Back to Archived Journals » Metalloproteinases In Medicine » Volume 2
Nonproteolytic functions of matrix metalloproteinases in pathology and insights for the development of novel therapeutic inhibitors
Authors García-Pardo A, Opdenakker G
Received 24 January 2015
Accepted for publication 24 February 2015
Published 6 May 2015 Volume 2015:2 Pages 19—28
DOI https://doi.org/10.2147/MNM.S63629
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor William Parks
Angeles García-Pardo,1 Ghislain Opdenakker2
1Department of Cellular and Molecular Medicine, Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain; 2Rega Institute for Medical Research, Department of Microbiology and Immunology, University of Leuven, Leuven, Belgium
Abstract: Structural selectivity – in fact, the lack thereof – has been invoked as an explanation for the failure of matrix metalloproteinase (MMP) inhibitors as oncology drugs. However, functional selectivity is needed to develop a good drug. In addition, many drugs (including in oncology) act by interfering with signaling functions. The present market of successful biologicals contains many monoclonal antibodies, such as signaling inhibitors, with antitumor necrosis factor (anti-TNF) being the flagship of an armada. However, aside from its many pathogenic functions, TNF also plays physiological (ie, beneficial) roles. As long as the inhibition of detrimental functions supersedes the negative side effects, anti-TNF will be used. For such reasons, it is critical to know all the functions of MMPs, ideally before inhibitors are used as drugs. Here, we briefly summarize the known catalytic MMP functions and focus on the noncatalytic roles of these proteins, with an emphasis on their signaling effects. Indeed, recent studies have addressed the biology of multimolecular signaling complexes containing MMPs and the tissue inhibitors of metalloproteinases. These complexes are observed in solution (eg, as heteromers or homomultimers) and at the cell surfaces (eg, as docking complexes and signaling receptors). Consequently, a good understanding of the broader contexts – from the molecular, to the cellular and tissue levels – in which such molecular complexes operate will provide essential insights into direct new drug developments. This is exemplified with clinical and recent preclinical successes.
Keywords: MMPs, signaling pathways, PEX domain, noncatalytic function
Introduction
The success of a molecule often depends on its name and, even with an attractive name, one can be wrong in terms of its applications. A few decades ago, two similar molecules with related receptor molecules, converging signal transduction cascades, and matching biological effects were identified and named tumor necrosis factor (TNF) and lymphotoxin (LT). “TNF” alluded to the beneficial effect of destroying cancer cells,1 whereas “LT” emphasized toxicity.2 Meanwhile, TNF has been cited about 150,000 times in the PubMed data library, whereas LT is creeping toward 4,000 citations. In addition, these names yielded the wrong perceptions. Indeed, it was the inhibition of TNF, in the form of neutralizing monoclonal antibodies and resulting in immunosuppression or toxicity toward autoantigen-specific T-lymphocytes, that entered clinical practice.3 Similar simple reasoning can be made for matrix metalloproteinases (MMPs). The medical literature contains an increasing number of manuscripts illustrating that the launched concepts and clinical uses of MMP inhibitors against cancer cell invasion and metastasis were too simple and, in fact, wrong.4–7
Although some activities of MMPs were reflected by their functional names, such as collagenases, stromelysins, gelatinases, and membrane-type (MT)-MMPs (Figure 1), we should humbly accept that their most recent nomenclature is not appealing, sometimes confusing for novices (where are MMP-4, MMP-5, and MMP-6?), and certainly not reflecting their noncatalytic functions. The main biochemical (domain structure, mechanism of substrate proteolysis, and regulation of catalysis at the protein level) and biological characteristics of human MMPs as enzymes have been the subject of excellent reviews on cancer8–10 and on inflammatory and vascular diseases.11–14 Catalytic functions are also illustrated by manuscripts on the processing of substrates by specific enzymes.15–17 In addition, useful reviews about membrane-bound and intracellular MMP substrates exist.18,19
 | Figure 1 Schematic drawing depicting the domain structure of MMPs. |
Recent developments of enzyme-based degradomics – ie, the definition of all substrates of one specific enzyme20,21 – and substrate-based reverse degradomics, in which all proteases acting on a single substrate are defined,22 illustrate renewed interests in proteolysis and its complexities. These approaches led to a conceptual change: from proteases acting in linear cascades (eg, clotting, fibrinolysis, and complement cascades)23 to the protease network, which was first elaborated in detail for all known interactions between serine proteases and MMPs in 200224 and further developed into the protease net17,21,25 or protease web.7 The network concept makes us better understand why the entire system is complex. However, such concepts are currently also used to develop signaling inhibitors by the definition of the most critical nodes and connections in signaling networks. Consequently, real hope exists that, by (reverse) degradomics analysis and by deciphering the MMP network nodes, inhibitors of specific key proteases may become useful drugs.
Here, however, the emphasis will be on the noncatalytic functions of mammalian MMPs. These functions are based on and include covalent and noncovalent interactions with heteromers and homomultimers, which bind onto cell surfaces and soluble molecular complexes, as well as interactions with cognate receptors with ensuing signaling events. Finally, we briefly address how “cascadic signaling” in life-threatening conditions involves MMPs and how such knowledge may give new turns towards the use of existing MMP inhibitors, developed against cancer, to become life-saving drugs for lethal inflammatory conditions, such as sepsis, endotoxinemia and superantigen-induced shock syndromes.
Structural features of MMPs – a brief overview
The primary structure of MMPs has been addressed in previous reviews.12,14,18,26 However, it is the tridimensional structure of glycoproteins that determines their functions. Although the crystal structures of some MMPs are known and were compiled long ago in a seminal review that also addressed molecular evolution,27 the determination of the secondary and tertiary structures of a number of MMPs remains a challenge in structural biology. For example, the full-size structures of MMP-9, as the most studied proteinase in the MMP family,28 and of all MT-MMPs, are not known. Therefore, theoretical models are presently used as a surrogate with the aim of understanding the interactions with substrates, inhibitors,27 receptors,29–31 proteoglycans,32 and other MMP ligands.33 As an example, full-size MMP-9 occurs in multiple forms: monomers; homomultimers; and heteromers.28 These molecular forms are endowed with different functions, as we recently demonstrated for the monomeric and trimeric form of MMP-9.34 In addition, when produced by specific cells, MMP-9 forms a covalent heteromer with neutrophil gelatinase B-associated lipocalin. This soluble form of MMP-9 links the functionalities of MMP-9 with those of the lipocalin.28
All MMPs are composed of several structural domains with distinct functions and are synthesized as catalytically inactive proforms (Figure 1). The simplest structure is represented by matrilysins (MMP-7, MMP-26), which contain the signal peptide and propeptide, as well as the catalytic domain. The rest of the MMPs contain a carboxy-terminal hemopexin domain (also known as PEX), which is bound to the catalytic part of the molecule by the so-called hinge region. Because of the focus of this review, we will dedicate a separate section to the hemopexin domain. MMP-2 and MMP-9 contain an additional region consisting of fibronectin-like repeats, responsible for the gelatin-binding activity of these MMPs. MMP-9 also contains a unique O-glycosylated domain,31 which confers flexibility to the molecule and appears to be indispensable for MMP-9 functions.28 The MT-MMPs contain either a glycosylphosphatidylinositol membrane anchor or a transmembrane and cytoplasmic domain (Figure 1). Both types of anchors serve to localize MT-MMPs to cell surfaces. The catalytic and carboxy-terminal domains (hemopexin and cytoplasmic) are therefore separate entities in the MMP molecule and, as we will define, they perform different and crucial functions in MMPs.
Targeting the catalytic domain of MMPs: from twice thinking small to integration and considering big
Originally, the catalytic function of proteases, such as plasminogen activators and MMPs, in cancer cell invasion and metastasis was viewed as the sole property of these glycoproteins. Hence, their inhibition was regarded as possible, preferably with orally active small drugs. This simple reasoning boosted enormous research on these proteases and generated excellent inhibitors. The integration of basic, preclinical, and clinical studies, however, showed that this view was narrow-sighted and that the developed drugs, by their small sizes, interacted with many proteases and hence possessed poor selectivity and yielded side effects during their use as cancer therapeutics.4–8,10–14 We and others tried to counter these negative results with a more optimistic and positive view because these small drugs may become excellent candidates for the treatment of life-threatening acute inflammation, in which side effects may be of secondary importance.12–14
The problem of low selectivity was considered, and large molecules – namely, inhibitory monoclonal antibodies – were also developed into inhibitors of MMP catalysis.35,36 It was demonstrated long ago that with such large-sized inhibitors, it is possible to reach high selectivity. For instance, the monoclonal antibody REGA-3G12 was the first reagent with selective inhibition of activated MMP-9 (and not of MMP-2).36 In a subsequent study, the interaction site between REGA-3G12 and its antigen MMP-9 was studied and found to consist of part of the catalytic site and an area aside this.37 These findings are in line with the concept of exosite interactions to obtain higher selectivity, as nicely promoted by several studies.38–40 These studies are examples of the integration of structural data and thinking beyond the small catalytic pocket of a proteolytic enzyme. In addition, the generation of activity-neutralizing monoclonal antibodies against MMPs has recently been further developed and the obtained reagents have been preclinically used to treat experimentally-induced colitis in mice.40 In this case, and if we think big about novel treatments of (invasive) cancer, the successful monoclonal antibody treatment of an inflammatory disease in a mouse model will hopefully pave the way to new successes when such, or other, catalytic inhibitors are tested in cancer models.41
Noncatalytic functions of MMPs
An increasing number of studies are demonstrating that MMPs can also perform functions independently of their proteolytic activity, thus providing additional ways by which MMPs may contribute to pathology. Nonproteolytic functions mostly rely on the localization of MMPs at the cell surface which, in the case of MT-MMPs, is mediated by their transmembrane and cytoplasmic domains or by their glycosylphosphatidylinositol membrane anchor (Figure 1). MMPs devoid of these structures can still bind to the cell surface via specific receptors; among these are the integrins and CD44 (Table 1).29–31,42,43 This receptor-binding function, which generally requires the MMP hemopexin domain, was thought to exclusively serve to activate/inhibit MMPs and to localize their catalytic activity to the pericellular environment. However, numerous recent evidences are changing this concept and supporting the fact that MMPs can also induce cell signaling upon interaction with their cell surface receptors.
 | Table 1 Molecular interactions and biological effects involving noncatalytic MMP domains |
Nonproteolytic functions of MMP-1, MMP-2, and MMP-3
Initial studies by Conant et al44 showed that MMP-1 binding to α2β1 integrin in neural cells and monocytes elicits a signaling pathway sensitive to pertussis toxin and results in the release of MMP-9. The same effect is observed when proMMP-1 is used or by inhibiting MMP-1 with GM-6001, indicating that the enzymatic activity of MMP-1 is not required. Additionally, the proMMP-1/α2β1 integrin interaction also induces Akt dephosphorylation and neuron cell death.45 This effect is blocked by an antibody against α2 integrin and it is independent of the MMP-1 proteolytic activity since 1) total Akt levels remain unchanged, and 2) batimastat, an inhibitor of the catalytic activity of MMP-1, does not prevent proMMP-1-induced Akt dephosphorylation. Similarly, proMMP-2 interaction with αVβ3 integrin on lung adenocarcinoma cells induces vascular endothelial growth factor (VEGF) expression via activation of PI3K/Akt/HIF-1α, leading to increased angiogenesis.46 In MMP-3, the hemopexin domain binds to the chaperone heat-shock protein 90β (HSP90β) extracellularly, and this is critical for mouse mammary epithelial cell invasion.47 Using several structural mutants of MMP-3, the authors demonstrate that the hemopexin domain is required for the invasive function of MMP-3 during branching morphogenesis. This is also demonstrated in primary organoids of the mammary gland. The mechanism by which MMP-3 overexpression induces mammary tumors in transgenic mice was elegantly demonstrated by Kessenbrock et al.48 Using lentiviral constructs containing MMP-3, a proteolytically inactive mutant, or the MMP-3 PEX domain, these authors demonstrate that these three proteins induce a hyperbranching phenotype equally well upon implanting into mammary fat pads. This was due to the specific interaction of the MMP-3 PEX domain with the extracellular noncanonical Wnt ligand Wnt5b, resulting in its inactivation. MMP-3 thus regulates Wnt signaling and adult epithelial stem cell function via the PEX domain in a noncatalytic manner.
Nonproteolytic functions of MMP-9
Several reports have addressed the notion of intracellular signaling induced upon MMP-9’s interaction with its cell surface receptors. Binding of MMP-9 or a fusion protein containing the hemopexin domain of MMP-9 (PEX9), coupled to GFP, to the low-density lipoprotein receptor-related protein (LRP-1) in Schwann cells activates extracellular signal-regulated kinase (ERK)1/2 and Akt, and it promotes cell migration.49 This effect is blocked by inhibiting MMP-9 binding either by LRP-1 gene silencing or by an antibody targeting PEX9. The related protein LRP-2/megalin is also a receptor for MMP-9 in epithelial cells and mediates its endocytosis and catabolism (Table 1).31 MMP-9 can also bind to insulin-like growth factor-1, ERbB, and platelet-derived growth factor receptors, resulting in the regulation of Ras/Raf/MEK–ERK pathways and the controlling of critical trophic signals and phenotypic remodeling of Schwann cells.50 Whether regulation of these pathways occurs via regulatory proteolysis or by direct receptor binding (nonproteolytic) was not determined. Dufour et al51 demonstrated that transfection of COS-1 cells with MMP-2, MMP-9, or a catalytically inactive mutant of MMP-9 enhances cell migration. This effect requires the PEX9 domain, as well as MAPK and PI3K activities, but the specific mechanism involved was not elucidated. proMMP-9 has also been shown to interact with the I domain of αLβ2 and αMβ2 integrins on leukocytes, and these proMMP-9/β2 integrin complexes are important for cell migration.52,53 This interaction involves the catalytic region of proMMP-9 rather than the hemopexin domain, and it is not known whether it results in intracellular signaling. Our group has shown that binding of proMMP-9 to α4β1 integrin and 190 kDa CD44v, its docking receptors in chronic lymphocytic leukemia (CLL) cells,54 induces a signaling pathway that leads to malignant cell survival.55 This pathway consists of Lyn kinase activation, STAT3 phosphorylation, and upregulation of the antiapoptotic protein Mcl-1, a member of the Bcl-2 protein family. The same effect can be elicited by a noncatalytic mutant of proMMP-9 and by the isolated PEX9 domain. Thus, proMMP-9 contributes to CLL survival by a nonproteolytic mechanism. Dufour et al56 showed that the interaction of the proMMP-9 PEX domain with CD44 activates the tyrosine kinase epidermal growth factor receptor (EGFR) and subsequent phosphorylation of its substrates, ERK, Akt, and focal adhesion kinase (FAK), resulting in breast carcinoma cell migration. Using xenograft models in NOD/SCID mice, we recently showed that the overexpression of proMMP-9 impairs CLL cell homing to bone marrow and spleen.57 A proteolytically inactive proMMP-9 mutant had a partial effect, indicating that both catalytic and noncatalytic functions were involved. Indeed, biochemical analyses demonstrated that proMMP-9, likely complexed to α4β1 integrin, downregulates the activation of RhoAGTPase, Akt, ERK, and FAK, while it increases p190RhoGAP (a RhoA inhibitor) and PTEN (an Akt, ERK, and FAK inhibitor).57 Modulation of these pathways by proMMP-9 may contribute to malignant cell retention in lymphoid organs and CLL progression. MMP-9 has also recently been shown to promote rounded amoeboid melanoma cell migration by a noncatalytic mechanism, consisting of the regulation of actomyosin contractility via CD44.58 Because actin is a substrate of MMP-9,16 a simple reasoning would be that this rounded amoeboid effect would be influenced by catalysis. However, this suggestion is not correct because the amoeboid cell migration was also observed when using a catalytically inactive MMP-9 mutant and also when inhibiting MMP-9 activity. This mechanism operates in vivo since MMP-9 expression increases during melanoma progression and is enriched at the invasive front of lesions, correlating with cell roundness. The authors propose that MMP-9 could be an amoeboid-selective marker for melanoma and that blocking this noncatalytic MMP-9 function could help to reduce melanoma cell invasion and metastasis.58 In another study, it was found that the PEX9 domain inhibits tumor angiogenesis and indirectly blocks the growth of orthotopic glioblastoma xenografts.59
Nonproteolytic functions of MMP-14
Novel functions of MMP-14 not requiring its proteolytic activity, but instead involving the hemopexin or cytoplasmic domain, have also been reported. Proteolytic and nonproteolytic roles of MMP-14 in different cell contexts have been reviewed.9,60 MMP-14 forms a complex at the cell surface with its physiological protein inhibitor, tissue inhibitor of metalloproteinase (TIMP)-2, and binding of TIMP-2 to MMP-14 induces cell proliferation and migration of MCF-7 breast carcinoma cells.61 This effect requires the MMP-14 cytoplasmic tail, but not its catalytic activity, and it involves activation of ERK1/2. Moreover, in xenograft models in mice, a proteolytically inactive MMP-14 mutant also promoted tumor growth in vivo, while a mutant lacking the cytoplasmic tail was ineffective.61 A similar MEK/ERK signaling cascade upon TIMP-2 binding to MMP-14 was observed in HT1080 fibrosarcoma cells, resulting in increased cell migration.62 These reports thus establish that the MMP-14–TIMP-2 interaction controls cell proliferation and migration by nonproteolytic mechanisms. Macrophages from MMP-14–/– mice have a reduced ability to invade basement membranes and to infiltrate into inflammatory sites, and these effects are restored by MMP-14 re-expression on these cells.63 Importantly, this activity requires the cytoplasmic tail of MMP-14, but not its catalytic or hemopexin domains, establishing that MMP-14 regulates macrophage invasion and migration by dual catalytic and noncatalytic functions. These authors also demonstrated that MMP-14–/– macrophages are characterized by a reduction in adenosine triphosphate (ATP) concentration and in glycolytic activity.64 The reason for this is that the cytoplasmic tail of MMP-14 binds to HIF-1α and releases it from its specific inhibitors, stimulating glycolysis and ATP production by macrophages. Other functions for the MMP-14 cytoplasmic domain have also been identified. Gonzalo et al65 reported yet another new function for MMP-14 consisting of the control of myeloid cell fusion. In this case, MMP-14 induces a signaling pathway in which its cytoplasmic tail binds to p130Cas and increases Rac1 activation and lamellipodia activity, directly impacting cell morphology, motility, and fusion by a nonproteolytic mechanism. In a more recent report, Mori et al66 demonstrated an association between MMP-14 and β1 integrin in mammary epithelial cells. This association modulates β1 integrin levels, activates ERK, and induces branching morphogenesis in collagen 1 gels and primary mammary organoids. Using several MMP-14 deletion mutants, these authors show that the transmembrane/cytoplasmic domain of MMP-14, but not its catalytic domain, is required for these functions. Collectively, these studies clearly expand the functions of MMPs beyond those involving substrate degradation. While the MMP enzymatic activity is certainly crucial to promote cell migration, invasion, and survival, we may conclude on the basis of various examples that MMPs also induce and/or modulate the signaling pathways necessary for these processes. This knowledge opens new avenues to explore and identify targets in MMPs outside the catalytic domain.
The hemopexin domain of MMPs as an emerging therapeutic target
The carboxy-terminal region of MMPs (hemopexin domain, PEX) has, for a long time, been recognized as an interesting module able to interact with several molecules and display multiple properties.67 Besides containing binding sites for TIMPs, gelatin, and other MMPs, PEX is required for MMP binding to cell surface receptors67 and plays an important role in the activation of signaling pathways, many of which have been described earlier. Because several of these signals are associated with pathological processes, the PEX domain – in particular, its interaction sites with cell surface receptors – has emerged as a novel therapeutic target in MMPs. This is further substantiated by the reported properties of the isolated PEX domain mentioned earlier for MMP-348 and MMP-9.55,59 The PEX domain from MMP-2 also inhibits tumor growth in an in vivo model of mouse glioma, together with a decrease in angiogenesis and cell proliferation.68 Additionally, the isolated murine PEX9 domain inhibits MMP-9 gelatin-binding activity and the invasion of melanoma cells,69 the adhesion and migration of colorectal cancer cells,70 as well as VEGF secretion, angiogenesis, and tumor growth in a glioblastoma animal model.59 These studies provide evidence that MMPs, through their PEX domain, can perform many nonproteolytic functions.
Crystal structure analyses71–75 were used to demonstrate that hemopexin domains consist of a four-bladed β-propeller structure (blades 1–4) (shown in Figure 2A for PEX9). The primary structure homology among the hemopexin domains of different MMPs is rather low (25%–30%),67 and this fact represents a major advantage when considering targeting this region in a particular MMP. This low homology contrasts with the existing higher resemblance among MMP catalytic domains.27 Strategies aimed to block the MMP hemopexin domain are already in progress. Using phage display analyses, Björklund et al76 identified a synthetic peptide (CRVYGPYLLC) that binds to the PEX9 domain and inhibits the association of MMP-9 with αVβ5 integrin in fibrosarcoma cells. This peptide also inhibits cell migration in vitro and tumor xenograft growth in vivo. Dufour et al56 designed the synthetic peptides SRPQGPFL and NQVDQVGY, mimicking motifs in the outermost strands of blades 1 and 4 of PEX9, respectively. According to the authors, these peptides inhibited MMP-9 dimerization and the migration of fibrosarcoma and carcinoma cells. Additionally, peptide SRPQGPFL blocked the interaction of MMP-9 with CD44, suggesting that this sequence was involved in MMP-9 docking at the cell surface. The same group reported similar findings for the hemopexin domain of MMP-14 (PEX14).77 In this case, the synthetic peptides, MVPDPMYG and GYPKSALR, containing sequences in the outermost strands of blade 1 and blade 4, respectively, inhibited carcinoma cell migration, tumor metastasis, and angiogenesis. By preparing truncated proteins containing GST fused to structural blades 1–2 or 3–4 of PEX9, and overlapping synthetic peptides corresponding to the entire PEX9 sequence, we have defined the PEX9 regions responsible for binding of proMMP-9 to CLL cells. Blades 1–2 are important for the interaction with CD44 and blades 3–4 for binding to α4β1 integrin.78,79 The specific binding sites involved in these interactions are the sequences FDAIAEIGNQLYLFKDGKYW, present in blade 1 and contained in peptide P6, and FPGVPLDTHDVFQYREKAYFC, present in blade 4 and contained in peptide P3.78,79 P6 and P3 bind, respectively, to CD44 and α4β1 integrin (Figure 2B) and partially inhibit CLL cell adhesion and transendothelial migration. P3 also blocks the Lyn/Mcl-1 survival pathway elicited by proMMP-9 or GST–PEX9 upon binding to CLL cells.55 Importantly, the combination of P3 and P6 is synergistic and results in the complete inhibition of cell adhesion and migration. This is in agreement with the spatial localization of both peptides within the central cavity of PEX9 (Figure 2B).78,79 The P3 and P6 sequences thus represent two potential targets to prevent proMMP-9 binding to CLL cells and subsequent pathological consequences. Peptide-based inhibitors targeting exosites and/or both exosites and active sites may therefore constitute an efficient way to block catalytic and noncatalytic MMP activities, such as the regulation of cell adhesion, migration, and signaling. The already identified peptides with inhibitory action mainly on MMP-9 and MMP-14 have been recently reviewed.80
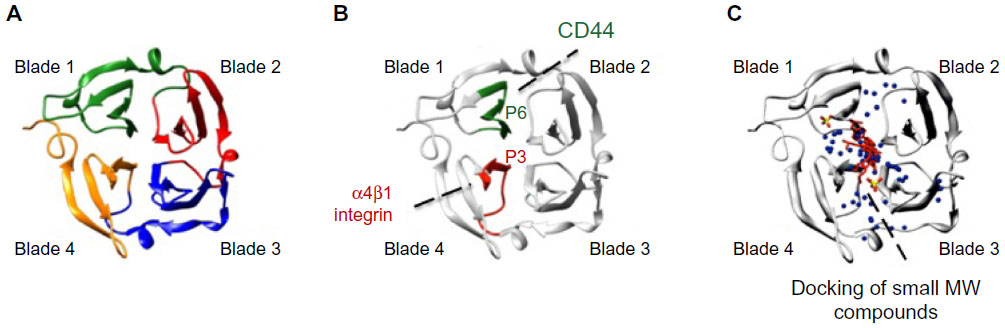 | Figure 2 Ribbon diagram of the monomeric hemopexin domain of MMP-9 (PEX9) showing the location of drug target sites. |
Besides the synthetic peptide strategies, in silico approaches are also being employed to target the MMP hemopexin domain. Using chemoinformatics-based analyses, Kothapalli et al81 found several molecules that specifically bind to the hemopexin domain of MMP-13. This information helped to design drugs to inhibit this MMP, particularly in inflammatory disorders. In silico docking approaches have also served to identify two small molecules that selectively bind to PEX9 and inhibit tumor growth and metastasis without affecting the MMP-9 catalytic activity.82 One of the compounds found in this study (compound 2) also blocks MMP-9 homodimerization and ERK1/2 phosphorylation, and the authors proposed that this is likely due to the inhibition of the binding of MMP-9 to CD44 and the subsequent activation of the EGFR–MAPK signaling pathway.82 A similar approach was employed by Remacle et al83 to identify a small compound that binds to the PEX domain of MMP-14 and represses the protumorigenic function of MMP-14 in a carcinoma xenograft model. Importantly, the docking analyses mapped the binding sites of these small molecule inhibitors to the central cavity of PEX9 and PEX14, respectively; in fact, they were in close proximity to the P3 and P6 sequences identified by us in PEX9 (Figure 2C). These findings highlight the central cavity of PEX as a prime target in future strategies aimed to overcome MMP pathologic signaling functions in cancer, inflammation, and other disorders.
Targeting the MMP hemopexin domain is therefore a promising alternative to previous attempts aimed at blocking the catalytic activity of MMPs and, in view of the lower homologies of various MMP hemopexin domains versus catalytic domains, this has the added advantage of higher selectivity. Another interesting advantage comes from studies in which targeting the hemopexin domain impairs the catalytic activity of MMPs. Using phage display techniques, Basu et al84 generated recombinant antibody fragments against PEX14 that significantly reduced the degradation of collagen type I by MMP-14 and inhibited CD44 shedding by MMP-14-expressing fibrosarcoma cells. These antibodies also inhibit invasion and angiogenesis in in vitro systems. Although it is not clear whether the antibody effect is directly affecting interactions with MMP-14 substrates or imposing structural restrictions that affect MMP-14 function, the results certainly highlight the hemopexin domain as a useful target to modulate MMP functions.
Conclusion
Previous attempts to block the pathological functions of MMPs by targeting the catalytic domain were disappointing in oncology studies and, therefore, the idea of targeting MMPs in invasive cancer was abandoned. The efforts to generate more selective inhibitors have been revitalized with the exosite concept in which larger areas of the catalytic domain are targeted with new drugs.85 Moreover, recent studies pointed to other regions of MMPs as promising targets to block the pathological activities of MMPs. In particular, the carboxy-terminal domains, both the hemopexin domain and the intracellular segments of MT-MMPs, have been studied as targets for inhibition. In these cases, the binding and signaling functions of MMPs, rather than their catalytic effects, are targeted. Two major directions are becoming clear in this area of research. First, membrane-bound MMPs (eg, MMP-14) signal through their intracellular domains by interaction with cytoplasmic adaptor molecules. Secondly, the hemopexin domain of secreted MMPs, such as MMP-347,48 and MMP-9,55,57 activates signaling cascades by interacting with signaling receptors (eg, integrins) or by being part of oligosaccharide lectin interactions (eg, CD44 [hyaluronan receptor] and chondroitin sulfate proteoglycans).32 For both types of signaling interactions, peptides and/or small molecule inhibitors have been developed. These studies generate the necessary insights for the development of novel therapeutics and form a complementary research route, alongside catalysis inhibitors, in the fight against cancer and other diseases.
Acknowledgments
We thank Irene Amigo-Jiménez for her valuable help with the preparation of the figures and table. Work from the authors’ laboratories was supported by grants from the Ministry of Economy and Competitiveness, Spain (SAF2009-07035; SAF2012-31613; RTICC RD12/0036/0061) and by the Comunidad de Madrid/European Union (S2010/BMD-2314-Neoplasbim) to AGP. GO is supported by the Concerted Research Actions (GOA/13/014) at KU Leuven, by the Fund for Scientific Research of Flanders (FWO-Vlaanderen), and by the European FP7/2007-2013 Cancer Research Project Save-Me under grant agreement number 263307.
Disclosure
The authors report no conflicts of interest in this work.
References
Helson L, Green S, Carswell E, Old LJ. Effect of tumour necrosis factor on cultured human melanoma cells. Nature. 1975;258(5537):731–732. | |
Oppenheim JJ, Blaese RM, Horton JE, Thor DE, Granger GA. Production of macrophage migration inhibitory factor and lymphotoxin by leukocytes from normal and Wiskott-Aldrich syndrome patients. Cell Immunol. 1973;8(1):62–70. | |
Sedger LM, McDermott MF. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants – past, present and future. Cytokine Growth Factor Rev. 2014;25(4):453–472. | |
Overall CM, Kleifeld O. Tumour microenvironment – opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6(3):227–239. | |
Martin MD, Matrisian LM. The other side of MMPs: protective roles in tumor progression. Cancer Metastasis Rev. 2007;26(3–4):717–724. | |
Fingleton B. MMPs as therapeutic targets – still a viable option? Semin Cell Dev Biol. 2008;19(1):61–68. | |
Krüger A, Kates RE, Edwards DR. Avoiding spam in the proteolytic internet: future strategies for anti-metastatic MMP inhibition. Biochim Biophys Acta. 2010;1803(1):95–102. | |
Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25(1):9–34. | |
Itoh Y, Seiki M. MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol. 2006;206(1):1–8. | |
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. | |
Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4(8):617–629. | |
Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6(6):480–498. | |
Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. 2013;13(9):649–665. | |
Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014;13(12):904–927. | |
Xu D, Suenaga N, Edelmann MJ, Fridman R, Muschel RJ, Kessler BM. Novel MMP-9 substrates in cancer cells revealed by a label-free quantitative proteomics approach. Mol Cell Proteomics. 2008;7(11):2215–2228. | |
Cauwe B, Martens E, Proost P, Opdenakker G. Multidimensional degradomics identifies systemic autoantigens and intracellular matrix proteins as novel gelatinase B/MMP-9 substrates. Integr Biol (Camb). 2009;1(5–6):404–426. | |
Prudova A, auf dem Keller U, Butler GS, Overall CM. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol Cell Proteomics. 2010; 9(5):894–911. | |
Cauwe B, Van den Steen PE, Opdenakker G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit Rev Biochem Mol Biol. 2007;42(3):113–185. | |
Cauwe B, Opdenakker G. Intracellular substrate cleavage: a novel dimension in the biochemistry, biology and pathology of matrix metalloproteinases. Crit Rev Biochem Mol Biol. 2010;45(5):351–423. | |
López-Otín C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. 2002;3(7):509–519. | |
Butler GS, Overall CM. Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry. 2009;48(46):10830–10845. | |
Piccard H, Hu J, Fiten P, et al. “Reverse degradomics”, monitoring of proteolytic trimming by multi-CE and confocal detection of fluorescent substrates and reaction products. Electrophoresis. 2009;30(13):2366–2377. | |
Cuzner ML, Opdenakker G. Plasminogen activators and matrix metalloproteases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J Neuroimmunol. 1999; 94(1–2):1–14. | |
Van den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit Rev Biochem Mol Biol. 2002;37(6):375–536. | |
Rodríguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803(1):39–54. | |
Murphy G, Nagase H. Progress in matrix metalloproteinase research. Mol Aspects Med. 2008;29(5):290–308. | |
Massova I, Kotra LP, Fridman R, Mobashery S. Matrix metalloproteinases: structures, evolution, and diversification. FASEB J. 1998;12(12):1075–1095. | |
Vandooren J, Van den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol. 2013;48(3):222–272. | |
Emonard H, Bellon G, Troeberg L, et al. Low density lipoprotein receptor-related protein mediates endocytic clearance of pro-MMP-2.TIMP-2 complex through a thrombospondin-independent mechanism. J Biol Chem. 2004;279(52):54944–54951. | |
Hahn-Dantona E, Ruiz JF, Bornstein P, Strickland DK. The low density lipoprotein receptor-related protein modulates levels of matrix metalloproteinase 9 (MMP-9) by mediating its cellular catabolism. J Biol Chem. 2001;276(18):15498–15503. | |
Van den Steen PE, Van Aelst I, Hvidberg V, et al. The hemopexin and O-glycosylated domains tune gelatinase B/MMP-9 bioavailability via inhibition and binding to cargo receptors. J Biol Chem. 2006;281(27):18626–18637. | |
Malla N, Berg E, Theocharis AD, Svineng G, Uhlin-Hansen L, Winberg JO. In vitro reconstitution of complexes between pro-matrix metalloproteinase-9 and the proteoglycans serglycin and versican. FEBS J. 2013;280(12):2870–2887. | |
Monferran S, Paupert J, Dauvillier S, Salles B, Muller C. The membrane form of the DNA repair protein Ku interacts at the cell surface with metalloproteinase 9. EMBO J. 2004;23(19):3758–3768. | |
Vandooren J, Born B, Solomonov I, et al. Circular trimers of gelatinase B/matrix metalloproteinase-9 constitute a distinct population of functional enzyme molecules differentially regulated by tissue inhibitor of metalloproteinases-1. Biochem J. 2015;465(2):259–270. | |
Ramos-DeSimone N, Moll UM, Quigley JP, French DL. Inhibition of matrix metalloproteinase 9 activation by a specific monoclonal antibody. Hybridoma. 1993;12(4):349–363. | |
Paemen L, Martens E, Masure S, Opdenakker G. Monoclonal antibodies specific for natural human neutrophil gelatinase B used for affinity purification, quantitation by two-site ELISA and inhibition of enzymatic activity. Eur J Biochem. 1995;234(3):759–765. | |
Martens E, Leyssen A, Van Aelst I, et al. A monoclonal antibody inhibits gelatinase B/MMP-9 by selective binding to part of the catalytic domain and not to the fibronectin or zinc binding domains. Biochim Biophys Acta. 2007;1770(2):178–186. | |
Sela-Passwell N, Rosenblum G, Shoham T, Sagi I. Structural and functional bases for allosteric control of MMP activities: can it pave the path for selective inhibition? Biochim Biophys Acta. 2010;1803(1):29–38. | |
Udi Y, Grossman M, Solomonov I, et al. Inhibition mechanism of membrane metalloprotease by an exosite-swiveling conformational antibody. Structure. 2015;23(1):104–115. | |
Sela-Passwell N, Kikkeri R, Dym O, et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat Med. 2012;18(1):143–147. | |
Fields GB. New strategies for targeting matrix metalloproteinases. Matrix Biol. Epub January 14, 2015. pii: S0945-053X(15)00003-7. | |
Fridman R, Toth M, Chvyrkova I, Meroueh SO, Mobashery S. Cell surface association of matrix metalloproteinase-9 (gelatinase B). Cancer Metastasis Rev. 2003;22(2–3):153–166. | |
Stefanidakis M, Koivunen E. Cell-surface association between matrix metalloproteinases and integrins: role of the complexes in leukocyte migration and cancer progression. Blood. 2006;108(5):1441–1450. | |
Conant K, Haughey N, Nath A, et al. Matrix metalloproteinase-1 activates a pertussis toxin-sensitive signaling pathway that stimulates the release of matrix metalloproteinase-9. J Neurochem. 2002;82(4):885–893. | |
Conant K, St Hillaire C, Nagase H, et al. Matrix metalloproteinase 1 interacts with neuronal integrins and stimulates dephosphorylation of Akt. J Biol Chem. 2004;279(9):8056–8062. | |
Chetty C, Lakka SS, Bhoopathi P, Rao JS. MMP-2 alters VEGF expression via alphaVbeta3 integrin-mediated PI3K/AKT signaling in A549 lung cancer cells. Int J Cancer. 2010;127(5):1081–1095. | |
Correia AL, Mori H, Chen EI, Schmitt FC, Bissell MJ. The hemopexin domain of MMP3 is responsible for mammary epithelial invasion and morphogenesis through extracellular interaction with HSP90β. Genes Dev. 2013;27(7):805–817. | |
Kessenbrock K, Dijkgraaf GJ, Lawson DA, et al. A role for matrix metalloproteinases in regulating stem cell function via the Wnt signaling pathway. Cell Stem Cell. 2013;13(3):300–313. | |
Mantuano E, Inoue G, Li X, et al. The hemopexin domain of matrix metalloproteinase-9 activates cell signaling and promotes migration of Schwann cells by binding to low-density lipoprotein receptor-related protein. J Neurosci. 2008;28(45):11571–11582. | |
Chattopadhyay S, Shubayev VI. MMP-9 controls Schwann cell proliferation and phenotypic remodeling via IGF-1 and ErbB receptor-mediated activation of MEK/ERK pathway. Glia. 2009;57(12):1316–1325. | |
Dufour A, Sampson NS, Zucker S, Cao J. Role of the hemopexin domain of matrix metalloproteinases in cell migration. J Cell Physiol. 2008;217(3):643–651. | |
Stefanidakis M, Bjorklund M, Ihanus E, Gahmberg CG, Koivunen E. Identification of a negatively charged peptide motif within the catalytic domain of progelatinases that mediates binding to leukocyte beta 2 integrins. J Biol Chem. 2003;278(36):34674–34684. | |
Stefanidakis M, Karjalainen K, Jaalouk DE, et al. Role of leukemia cell invadosome in extramedullary infiltration. Blood. 2009;114(14):3008–3017. | |
Redondo-Muñoz J, Ugarte-Berzal E, García-Marco JA, et al. Alpha4beta1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood. 2008;112(1):169–178. | |
Redondo-Muñoz J, Ugarte-Berzal E, Terol MJ, et al. Matrix metalloproteinase-9 promotes chronic lymphocytic leukemia b cell survival through its hemopexin domain. Cancer Cell. 2010;17(2):160–172. | |
Dufour A, Zucker S, Sampson NS, Kuscu C, Cao J. Role of matrix metalloproteinase-9 dimers in cell migration: design of inhibitory peptides. J Biol Chem. 2010;285(46):35944–35956. | |
Bailón E, Ugarte-Berzal E, Amigo-Jiménez I, et al. Overexpression of progelatinase B/proMMP-9 affects migration regulatory pathways and impairs chronic lymphocytic leukemia cell homing to bone marrow and spleen. J Leukoc Biol. 2014;96(2):185–199. | |
Orgaz JL, Pandya P, Dalmeida R, et al. Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat Commun. 2014;5:4255. | |
Ezhilarasan R, Jadhav U, Mohanam I, Rao JS, Gujrati M, Mohanam S. The hemopexin domain of MMP-9 inhibits angiogenesis and retards the growth of intracranial glioblastoma xenograft in nude mice. Int J Cancer. 2009;124(2):306–315. | |
Strongin AY. Proteolytic and non-proteolytic roles of membrane type-1 matrix metalloproteinase in malignancy. Biochim Biophys Acta. 2010; 1803(1):133–141. | |
D’Alessio S, Ferrari G, Cinnante K, et al. Tissue inhibitor of metalloproteinases-2 binding to membrane-type 1 matrix metalloproteinase induces MAPK activation and cell growth by a non-proteolytic mechanism. J Biol Chem. 2008;283(1):87–99. | |
Sounni NE, Rozanov DV, Remacle AG, Golubkov VS, Noel A, Strongin AY. Timp-2 binding with cellular MT1-MMP stimulates invasion-promoting MEK/ERK signaling in cancer cells. Int J Cancer. 2010;126(5):1067–1078. | |
Sakamoto T, Seiki M. Cytoplasmic tail of MT1-MMP regulates macrophage motility independently from its protease activity. Genes Cells. 2009;14(5):617–626. | |
Sakamoto T, Seiki M. A membrane protease regulates energy production in macrophages by activating hypoxia-inducible factor-1 via a non-proteolytic mechanism. J Biol Chem. 2010;285(39):29951–29964. | |
Gonzalo P, Guadamillas MC, Hernández-Riquer MV, et al. MT1-MMP is required for myeloid cell fusion via regulation of Rac1 signaling. Dev Cell. 2010;18(1):77–89. | |
Mori H, Lo AT, Inman JL, et al. Transmembrane/cytoplasmic, rather than catalytic, domains of Mmp14 signal to MAPK activation and mammary branching morphogenesis via binding to integrin β1. Development. 2013;140(2):343–352. | |
Piccard H, Van den Steen PE, Opdenakker G. Hemopexin domains as multifunctional liganding modules in matrix metalloproteinases and other proteins. J Leukoc Biol. 2007;81(4):870–892. | |
Kim SK, Cargioli TG, Machluf M, et al. PEX-producing human neural stem cells inhibit tumor growth in a mouse glioma model. Clin Cancer Res. 2005;11(16):5965–5970. | |
Roeb E, Schleinkofer K, Kernebeck T, et al. The matrix metalloproteinase 9 (mmp-9) hemopexin domain is a novel gelatin binding domain and acts as an antagonist. J Biol Chem. 2002;277(52):50326–50332. | |
Burg-Roderfeld M, Roderfeld M, Wagner S, Henkel C, Grötzinger J, Roeb E. MMP-9-hemopexin domain hampers adhesion and migration of colorectal cancer cells. Int J Oncol. 2007;30(4):985–992. | |
Libson AM, Gittis AG, Collier IE, Marmer BL, Goldberg GI, Lattman EE. Crystal structure of the haemopexin-like C-terminal domain of gelatinase A. Nat Struct Biol. 1995;2(11):938–942. | |
Gohlke U, Gomis-Rüth FX, Crabbe T, Murphy G, Docherty AJ, Bode W. The C-terminal (haemopexin-like) domain structure of human gelatinase A (MMP2): structural implications for its function. FEBS Lett. 1996; 378(2):126–130. | |
Gomis-Rüth FX, Gohlke U, Betz M, et al. The helping hand of collagenase-3 (MMP-13): 2.7 A crystal structure of its C-terminal haemopexin-like domain. J Mol Biol. 1996;264(3):556–566. | |
Cha H, Kopetzki E, Huber R, Lanzendörfer M, Brandstetter H. Structural basis of the adaptive molecular recognition by MMP9. J Mol Biol. 2002;320(5):1065–1079. | |
Tochowicz A, Goettig P, Evans R, et al. The dimer interface of the membrane type 1 matrix metalloproteinase hemopexin domain: crystal structure and biological functions. J Biol Chem. 2011;286(9):7587–7600. | |
Björklund M, Heikkilä P, Koivunen E. Peptide inhibition of catalytic and noncatalytic activities of matrix metalloproteinase-9 blocks tumor cell migration and invasion. J Biol Chem. 2004;279(28):29589–29597. | |
Zarrabi K, Dufour A, Li J, et al. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J Biol Chem. 2011;286(38):33167–33177. | |
Ugarte-Berzal E, Bailón E, Amigo-Jiménez I, et al. A 17-residue sequence from the matrix metalloproteinase-9 (MMP-9) hemopexin domain binds α4β1 integrin and inhibits MMP-9-induced functions in chronic lymphocytic leukemia B cells. J Biol Chem. 2012;287(33):27601–27613. | |
Ugarte-Berzal E, Bailón E, Amigo-Jiménez I, Albar JP, García-Marco JA, García-Pardo A. A novel CD44-binding peptide from the pro-matrix metalloproteinase-9 hemopexin domain impairs adhesion and migration of chronic lymphocytic leukemia (CLL) cells. J Biol Chem. 2014;289(22):15340–15349. | |
Ndinguri MW, Bhowmick M, Tokmina-Roszyk D, Robichaud TK, Fields GB. Peptide-based selective inhibitors of matrix metalloproteinase-mediated activities. Molecules. 2012;17(12):14230–14248. | |
Kothapalli R, Khan AM, Basappa, Gopalsamy A, Chong YS, Annamalai L. Cheminformatics-based drug design approach for identification of inhibitors targeting the characteristic residues of MMP-13 hemopexin domain. PLoS One. 2010;5(8):e12494. | |
Dufour A, Sampson NS, Li J, et al. Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res. 2011;71(14):4977–4988. | |
Remacle AG, Golubkov VS, Shiryaev SA, et al. Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res. 2012;72(9):2339–2349. | |
Basu B, Correa de Sampaio P, Mohammed H, et al. Inhibition of MT1-MMP activity using functional antibody fragments selected against its hemopexin domain. Int J Biochem Cell Biol. 2012;44(2):393–403. | |
Sela-Passwell N, Trahtenherts A, Krüger A, Sagi I. New opportunities in drug design of metalloproteinase inhibitors: combination between structure-function experimental approaches and systems biology. Expert Opin Drug Discov. 2011;6(5):527–542. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.