulocytes to endothelial cells in the airways.73 Therefore, targeting these molecules may provide another approach to reducing inflammation in the lungs of patients with COPD.
Inhaled bimosiamose, a synthetic pan-selectin antagonist, decreased airway inflammation and improved lung function in patients with COPD, confirming the critical function of selectins in the movement of inflammatory cells.74 Also, rivipansel, a pan-selectin antagonist, and uproleselan, a selective inhibitor of E-selectin, might be evaluated in the treatment of COPD.6
Developing inhibitors of selectin-ligand interactions in vivo is tricky because extremely potent inhibitors could have undesirable effects on healing processes, and weak inhibitors may not sufficiently act on the pathological processes of serious inflammatory diseases.75 It has been suggested that a clinically effective inhibitor is likely to inhibit at least two selectins.75
Biologic Therapies
Anti-IL-17A mAbs, collectins, anti-IL-5 mAbs, anti-IL13/IL4 mAbs, anti-TSLP mAbs, and anti-IL-33 mAbs are among the biological therapies that have the potential to treat COPD.24,76
Anti-IL-17A mAbs
Along with IL-22, IL-17A and IL-17F are linked to an increase in neutrophils in the airways, which results in chronic inflammation and increased mucus production and smooth muscle mass in the airways.76 To reduce neutrophil recruitment and airway inflammation, many anti-IL-17A and anti-IL-17 receptor A (IL-17RA) mAbs are being developed,77 but CNTO 6785 is the only anti-IL-17A mAb that has been investigated in patients with COPD. It caused a small improvement in FEV1 but did not interfere with the other primary or secondary endpoints.78 Furthermore, more AECOPDs appeared in the CNTO 6785 arm than in the placebo arm at week 16. Brodalumab, which is a mAb that targets IL-17RA, secukinumab, a fully human anti-IL-17A of the immunoglobulin (Ig)G1κ isotype, and CCJM112, a novel fully human anti-IL-17A IgG1κ mAb that binds with similar affinity to both human IL-17A and IL-17AF, have not yet been tested in patients with COPD.76
Collectins
SP-A and SP-D are termed collectins because they contain collagen and are functional lectins.79 By competing with LPS for CD14 binding on alveolar macrophages, SP-A and SP-D decrease TNF-α production.79 Furthermore, they modify inflammatory cytokine production following macrophage activation by cytokines or pathogen-associated molecular patterns.17 Pulmonary SP-D levels in COPD are lower than in smokers.80 Supplementation with recombinant forms of SP-A and SP-D might correct collectin deficiencies in inflammatory respiratory disorders. The trimeric proteins SP-A and SP-D are available as small recombinant forms of human fragments (rfhSP-A and rfhSP-D).78 AT-100 is an engineered version of the endogenous human protein rhSP-D. Local treatment of rfhSP-D reduces inflammation, ceramide production, and epithelial cell death caused by cigarette smoke in mice,81 but it has not yet been investigated in patients with COPD.
Targeting IL-5
As already mentioned, a subgroup of patients with COPD has elevated blood and airway eosinophils and elevated sputum IL-5 concentrations.82 There is a relationship between IL-5 levels and the quantity of eosinophils in the sputum of COPD patients.83 Moreover, there is proof that virus-induced AECOPD causes a rise in soluble IL-5Rα.84 Blocking IL-5 may likely prevent or decrease eosinophil-mediated inflammation due to the fast death of eosinophils in the absence of IL-5.76 Mepolizumab, a mAb that blocks IL-5, and benralizumab, an anti-IL-5Rα mAb, lower the rate of moderate and severe exacerbations in the highly selected population of individuals with COPD and higher levels of blood eosinophils85,86 (Figure 3). However, substantial eosinophil clearance from the airways in many COPD patients does not result in appreciable clinical improvement.87 This is probably because human lung-resident eosinophils are present independently of IL-5.88 Nonetheless, both mepolizumab (NCT04133909 and NCT04075331) and benralizumab (NCT04053634 and NCT04098718) are still being investigated in patients having COPD exacerbations and higher levels of blood eosinophils.
 |
Figure 3 Monoclonal antibodies targeting T2 cytokines in COPD. Abbreviations: ICL2, group 2 innate lymphoid cells; IL-4Rα, interleukin-4 receptor subunit α; IL-5Rα, interleukin-5 receptor subunit α; IL-13Rα, interleukin-13 receptor subunit α1. |
Targeting IL13/IL4
IL-4 and IL-13 are cytokines that promote eosinophilic recruitment, IgE production, mucus hypersecretion, and airway fibrosis and remodeling.89 They signal through a common receptor, IL-4Rα, expressed by airway epithelial cells76 (Figure 3). IL-4 and IL-13 can drive eosinophilic inflammation through the production of chemoattractants such CC-chemokine ligand 26 from airway epithelial cells.89 Patients with eosinophilic COPD and concomitant emphysema had significant levels of sputum matrix metalloprotease-12 (MMP-12), which is produced by alveolar macrophages when IL-13 is present.76 Furthermore, it has been demonstrated that individuals with stable COPD or during an AECOPD have greater levels of group 2 innate lymphoid cells, which can produce IL-13.90
Lebrikizumab, a humanized mAb that binds to soluble IL-13 and inhibits activation of IL-4Rα and IL-13Rα1 heterodimers, has been examined in patients with COPD with a history of exacerbations despite using ICS and at least one long-acting bronchodilator inhaler drug (NCT02546700).76 Although the complete results have not yet been made public, early data indicates that lebrikizimab did not affect the AECOPD rate compared to placebo. Consequently, the clinical development of lebrikizumab for the treatment of patients with COPD has been halted indefinitely. Nevertheless, since IL-13 directly affects airway contractility and is critical for mucus production and remodeling24 and airflow limitation and mucus hypersecretion are commonly treatable features in COPD, it would be interesting to conduct an RCT with lebrikizimab in patients belonging to the GOLD Group B, i.e. with COPD that is symptomatic but only slightly exacerbating.
A pivotal 52-week study, in which 939 adults aged 40 to 80 years with moderate-to-severe COPD and T2 inflammation, as measured by BECs ≥300 cells/µL, received dupilumab (n = 468) or placebo (n = 471), added to standard-of-care inhaled therapy, has shown that this biologic, a mAb that binds to IL4Rα and inhibits both IL-4 and IL-13 signaling, cut exacerbations by 30% compared with placebo (p<0.001).91 The annualized rate of moderate or severe exacerbations was 0.78 with dupilumab and 1.10 with placebo. It also caused an increase in FEV1 from baseline of 160 mL at 12 weeks compared with 77 mL for placebo (p<0.001), with the benefit versus placebo sustained through week 52. Furthermore, it improved patient-reported HRQoL as measured by SGRQ and reduced the severity of respiratory symptoms of COPD as measured by Evaluation Respiratory Symptoms: COPD (E-RS: COPD) Scale. Another phase 3 pivotal trial (NCT04456673) is also assessing the efficacy, safety, and tolerability of dupilumab in COPD patients with evidence of T2 inflammation, with data expected in 2024.
Targeting Thymic Stromal Lymphopoietin (TSLP)
TSLP, an IL-7-like cytokine, is an upstream epithelial alarmin that plays an important role in the regulation of T2 immunity.76 TSLP operates on a variety of cells, including dendritic cells, T-cells, mast cells, innate lymphoid cells, and eosinophils, causing the production of a variety of interleukins, including IL-4, IL-5, and IL-13, which leads to airway eosinophilia and hyperresponsiveness. It is also capable of exerting multipotential pathogenic effects beyond T2 inflammation76 (Figure 4).
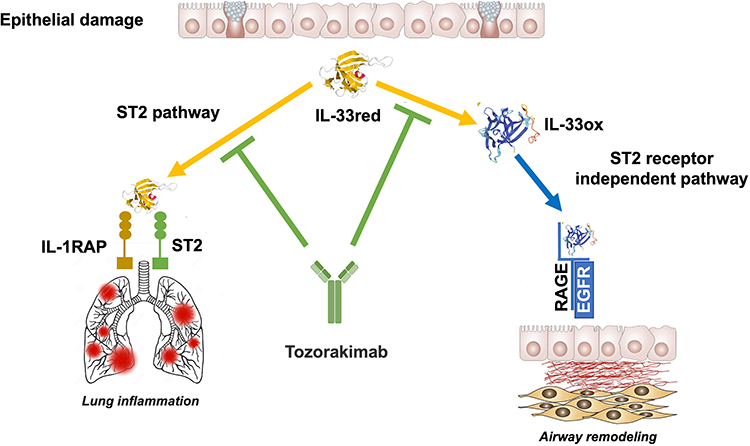 |
Figure 4 Targeting alarmins in COPD. Abbreviations: DC, dendritic cell; EGFR, epidermal growth factor receptor; ILC2, group 2 innate lymphoid cells; IL-1RAP, IL-1 receptor accessory protein; IL-33red, reduced IL-33; IL-33ox, oxidated IL-33; MC, mast cell; RAGE, receptor for advanced glycation end products; ST2, suppression of tumorigenicity 2; TSLP, thymic stromal lymphopoietin; TSLPR, TSLP receptor. |
TSLP is constitutively expressed in the airway smooth muscle of patients with COPD, where it probably interacts with and influences local immune cells.92 In addition, viruses and T1 cytokines can induce COPD epithelial cells to overproduce TSLP,93 suggesting that TSLP may play a role in AECOPD. However, it has been hypothesized that TSLP produced by DCs serves as a crucial molecular checkpoint to limit IL-1β-mediated effector responses through a negative feedback loop, potentially reducing the severity of the inflammatory response to injury.94
Tezepelumab, a first-in-class human IgG2/λ mAb that binds to TSLP and inhibits its interaction with the TSLP receptor complex,76 is currently under investigation as adjunctive therapy in preventing AECOPD (NCT04039113) and in reducing airway inflammation in patients with COPD (NCT05507242). Ecleralimab, an antibody fragment that is part of the G1/λ immunoglobulin isotype subclass and binds to TSLP, is being evaluated in a Phase 2 study in patients with COPD (NCT04882124).
Targeting IL-33
IL-33, a member of the IL-1 superfamily, is released in a reduced form (IL-33red) from damaged epithelial cells and acts as an alarmin to notify the immune system of harm, thereby promoting the inflammatory response.95 It exerts its biologically pro-inflammatory effects by activating its receptor, a heterodimeric complex composed of the IL-1 receptor accessory protein and tumor suppressor protein 2 (ST2)96 (Figure 4).
IL-33 stimulates lung epithelial and endothelial cells to produce and release IL-6 and IL-8, resulting in an influx of neutrophils that damage lung tissue by producing elastases and proteases.97 This, in turn, promotes pulmonary fibrosis and impairs lung function. Additionally, in non-atopic patients with COPD, IL-33 has been connected to the emergence of eosinophilic airway inflammation.98
Itepekimab, a human IgG4 mAb targeting IL-33, added to the standard of care, did not significantly reduce the annualized rate of moderate-to-severe AECOPDs compared to placebo after 52 weeks of treatment.99 However, it decreased the AECOPD rate and improved lung function in former COPD smokers. Itepekimab is under investigation in two Phase 3 trials to compare its efficacy to placebo on the annualized rate of mild to moderately severe AECOPDs in a 52-week placebo-controlled trial in former smokers with mild to moderately severe COPD (NCT04701983 and NCT04751487).
Astegolimab, an anti-ST2 mAb, did not significantly reduce the AECOPD rate but improved health status compared with placebo in patients with moderate-to-very severe COPD who were administered this drug subcutaneously in a 48-week RCT.100 Two ongoing trials are evaluating the efficacy and safety of astegolimab compared with placebo in participants with COPD who are former or current smokers and have a history of frequent exacerbations (NCT05037929 and NCT05595642).
Tozorakimab (MEDI3506) is a human IgG1 mAb that prevents IL-33 signaling. It possesses a higher affinity for the reduced form (IL-33red) than soluble version of ST2 (sST2) and a rapid association rate comparable to that of sST2.101 In primary human cells and in a humanized IL-33 murine model of airway inflammation, tozorakimab suppressed ST2-dependent inflammation.100 It binds IL-33red, blocking oxidation and, consequently, the action of the oxidated form (IL-33ox) via receptor for advanced glycation end products/epidermal growth factor receptor.102 Elimination of IL-33ox enhanced wound healing in vitro following epithelial injury. Tozorakimab is being investigated in several RCTs in patients with COPD. One of them (NCT04631016) will assess its efficacy and safety in patients with moderate to severe COPD and chronic bronchitis. Furthermore, two identical global Phase 3 studies (NCT05166889 and NCT05158387) are designed to evaluate the efficacy and safety of two different doses of tozorakimab administered for 52 weeks in patients with symptomatic COPD and a history of ≥2 moderate exacerbations or ≥1 severe AECOPD in the previous 12 months, despite receiving optimized treatment (triple therapy or dual therapy if triple therapy is not indicated or contraindicated) at a stable dose for at least 3 months before enrolment.
Targeting the Products of the Cellular Components of Inflammation
Several attempts have been made to develop therapies that target the products of the cellular components of inflammation. Some of these have been evaluated in humans (Table 2).
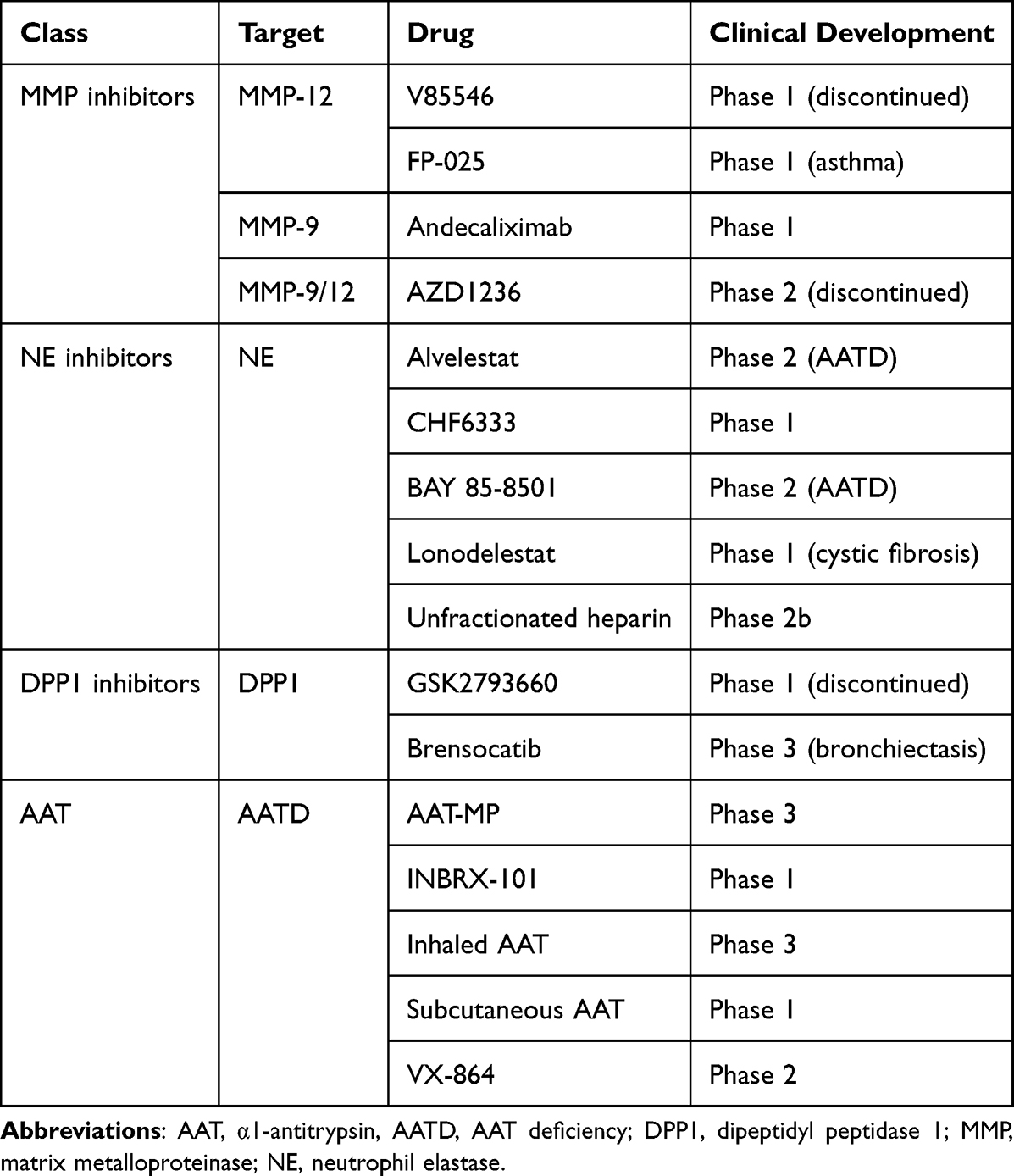 |
Table 2 Therapies That Antagonize the Products of the Cellular Components of Inflammation in COPD |
Matrix Metalloproteinase Inhibitors
MMPs are proteolytic enzymes that break down basement membrane and ECM proteins.103 Consequently, these enzymes control airway remodelling, a critical pathogenetic aspect of COPD. Furthermore, proteolytic damage in the lungs can lead to elastin loss and emphysema formation, which are linked to reduced lung function in COPD patients. MMPs, particularly those derived from macrophages, have been linked to the development of COPD in several human and animal studies. MMPs have been shown to influence macrophage activation rather than working directly to destroy elastin, promoting inflammation and the disease course.104,105 In addition to their activity on the ECM, MMPs can act on various other substrates.
In humans, there are 23 different MMPs.103 MMP-9 and MMP-12 are those particularly involved in tissue remodelling and degradation of the ECM components and likely perpetuate and regulate lung inflammation when released by neutrophils and macrophages.6 There is currently limited research investigating the use of MMP inhibitors in patients with COPD.6 While V85546, a selective MMP-12 inhibitor, has completed Phase 1 clinical testing, AZD1236, a dual MMP-9/12 inhibitor, was ineffective in patients with moderate-to-severe COPD.106 Andecaliximab, a recombinant chimeric IgG4 mAb, has completed a Phase 1 clinical trial and is now being investigated in patients with COPD (NCT02077465). FP-025, an inhibitor of MMP-12, is currently being tested for efficacy in patients with asthma (NCT03858686), but to date no studies have been described in patients with COPD.
A novel series of compounds with single-digit nanomolar inhibition against MMP-12 have been discovered using the high-throughput screening by nuclear magnetic resonance technique.107 Animals exposed to porcine pancreatic elastase but given MMP-12 inhibitors (MMP408, or lead agents compound 25, and 26) showed a substantial reduction in emphysema-like pathologies.
Drug repurposing might be an appealing technique for identifying current medicines with unique MMP inhibitory action. For example, doxycycline, a broad-spectrum antibiotic, has been shown to reduce MMP12 activity in vitro.108 Despite its safety profile, antibiotic medication repurposing requires close monitoring to prevent the possibility of antibiotic resistance.
Neutrophil Elastase Inhibitors
NE is stored in neutrophil azurophilic granules and is released when neutrophils are activated by exposure to cigarette smoke or inflammation107 It causes alveolar expansion and disintegration by proteolyzing lung tissues. Furthermore, NE induces airway epithelial cells to express MUC5AC, causing secretory cell hyperplasia and mucin synthesis.109 Finally, NE stimulates macrophages, causing them to release active MMPs, which cause lung damage and emphysema, and proinflammatory cytokines, which cause inflammation.110
In individuals with COPD who have an overexpression of this enzyme, restoring the balance between NE and endogenous antiproteases by blocking NE might be a treatment option.
However, alvelestat, an orally available, potent, and selective inhibitor of human NE, did not induced changes in lung function or inflammation when added to budesonide/formoterol maintenance therapy in patients with COPD.111 Furthermore, combined with tiotropium in patients with COPD, it failed to demonstrate a clinical benefit or an impact on biomarkers of tissue deterioration.112 Nevertheless, a trial is investigating the effects of alvelestat on blood and sputum biomarkers in patients with PiZZ α1-antitrypsin deficiency (AATD) (NCT03636347).
Sivelestat is a NE inhibitor available only in Japan and South Korea to treat acute lung damage and acute respiratory distress syndrome in individuals with a systemic inflammatory response. Other NE inhibitors, such as BAY 85–8501, CHF6333, and lonodelestat, which could be useful in COPD, are currently in different stages of clinical development, but there is still no information on their effects on COPD.6
Interestingly, polysulfated glycosaminoglycans (GAGs) are potent anti-elastase drugs with multiple anti-inflammatory properties.113 In one double-blind, placebo-controlled pilot research, inhaling unfractionated heparin, the prototypical drug in this class, as a COPD treatment resulted in enhanced lung function, highlighting the potential of GAG therapy for the treatment of chronic lung disorders.114
However, focusing solely on anti-NE activity does not appear to be sufficient to change the ongoing inflammatory state in the airways in a way that would alter the trajectory of clinical outcomes.115 It has been suggested that employing combination therapy and/or multi-function drugs that have antiprotease and anti-inflammatory properties115 or, alternatively developing an airway delivery system for these therapeutics, allowing activity not only on the free NE but also on the form bound to the extracellular vesicles found in the BALF of COPD patients116 may be a more successful strategy.
Dipeptidyl Peptidase 1 Inhibitors
DPP1 transforms the neutrophil serine protease pool, including NE, cathepsin G, proteinase 3, into catalytically active forms and activates proteases in other immune cell types, such as chymases in mast cells.117,118 Therefore, it may be a promising target for new drugs to treat COPD. GSK2793660, an irreversible DPP1 inhibitor, was stopped after a Phase 1 clinical study due to drug-related side effects and lack of impact on biomarkers.119 This drug only decreased the activity of downstream serine proteases in 20% of the cases, although it suppressed most of DPP1. Brensocatib, an oral reversible DPP1 inhibitor, has been investigated for its effects in adult patients with non-cystic fibrosis bronchiectasis,120 but its effects on COPD are unknown. Nevertheless, it has been proposed that limiting neutrophil protease activity before cell discharge into the circulation rather than at the inflammatory site, as well as blocking several inflammatory targets such as elastase, but also cathepsin G and proteinase-3, may be a crucial driver of success.121
Granzyme B Inhibitors
When released from the cytoplasmic secretory granules of CD8+ T lymphocytes, granzyme B, which is a multifunctional serine protease and is also called α-1-antichymotrypsin (ACT), can start a cascade of DNA disruption that causes the apoptotic death of target cells, such as bronchial epithelial cells, which may lead to tissue damage and remodelling.122 Furthermore, it increases inflammation by enhancing cytokine activity or sequestered growth factor release.123 Thus, using targeted granzyme B inhibitors to treat COPD seems plausible. Serpina3n, a serine protease inhibitor that inhibits granzyme B,124 an engineered extracellular granzyme B inhibitor in which the reactive center loop of human extracellular ACT is replaced with serpina3n,125 VTI-1002, a first-in-class potent small-molecule inhibitor of granzyme B,126 proteinase inhibitor 9 (PI-9), an endogenous GZMB inhibitor that can bind to GZMB in the cytosol of activated CD8+ T cells,127 serine protease inhibitor 6 (SPI-6), the mouse homolog of PI-9128 are potentially interesting molecules but have not yet been investigated on humans. Furthermore, cefpiramide, a third-generation semi-synthetic cephalosporin antibiotic, and mupirocin, produced by Pseudomonas fluorescens, have the propensity to inhibit the active site of granzyme B129 and may therefore be of value in treating COPD.
α1-Antitrypsin Replacement Therapy
AAT modulates neutrophilic inflammation and inhibits proteases.130 There is a proteolytic interplay between AAT, MMP and neutrophil elastase (NE) (Figure 5). In patients with AATD, augmentation treatment with pure intravenous AAT is essential, although it reaches the lung in a somewhat inactive state.130 New intravenous formulations could solve this problem. AAT Modified Process (AAT-MP), with improved AAT purity, is in a 3-year Phase 3 trial (NCT01983241). INBRX-101, a recombinant human AAT Fc fusion protein that extends the half-life of AAT in blood by enabling it to be recycled, is under Phase 1 study (NCT03815396).
 |
Figure 5 Proteolytic interplay between α1-antitrypsin (AAT), matrix metalloproteinases (MMPs) and neutrophil elastase (NE). Several proteases, including neutrophil elastase (NE) and MMP-9 and MMP-12, are involved in COPD. Therefore, inhibiting a single enzyme with a NE inhibitor or a MMP inhibitor may not have a significant therapeutic impact. AAT inhibits NE and reduces macrophage MMP-12 synthesis. |
The inhaled route for AAT is also of interest as it can act directly on the target organ when administered in this way.130 Inhaled AAT administered for 50 weeks significantly reduced the number of symptomatic exacerbations of Anthonisen type I, with a propensity to improve FEV1 but without affecting the time to first exacerbation.131 A trial is evaluating inhaled AAT in patients with AATD who have moderate or severe airflow limitation and have not experienced two or more moderate or one or more severe exacerbations of COPD during the past year (NCT04204252). Developing gene therapy to express AAT using various vector systems130 or administering AAT treatment via the transepidermal route132 are intriguing alternative approaches currently under investigation. A study is evaluating the safety and tolerability of subcutaneous AAT (NCT04722887).
Newer approaches that are predominantly in the preclinical phase tend to silence AAT production, enhance protein folding and secretion, or promote AAT degradation.133 A folding corrector called VX-864 aims to increase Z-AAT secretion and activity. It is starting a Phase 2 study (NCT04474197) to show its safety and effectiveness in PiZZ patients.
Targeting Neutrophil Extracellular Traps
Great attention has been given to the dysregulation of NETs that play an essential role in the innate immune system against infections. NETs have been identified in the airways of patients with COPD with chronic neutrophilic inflammation.134 NETs result from the suicidal death of neutrophils during the attack of a pathogen or lesion, with the release of decondensed chromatin entangled with antimicrobial peptides to trap and capture pathogens to be an effective mechanism against invading microorganisms thus controlling overwhelming infections.135 However, an aberrant and massive NET formation can directly induce epithelial and endothelial cell death, impairing lung function and accelerating disease progression.6,136
Interfering with the NETopathic inflammation pathways could help develop innovative therapeutics for COPD.135 Heparin, which interferes with neutrophil autophagy, suppresses histones, prevents platelets-histone interaction, and blocks high mobility group box-1, deoxyribonuclease, which acts on and cleaves DNA matrices, and reduces the infiltration of neutrophils hence playing role in inhibition/reduction of NETs, and hydroxychloroquine, which mediates MMPs-TIMPs interaction that helps in maintaining homeostasis of extracellular matrix, are potential anti-NET therapeutics.6,135 However, also protease inhibitors, CXCR2 antagonists, macrolides, and DPP1 inhibitors are drug classes that can specifically or non-specifically target NET formation,137 although a pilot study did not find an effect of the reversible CXCR2 antagonist danirixin on NET formation.49 Blockade of IL-1β and IL-17 could also be a valid strategy for interfering with the NETopathic inflammation pathways.135
Conclusion
Finding therapies that can reduce COPD-related inflammation and prevent the disorder from becoming worse is crucial. Recent discoveries have resulted in developing novel drugs for several novel potential targets directly involved in the inflammatory process.6,14 To date, most of these new therapies are in preclinical or early clinical development and the results from clinical trials in patients with COPD are eagerly awaited.138 However, the redundancy of the actions of signal-transmitting mediators involved in the complexity of the inflammatory response in COPD suggests that there will likely be several failures of the different approaches discussed above.139
Since COPD is heterogeneous, with many endotypes and phenotypes reflecting different pathophysiological mechanisms, the endo/phenotypic characterization of the inflammatory profile of patients with COPD may allow the identification of a reasonably homogeneous population such that there is a high possibility that the disease being treated is driven by the specific target pathway of the drug being tested. This strategy involves defining more specific management approaches.1 As recently highlighted by the Lancet commission, the new COPD therapies must be specific and target the exact biological pathways or endotypes responsible for disease manifestation to have the greatest impact and eventually abolish the disease.1 Characterization of the heterogeneity of the inflammatory signature associated with COPD could pave the way for personalized medicine by identifying new and effective therapeutic approaches for COPD.140 For this reason, many clinical trials are underway to characterize the impact of some novel molecules that act directly against the inflammatory process in treating distinct subgroups of COPD patients.6
Nevertheless, other possible approaches, such as stem cell-based regenerative therapy and derivative products141 and modulators of cystic fibrosis transmembrane conductance regulator,142 may indirectly modulate inflammation in COPD and do not require accurate endotypic precision. The scientific community eagerly awaits the results of this experimental and mainly clinical research in the hope of improving treatments for such a challenging disease.
Abbreviations
AAT, α1-antitrypsin, AATD, AAT deficiency; ACT, α-1-antichymotrypsin; AECOPD, acute exacerbations of COPD; AR, adrenoceptor; BALF, bronchoalveolar lavage fluid; BEC, blood eosinophil count; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; CS corticosteroid; COPD, chronic obstructive pulmonary disease; CXCR, CXC chemokine receptor; DC, dendritic cell; DPP1, dipeptidyl peptidase 1; ECM, extracellular matrix; FEV1, forced expiratory volume in the first second; GR, glucocorticoid receptor; HDAC. Histone deacetylase; HRQoL, health-related quality of life; ICS, inhaled corticosteroid; Ig, immunoglobulin; IL, interleukin; IL-4Rα, interleukin-4 receptor subunit α; IL-5Rα, interleukin-5 receptor subunit α; IL-17RA, IL-17 receptor A; IL-33ox, IL-33 oxidated form; IL-33red, IL-33 reduced form; LABA, long-acting β2-agonist; LAMA, long-acting muscarinic antagonist; LPS, lipopolysaccharide; mAb, monoclonal antibody; MAPK, mitogen-activated protein kinase; MMP, matrix metalloprotease; NE, neutrophil elastase; NETs, neutrophil extracellular traps; PDE, phosphodiesterase; PI3K, phosphoinositide 3-kinase; RCT, randomized controlled trial; Rfh, recombinant form of human fragments; sAC, soluble adenylyl cyclase; SP, surfactant protein; Syk, spleen tyrosine kinase; ST2, tumor suppressor protein 2; sST2, soluble version of ST2; T, type; TNF-α, tumor necrosis factor-α; TSLP, thymic stromal lymphopoietin.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
M.C. participated as a faculty member and advisor in scientific meetings and courses under the sponsorship of Abdi Ibrahim, Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi Farmaceutici, Cipla, Edmond Pharma, GlaxoSmithKline, Glenmark, Lallemand, Mankind Pharma, Menarini Group, Mundipharma, Novartis, Pfizer, Sanofi, Teva, Verona Pharma, and Zambon and is or was a consultant to ABC Farmaceutici, AstraZeneca, Chiesi Farmaceutici, Edmond Pharma, Lallemand, Novartis, Ockham Biotech, Verona Pharma, and Zambon. N.A.H. received honoraria for serving as advisor or consultant for GSK, AstraZeneca, Sanofi, Regeneron, Boehringer Ingelheim, Verona, Amgen, Genentech, Novartis and Teva. His institution received research grant support of his behalf from GSK, Genentech, Sanofi, Teva, Novartis, and Astra Zeneca. C.P.P. has acted as a consultant to Eurodrug, Recipharm, Glycosynnovation and PrEP Biopharma. C.P.P. also holds equity in Verona Pharma. M.G.M. participated as a faculty member and advisor in scientific meetings and courses under the sponsorship of ABC Farmaceutici, Almirall, AstraZeneca, Chiesi Farmaceutici, GlaxoSmithKline, and Novartis and was a consultant to Chiesi Farmaceutici and GlaxoSmithKline. Her department was funded by GlaxoSmithKline and Novartis. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in, or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
1. Stolz D, Mkorombindo T, Schumann DM, et al. Towards the elimination of chronic obstructive pulmonary disease: a lancet commission. Lancet. 2022;400(10356):921–972. doi:10.1016/S0140-6736(22)01273-9
2. Gamble E, Qiu Y, Wang D, et al. Variability of bronchial inflammation in chronic obstructive pulmonary disease: implications for study design. Eur Respir J. 2006;27(2):293–299. doi:10.1183/09031936.06.00027705
3. Deng F, Zhong S, Yu C, et al. Abnormal neutrophil polarization in chronic obstructive pulmonary disease and how cigarette smoke extracts attract neutrophils. Ann Transl Med. 2022;10(8):472. doi:10.21037/atm-22-1480
4. David B, Bafadhel M, Koenderman L, De Soyza A. Eosinophilic inflammation in COPD: from an inflammatory marker to a treatable trait. Thorax. 2021;76(2):188–195. doi:10.1136/thoraxjnl-2020-215167
5. Burgoyne RA, Fisher AJ, Borthwick LA. The role of epithelial damage in the pulmonary immune response. Cells. 2021;10(10):2763. doi:10.3390/cells10102763
6. Matera MG, Cazzola M, Page C. Prospects for COPD treatment. Curr Opin Pharmacol. 2021;56:74–84. doi:10.1016/j.coph.2020.11.003
7. Cazzola M, Ora J, Calzetta L, Rogliani P, Matera MG. Advances in inhaled corticosteroids for the treatment of chronic obstructive pulmonary disease: what is their value today? Expert Opin Pharmacother. 2022;23(8):917–927. doi:10.1080/14656566.2022.2076592
8. Barnes PJ. Glucocorticosteroids. Handb Exp Pharmacol. 2017;237:93–115. doi:10.1007/164_2016_62
9. Adcock IM, Bhatt SP, Balkissoon R, Wise RA. The use of inhaled corticosteroids for patients with COPD who continue to smoke cigarettes: an evaluation of current practice. Am J Med. 2022;135(3):302–312. doi:10.1016/j.amjmed.2021.09.006
10. Plumb J, Gaffey K, Kane B, et al. Reduced glucocorticoid receptor expression and function in airway neutrophils. Int Immunopharmacol. 2012;12(1):26–33. doi:10.1016/j.intimp.2011.10.006
11. Milara J, Lluch J, Almudever P, Freire J, Xiaozhong Q, Cortijo J. Roflumilast N-oxide reverses corticosteroid resistance in neutrophils from patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2014;134(2):314–322. doi:10.1016/j.jaci.2014.02.001
12. Li LB, Leung DY, Martin RJ, Goleva E. Inhibition of histone deacetylase 2 expression by elevated glucocorticoid receptor beta in steroid-resistant asthma. Am J Respir Crit Care Med. 2010;182(7):877–883. doi:10.1164/rccm.201001-0015OC
13. Pavord ID, Lettis S, Anzueto A, Barnes N. Blood eosinophil count and pneumonia risk in patients with chronic obstructive pulmonary disease: a patient-level meta-analysis. Lancet Respir Med. 2016;4(9):731–741. doi:10.1016/S2213-2600(16)30148-5
14. Matera MG, Calzetta L, Annibale R, Russo F, Cazzola M. Classes of drugs that target the cellular components of inflammation under clinical development for COPD. Expert Rev Clin Pharmacol. 2021;14(8):1015–1027. doi:10.1080/17512433.2021.1925537
15. Barnes PJ. Identifying molecular targets for new drug development for chronic obstructive pulmonary disease: what does the future hold? Semin Respir Crit Care Med. 2015;36(4):508–522. doi:10.1055/s-0035-1555611
16. Barnes PJ. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2018;18(7):454–466. doi:10.1038/s41577-018-0006-6
17. Cazzola M, Page CP, Calzetta L, Matera MG. Emerging anti-inflammatory strategies for COPD. Eur Respir J. 2012;40(3):724–741. doi:10.1183/09031936.00213711
18. Prenner BM, Bukofzer S, Behm S, Feaheny K, McNutt BE. A randomized, double-blind, placebo-controlled study assessing the safety and tolerability of regadenoson in subjects with asthma or chronic obstructive pulmonary disease. J Nucl Cardiol. 2012;19(4):681–692. doi:10.1007/s12350-012-9547-4
19. Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–770. doi:10.1038/nrd2638
20. Mahler DA, Huang S, Tabrizi M, Bell GM. Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in COPD: a pilot study. Chest. 2004;126(3):926–934. doi:10.1378/chest.126.3.926
21. van der Vaart H, Koëter GH, Postma DS, Kauffman HF, ten Hacken NH. First study of infliximab treatment in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(4):465–469. doi:10.1164/rccm.200501-147OC
22. Rennard SI, Fogarty C, Kelsen S, et al. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(9):926–934. doi:10.1164/rccm.200607-995OC
23. Aaron SD, Vandemheen KL, Maltais F, et al. TNFα antagonists for acute exacerbations of COPD: a randomised double-blind controlled trial. Thorax. 2013;68(2):142–148. doi:10.1136/thoraxjnl-2012-202432
24. Cazzola M, Ora J, Cavalli F, Rogliani P, Matera MG. An overview of the safety and efficacy of monoclonal antibodies for the chronic obstructive pulmonary disease. Biologics. 2021;15:363–374. doi:10.2147/BTT.S295409
25. Rogliani P, Matera MG, Puxeddu E, et al. Emerging biological therapies for treating chronic obstructive pulmonary disease: a pairwise and network meta-analysis. Pulm Pharmacol Ther. 2018;50:28–37. doi:10.1016/j.pupt.2018.03.004
26. Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. 2014;13(4):290–314. doi:10.1038/nrd4228
27. Jin SL, Goya S, Nakae S, et al. Phosphodiesterase 4B is essential for TH2-cell function and development of airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol. 2010;126(6):1252–9.e12. doi:10.1016/j.jaci.2010.08.014
28. Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123(20):2263–2273. doi:10.1161/CIRCULATIONAHA.110.981738
29. Lam M, Bourke JE. A new pathway to airway relaxation: targeting the “other” cyclase in asthma. Am J Respir Cell Mol Biol. 2020;62(1):3–4. doi:10.1165/rcmb.2019-0274ED
30. Page CP. Phosphodiesterase inhibitors for the treatment of asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol. 2014;165(3):152–164. doi:10.1159/000368800
31. Cazzola M, Calzetta L, Rogliani P, Matera MG. The discovery of roflumilast for the treatment of chronic obstructive pulmonary disease. Expert Opin Drug Discov. 2016;11(7):733–744. doi:10.1080/17460441.2016.1184642
32. Rogliani P, Calzetta L, Cazzola M, Matera MG. Drug safety evaluation of roflumilast for the treatment of COPD: a meta-analysis. Expert Opin Drug Saf. 2016;15(8):1133–1146. doi:10.1080/14740338.2016.1199683
33. Agustí A, Celli BR, Criner GJ, et al. Global Initiative for Chronic Obstructive Lung Disease 2023 report: GOLD executive summary. Am J Respir Crit Care Med. 2023;207(7):819–837. doi:10.1164/rccm.202301-0106PP
34. Matera MG, Rogliani P, Calzetta L, Cazzola M. Phosphodiesterase inhibitors for chronic obstructive pulmonary disease: what does the future hold? Drugs. 2014;74(17):1983–1992. doi:10.1007/s40265-014-0303-8
35. Matera MG, Ora J, Cavalli F, Rogliani P, Cazzola M. New avenues for phosphodiesterase inhibitors in asthma. J Exp Pharmacol. 2021;13:291–302. doi:10.2147/JEP.S242961
36. Phillips JE. Inhaled phosphodiesterase 4 (PDE4) inhibitors for inflammatory respiratory diseases. Front Pharmacol. 2020;11:259. doi:10.3389/fphar.2020.00259
37. Singh D, Emirova A, Francisco C, Santoro D, Govoni M, Nandeuil MA. Efficacy and safety of CHF6001, a novel inhaled PDE4 inhibitor in COPD: the PIONEER study. Respir Res. 2020;21(1):246. doi:10.1186/s12931-020-01512-y
38. Nguyen HO, Salvi V, Tiberio L, et al. The PDE4 inhibitor tanimilast shows distinct immunomodulatory properties associated with a type 2 endotype and CD141 upregulation. J Transl Med. 2022;20(1):203. doi:10.1186/s12967-022-03402-x
39. Li G, He D, Cai X, et al. Advances in the development of phosphodiesterase-4 inhibitors. Eur J Med Chem. 2023;250:115195. doi:10.1016/j.ejmech.2023.115195
40. Paes D, Hermans S, van den Hove D, Vanmierlo T, Prickaerts J, Carlier A. Computational investigation of the dynamic control of cAMP signaling by PDE4 isoform types. Biophys J. 2022;121(14):2693–2711. doi:10.1016/j.bpj.2022.06.019
41. Page C, Cazzola M. Bifunctional drugs for the treatment of respiratory diseases. Handb Exp Pharmacol. 2017;237:197–212. doi:10.1007/164_2016_69
42. Abbott-Banner KH, Page CP. Dual PDE3/4 and PDE4 inhibitors: novel treatments for COPD and other inflammatory airway diseases. Basic Clin Pharmacol Toxicol. 2014;114(5):365–376. doi:10.1111/bcpt.12209
43. Cazzola M, Calzetta L, Rogliani P, Matera MG. Ensifentrine (RPL554): an investigational PDE3/4 inhibitor for the treatment of COPD. Expert Opin Investig Drugs. 2019;28(10):827–833. doi:10.1080/13543784.2019.1661990
44. Barjaktarevic IZ, Rheault T, Bengtsson T, Rickard K. Ensifentrine, a novel dual phosphodiesterase (PDE) 3 and 4 inhibitor, significantly reduces annualized exacerbations and delays the time to first exacerbation in COPD: pooled sub-group analyses of ENHANCE-1 and ENHANCE-2 phase 3 trials. Am J Respir Crit Care Med. 2023;207:A5008. doi:10.1164/ajrccm-conference.2023.207.1_MeetingAbstracts.A5008
45. Sciurba F, Anzueto A, Rheault T, Bengtsson T, Rickard K. Ensifentrine, a novel dual phosphodiesterase (PDE) 3 and 4 inhibitor, improves lung function, symptoms, quality of life and reduces exacerbation rate and risk in patients with COPD: results from replicate phase 3 trials. Am J Respir Crit Care Med. 2023;207:A5005. doi:10.1164/ajrccm-conference.2023.207.1_MeetingAbstracts.A5005
46. Anzueto A, Rheault T, Bengtsson T, Rickard K. Treatment with ensifentrine, a dual PDE3 and PDE4 inhibitor, significantly reduced exacerbation rate and risk in subjects with COPD: sub-group results from the phase 3 trial, ENHANCE-2. Am J Respir Crit Care Med. 2023;207:A4998. doi:10.1164/ajrccm-conference.2023.207.1_MeetingAbstracts.A4998
47. Chapman RW, Phillips JE, Hipkin RW, Curran AK, Lundell D, Fine JS. CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol Ther. 2009;121(1):55–68. doi:10.1016/j.pharmthera.2008.10.005
48. Thatcher TH, McHugh NA, Egan RW, et al. Role of CXCR2 in cigarette smoke-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2005;289(2):L322–L328. doi:10.1152/ajplung.00039.2005
49. Rennard SI, Dale DC, Donohue JF, et al. CXCR2 antagonist MK-7123. A phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;191(9):1001–1011. doi:10.1164/rccm.201405-0992OC
50. Lazaar AL, Miller BE, Donald AC, et al. CXCR2 antagonist for patients with chronic obstructive pulmonary disease with chronic mucus hypersecretion: a phase 2b trial. Respir Res. 2020;21(1):149. doi:10.1186/s12931-020-01401-4
51. Keir HR, Richardson H, Fillmore C, et al. CXCL-8-dependent and -independent neutrophil activation in COPD: experiences from a pilot study of the CXCR2 antagonist danirixin. ERJ Open Res. 2020;6(4):00583–2020. doi:10.1183/23120541.00583-2020
52. Mattos MS, Ferrero MR, Kraemer L, et al. CXCR1 and CXCR2 inhibition by ladarixin improves neutrophil-dependent airway inflammation in mice. Front Immunol. 2020;11:566953. doi:10.3389/fimmu.2020.566953
53. Pelaia C, Vatrella A, Sciacqua A, Terracciano R, Pelaia G. Role of p38-mitogen-activated protein kinase in COPD: pathobiological implications and therapeutic perspectives. Expert Rev Respir Med. 2020;14(5):485–491. doi:10.1080/17476348.2020.1732821
54. Pascoe S, Costa M, Marks-Konczalik J, McKie E, Yang S, Scherbovsky PS. Biological effects of p38 MAPK inhibitor losmapimod does not translate to clinical benefits in COPD. Respir Med. 2017;130:20–26. doi:10.1016/j.rmed.2017.07.002
55. Higham A, Karur P, Jackson N, Cunoosamy DM, Jansson P, Singh D. Differential anti-inflammatory effects of budesonide and a p38 MAPK inhibitor AZD7624 on COPD pulmonary cells. Int J Chron Obstruct Pulmon Dis. 2018;13:1279–1288. doi:10.2147/COPD.S159936
56. Patel NR, Cunoosamy DM, Fagerås M, et al. The development of AZD7624 for prevention of exacerbations in COPD: a randomized controlled trial. Int J Chron Obstruct Pulmon Dis. 2018;13:1009–1019. doi:10.2147/COPD.S150576
57. Charron CE, Russell P, Ito K, et al. RV568, a narrow-spectrum kinase inhibitor with p38 MAPK-α and -γ selectivity, suppresses COPD inflammation. Eur Respir J. 2017;50(4):1700188. doi:10.1183/13993003.00188-2017
58. Strâmbu IR, Kobalava ZD, Magnusson BP, MacKinnon A, Parkin JM. Phase II study of single/repeated doses of acumapimod (BCT197) to treat acute exacerbations of COPD. COPD. 2019;16(5–6):344–353. doi:10.1080/15412555.2019.1682535
59. Wedzicha JA, MacKinnon A, Parkin JM. Effectiveness of acumapimod oral p38 inhibitor in the treatment of acute severe exacerbations of COPD: results of the AETHER phase II trial. Am J Respir Crit Care Med. 2018;197:A7710.
60. Yu H, Su X, Lei T, et al. Safety and efficacy of p38 mitogen-activated protein kinase inhibitors (MAPKIs) in COPD. Front Pharmacol. 2022;13:950035. doi:10.3389/fphar.2022.950035
61. Wang C, Zhou J, Wang J, et al. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct Target Ther. 2020;5(1):248. doi:10.1038/s41392-020-00345-x
62. Onions ST, Ito K, Charron CE, et al. Discovery of narrow spectrum kinase inhibitors: new therapeutic agents for the treatment of COPD and steroid-resistant asthma. J Med Chem. 2016;59(5):1727–1746. doi:10.1021/acs.jmedchem.5b01029
63. Curran A, Charron C, Russell P, Hava D. PUR1800 (RV1162), A novel narrow spectrum kinase inhibitor, but not fluticasone, reduces TNFa-induced cytokine release by primary bronchial epithelial cells from healthy volunteers and COPD patients. Eur Respir J. 2018;52:A5247. doi:10.1183/13993003.congress-2018.PA5247
64. Wasilewski M, Clayton R, Singh D, Perry J, Curran A. Safety and tolerability of PUR1800, an orally inhaled narrow spectrum kinase inhibitor, in patients with stable chronic obstructive pulmonary disease (COPD). J Allergy Clin Immunol. 2023;151(2 suppl):AB10. doi:10.1016/j.jaci.2022.12.062
65. Gupta V, Khan A, Higham A, et al. The effect of phosphatidylinositol-3 kinase inhibition on matrix metalloproteinase-9 and reactive oxygen species release from chronic obstructive pulmonary disease neutrophils. Int Immunopharmacol. 2016;35:155–162. doi:10.1016/j.intimp.2016.03.027
66. Sapey E, Stockley JA, Greenwood H, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(9):1176–1186. doi:10.1164/rccm.201008-1285OC
67. Stark AK, Chandra A, Chakraborty K, et al. PI3Kδ hyper-activation promotes development of B cells that exacerbate Streptococcus pneumoniae infection in an antibody-independent manner. Nat Commun. 2018;9(1):3174. doi:10.1038/s41467-018-05674-8
68. Cahn A, Hamblin JN, Robertson J, et al. An inhaled PI3Kδ inhibitor improves recovery in acutely exacerbating COPD patients: a randomized trial. Int J Chron Obstruct Pulmon Dis. 2021;16:1607–1619. doi:10.2147/COPD.S309129
69. Fahy WA, Homayoun-Valiani F, Cahn A, et al. Nemiralisib in patients with an acute exacerbation of COPD: placebo-controlled, dose-ranging study. Int J Chron Obstruct Pulmon Dis. 2021;16:1637–1646. doi:10.2147/COPD.S309320
70. Begg M, Hamblin JN, Jarvis E, et al. Exploring PI3Kδ molecular pathways in stable COPD and following an acute exacerbation, two randomized controlled trials. Int J Chron Obstruct Pulmon Dis. 2021;16:1621–1636. doi:10.2147/COPD.S309303
71. Sadiq MW, Asimus S, Belvisi MG, et al. Characterisation of pharmacokinetics, safety, and tolerability in a first-in-human study for AZD8154, a novel inhaled selective PI3Kγδ dual inhibitor targeting airway inflammatory disease. Br J Clin Pharmacol. 2022;88(1):260–270. doi:10.1111/bcp.14956
72. Di Stefano A, Maestrelli P, Roggeri A, et al. Upregulation of adhesion molecules in the bronchial mucosa of subjects with chronic obstructive bronchitis. Am J Respir Crit Care Med. 1994;149(3 Pt 1):803–810. doi:10.1164/ajrccm.149.3.7509705
73. Blidberg K, Palmberg L, James A, et al. Adhesion molecules in subjects with COPD and healthy non-smokers: a cross sectional parallel group study. Respir Res. 2013;14(1):47. doi:10.1186/1465-9921-14-47
74. Watz H, Bock D, Meyer M, et al. Inhaled pan-selectin antagonist Bimosiamose attenuates airway inflammation in COPD. Pulm Pharmacol Ther. 2013;26(2):265–270. doi:10.1016/j.pupt.2012.12.003
75. Tvaroška I, Selvaraj C, Koča J. Selectins - The two Dr. Jekyll and Mr. Hyde faces of adhesion molecules - A review. Molecules. 2020;25(12):2835. doi:10.3390/molecules25122835
76. Matera MG, Calzetta L, Cazzola M, Ora J, Rogliani P. Biologic therapies for chronic obstructive pulmonary disease. Expert Opin Biol Ther. 2023;23(2):163–173. doi:10.1080/14712598.2022.2160238
77. Higham A, Beech A, Wolosianka S, et al. Type 2 inflammation in eosinophilic chronic obstructive pulmonary disease. Allergy. 2021;76(6):1861–1864. doi:10.1111/all.14661
78. Eich A, Urban V, Jutel M, et al. A randomized, placebo-controlled phase 2 trial of CNTO 6785 in chronic obstructive pulmonary disease. COPD. 2017;14(5):476–483. doi:10.1080/15412555.2017.1335697
79. Murugaiah V, Tsolaki AG, Kishore U. Collectins: innate Immune pattern recognition molecules. Adv Exp Med Biol. 2020;1204:75–127. doi:10.1007/978-981-15-1580-4_4
80. Watson A, Madsen J, Clark HW. SP-A and SP-D: dual functioning immune molecules with antiviral and immunomodulatory properties. Front Immunol. 2021;11:622598. doi:10.3389/fimmu.2020.622598
81. Pilecki B, Wulf-Johansson H, Støttrup C, et al. Surfactant protein D deficiency aggravates cigarette smoke-induced lung inflammation by upregulation of ceramide synthesis. Front Immunol. 2018;9:3013. doi:10.3389/fimmu.2018.03013
82. Bafadhel M, Saha S, Siva R, et al. Sputum IL-5 concentration is associated with a sputum eosinophilia and attenuated by corticosteroid therapy in COPD. Respiration. 2009;78(3):256–262. doi:10.1159/000221902
83. Eltboli O, Mistry V, Barker B, Brightling CE. Relationship between blood and bronchial submucosal eosinophilia and reticular basement membrane thickening in chronic obstructive pulmonary disease. Respirology. 2015;20(4):667–670. doi:10.1111/resp.12475
84. Caramori G, Adcock IM, Di Stefano A, Chung KF. Cytokine inhibition in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:397–412. doi:10.2147/COPD.S42544
85. Pavord ID, Chapman KR, Bafadhel M, et al. Mepolizumab for eosinophil-associated COPD: analysis of METREX and METREO. Int J Chron Obstruct Pulmon Dis. 2021;16:1755–1770. doi:10.2147/COPD.S294333
86. Criner GJ, Celli BR, Singh D, et al. Predicting response to benralizumab in chronic obstructive pulmonary disease: analyses of GALATHEA and TERRANOVA studies. Lancet Respir Med. 2020;8(2):158–170. doi:10.1016/S2213-2600(19)30338-8
87. Brightling CE, Bleecker ER, Panettieri RA, et al. Benralizumab for chronic obstructive pulmonary disease and sputum eosinophilia: a randomised, double-blind, placebo-controlled, phase 2a study. Lancet Respir Med. 2014;2(11):891–901. doi:10.1016/S2213-2600(14)70187-0
88. Mesnil C, Raulier S, Paulissen G, et al. Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J Clin Invest. 2016;126(9):3279–3295. doi:10.1172/JCI85664
89. Lange P, Ahmed E, Lahmar ZM, Martinez FJ, Bourdin A. Natural history, and mechanisms of COPD. Respirology. 2021;26(4):298–321. doi:10.1111/resp.14007
90. Sun J, Liu T, Yan Y, et al. The role of Th1/Th2 cytokines played in regulation of specific CD4+ Th1 cell conversion and activation during inflammatory reaction of chronic obstructive pulmonary disease. Scand J Immunol. 2018;88(1):e12674. doi:10.1111/sji.12674
91. Bhatt SP, Rabe KF, Hanania NA, et al. Dupilumab for COPD with type 2 inflammation indicated by eosinophil counts. N Engl J Med. 2023. doi:10.1056/NEJMoa2303951
92. Zhang K, Shan L, Rahman MS, Unruh H, Halayko AJ, Gounni AS. Constitutive and inducible thymic stromal lymphopoietin expression in human airway smooth muscle cells: role in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2007;293(2):L375–L382. doi:10.1152/ajplung.00045.2007
93. Calvén J, Yudina Y, Hallgren O, et al. Viral stimuli trigger exaggerated thymic stromal lymphopoietin expression by chronic obstructive pulmonary disease epithelium: role of endosomal TLR3 and cytosolic RIG-I-like helicases. J Innate Immun. 2012;4(1):86–99. doi:10.1159/000329131
94. Elder MJ, Webster SJ, Williams DL, Gaston JS, Goodall JC. TSLP production by dendritic cells is modulated by IL-1β and components of the endoplasmic reticulum stress response. Eur J Immunol. 2016;46(2):455–463. doi:10.1002/eji.201545537
95. Cohen ES, Scott IC, Majithiya JB, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. 2015;6(1):8327. doi:10.1038/ncomms9327
96. Cayrol C. IL-33, an Alarmin of the IL-1 Family involved in allergic and non allergic inflammation: focus on the mechanisms of regulation of its activity. Cells. 2021;11(1):107. doi:10.3390/cells11010107
97. Gabryelska A, Kuna P, Antczak A, Białasiewicz P, Panek M. IL-33 mediated inflammation in chronic respiratory diseases-understanding the role of the member of IL-1 superfamily. Front Immunol. 2019;10:692. doi:10.3389/fimmu.2019.00692
98. Tworek D, Majewski S, Szewczyk K, et al. The association between airway eosinophilic inflammation and IL-33 in stable non-atopic COPD. Respir Res. 2018;19(1):108. doi:10.1186/s12931-018-0807-y
99. Rabe KF, Celli BR, Wechsler ME, et al. Safety and efficacy of itepekimab in patients with moderate-to-severe COPD: a genetic association study and randomised, double-blind, phase 2a trial. Lancet Respir Med. 2021;9(11):1288–1298. doi:10.1016/S2213-2600(21)00167-3
100. Yousuf AJ, Mohammed S, Carr L, et al. Astegolimab, an anti-ST2, in chronic obstructive pulmonary disease (COPD-ST2OP): a phase 2a, placebo-controlled trial. Lancet Respir Med. 2022;10(5):469–477. doi:10.1016/S2213-2600(21)00556-7
101. Cazzola M, Rogliani P, Calzetta L, Matera MG. Tozorakimab. Anti-IL-33 monoclonal antibody, Treatment of COPD and asthma, Treatment of atopic dermatitis, Treatment of diabetic kidney disease. Drugs Future. 2023;48(2):101–109. doi:10.1358/dof.2023.48.2.3474010
102. Scott IC, England E, Rees DG, et al. Tozorakimab: a dual-pharmacology anti-IL-33 antibody that inhibits IL-33 signalling via ST2 and RAGE/EGFR to reduce inflammation and epithelial dysfunction. Eur Respir J. 2022;60:2467. doi:10.1183/13993003.congress-2022.246
103. Christopoulou ME, Papakonstantinou E, Stolz D. Matrix metalloproteinases in chronic obstructive pulmonary disease. Int J Mol Sci. 2023;24(4):3786. doi:10.3390/ijms24043786
104. Fingleton B. Matrix metalloproteinases as regulators of inflammatory processes. Biochim Biophys Acta Mol Cell Res. 2017;1864(11 Pt A):2036–2042. doi:10.1016/j.bbamcr.2017.05.010
105. Gharib SA, Manicone AM, Parks WC. Matrix metalloproteinases in emphysema. Matrix Biol. 2018;73:34–51. doi:10.1016/j.matbio.2018.01.018
106. Dahl R, Titlestad I, Lindqvist A, et al. Effects of an oral MMP-9 and −12 inhibitor, AZD1236, on biomarkers in moderate/severe COPD: a randomised controlled trial. Pulm Pharmacol Ther. 2012;25(2):169–177. doi:10.1016/j.pupt.2011.12.011
107. Baggio C, Velazquez JV, Fragai M, Nordgren TM, Pellecchia M. Therapeutic targeting of MMP-12 for the treatment of chronic obstructive pulmonary disease. J Med Chem. 2020;63(21):12911–12920. doi:10.1021/acs.jmedchem.0c01285
108. Li H, Ezra DG, Burton MJ, Bailly M. Doxycycline prevents matrix remodeling and contraction by trichiasis-derived conjunctival fibroblasts. Invest Ophthalmol Vis Sci. 2013;54(7):4675–4682. doi:10.1167/iovs.13-11787
109. Ham J, Kim J, Ko YG, Kim HY. The dynamic contribution of neutrophils in the chronic respiratory diseases. Allergy Asthma Immunol Res. 2022;14(4):361–378. doi:10.4168/aair.2022.14.4.361
110. Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–2004. doi:10.1126/science.277.5334.2002
111. Kuna P, Jenkins M, O’Brien CD, Fahy WA. AZD9668, a neutrophil elastase inhibitor, plus ongoing budesonide/formoterol in patients with COPD. Respir Med. 2012;106(4):531–539. doi:10.1016/j.rmed.2011.10.020
112. Vogelmeier C, Aquino TO, O’Brien CD, Perrett J, Gunawardena KA. A randomised, placebo-controlled, dose-finding study of AZD9668, an oral inhibitor of neutrophil elastase, in patients with chronic obstructive pulmonary disease treated with tiotropium. COPD. 2012;9(2):111–120. doi:10.3109/15412555.2011.641803
113. Voynow JA, Zheng S, Kummarapurugu AB. Glycosaminoglycans as multifunctional anti-elastase and anti-inflammatory drugs in cystic fibrosis lung disease. Front Pharmacol. 2020;11:1011. doi:10.3389/fphar.2020.01011
114. Voynow JA, Shinbashi M. Neutrophil elastase and chronic lung disease. Biomolecules. 2021;11(8):1065. doi:10.3390/biom11081065
115. Shute JK, Calzetta L, Cardaci V, Di Toro S, Page CP, Cazzola M. Inhaled nebulised unfractionated heparin improves lung function in moderate to very severe COPD: a pilot study. Pulm Pharmacol Ther. 2018;48:88–96. doi:10.1016/j.pupt.2017.10.001
116. Margaroli C, Madison MC, Viera L, et al. An in vivo model for extracellular vesicle-induced emphysema. JCI Insight. 2022;7(4):e153560. doi:10.1172/jci.insight.153560
117. Crocetti L, Quinn MT, Schepetkin IA, Giovannoni MP. A patenting perspective on human neutrophil elastase (HNE) inhibitors (2014–2018) and their therapeutic applications. Expert Opin Ther Pat. 2019;29(7):555–578. doi:10.1080/13543776.2019.1630379
118. Palmér R, Mäenpää J, Jauhiainen A, et al. Dipeptidyl peptidase 1 inhibitor AZD7986 induces a sustained, exposure-dependent reduction in neutrophil elastase activity in healthy subjects. Clin Pharmacol Ther. 2018;104(6):1155–1164. doi:10.1002/cpt.1053
119. Miller BE, Mayer RJ, Goyal N, et al. Epithelial desquamation observed in a Phase I study of an oral cathepsin C inhibitor (GSK2793660). Br J Clin Pharmacol. 2017;83(12):2813–2820. doi:10.1111/bcp.13398
120. Chalmers JD, Haworth CS, Metersky ML, et al. Phase 2 trial of the DPP-1 inhibitor brensocatib in bronchiectasis. N Engl J Med. 2020;383(22):2127–2137. doi:10.1056/NEJMoa2021713
121. Long MB, Chalmers JD. Treating neutrophilic inflammation in airways diseases. Arch Bronconeumol. 2022;58(6):463–465. doi:10.1016/j.arbres.2021.11.003
122. Kim WD, Chi HS, Choe KH, Kim WS, Hogg JC, Sin DD. The role of granzyme B containing cells in the progression of chronic obstructive pulmonary disease. Tuberc Respir Dis. 2020;83(Supple 1):S25–S33. doi:10.4046/trd.2020.0089
123. Afonina IS, Tynan GA, Logue SE, et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol Cell. 2011;44(2):265–278. doi:10.1016/j.molcel.2011.07.037
124. Aslam MS, Yuan L. Serpina3n: potential drug and challenges, mini review. J Drug Target. 2020;28(4):368–378. doi:10.1080/1061186X.2019.1693576
125. Marcet-Palacios M, Ewen C, Pittman E, et al. Design and characterization of a novel human Granzyme B inhibitor. Protein Eng Des Sel. 2015;28(1):9–17. doi:10.1093/protein/gzu052
126. Shen Y, Zeglinski MR, Turner CT, et al. Topical small molecule granzyme B inhibitor improves remodeling in a murine model of impaired burn wound healing. Exp Mol Med. 2018;50(5):1–11. doi:10.1038/s12276-018-0095-0
127. Hirst CE, Buzza MS, Bird CH, et al. The intracellular granzyme B inhibitor, proteinase inhibitor 9, is up-regulated during accessory cell maturation and effector cell degranulation, and its overexpression enhances CTL potency. J Immunol. 2003;170(2):805–815. doi:10.4049/jimmunol.170.2.805
128. Du W, Mohammadpour H, O’Neill RE, et al. Serine protease inhibitor 6 protects alloreactive T cells from Granzyme B-mediated mitochondrial damage without affecting graft-versus-tumor effect. Oncoimmunology. 2017;7(3):e1397247. doi:10.1080/2162402X.2017.1397247
129. Ikram S, Ahmad J, Durdagi S. Screening of FDA approved drugs for finding potential inhibitors against Granzyme B as a potent drug-repurposing target. J Mol Graph Model. 2020;95:107462. doi:10.1016/j.jmgm.2019.107462
130. Cazzola M, Stolz D, Rogliani P, Matera MG. α1-Antitrypsin deficiency and chronic respiratory disorders. Eur Respir Rev. 2020;29(155):190073. doi:10.1183/16000617.0073-2019
131. Stolk J, Tov N, Chapman KR, et al. Efficacy and safety of inhaled α1-antitrypsin in patients with severe α1-antitrypsin deficiency and frequent exacerbations of COPD. Eur Respir J. 2019;54(5):1900673. doi:10.1183/13993003.00673-2019
132. Tumpara S, Martinez-Delgado B, Gomez-Mariano G, et al. The delivery of α1-antitrypsin therapy through transepidermal route: worthwhile to explore. Front Pharmacol. 2020;11:983. doi:10.3389/fphar.2020.00983
133. Remih K, Amzou S, Strnad P. Alpha1-antitrypsin deficiency: new therapies on the horizon. Curr Opin Pharmacol. 2021;59:149–156. doi:10.1016/j.coph.2021.06.001
134. Zou Y, Chen X, He B, et al. Neutrophil extracellular traps induced by cigarette smoke contribute to airway inflammation in mice. Exp Cell Res. 2020;389(1):111888. doi:10.1016/j.yexcr.2020.111888
135. Trivedi A, Khan MA, Bade G, Talwar A. Orchestration of neutrophil extracellular traps (NETs), a unique innate immune function during chronic obstructive pulmonary disease (COPD) development. Biomedicines. 2021;9(1):53. doi:10.3390/biomedicines9010053
136. Grabcanovic-Musija F, Obermayer A, Stoiber W, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res. 2015;16(1):59. doi:10.1186/s12931-015-0221-7
137. Keir HR, Chalmers JD. Neutrophil extracellular traps in chronic lung disease: implications for pathogenesis and therapy. Eur Respir Rev. 2022;31(163):210241. doi:10.1183/16000617.0241-2021
138. Tanner L, Single AB. Animal Models Reflecting chronic obstructive pulmonary disease and related respiratory disorders: translating pre-clinical data into clinical relevance. J Innate Immun. 2020;12(3):203–225. doi:10.1159/000502489
139. Segreti A, Stirpe E, Rogliani P, Cazzola M. Defining phenotypes in COPD: an aid to personalized healthcare. Mol Diagn Ther. 2014;18(4):381–388. doi:10.1007/s40291-014-0100-9
140. Xu X, Huang K, Dong F, et al. The heterogeneity of inflammatory response and emphysema in chronic obstructive pulmonary disease. Front Physiol. 2021;12:783396. doi:10.3389/fphys.2021.783396
141. Calzetta L, Aiello M, Frizzelli A, et al. Stem cell-based regenerative therapy and derived products in COPD: a systematic review and meta-analysis. Cells. 2022;11(11):1797. doi:10.3390/cells11111797
142. Martinez-Garcia MA, Sierra-Párraga JM, Quintana E, López-Campos JL. CFTR dysfunction and targeted therapies: a vision from non-cystic fibrosis bronchiectasis and COPD. J Cyst Fibros. 2022;21(5):741–744. doi:10.1016/j.jcf.2022.04.018
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.