")
Back to Journals » OncoTargets and Therapy » Volume 9
Noninvasive monitoring of the genetic evolution of EGFR-mutant non-small-cell lung cancer by analyzing circulating tumor DNA during combination chemotherapy with gefitinib and pemetrexed or S-1
Authors Nakahara Y , Takagi Y, Hosomi Y , Kagei A, Yamamoto T, Sawada T, Yomota M, Okuma Y , Mikura S, Okamura T
Received 6 February 2016
Accepted for publication 16 May 2016
Published 24 August 2016 Volume 2016:9 Pages 5287—5295
DOI https://doi.org/10.2147/OTT.S105976
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Min Li
Yoshiro Nakahara,1,2 Yusuke Takagi,1,3 Yukio Hosomi,1 Akiko Kagei,4 Tomohiro Yamamoto,4 Takeshi Sawada,5 Makiko Yomota,1 Yusuke Okuma,1 Shinichiro Mikura,1,6 Tatsuru Okamura1
1Department of Thoracic Oncology and Respiratory Medicine, Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital, Tokyo, 2Department of Respiratory Medicine, Kitasato University School of Medicine, Sagamihara, 3Oncology Scientific Affairs, Merck Sharp & Dohme Corp, 4GeneticLab Co., Ltd., Sapporo, 5Department of Medical Oncology, Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital, Tokyo, 6Department of Respiratory Medicine, Fujieda Municipal General Hospital, Fujieda, Japan
Background: Repetitive genotyping is useful to assess the genetic evolution of non-small-cell lung cancer (NSCLC) during treatment, but the need for sampling by biopsy is a major obstacle. Digital polymerase chain reaction (PCR) is a promising procedure for the detection of mutant alleles in plasma of cancer patients.
Methods: This prospective study enrolled patients with NSCLC and known epidermal growth factor receptor (EGFR) mutations and who had experienced disease progression during ongoing EGFR-tyrosine kinase inhibitor (TKI) therapy. Eligible patients received daily gefitinib and either pemetrexed or S-1 every 3 weeks until disease progression or the development of unacceptable toxicity. Peripheral blood was collected before and after the combination therapy for digital PCR and hepatocyte growth factor measurement.
Results: From May 2012 to January 2014, nine patients with a median age of 67 (range 52–80) years were enrolled. Patterns of disease progression during adjacent EGFR-TKI therapy were acquired resistance, observed in seven patients, and primary resistance, observed in two patients. Known EGFR mutations were detected in plasma samples of six (67%) patients at study enrollment. Of these, T790M mutation was concurrently detected in three (50%) patients. Four patients underwent gefitinib plus pemetrexed therapy, and five patients underwent gefitinib and S-1 therapy. The median number of cycles delivered was five, and the median progression-free survival was 5.7 months. Efficacy outcomes did not differ between treatments. After the combination therapy, plasma T790M status changed to positive in two patients. Hepatocyte growth factor level did not significantly change through the combination therapy.
Conclusion: The usefulness of monitoring the genetic evolution of EGFR-driven tumors using noninvasive procedures was demonstrated. Since continuation of EGFR-TKI therapy with cytotoxic agents has an acceptable tolerability and a possibility of inducing T790M mutation, the combination therapy may be useful for EGFR-mutant NSCLC resistant to EGFR-TKI therapy without T790M mutation.
Keywords: circulating tumor DNA, epidermal growth factor receptor, gefitinib, S-1, pemetrexed
Introduction
Recent studies have discovered several driver mutations of non-small-cell lung cancer (NSCLC) with therapeutic implications.1 Epidermal growth factor receptor (EGFR) gene mutation is the most common driver mutation found in NSCLC patients in East Asian countries2 and the second most common in Western countries.1 EGFR tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib, are considerably effective against EGFR-driven NSCLC; EGFR-TKI therapy improves response rate, progression-free survival, overall survival, and quality of life compared with cytotoxic chemotherapies.3–5 However, the median progression-free survival of EGFR-TKI therapy monotherapy is ~1 year, and a 2-year course of EGFR-TKI therapy leads to acquired resistance in ~90% of the patients.3
Resistance mechanisms to EGFR-TKI therapy in EGFR-mutated NSCLC include secondary EGFR T790M mutation, c-Met amplification, PIK3CA mutation, and transformation to small-cell lung cancer.6 A number of reports indicated that T790M mutation accounts for about a half of the acquired resistance to EGFR-TKI therapy.6,7 Approximately, 20% of the NSCLC cases showing progression after EGFR-TKI therapy presented c-Met amplification.6 A recent study reported that overexpression of hepatocyte growth factor (HGF), the ligand of c-Met, occurs in 61% of patients with TKI-resistant NSCLC, and HGF overexpression can be found in parallel with T790M mutation.8 Additionally, high HGF levels in plasma correlate with the decreased efficacy of EGFR-TKI therapy.9
The optimal treatment for NSCLC with acquired resistance to EGFR-TKI therapy varies by resistance mechanisms. Early study of mutant-selective EGFR-TKI therapy indicated that the third-generation EGFR-TKIs are much more effective for T790M-positive NSCLCs than tumors without T790M mutation.10 Conventional chemotherapy is effective for tumors that transformed to small-cell lung cancer.11 Preclinical studies suggested that gefitinib downregulates thymidylate synthase,12 the predictive marker of pemetrexed (PEM) therapy.13 Combination therapy with gefitinib and S-1 is effective for TKI-resistant tumors with c-Met amplification.12 Thus, repeated evaluations of the genetic profile of EGFR-mutated NSCLC are highly useful for appropriate treatment selection.
Biopsy (such as bronchoscopy or core needle biopsy) is the most precise evaluation method for determining acquired resistance to EGFR-TKI therapy. However, these invasive procedures cannot necessarily be applied for patients with progressive disease. A noninvasive procedure with a prompt result is desirable for repetitive evaluations of EGFR-driven tumors. A previous report indicated that T790M mutation can be detected in circulating tumor DNA (ctDNA).14 Digital polymerase chain reaction (PCR) is a highly sensitive method that can detect single template molecules by dividing samples into nanoliter partitions.15
Continuation of EGFR-TKI therapy in addition to chemotherapy was proven to be ineffective for TKI-resistant NSCLC, but the biological mechanism is yet to be clarified.16 To our knowledge, the genetic change in EGFR-mutated NSCLC during combination chemotherapy with post-progression TKI has not been prospectively studied. Here, we examined the utility of noninvasive monitoring of the genetic evolution of EGFR-mutated NSCLC treated with combination therapy of EGFR-TKI therapy and cytotoxic agents.
Methods
The work described here was conducted in accordance with the Declaration of Helsinki and the Ethical Guidelines for Clinical Research issued by the Japanese Ministry of Health, Labour and Welfare. The protocol was approved by the Institutional Review Board of Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital (Tokyo, Japan). All patients gave their written informed consent. The clinical trial registry number is UMIN000007817.
Study participants
This study enrolled patients with advanced NSCLC harboring EGFR exon 19 deletions or L858R with disease progression during EGFR-TKI therapy. Eligible patients had undergone EGFR-TKI therapy treatment for 28 days or longer and continued with the EGFR-TKI therapy. The other criteria for inclusion in the study were as follows:Eastern Cooperative Oncology Group performance status of 0–2; pretreatment with platinum-based chemotherapy or no indication of platinum use; measurable lesions defined by Response Evaluation Criteria in Solid Tumors, version 1.1 (RECIST 1.1);17 and having adequate organ function (neutrophil count ≥2,000 cells/μL, hemoglobin ≥9.0 g/dL, platelet count ≥100,000 μL, aminotransferase ≤2.5× the upper limit of normal, total bilirubin ≤1.5× the upper limit of normal, creatinine clearance ≥45 mL/min, and oxygen saturation by pulse oximetry ≥95%). Patients were excluded if they were treated with both PEM and S-1, had a history of interstitial lung disease, severe or uncontrollable comorbidities, a malignancy that required treatment within 6 months after enrollment, symptomatic central nervous system metastases, or massive pleural effusion or ascites. Patients who were pregnant or nursing women were also excluded.
Treatment
Eligible patients received daily gefitinib (250 mg) and either PEM (500 mg/m2, day 1) or S-1 (80 mg/m2, days 1–14). If a patient had received chemotherapy with neither PEM nor S-1, the regimen administered in this study was chosen by each investigator. The actual dose of S-1 was 120 mg/day for patients with a body surface area (BSA) ≥1.5 m2, 100 mg/day for 1.25 m2 ≤ BSA <1.5 m2, and 80 mg/day for BSA <1.25 m2. The treatment was repeated every 3 weeks until disease progression or the development of unacceptable toxicity. If the creatinine clearance was below 60 mL/min, S-1 was started with a decreased dose: 100 mg/day for patients with BSA ≥1.5 m2, 80 mg/day for 1.25 m2 ≤ BSA <1.5 m2, and 50 mg/day for BSA <1.25 m2.
Subsequent cycles were started if the performance status was 0–2, neutrophil count ≥1,500 cells/μL, platelet count ≥75,000 cells/μL, aminotransferase ≤2.5× the upper limit of normal, total bilirubin ≤1.5× the upper limit of normal, creatinine clearance ≥45 mL/min, nonhematological toxicities (except rash, weight loss, and electrocyte abnormality) ≤ grade 2, and diarrhea/vomiting ≤ grade 1. If a patient experienced neutrophil count <500 cells/μL, platelet count <50,000 cells/μL, creatinine ≥1.5 mg/mL, grade 2 diarrhea/vomiting lasting 2 days, or grade 3 nonhematologic toxicities (other than rash, weight loss, and electrolyte abnormalities), a dose reduction of PEM/S-1 was required. Toxicities such as grade 4 nonhematological toxicities, pneumonitis of any grade, treatment delay >21 days, and other conditions unsuitable for continuing chemotherapy were considered for termination of the study treatment.
Assessments
After enrollment, patient plasma samples were collected and further analyzed using a first digital PCR. Serum HGF concentration was measured before the initiation of combination chemotherapy. A second digital PCR was performed upon disease progression, 6 months after the initiation of combination chemotherapy, or termination of the study. Serum HGF concentration was measured again at the termination of study treatment. Tumor response to chemotherapy was assessed using RECIST 1.1. After baseline evaluation, tumor status was assessed every 6 weeks (two cycles). Toxicities were evaluated according to the National Cancer Institute’s Common Toxicity Criteria for Adverse Events, version 4.0.18
Sample processing and digital PCR
For plasma samples, 7–14 mL of peripheral blood was collected in ethylene diaminetetraacetic acid tubes and centrifuged at 1,000× g for 10 minutes to separate plasma from the peripheral blood cells. The plasma was then stored at −80°C until DNA extraction. All samples were centrifuged and frozen within 30 minutes after the collection. DNA was extracted from plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. DNA was eluted into water containing 0.04% sodium azide and stored at −20°C. DNA concentration was measured using NanoDrop 1,000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) Table S1.
Fluidigm 48.770 Digital-Array based digital PCR was carried out as described previously (Fluidigm, San Francisco, CA, USA).19 Samples were mixed with sequence-specific TaqMan probes (MGB SNP detection kit for exon 19 deletions and L858R, and Mutation Detection Assays for T790M), Master Mix, and Gene Expression Sample Loading Reagent (Thermo Fisher Scientific). Six microliters of this mixture was added to the 48.770 Digital Array Chip (Fluidigm) where it was partitioned into 770 separate PCR reactions. PCR cycle was performed by Fluidigm BioMark Genetic Analysis according to the manufacturer’s instructions and all positive reaction chambers were counted. Allele fraction was defined as the ratio of the mutant copy number divided by the wild-type copy number of each locus. Data were analyzed using a Poisson correction by BioMark Digital PCR Analysis Software v3.0 (Fluidigm).
Statistical methods
The primary end point of this study was the detection rate of known EGFR mutation from the plasma collected before combination therapy. To detect the difference between the expected 73% of samples and the null hypothesis of 50% using a two-stage design with 80% power and 10% alpha (one-sided), 20 patients were to be enrolled. Secondary end points were detection rate of EGFR T790M mutation, detection rate of known EGFR mutation after combination therapy, serum HGF concentrations, progression-free survival (PFS), and chemotherapy toxicity. The number of months that elapsed between the enrollment and the date of disease progression or death was defined as PFS. Patients who remained alive without disease progression at the end of follow-up and patients who started subsequent chemotherapy without disease progression were censored. Time-to-event data were estimated using the Kaplan–Meier method. All tests were two-sided, and the significance level was set at 0.05. All data were analyzed using JMP version 9.0 software (SAS Institute Inc., Cary, NC, USA).
Results
Patient characteristics
From May 2012 to January 2014, nine patients with EGFR-mutated NSCLC and progression during EGFR-TKI therapy monotherapy were enrolled. Patient enrollment was discontinued in February 2014 because of slow accrual due to a competing Phase III trial. The median age of enrolled patients was 67 (range 52–80) years, six patients (67%) were female and seven patients (78%) were nonsmokers. All patients had adenocarcinoma, and known EGFR mutation was L858R in six patients and exon 19 deletions in three patients. Performance status was 0 in two patients, 1 in six patients, and 2 in one patient (Table 1).
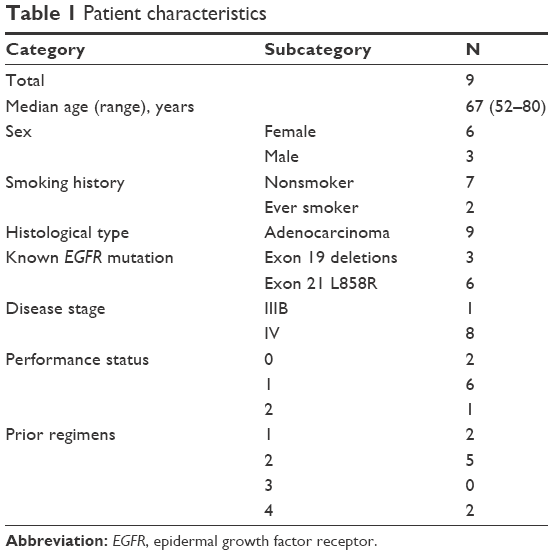 | Table 1 Patient characteristics |
The median number of previous chemotherapy regimens was two (range 1–4). Two patients experienced only one regimen (EGFR-TKI therapy monotherapy) before enrollment, because they were both 80 years old and were considered unfit for platinum-based chemotherapy by the attending physician. Seven patients (78%) fulfilled Jackman’s criteria20 of acquired resistance to EGFR-TKI therapy, and the remaining two patients had experienced disease progression within 6 months of adjacent EGFR-TKI therapy without achieving partial response. The EGFR-TKIs used immediately prior to enrollment were gefitinib in eight patients and erlotinib in one patient. The median interval from the initiation of systemic chemotherapy to study enrollment was 20.6 (range 4.2–54.9) months.
Treatment
Regimens of combination chemotherapy were gefitinib plus PEM and gefitinib plus S-1 in four and five patients, respectively. The median number of cycles delivered was five (range 2–12). Toxicity in one patient prompted the discontinuation of gefitinib plus PEM therapy, whereas the remaining eight patients underwent combination chemotherapy until disease progression. PEM dose was reduced in two (50%) patients because of febrile neutropenia and prolonged grade 2 diarrhea and patient’s request associated with grade 2 fatigue, respectively. Dose reduction of S-1 was required in three (60%) patients because of diarrhea (grade 3 in one patient and prolonged grade 2 in another patient) and reduced creatinine clearance. Delivered dose intensities were 95.8%, 93.2%, and 82.6% for gefitinib, PEM, and S-1, respectively.
Detection of mutated EGFR from plasma
Digital PCR detected known EGFR mutation in six (67%) initial plasma samples (Table 2). EGFR L858R mutation was not detected from the plasma of patients with exon 19 deletions, and exon 19 deletions were not detected from the plasma of patients with L858R; therefore, the specificity before combination therapy was 100%. EGFR T790M mutation in plasma was detected from three (50%) patients, and all of them had NSCLC with EGFR L858R mutation. Samples after initiation of combination chemotherapy were obtained from all nine patients, six samples at disease progression and termination of combination therapy, one at 6 months after enrollment but still under treatment, one sample at termination of therapy because of toxicity, and one last sample at termination of this trial and still under treatment (Figure 1). Known EGFR mutation was detected from five patients, and four (67%) of the six samples were obtained at disease progression. Exon 19 deletions were detected from two patients with a tumor harboring L858R mutation. Notably, plasma T790M status changed to positive in two patients at the second evaluation. Of the three patients with positive T790M status initially, two samples at the second evaluation also presented T790M mutation, although one sample contained neither known EGFR mutation nor T790M mutation (Table 2).
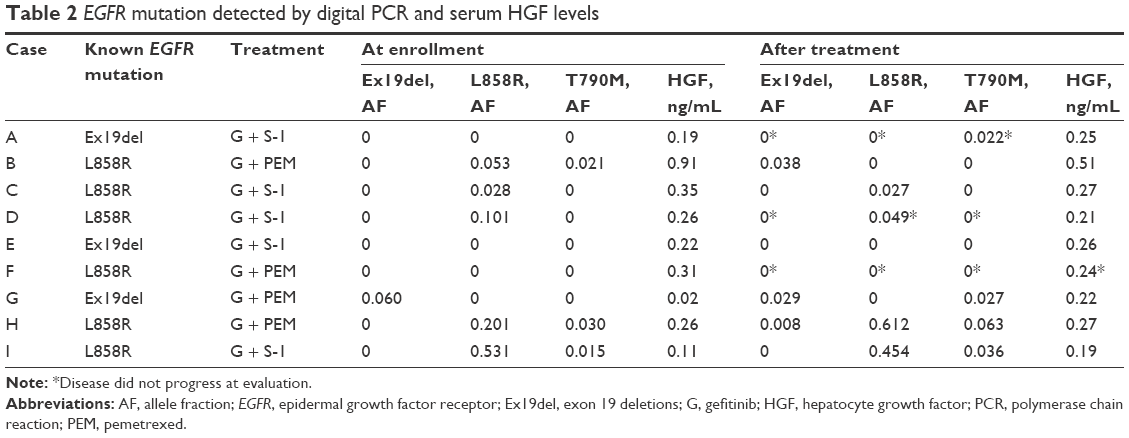 | Table 2 EGFR mutation detected by digital PCR and serum HGF levels |
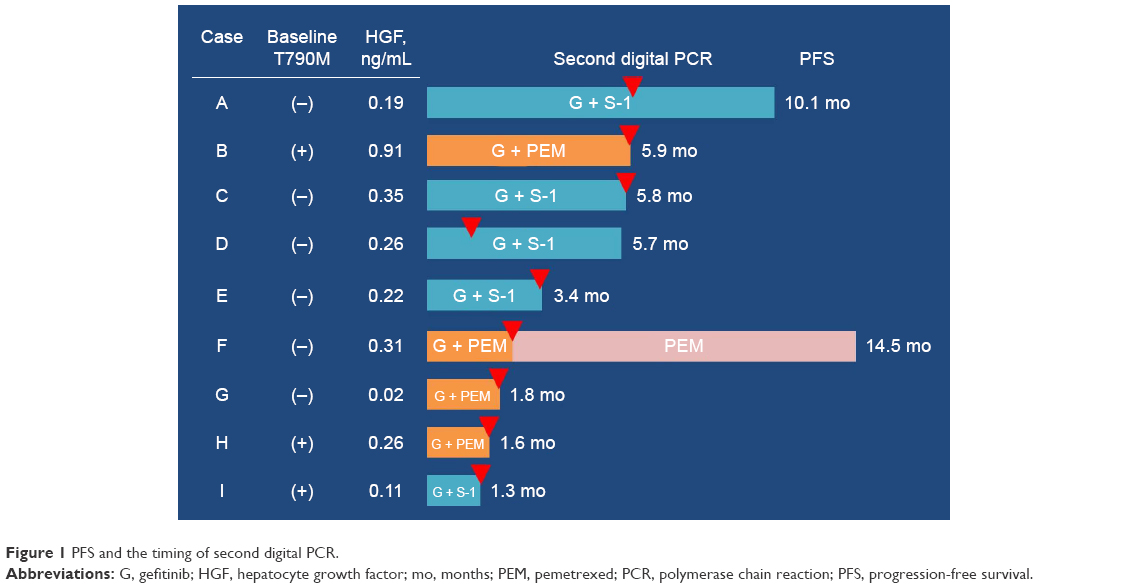 | Figure 1 PFS and the timing of second digital PCR. |
Hepatocyte growth factor levels
Mean baseline HGF level was 0.29 ng/mL (range 0.02–0.91; median 0.26; standard deviation [SD] 0.25). At the termination of protocol treatment, the mean HGF level was 0.28 ng/mL (range 0.19–0.51; median 0.26; SD 0.10). Of the four patients who underwent gefitinib plus PEM therapy, baseline HGF levels were 0.38±0.38 ng/mL initially and 0.31±0.13 ng/mL at the end of the study treatment. Five patients in the gefitinib plus S-1 cohort presented baseline HGF levels of 0.23±0.09 ng/mL initially and 0.24±0.04 ng/mL at disease progression. Serum HGF levels did not change significantly through combination therapy (Table 2).
Efficacy
Partial response to combination therapy was observed in one patient each (25% and 20%) in gefitinib plus PEM and gefitinib plus S-1 cohorts. Overall response rate was 22% (95% confidence interval [CI] 6%–55%) and disease control rate was 67% (95% CI 35%–88%). The median PFS was 5.7 months (95% CI 1.3–5.9) and the efficacy outcomes did not differ between the treatment regimens (Figure 1). PFS of three patients with plasma T790M before combination therapy was 1.3, 1.6, and 5.9 months. PFS of three patients with baseline serum HGF concentrations higher than the median (0.26 ng/mL) was 5.8, 5.9, and 14.5 months. The median overall survival was 16.2 months (95% CI 4.7–19.9, Figure 2).
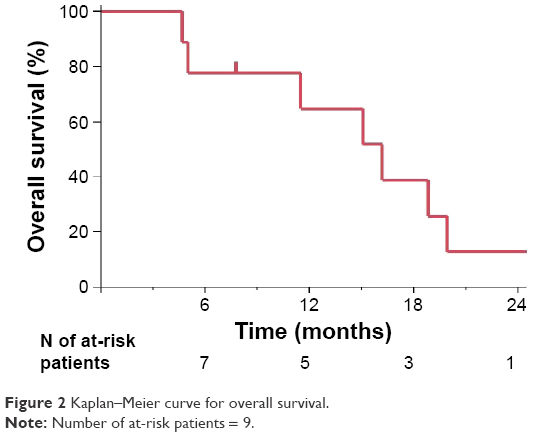 | Figure 2 Kaplan–Meier curve for overall survival. |
Safety
No treatment-related deaths or grade 4 toxicity occurred. Grade 3 toxicity was observed in two (22%) patients. One patient in gefitinib plus PEM cohort developed grade 3 leukopenia, neutropenia, and febrile neutropenia. Another patient in gefitinib plus S-1 cohort experienced grade 3 diarrhea. Grade 2 diarrhea lasting for 2 days was observed in one patient each in gefitinib plus PEM and gefitinib plus S-1 cohorts. Other grade 1 and 2 toxicities are described in Table 3.
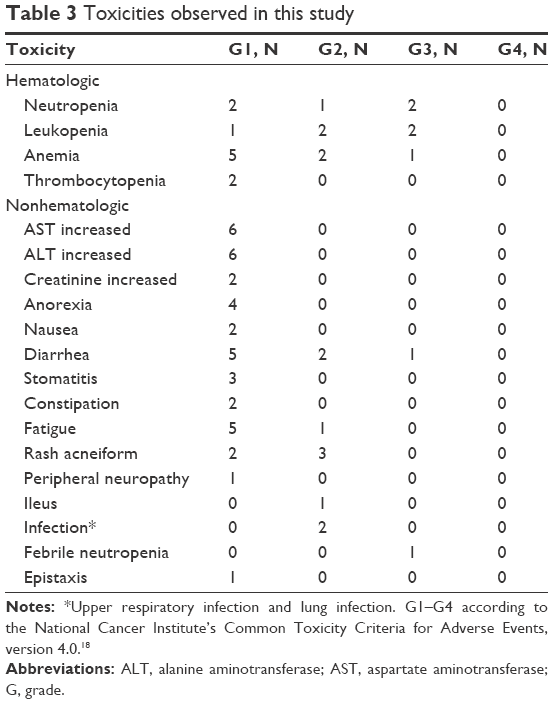 | Table 3 Toxicities observed in this study |
Discussion
In this study, we analyzed ctDNA using digital PCR and gained a favorable detection rate of known EGFR mutations. The number of patients with EGFR T790M mutation in plasma increased after combination chemotherapy. Combination chemotherapy with EGFR-TKI therapy beyond progression, and cytotoxic agents has a satisfactory activity with acceptable toxicity.
Previous studies reported various procedures for detecting ctDNA. Detection rates of mutated EGFR from untreated, EGFR-mutant NSCLC patients were reported to be 75% and 82% for the Scorpion amplification refractory mutation system21 and denaturing high-performance liquid chromatography, respectively.22 An analysis using BEAMing to detect mutated EGFR from plasma reported a detection rate of 83% before the treatment, and it decreased to 62% after EGFR-TKI therapy.23 A meta-analysis investigating the diagnostic value of ctDNA for the identification of EGFR mutations has been published.
According to the analysis, the pooled sensitivity and specificity were 0.620 (95% CI 0.513–0.716) and 0.959 (95% CI 0.929–0.977), respectively.24
We detected known EGFR mutation from 67% of patients who had undergone EGFR-TKI therapy, and the sensitivity is comparable to previous studies. The amount of ctDNA correlates with tumor burden.25 EGFR-TKI therapy is highly effective for EGFR-mutated NSCLC; thus, the tumor burden at stages of acquired resistance to EGFR-TKI therapy is often less than before the initiation of EGFR-TKI therapy. The decreased detection rate of known EGFR mutation from plasma after EGFR-TKI therapy may be explained by the decrease of tumor burden.
After combination chemotherapy, the number of patients with EGFR T790M mutations in plasma increased. To date, genetic change in patients who underwent combination therapy with EGFR-TKI therapy beyond progression is unknown. Preclinical models suggested that the combination therapy increases the population of T790M-positive cells,26 and our clinical study confirmed the prediction. Although lung cancer cells with sensitizing and T790M mutation show slower growth compared with cells without T790M mutation,26 a prospective study found no difference in post-progression survival between T790M-positive and -negative patients (median 14.7 vs 14.1 months, respectively).27 Thus, the prognostic impact of T790M mutation in TKI-resistant NSCLC patients is unknown.
In this study, we also evaluated serum HGF levels before and after combination chemotherapy. We employed a treatment regimen effective on c-Met-amplified lung cancer cells with EGFR sensitizing mutation.12 Although we cannot draw a conclusion because of the overly small sample size, relatively longer PFS achieved in patients with higher HGF levels at baseline may be associated with the preclinical findings.12,13 Further investigation to identify predictive markers for the treatment of TKI-resistant NSCLC not driven by T790M mutation is needed.
The key limitation of this nonrandomized study is the small sample size. Although 20 patients were required to prove a detection rate above 50%, we could enroll only nine patients because of slow accrual. We could not prove the hypothesis because the primary end point was not met. However, genetic change observed in EGFR-mutated NSCLC before and after combination therapy with EGFR-TKI therapy and cytotoxic agents had not been prospectively studied, thus our findings have some implications. Another limitation is the low detection rate in three patients with exon 19 deletions, although we employed a commercially available method. Since the previous retrospective study did not indicate a difference in sensitivity between two mutations,19 the results might be obtained by chance and need further validation.
Third-generation EGFR-TKIs are highly effective for NSCLC with sensitizing EGFR mutation and T790M mutation, whereas activity for T790M-negative cells was limited. Combination chemotherapy evaluated in our study was effective for patients without T790M mutation and induced an increase of T790M-positive patients. Therefore, a strategy to induce T790M mutation by continuation of EGFR-TKI therapy combined with cytotoxic agents may be useful for TKI-resistant NSCLC not driven by T790M mutation. Because the treatment strategy for EGFR-mutated NSCLC is dependent on the resistance mechanisms, sequential genetic profiling before treatment change will become increasingly important. Our study showed that noninvasive methods targeting ctDNA are useful in monitoring the genetic evolution of EGFR-driven tumors. However, challenges remain, such as detection in patients with low tumor burden. Further improvement of the methodology is warranted.
Conclusion
In conclusion, we demonstrated the usefulness of monitoring the genetic evolution of EGFR-driven tumors using noninvasive procedures. Since continuation of EGFR-TKI therapy with cytotoxic agents has an acceptable tolerability and a possibility of inducing T790M mutation, the combination therapy may be useful as a part of treatment strategy for EGFR-mutant NSCLC resistant to EGFR-TKI therapy without T790M mutation.
Acknowledgments
This study was supported by Clinical Research Fund of Tokyo Metropolitan Government: H240301. We acknowledge the contributions of Kan Kato, Shingo Miyamoto, and Masahisa Kudo as members comprising the Data and Safety Monitoring Board.
Disclosure
YT is an employee of MSD KK and has received research funding from Taiho Pharmaceutical. YH has received payment for lectures from AstraZeneca, Eli Lilly, and Taiho Pharmaceutical and has received research funding from Eli Lilly. AK and TY are employees of GeneticLab Co., Ltd. The other authors report no conflicts of interest in this work.
References
Gerber DE, Gandhi L, Costa DB. Management and future directions in non-small cell lung cancer with known activating mutations. Am Soc Clin Oncol Educ Book. 2014;e353–e365. | ||
Broët P, Dalmasso C, Tan EH, et al. Genomic profiles specific to patient ethnicity in lung adenocarcinoma. Clin Cancer Res. 2011;17(11):3542–3550. | ||
Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. | ||
Takano T, Fukui T, Ohe Y, et al. EGFR mutations predict survival benefit from gefitinib in patients with advanced lung adenocarcinoma: a historical comparison of patients treated before and after gefitinib approval in Japan. J Clin Oncol. 2008;26(24):5589–5595. | ||
Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. | ||
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | ||
Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. | ||
Yano S, Yamada T, Takeuchi S, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6(12):2011–2017. | ||
Kasahara K, Arao T, Sakai K, et al. Impact of serum hepatocyte growth factor on treatment response to epidermal growth factor receptor tyrosine kinase inhibitors in patients with non-small cell lung adenocarcinoma. Clin Cancer Res. 2010;16(18):4616–4624. | ||
Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. | ||
Takagi Y, Nakahara Y, Hosomi Y, Hishima T. Small-cell lung cancer with a rare epidermal growth factor receptor gene mutation showing “wax-and-wane” transformation. BMC Cancer. 2013;13:529. | ||
Okabe T, Okamoto I, Tsukioka S, et al. Addition of S-1 to the epidermal growth factor receptor inhibitor gefitinib overcomes gefitinib resistance in non-small cell lung cancer cell lines with MET amplification. Clin Cancer Res. 2009;15(3):907–913. | ||
Ozasa H, Oguri T, Uemura T, et al. Significance of thymidylate synthase for resistance to pemetrexed in lung cancer. Cancer Sci. 2010;101(1): 161–166. | ||
Kuang Y, Rogers A, Yeap BY, et al. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res. 2009;15(8):2630–2636. | ||
Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96(16):9236–9241. | ||
Yoshimura N, Okishio K, Mitsuoka S, et al. Prospective assessment of continuation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of pemetrexed. J Thorac Oncol. 2013;8(1):96–101. | ||
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009(1);45(2):228–247. | ||
National Institutes of Health. Common Terminology Criteria for Adverse Events (CTCAE) v4.0. Bethesda, MD: National Institutes of Health; 2009. Available from: http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed August 1, 2016. | ||
Yung TK, Chan KC, Mok TS, Tong J, To KF, Lo YM. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin Cancer Res. 2009;15(6):2076–2084. | ||
Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28(2):357–360. | ||
Kimura H, Suminoe M, Kasahara K, et al. Evaluation of epidermal growth factor receptor mutation status in serum DNA as a predictor of response to gefitinib (IRESSA). Br J Cancer. 2007;97(6):778–784. | ||
Bai H, Mao L, Wang HS, et al. Epidermal growth factor receptor mutations in plasma DNA samples predict tumor response in Chinese patients with stages IIIB to IV non-small-cell lung cancer. J Clin Oncol. 2009;27(16):2653–2659. | ||
Taniguchi K, Uchida J, Nishino K, et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res. 2011;17(24):7808–7815. | ||
Qiu M, Wang J, Xu Y, et al. Circulating tumor DNA is effective for the detection of EGFR mutation in nonsmall cell lung cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2015;24(1):206–212. | ||
Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136):136ra68. | ||
Chmielecki J, Foo J, Oxnard GR, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011;3(90):90ra59. | ||
Sun JM, Ahn MJ, Choi YL, Ahn JS, Park K. Clinical implications of T790M mutation in patients with acquired resistance to EGFR tyrosine kinase inhibitors. Lung Cancer. 2013;82(2):294–298. |
Supplementary material
 | Table S1 Total extracted DNA and DNA used for each reaction chip |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.