Back to Journals » Biologics: Targets and Therapy » Volume 8
Non-tumor necrosis factor-based biologic therapies for rheumatoid arthritis: present, future, and insights into pathogenesis
Received 16 September 2013
Accepted for publication 11 October 2013
Published 9 December 2013 Volume 2014:8 Pages 1—12
DOI https://doi.org/10.2147/BTT.S35475
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Filipe Seguro Paula,1 José Delgado Alves1,2
1Immunomediated Systemic Diseases Unit, Department of Medicine 4, Fernando Fonseca Hospital, Amadora, 2Center for the Study of Chronic Diseases, Department of Pharmacology, Faculty of Medical Sciences, Lisbon, Portugal
Abstract: The way rheumatoid arthritis is treated has changed dramatically with the introduction of anti-tumor necrosis factor (anti-TNF) biologics. Nevertheless, many patients still have less than adequate control of their disease activity even with these therapeutic regimens, and current knowledge fails to explain all the data already gathered. There is now a wide range of drugs from different classes of biologic disease-modifying anti-rheumatic drugs available (and soon this number will increase significantly), that provides the opportunity to address each patient as a particular case and thereby optimize medical intervention. Currently available biologics for the treatment of rheumatoid arthritis apart from anti-TNF-based therapies are reviewed, along with an analysis of the new insights they provide into the pathogenesis of the disease and a discussion of future prospects in the area.
Keywords: rheumatoid arthritis, non-anti-tumor necrosis factor, treatment, pathogenesis
Introduction
Rheumatoid arthritis (RA) is a chronic, systemic, inflammatory disease of adulthood. Although both sexes and every age group may be affected, it is more frequent in women in the fourth and fifth decades of life. The disease may cause damage to various organs and tissues, including the lungs, skin, and heart, but predominantly affects the joints. Further, there is a strong association with increased cardiovascular risk, which ultimately leads to premature death.1 Arthritis is the most recognized and studied manifestation of the disease, being the area in which the most extensive data have been gathered from a pathophysiologic point of view, and the one that is evaluated in the large majority of clinical trials.
The way RA is treated has changed dramatically in the last 15 years with the introduction of biologics, namely anti-tumor necrosis factor (TNF) therapy. With the use of these molecular targeted therapies, the standard of care for patients with RA has improved, better clinical results have been achieved, and knowledge about the pathogenesis of RA has grown exponentially. Nevertheless, many patients still have less than adequate control of their disease activity even with these therapeutic regimens or become resistant to their beneficial effects. This imposes the need for other treatment options, and suggests that current theories about RA may be only partially correct. In recent years, other drugs have been produced that target other components of the inflammatory process in RA, with good efficacy and safety profiles, which eventually led to their approval and widespread use. It is the aim of this paper to review the biologics currently available for the treatment of RA apart from anti-TNF-based therapies, to analyze the new insights they provide into the pathogenesis of the disease, and to discuss future prospects in the area.
Etiology and pathogenesis
A clear etiology for RA has never been identified. Genetic factors clearly play a role, as revealed by an up to 30% concordance rate in monozygotic twins,2,3 and most genomic association studies point to the major histocompatibility complex locus as the one with the highest relative risk.4 The most important environmental risk factor is smoking, and some microbes (such as Porphyromonas gingivalis infection in periodontal tissue) have been implicated, but this has been regarded as of uncertain value.5
RA is characterized by activation of resident synovial inflammatory cells, mainly macrophages, infiltration of lymphocytes and neutrophils in the synovium, and the production of an inflammatory milieu, which in turn promotes proliferation of synoviocytes and fibroblasts, and neoangiogenesis. The presence of autoantibodies (rheumatoid factor and anti-cyclic citrullinated peptide antibodies), a reflection of the role of B-cells, is one of the laboratory hallmarks of RA, in some cases being detected more than 10 years before clinical onset.6 All this leads to an aberrant, hyperplastic architecture of the synovial membrane, the rheumatoid pannus, and to the differentiation and activation of osteoclasts and subsequent bone destruction. Chondrocyte function is also altered, leading to degradation of cartilage and biomechanical derangement of normal articular function. Periarticular structures such as ligaments and tendons are also eventually involved in the inflammatory process, culminating in further dysfunction and production of the typical clinical and radiologic picture of RA.
As such, apart from macrophages and other “effector” cell types (dendritic cells, neutrophils, synoviocytes, osteoblasts, osteoclasts, and chondrocytes), three components of RA pathogenesis have become recognized as major players based on both basic and clinical research, ie, B-cells, T-cells, and a wide range of inflammatory cytokines and growth factors that, acting as an intricate and redundant network both systemically and locally, shift the balance towards a proinflammatory state. Accumulating evidence shows that all these players act interdependently, and have continuously challenged our understanding of immune physiology and pathology.
After TNF blockers were introduced in the clinical management of RA, two types of clinical picture have emerged in daily practice, ie, an inadequate response and/or existence of contraindications or intolerance, precluding the use of these agents and raising the need to find alternatives. In fact, anti-TNF therapy achieves a 20% improvement in American College of Rheumatology response criteria (ACR20) in about 42%–85% of patients, and an ACR50 response in only 21%–69%,7,8 with secondary failure rates of up to 50% during the first year.9
Current molecular targeted strategies to control RA (beyond TNF) have tried to block at least one of the three components mentioned, and the most relevant of those are reviewed here.
B-cell-targeted therapies
Role of B-cells in RA
Improvement in RA through B-cell depletion has highlighted the importance of B-cells in the pathogenesis of the disease. The presence of rheumatoid factor is related to disease severity and the frequency of extra-articular manifestations,10 and anti-cyclic citrullinated peptide antibodies are related to aggressiveness of the disease.11 Moreover, baseline rheumatoid factor seropositivity seems to be related to the response to rituximab.12–14 However, CD20 is lacking in antibody-producing plasmablasts and plasma cells; the response to rituximab is related to the level of B-cell depletion in peripheral blood15,16 and synovial tissue,17 and is coincident with a reduction in the number of peripheral memory B-cells (CD19+/CD27+),18 and not with the degree of reduction in plasma immunoglobulins.19 Further, relapse is also related to B-cell repopulation,20,21 and non-antibody-producing B-cells are able to activate T-cells and produce articular disease.22 All this reinforces the idea of an important role of B-cells beyond antibody production.
B-cells are potent antigen-presenting cells22 in the context of multiple diseases.23,24 They are able to activate CD4+ T-cells, and their presence is necessary for T-cell activation in synovial tissue.25 B-cells are also capable of enhancing the differentiation of T-cells into the inflammatory T-helper (Th)17 phenotype.26 Further, B-cells are potent cytokine producers that act not only in an autocrine manner (interleukin [IL]-10) but also activate other immune cells, including macrophages, neutrophils, and dendritic cells (IL-6, TNF-α, IL-1, and IL-10).27 They are also important sources of potent chemotactic molecules that are crucial to pannus development.28,29 These findings add a twist to the classical view of T-helper cells ensuring activation and maturation of B-cells and innate immunity activating adaptive immunity. They also suggest that the presence of autoantibodies may be interpreted in RA as a manifestation of loss of tolerance and of the presence of autoreactive B-cells that are themselves pathogenic, even before differentiating into permanent antibody-producing plasma cells.
The case for rituximab
Blocking the contribution of B-cells to disease activity has been achieved by B-cell-depleting therapies with great success. Rituximab, a chimeric monoclonal anti-CD20 antibody originally used in non-Hodgkin’s lymphoma, induces antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and apoptosis of B-cells in various stages of development,27,30,31 leading to their transient but almost complete depletion in peripheral blood, although only partially in the bone marrow and synovial tissue niches.32,33
The beneficial effect of rituximab-induced B-cell depletion has been substantiated in various major randomized clinical trials in RA, as well as in other immune-mediated diseases.34 The benefit is reflected in a reduction of clinical disease activity and laboratory inflammation markers, halted radiographic progression, and better functional ability and quality of life scores. The effect consistently lasts for more than 6 months after a single course of two administrations of the drug, separated by 15 days, with clear benefit when compared with placebo, and with a good safety profile. This has been observed in methotrexate-naïve patients (anti-TNF-naïve) in IMAGE (International Study in Methotrexate-naïve Subjects Investigating Rituximab’s Efficacy),13 and in methotrexate-inadequate responders in SERENE40 (Study Evaluating Rituximab’s Efficacy in MTX iNadequate rEsponders) and MIRROR35 (Methotrexate Inadequate Responders Randomized study Of Rituximab), with comparable results. In SERENE,40 ACR20, ACR50, and ACR70 responses at 24 weeks were 51%, 26%, and 10%, respectively, with a clear-cut difference from placebo (23%, 9%, and 5%). Importantly, in REFLEX36 (Randomized Evaluation oF Long-term Efficacy of rituXimab in RA), rituximab again produced similar results in anti-TNF-inadequate responders, with ACR20, ACR50, and ACR70 responses at 24 weeks of 51%, 27%, and 12%, respectively. A benefit from placebo has been shown for up to 5 years of continued rituximab retreatment in a post hoc open-label extension of REFLEX.37 The MIRAR trial focused on an important issue directly comparing the use of an alternative anti-TNF versus rituximab after a first anti-TNF failure, with both treatments producing comparable benefits.38 Conversely, several observational studies have suggested that rituximab may be more effective than a second anti-TNF in some subgroups,39 although clinical trials confirming this are lacking.
Rituximab has shown to be effective in monotherapy, but considerably less so when used in combination with methotrexate, both in the magnitude and duration of benefit.12,40 This is one of the major caveats of using rituximab in RA because tocilizumab is still preferred as an alternative to anti-TNF agents for methotrexate-intolerant patients or when methotrexate is contraindicated, such as in severe pulmonary disease. The use of rituximab with other synthetic disease-modifying antirheumatic drugs (DMARDs), mainly leflunomide, has been evaluated as producing similar results,41 but large head-to-head randomized trials are still lacking.
In light of these accumulated data, there seems to be no reason for rituximab not being recommended as a first-line agent. However, rituximab’s debut in the treatment of RA was somewhat obscured by the initial enthusiasm for anti-TNF agents, and its initial use in malignancies may have raised some concern regarding its widespread use. In Europe, rituximab was approved in RA patients with severe disease, but only after failure of at least one anti-TNF agent (and always in association with methotrexate).42 However, in the more recent 2012 ACR guidelines, rituximab has already been recommended as a first-line biologic agent after synthetic DMARD failure for patients with moderate to severe disease activity and for those with low disease activity and poor prognosis markers.43
Some controversy still exists regarding the optimal dosage of rituximab. The evidence suggests that a 2 × 500 mg regimen has almost similar efficacy to the 2 × 1 g regimen used in the majority of trials, with a reduced rate of infusion reactions and adverse events.44 However, the full-dose regimen seems to result in better ACR70 response rates, and better radiographic outcomes were produced in the IMAGE trial.13 As such, the approved dose in most consensus guidelines is 2 × 1 g.
Beyond rituximab
CD20 is absent in long-lived plasma cells and plasmablasts and is not expressed until after the pre-B stage of B-cell development. As such, rituximab therapy is not expected to interfere directly with permanent antibody-producing cells, but rather with their precursors. Also, the bone marrow consists of a protective niche, being largely refractory to B-cell depletion after administration of rituximab.32,33,45 Importantly, synovial tissue seems to be also relatively resistant to the action of rituximab.32,33 In RA, the synovial tissue is infiltrated with B-cells and T-cells, producing three types of synovitis: diffuse synovitis, in which both types of cells are interspersed with no apparent spatial organization; follicular synovitis, in which ectopic germinal centers form, with a nucleus of B-cells surrounded by T-cells and with autoantibody-secreting capacity;46 and granulomatous synovitis. Sex, age, disease duration, and previous treatment do not correlate with the type of synovitis found, and each patient shows only one type. This suggests that there may be different subtypes of the disease. Accordingly, seronegative RA patients tend to have a diffuse pattern, and all patients with follicular synovitis were seropositive in a cohort of 21 cases.47 Additionally, rheumatoid nodules, a common extra-articular manifestation of the disease, seem to be closely associated with granulomatous synovitis. Interestingly, rheumatoid factor levels tend to be higher and the effect of rituximab on immunoglobulin levels in synovial fluid tends to be more pronounced in follicular synovitis.32 In one study, the diameter of follicular aggregates was significantly associated with disease severity,48 but no comparison was made between the different types of synovitis. The clinical efficacy of rituximab or even baseline disease activity in each subgroup was never evaluated. Nevertheless, these studies show that B-cells in local synovial tissue are crucial determinants of the disease, and a more potent way of inhibiting B-cell function in the synovium could be interesting.
CD19, a membrane-associated molecule involved in modulation of the threshold of B-cell activation, is present in a larger range of B-cells, including plasmablasts, plasma cells, and pre-B cells,49 and is also specific for B-cells. In contrast with rituximab, administration of anti-CD19 monoclonal antibody depletes the majority of B-cells in the bone marrow, lymphoid organs, and synovial tissue,50 as well as some anti-CD20-resistant subtypes resident in the peritoneal cavity (B1a, B1b),50,51 potentially leading to an increased clinical efficacy, as was observed in animal models of RA. There is, however, some concern that depletion of regulating B10 (CD20-) cells could induce autoimmunity.52 Depletion of plasma cells also induces at least a partial loss of protective immunity, which could lead to increased safety issues concerning the risk of infection. These issues have not been fully addressed in humans, with the only experience having been gathered in patients with hematologic malignancies, first with a mouse anti-hCD19,53 then with a humanized version,54 with both having shown an acceptable safety profile.
Other strategies to block the B-cell contribution to RA have been envisaged, including blocking B-cell activation with an anti-BAFF/BLyS (B-cell activating factor/B-lymphocyte stimulator) antibody (belimumab)55 or with a TACI-immunoglobulin fusion protein (atacicept) which blocks both BLyS and APRIL (A PRoliferation Inducing Ligand) costimulators.56 Belimumab has shown modest efficacy (only ACR20 responses different from placebo), although with a good safety profile, mainly in moderate to severe, seropositive, anti-TNF-naïve RA patients who have failed methotrexate therapy.57 Atacicept did not meet expectations, producing no clinical improvement, but is safe to use in methotrexate-inadequate responders58 or after antiTNF failure.56 It is interesting to note that atacicept fulfilled its expected biological effect,59 reducing circulating immunoglobulin levels and autoantibody titers,58 which reinforces the notion of autoantibodies being markers of disease and not effectors per se.
T-cell-targeted therapies
Role of T-cells in RA
T-cells are found within the synovial tissue in RA, and play a major role in perpetuating inflammation and producing tissue damage. They are activated by antigen-presenting cells, including dendritic cells, macrophages, and even B-cells, and subsequently secrete various cytokines (which contribute to inflammation in synovial tissue), activate innate immune cells, support B-cell activation, and induce destructive chondrocyte and osteoclast activation. Various subtypes of T-helper cells have been described, and modulation of the differentiation of naïve T-cells into each of those types is being increasingly studied, and proving to be central in RA.
The inflammatory pattern in RA has long been characterized by a shift towards a Th1 phenotype in CD4+ T-cell differentiation. This leads to a predominant production of Th1-associated cytokines such as interferon-gamma, with Th2-associated cytokines such as IL-4 and IL-13 found to be nearly absent in the synovial tissue.60 The Th17 subtype of CD4+ T-cells is today recognized as a major player in synovial inflammation and bone erosion, mainly by the production of IL-17 and IL-23.61 In fact, the pivotal role of Th1 in the pathogenesis of RA has been increasingly recognized as taking place via actions ascribed to Th17, and it is not clear whether Th17 cells consist of a specialized Th1 subtype or a different phenotype that would act in concert with Th1 cells. Th17 differentiation from naïve T-cells seems to require IL-6 and transforming growth factor-beta concomitant signaling, and their proliferation is dependent on IL-23, produced by antigen-presenting cells.62,63 In any case, both subtypes seem to have an important role in the regulation of inflammation in RA. The relative weight of each one is still under discussion.
Conversely, inhibition of regulatory T-cell (Treg) differentiation seems to be important in the perpetuation of autoimmune diseases,64 and their numbers and activation status are associated with RA activity in general65,66 and with the response to treatment with either synthetic or biologic DMARDs.67,68
Case for abatacept
Efforts to block T-cell activity in RA have produced important results.69 Abatacept, a human CTLA4-IgFc (CTLA, cytotoxic T-lymphocyte antigen) fusion protein, binds to CD80/86 on antigen-presenting cells and competitively prevents its binding to the T-cell CD28 molecule (CTLA). CD80/CD86-CD28 interaction produces a crucial costimulatory signal upon major histocompatibility complex-T-cell receptor interaction, and its blocking inhibits T-cell activation. Administration of abatacept subsequently leads to impaired B-cell activation and reduced levels of autoantibodies, together with a reduction in T-cell-mediated activation of osteoclasts and diminished cytokine release from T-cells, B-cells, and macrophages.70,71 This translates into reduced disease activity, a decrease in inflammatory markers and halted bone erosion, as seen in various randomized clinical trials supporting its use, with benefits being present as early as 1–4 months of continued administration.72 Abatacept is more effective in anticyclic citrullinated peptide antibody-positive patients.73 The reason for this is unknown, and emphasizes that RA is a syndrome with multiple subtypes that may respond differently to different therapies.
The efficacy and safety of abatacept was consistently shown in biologic-naïve methotrexate-inadequate responders in the AIM (Abatacept in Inadequate responders to Methotrexate) trial,74 with ACR20, ACR50, and ACR70 responses of 68%, 40%, and 20%, respectively, compared with 40%, 17%, and 6.5% with placebo, at 24 weeks. In the same group of patients, abatacept produced similar results as a first-line biologic agent when compared with the anti-TNF agent infliximab in ATTEST (Abatacept or infliximab versus placebo, a Trial for Tolerability, Efficacy and Safety in Treating RA).75 In this trial, it was even suggested that abatacept could be more effective than infliximab in sustaining clinical improvement and remission rates after 12 months of therapy.
Abatacept also proved to be an effective therapeutic option after failure of an anti-TNF agent in ATTAIN (Abatacept Trial in Treatment of Anti-TNF INadequate responders)76 (ACR20, ACR50, and ACR70 responses of 50%, 20%, and 10%, respectively, versus 19%, 4%, and 1% in controls), and also in the open-label extension of the ATTEST trial, with infliximab-inadequate responders being rescued by abatacept.77 Additionally, no washout period is necessary before abatacept treatment after anti-TNF failure.78
Because clinical guidelines increasingly value the recognition of poor prognostic markers to justify the immediate beginning of biologic DMARDs, abatacept also produced a good clinical and radiographic response in methotrexate-naïve patients with early erosive RA in AGREE (Abatacept trial to Gauge Remission and joint damage progression in methotrexate-naïve patients with Early Erosive rheumatoid arthritis).79
Administration of abatacept in the majority of clinical trials refers to the 20 mg/kg monthly intravenous regimen, on background methotrexate. It has become increasingly clear that subcutaneous administration is equally effective and safe.80 Subcutaneous injection requires a much smaller dose (125 mg weekly injections, 500 mg per month), which has a clear advantage when considering cost-effectiveness.
Abatacept monotherapy (without methotrexate) has not been sufficiently studied. In the ACCOMPANY (Abatacept in Subjects with Rheumatoid Arthritis Administered Plus or Minus Background Methotrexate Subcutaneously) trial, subcutaneous abatacept showed similar efficacy and immunogenicity whether with or without methotrexate.81 Data regarding combination regimens with other synthetic DMARDs are sparse.
Abatacept is presently included in clinical consensus and guidelines in Europe as an option after anti-TNF failure, always in combination with methotrexate.42 In Switzerland, it has already been adopted as a first-line biologic agent, in concordance with the results referred to above.72 In the US, like rituximab, it has also been included in the recommendation guidelines43 as a first-line biologic agent for all patients with moderate to severe disease activity and for patients with low activity and poor prognostic markers who have failed synthetic DMARDs.
Beyond abatacept
Abatacept, as a costimulation blocker, inhibits the activation and proliferation of all T-cell subtypes,82 including Tregs. This is in contrast with the mechanism of action of other biologic agents such as anti-IL-6R, anti-CD20, or anti-TNF, which enhance Treg regulatory function, thereby ameliorating disease. Maintaining Treg function by selectively inhibiting other T-cell subtypes would imply a different molecular target, as CTLA-IgFc is nonspecific. More precise modulation of the T-cell activation pattern, selectively inhibiting Th1 and Th17 subtypes, would theoretically result in a more effective way of controlling disease activity, maybe even with fewer adverse effects with respect to susceptibility to infection.
Combination therapy with other biologics has been tried. Association with an anti-TNF agent (etanercept) did not produce significant clinical benefits, but induced a serious rise in adverse event rates.83 Combination regimens with other biologics such as tocilizumab remain to be tested.
Anti-cytokine agents
Targeting specific cytokines involved in the inflammation process in the joints and systemically has been a logical strategy for controlling disease activity.84 It modulates not only the direct effects of these cytokines on critical “effector” cells such as osteoclasts, chondrocytes, or macrophages, but also has indirect cell-mediated effects subsequent to the differential pattern of activation and differentiation of T-cells and B-cells.85 Anti-TNF agents were the first successful case, leading to the most impressive improvement in the care of patients with RA. Targeting IL-6 signaling was the next successful step, leading to widespread use of the anti-IL-6 agent tocilizumab. In the future, drugs inhibiting the signaling of other cytokines such as IL-12, IL-13, and IL-17 may be promising strategies in the treatment of RA.
IL-6 in RA
IL-6 is produced by several cell types, including dendritic cells, B-cells, macrophages, and endothelial cells, upon their induction by inflammatory stimuli such as TNF-α, and is present in large quantities in the synovial fluid of RA-affected joints and in serum. It has a central role in the pathogenesis of RA, inducing endothelial activation, neutrophil activation and chemotaxis, directly activating osteoclasts and chondrocytes and stimulating matrix turnover and bone erosion.86 IL-6 is also responsible for important biases in T-cell development.62 Through inhibition of interferon-gamma and enhancing IL-4 signaling, IL-6 produces a shift towards a Th2 phenotype of CD4+ T-cells, which is in contrast with the main Th1 response seen in RA. An explanation for this contradiction may come from the discovery that IL-6 is central to the Th17 differentiation of naïve T-cells when in the presence of transforming growth factor-beta.62 Th17 T-cells are known to be potent inflammatory cells in the synovium, mainly through the production of IL-17, which inhibits Th2 differentiation in favor of the Th1 phenotype; this could explain the contradictory isolated actions of IL-6, which also inhibits Treg differentiation, contributing to inflammation and loss of tolerance.
Further, IL-6 is largely responsible for the systemic effects of RA.87 By inducing the expression of hepcidin, IL-6 contributes to the inflammatory anemia seen in many patients. Fatigue in RA is closely correlated with IL-6 concentrations in serum. IL-6 induces the production of acute-phase reactants in hepatocytes via the JAK/STAT signaling pathway, including C-reactive protein and serum amyloid A. C-reactive protein binds to apolipoprotein B-containing lipoproteins88 and increases the uptake of low-density lipoprotein in atheromatous lesions.89 Serum amyloid A, in turn, was shown to be introduced into cholesterol-containing high-density lipoprotein particles, impairing protective reverse cholesterol transport90,91 and inducing formation of foam cells.92
The case for tocilizumab
Blocking IL-6 signaling by administration of tocilizumab has produced consistent results whether in combination with methotrexate or in monotherapy.93 In OPTION94 (Tocilizumab Pivotal Trial in Methotrexate Inadequate RespONders), the superiority of tocilizumab with methotrexate was shown in methotrexate-inadequate responders, when compared with methotrexate alone, for both clinical response and inflammatory markers (ACR20, ACR50, and ACR70 responses of 59%, 44%, and 22%, respectively, at 24 weeks versus 26%, 22%, and 4% with placebo). The LITHE (Tocilizumab Safety and the Prevention of Structural Joint Damage)95 study confirmed a benefit in radiographic outcomes for tocilizumab. In the TOWARD96 (Tocilizumab in Combination With Traditional DMARD Therapy) trial, a significant response was also seen when in combination with any synthetic DMARD versus any DMARD in monotherapy in patients with an inadequate response to synthetic DMARD therapy. After failure of one anti-TNF agent, tocilizumab also proved to be beneficial in combination with methotrexate (ACR20, ACR50, and ACR70 responses of 50%, 29%, and 12% at 24 weeks versus 10%, 3.8%, and 1.3% with methotrexate only) in the RADIATE97 (Research on Tocilizumab Determining Efficacy after Anti-TNF Failures) trial.
The efficacy of tocilizumab in monotherapy has been increasingly substantiated. In the ACT-RAY98 (Adding tocilizumab or switching to tocilizumab monotherapy in methotrexate inadequate responders: 24-week symptomatic and structural results of a 2-year randomised controlled strategy trial in rheumatoid arthritis) study, addition of methotrexate to tocilizumab therapy in methotrexate-inadequate responders brought no benefit, and the AMBITION99 (Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis) trial showed clear superiority of tocilizumab in monotherapy versus methotrexate alone as soon as 6 months in patients naïve to any biologic or methotrexate. These data make tocilizumab an excellent choice (if not the only one) in the large group of patients who are intolerant or have contraindications to methotrexate (about 30%).100
An important advantage of tocilizumab is its ability to control the extra-articular manifestations of RA, namely chronic inflammatory anemia, asthenia, and cardiovascular risk. Tocilizumab was seen to raise hemoglobin levels and to alleviate asthenia.87 Initially, the impact of tocilizumab on the lipid profile, ie, increasing total cholesterol levels, raised concern that it could increase cardiovascular risk. However, total cholesterol levels are increased at the expense of both low-density and high-density lipoprotein particles, in a ratio that may actually confer a protective effect on atherogenesis.101 This, in parallel with the benefit conferred by reducing atherogenic acute-phase proteins (see above), suggests that tocilizumab may be an excellent drug for preventing cardiovascular comorbidity in these patients.
In Europe, tocilizumab is recommended in the 2010 European League Against Rheumatism guidelines only after failure of at least one anti-TNF agent, with or without a synthetic DMARD.42 In the US, tocilizumab is suggested in the 2012 ACR guidelines as an option only after failure of an anti-TNF, abatacept or rituximab,43 although it was approved by the US Food and Drug Administration for moderate to severe RA with inadequate response to any DMARD. Also, in the US the initial recommended dose of tocilizumab is 4 mg/kg while in Europe it is 8 mg/kg. In the large majority of clinical trials, tocilizumab was tested with the 8 mg/kg dose, making this regimen the one most supported by the available evidence. Nevertheless, there have been some reports and registry data supporting the use of the 4 mg/kg dose, especially when there are concerns about toxicity and the patient has achieved remission.102
Beyond tocilizumab
IL-6 signaling has an important singularity: the membrane-bound IL-6 receptor is a hexameric complex composed of a nonsignaling IL-6 binding molecule and two signaling molecules called gp130 (shared by all members of the IL-6 family of receptors).63 Many cell types, upon IL-6 signaling or during apoptosis, shed cleaved soluble molecules of the IL-6 receptor (IL-6R). This soluble version of IL-6R may thereby interact with membrane-bound gp130 in cell types which do not normally express the IL-6R molecule, conferring on them the capacity to be responsive to IL-6 signaling. This process, called trans-signaling,103 is responsible for a fair amount of the recognized actions of IL-6 including its systemic effects, T-cell induction, and chemotaxis. In animal models, blockade of IL-6 trans-signaling with a gp130-Fc fusion protein produced interesting results, alleviating arthritis104 and even reducing atherogenesis. Most importantly, it did better than IL-6R blockade in a sepsis model, conferring 100% survival after cecal puncture.105 The use of trans-signaling-selective blocking agents has not yet been tested in humans, but they could serve as a valid strategy to maintain the major efficacy of tocilizumab with much less toxicity and fewer side effects. Human trials are programmed for 2013, and the results are awaited with some enthusiasm.
On the other hand, inhibition of one of the major intracellular effectors of IL-6 signaling, ie, the JAK-STAT pathway, has been given much attention, and preliminary results are being received with enthusiasm. JAK and STAT molecules have several isoforms which are expressed differently in multiple cell types. They are intracellular messengers for a multitude of kinase cell receptors, including erythropoietin, interferon-gamma, IL-4, and IL-23, apart from IL-6.106 Given its pleiotropic use throughout development, maintenance and regeneration of adult tissue, overt blocking of JAK-STAT signaling would be necessarily fatal, or at least related to serious adverse effects. However, selective inhibition of specific isotypes of JAK or STAT could have a different profile. Inhibition of JAK3 by tofacitinib seems to be almost selective to lymphocytes, reducing its toxicity, and has a major impact on several IL-6 actions (including systemic and cardiovascular effects), inhibiting Th1 and Th17 differentiation and enhancing Treg expansion.107 Phase III trials have already been conducted with an orally available formulation of tofacitinib, which has proved to be successful in reducing clinical and laboratory disease activity and radiographic progression compared with placebo, with a good safety profile. This has been shown at least in methotrexate-inadequate responders (ORAL Scan,108 ORAL Solo,109 and ORAL Standard studies)110 and in other DMARD-inadequate responders (ORAL Sync study),111 and tofacitinib has shown an efficacy and safety profile similar to that of adalimumab, an anti-TNF agent, in association with methotrexate, in the ORAL Standard study. In the ORAL Solo trial, it even demonstrated effectiveness in reaching clinical endpoints in monotherapy.
The forgotten case of anakinra
The IL-1 inhibitor anakinra is a recombinant nonglycosylated form of the human IL-1 receptor antagonist. It has proven to be superior to placebo in RA patients with moderate to severe disease activity in three randomized clinical trials,112–114 with differences of up to 42% versus 23% on ACR20 responses (although more modest in ACR50 responses). However, no head-to-head studies have been performed comparing anakinra with other biologics. In a meta-analysis by Thaler et al,115 anakinra seemed to confer no clinical benefit over anti-TNF therapy, and had a worse safety profile, with a greater incidence of infusion reactions (adverse events in 67.2% with anakinra versus 17.5%–22.4% with an anti-TNF). Nevertheless, the data on anakinra are sparse, and for that reason it was not included in the 2012 ACR guidelines for treatment of RA. It is plausible that anakinra could be of interest in specific populations, namely after failure of other biologics, but no studies addressing this have been published.
Conclusion
A fair amount of exciting information has been gathered over the past two decades about the treatment of RA. New biologic agents approved after anti-TNF therapy (Table 1) have continued to improve the prognosis, quality of life, and life expectancy of RA patients, and a huge number of new agents are being tested in Phase II and III clinical trials, whether targeting new molecules (eg, sarilumab, a monoclonal antibody directed against the alpha subunit of IL-6 receptor, or secukinumab, an anti-IL-17 monoclonal antibody)116 or improving existing drugs (such as ocrelizumab, a humanized version of rituximab).117,118
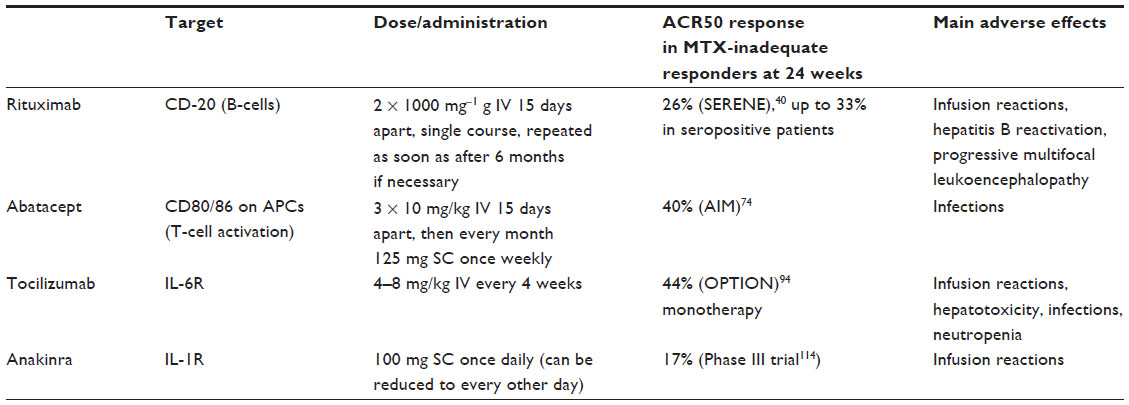 | Table 1 Currently approved molecular targeted strategies to control RA disease activity |
Even though great efforts are being undertaken to deepen our understanding of the pathobiology of RA, controversy still exists, and current knowledge fails to explain all the data already gathered. RA is clearly a disease with many subentities, with different outcomes and different potential “optimal therapies”. Synovial tissue histology is a good example, as each different histopathology pattern is correlated with particular treatment responses and clinical characteristics. Subgroup-based studies in RA are still in their infancy, but could provide a means to identify variables predictive of a good biologic treatment response in particular patients.
The outcomes measured in the large majority of clinical trials, and even in basic research, are hitherto centered on arthritis and quality of life scores. Not taking into account all the frequent extra-articular manifestations of RA underestimates the systemic burden of disease, and therefore, it is possible that the results of these trials could be different if a more holistic approach for outcome evaluation had been used. Further, disease activity and bone damage seem to be unlinked during biologic therapy (at least with rituximab119 and tocilizumab120), and this may make the use of current disease activity scores to evaluate the efficacy of such biologic therapies even less adequate. On the other hand, some extra-articular manifestations are often major variables in clinical decision-making (eg, pulmonary disease), and thus should be more extensively studied and considered; present data and guidelines are based on the belief that whatever action improves articular disease will improve other manifestations, which is not necessarily true. Cardiovascular risk, being the major cause of death in this context, has come to attention recently, but the impact of each biologic therapy in relation to others is still understudied.
There is now a wide range of different drugs from different classes of biologic DMARDs available (and soon this number will increase significantly), providing the opportunity to address each patient as a particular case and thereby optimize medical intervention. A strong knowledge of the pharmacologic characteristics of all the drugs, together with a comprehensive approach to each patient, will allow a true informed choice of the best treatment for each case and better overall population-based control of the disease.
Disclosure
The authors report no conflicts of interest in this work.
References
Meune C, Tousé E, Trinquart L, Allanore Y. Trends in cardiovascular mortality in patients with rheumatoid arthritis over 50 years: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford). 2009;48:1309–1313. | |
MacGregor AJ, Snieder H, Rigby AS, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000;43:30–37. | |
Silman AJ, MacGregor AJ, Thomson W, et al. Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol. 1993;32:903–907. | |
Boissier MC, Semerano L, Challal S, Saidenberg-Kermanac KN, Falgarone G. Rheumatoid arthritis: from autoimmunity to synovitis and joint destruction. J Autoimmun. 2012;39:222–228. | |
Arvikar SL, Collier DS, Fisher MC, et al. Clinical correlations with Porphyromonas gingivalis antibody responses in patients with early rheumatoid arthritis. Arthritis Res Ther. 2013;15:R109. | |
Rantapaa-Dahlqvist S, de Jong BA, Berglin E, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48: 2741–2749. | |
Smolen J, Landewé RB, Mease P, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis. 2009;68: 797–804. | |
Keystone EC, Genovese MC, Klareskog L, et al. Golimumab, a human antibody to tumour necrosis factor {alpha} given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: the GO-FORWARD Study. Ann Rheum Dis. 2009;68:789–796. | |
Buch MH, Bingham SJ, Bryer D, Emery P. Long-term infliximab treatment in rheumatoid arthritis: subsequent outcome of initial responders. Rheumatology (Oxford). 2007;46:1153–1156. | |
Pai S, Pai L, Birkenfeldt R. Correlation of serum IgA rheumatoid factor levels with disease severity in rheumatoid arthritis. Scand J Rheumatol. 1998;27:252–256. | |
van der Helm-van Mil AH, Verpoort KN, Breedveld FC, Toes RE, Huizinga TW. Antibodies to citrullinated proteins and differences in clinical progression of rheumatoid arthritis. Arthritis Res Ther. 2005;7: R949–R958. | |
Emery P, Fleischmann R, Filipowicz-Sosnowska A, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54:1390–1400. | |
Tak PP, Rigby WF, Rubbert-Roth A, et al. Inhibition of joint damage and improved clinical outcomes with rituximab plus methotrexate in early active rheumatoid arthritis: the IMAGE trial. Ann Rheum Dis. 2011;70:39–46. | |
Mease PJ, Cohen S, Gaylis NB, et al. Efficacy and safety of retreatment in patients with rheumatoid arthritis with previous inadequate response to tumor necrosis factor inhibitors: results from the SUNRISE trial. J Rheumatol. 2010;37:917–927. | |
Dass S, Rawstron AC, Vital EM, Henshaw K, McGonagle D, Emery P. Highly sensitive B cell analysis predicts response to rituximab therapy in rheumatoid arthritis. Arthritis Rheum. 2008;58:2993–2999. | |
Vital EM, Rawstron AC, Dass S, et al. Reduced-dose rituximab in rheumatoid arthritis. Arthritis Rheum. 2011;63:603–608. | |
Teng YK, Levarht EW, Toes RE, Huizinga TW, van Laar JM. Residual inflammation after rituximab treatment is associated with sustained synovial plasma cell infiltration and enhanced B cell repopulation. Ann Rheum Dis. 2009;68:1011–1016. | |
Sellam J, Rouanet S, Hendel-Chavez H, et al. Blood memory B cells are disturbed and predict the response to rituximab in patients with rheumatoid arthritis. Arthritis Rheum. 2011;63:3692–3701. | |
Pisetsky DS, Grammer AC, Ning TC, Lipsky PE. Are autoantibodies the targets of B-cell-directed therapy? Nat Rev Rheumatol. 2011;7: 551–556. | |
Leandro MJ, Cambridge G, Ehrenstein MR, Edwards JCW. Reconstitution of peripheral blood B cells after depletion with rituximab in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:613–620. | |
Roll P, Dörner T, Tony HP. Anti-CD20 therapy in patients with rheumatoid arthritis: predictors of response and B cell subset regeneration after repeated treatment. Arthritis Rheum. 2008;58:1566–1575. | |
O’Neill SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis. J Immunol. 2005;174:3781–3788. | |
Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189:1639–1648. | |
Tanaka-Watanabe Y, Matsumoto I, Iwanami K, et al. B cells play a crucial role as antigen-presenting cells and collaborate with inflammatory cytokines in glucose-6-phosphate isomerase-induced arthritis. Clin Exp Immunol. 2009;155:285–294. | |
Takemura S, Klimiuk PA, Brown A, Goronzy JJ, Weyand CM. T cell activation in rheumatoid synovium is B cell dependent. J Immunol. 2001;167:4710–4718. | |
van de Veerdonk FL, Lauwerys B, Marijnissen RJ, et al. The anti-CD20 antibody rituximab reduces the Th17 cell response. Arthritis Rheum. 2011;63:1507–1516. | |
Moura RA, Graça L, Fonseca JE. To B or not to B: the conducter of rheumatoid arthritis orchestra. Clin Rev Allergy Immunol. 2012;43: 281–291. | |
Matsumoto M, Lo SF, Carruthers CJ, et al. Affinity maturation without germinal centres in lymphotoxin-alpha-deficient mice. Nature. 2006;382:462–466. | |
Rennert PD, James D, Mackay F, Browning JL, Hochman PS. Lymph node genesis is induced by signaling through the lymphotoxin beta receptor. Immunity. 1998;9:71–79. | |
Buch MH, Smolen JS, Betteridge N, et al. Updated consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70:909–920. | |
Martinez-Gamboa L, Brezinschek HP, Burmester GR, Dörner T. Immunopathologic role of B lymphocytes in rheumatoid arthritis: rational of B cell-directed therapy. Autoimmun Rev. 2006;5: 437–442. | |
Nakou M, Katsikas G, Sidiropoulos P, et al. Rituximab therapy reduces antivated B cells in both the peripheral blood and bone marrow of patients with rheumatoid arthritis: depletion of memory B cells correlates with clinical response. Arthritis Res Ther. 2009;11:R131. | |
Kavanaugh A, Rosengren S, Lee SJ, et al. Assessment of rituximab’s immunomodulatory synovial effects (ARISE trial). 1: clinical and synovial biomarker results. Ann Rheum Dis. 2008;67:402–408. | |
Gheita TA, Gheita HA, Kenawy SA. Rituximab restored the muscle power and rescued from a refractory fatal respiratory failure in a patient with elderly-onset polymyositis. Joint Bone Spine. 2012;79: 101–102. | |
Rubbert-Roth A, Tak PP, Zerbini C, et al. Efficacy and safety of various repeat treatment dosing regimens of rituximab in patients with active rheumatoid arthritis: results of a phase III randomized study (MIRROR). Rheumatology (Oxford). 2010;49:1683–1693. | |
Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–2806. | |
Keystone EC, Cohen SB, Emery P, et al. Multiple courses of rituximab produce sustained clinical and radiographic efficacy and safety in patients with rheumatoid arthritis and an inadequate response to 1 or more tumor necrosis factor inhibitors: 5-year data from the REFLEX study. J Rheumatol. 2012;39:2238–2246. | |
Gomez-Reino JJ, Maneiro JR, Ruiz J, et al. Comparative effectiveness of switching to alternative tumour necrosis factor (TNF) antagonists versus switching to rituximab in patients with rheumatoid arthritis who failed previous TNF antagonists: the MIRAR Study. Ann Rheum Dis. 2012;71:1861–1864. | |
Soliman MM, Hyrich KL, Lunt M, Watson KD, Symmons DP, Ashcroft DM. Rituximab or a second anti-tumor necrosis factor therapy for rheumatoid arthritis patients who have failed their first anti-tumor necrosis factor therapy? Comparative analysis from the British Society for Rheumatology Biologics Register. Arthritis Care Res. 2012;64:1108–1115. | |
Emery P, Deodhar A, Rigby WF, et al. Efficacy and safety of different doses and retreatment of rituximab: a randomised, placebo-controlled trial in patients who are biological naive with active rheumatoid arthritis and an inadequate response to methotrexate (Study Evaluating Rituximab’s Efficacy in MTX iNadequate rEsponders (SERENE). Ann Rheum Dis. 2010;69:1629–1635. | |
Chatzidionysiou K, Lie E, Nasonov E, et al. Effectiveness of disease-modifying antirheumatic drug co-therapy with methotrexate and leflunomide in rituximab-treated rheumatoid arthritis patients: results of a 1-year follow-up study from the CERERRA collaboration. Ann Rheum Dis. 2012;71:374–377. | |
Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatid drugs. Ann Rheum Dis. 2010;69:964–975. | |
Singh JA, Furst DE, Bharat A, et al. 2012 Update of the 2008 American College of Rheumatology recommendations for the use of diseas-modifying antirheumatic drugs and biologic agents in the treatment of theumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64:625–639. | |
Cantaert T, Kolln J, Timmer T, et al. B lymphocyte autoimmunity in rheumatoid synovitis is independent of ectopic lymphoid neogenesis. J Immunol. 2008;181:785–794. | |
Neves M, Alves JD. Factors implicated in the generation and persistence of long-lived plasma cell-mediated autoimmunity. Autoimmun Rev. 2011;10:375–382. | |
Humby F, Bombardieri M, Manzo A, et al. Ectopic lymphoid structures support ongoing production of class-switch autoantibodies in rheumatoid synovium. PLoS Med. 2009;6:59–75. | |
Klimiuk PA, Goronzy JJ, Björnsson J, Beckenbaugh RD, Weyand CM. Tissue cytokine patterns distinguishes variants of rheumatoid synovitis. Am J Pathol. 1997;151:1311–1319. | |
Fonseca JE, Canhão H, Resende C, et al. Histology of the synovium tissue: value of semi-quantitative analysis for the prediction of joint erosions in rheumatoid arthritis. Clin Exp Rheumatol. 2000;18:559–564. | |
Mei HE, Schmidt S, Dörner T. Rational of anti-CD19 immunotherapy: an option to target autoreactive plasma cells in autoimmunity. Arthritis Res Ther. 2012;14 Suppl 5:S1. | |
Yazawa N, Hamaguchi Y, Poe JC, Tedder TF. Immunotherapy using unconjugated CD19 monoclonal antibodies in animal models for B lymphocyte malignancies and autoimmune disease. Proc Natl Acad Sci U S A. 2005;102:15178–15183. | |
Hamaguchi Y, Uchida J, Cain DW, et al. The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J Immunol. 2005;174:4389–4399. | |
Tedder TF. CD19: a promising B cell target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:572–577. | |
Stone MJ, Sausville EA, Fay JW, et al. A Phase I study of bolus versus continuous infusion of the anti-CD19 immunotoxin, IgG-HD37-dgA, in patients with B-cell lymphoma. Blood. 1996;88:1188–1197. | |
Younes A, Kim S, Romaguera J, et al. Phase I multidose-escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous infusion every 3 weeks to patients with relapsed/refractory B-cell lymphoma. J Clin Oncol. 2012;30:2776–2782. | |
Jin X, Ding C. Belimumab – an anti-BLyS human monoclonal antibody for rheumatoid arthritis. Expert Opin Biol Ther. 2013;13:315–322. | |
Genovese MC, Kinnman N, de La Bourdonnaye G, Pena Rossi C, Tak PP. Atacicept in patients with rheumatoid arthritis and an inadequate response to tumor necrosis factor antagonist therapy: results of a Phase II, randomized, placebo-controlled, dose-finding trial. Arthritis Rheum. 2011;63:1793–1803. | |
Stohl W, Merrill JT, McKay JD, et al. Efficacy and safety of belimumab in patients with rheumatoid arthritis: a Phase II, randomized, double-blind, placebo-controlled, dose-ranging study. J Rheumatol. 2013;40: 579–589. | |
van Vollenhoven RF, Kinnman N, Vincent E, Wax S, Bathon J. Atacicept in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheum. 2011;63:1782–1792. | |
Dillon SR, Harder B, Lewis KB, et al. B-lymphocyte stimulator/a proliferation-inducing ligand heterotrimers are elevated in the sera of patients with autoimmune disease and are neutralized by atacicept and B-cell maturation antigen-immunoglobulin. Arthritis Res Ther. 2010;12: R48. | |
Firestein GS. Etiology and pathogenesis of rheumatoid arthritis. In: Firestein GS, Budd RC, Harris EDJ, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s Textbook of Rheumatology. 8th ed. Philadelphia, PA: Saunders; 2009. | |
Koenders MI, Lubberts E, van de Loo FAJ, et al. Interleukin-17 acts independently of TNF-alpha under arthritic conditions. J Immunol. 2006;176:6262–6269. | |
Dienz O, Rincon M. The effects of IL-6 on CD4 T cell responses. Clin Immunol. 2009;130:27–33. | |
Kishimoto T. IL-6: from its discovery to clinical applications. Int Immunol. 2010;22:347–352. | |
Szodoray P, Nakken B, Barath S, et al. Altered Th17 cells and Th17/regulatory T-cell ratios indicate the subsequent conversion from undifferentiated connective tissue disease to definitive systemic autoimmune disorders. Hum Immunol. August 20, 2013. [Epub ahead of print.] | |
Matsuki F, Saegusa J, Miyamoto Y, Misaki K, Kumagai S, Morinobu A. CD45RA-Foxp3high activated/effector regulatory T cells in the CCR7+CD45RA-CD27+CD28+ central memory subset are the decreased in peripheral blood from patients with rheumatoid arthritis. Biochem Biophys Res Commun. 2013;438:778–783. | |
Esensten JH, Wofsy D, Bluestone JA. Regulatory T cells as therapeutic targets in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:560–565. | |
Ceeraz S, Hall C, Choy E, Spencer J, Corrigall VM. Defective CD8+CD28- regulatory T cell suppressor function in rheumatoid arthritis is restored by TNF inihibitor therapy. Clin Exp Immunol. 2013;174: 18–26. | |
Szalay B, Vásárhelyi B, Cseh A, et al. The impact of conventional DMARD and biological therapies on CD4+ cell subsets in rheumatoid arthritis: a follow-up study. Clin Rheumatol. August 11, 2013. [Epub ahead of print.] | |
Solomon GE. T-cell agents in the treatment of rheumatoid arthritis. Bull NYU Hosp Jt Dis. 2012;70:191–194. | |
Buch MH, Boyle DL, Rosengren S, et al. Mode of action of abatacept in rheumatoid arthritis patients having failed tumour necrosis factor blockade: a histological, gene expression and dynamic magnetic resonance imaging pilot study. Ann Rheum Dis. 2009;68:1220–1227. | |
Weisman MH, Durez P, Hallegua D, et al. Reduction of inflammatory biomarker response by abatacept in treatment of rheumatoid arthritis. J Rheumatol. 2006;33:2162–2166. | |
von Kempis J, Dudler J, Hasler P, et al. Use of abatacept in rheumatoid arthritis. Swiss Med Wkly. 2012;142:w13581. | |
Gottenberg JE, Ravaud P, Cantagrel A, et al. Positivity for anti-cyclic citrullinated peptide is associated with a better response to abatacept: data from the ‘Orencia and Rheumatoid Arthritis’ registry. Ann Rheum Dis. 2012;71:1815–1819. | |
Kremer JM, Genant HK, Moreland LW, et al. Effects of abatacept in patients with methotrexate-resistant active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2006;144:865–876. | |
Schiff M, Keiserman M, Codding C, et al. Efficacy and safety of abatacept or infliximab vs placebo in ATTEST: a phase III, multi-centre, randomised, doubleblind, placebo-controlled study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Ann Rheum Dis. 2008;67:1096–1103. | |
Genovese MC, Becker JC, Schiff M, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med. 2005;353:1114–1123. | |
Schiff M, Keiserman M, Codding C, et al. Clinical response and tolerability to abatacept in patients with rheumatoid arthritis previously treated with infliximab or abatacept: open-label extension of the ATTEST Study. Ann Rheum Dis. 2011;70:2003–2007. | |
Schiff M, Pritchard C, Huffstutter JE, et al. The 6-month safety and efficacy of abatacept in patients with rheumatoid arthritis who underwent a washout after anti-tumour necrosis factor therapy or were directly switched to abatacept: the ARRIVE trial. Ann Rheum Dis. 2009;68: 1708–1714. | |
Westhovens R, Robles M, Ximenes AC, et al. Clinical efficacy and safety of abatacept in methotrexate naive patients with early rheumatoid arthritis and poor prognostic factors. Ann Rheum Dis. 2009;68: 1870–1877. | |
Schiff M. Subcutaneous abatacept for the treatment of rheumatoid arthritis. Rheumatology (Oxford). 2013;52:986–997. | |
Nash P, Nayiager S, Genovese MC, et al. Immunogenicity, safety, and efficacy of abatacept administered subcutaneously with or without background methotrexate in patients with rheumatoid arthritis: results from a phase III, international, multicenter, parallel-arm, open-label study. Arthritis Care Res (Hoboken). 2013;65:718–728. | |
Pieper J, Herrath J, Raghavan S, Muhammad K, van Vollenhoven R, Malmström V. CTLA4-Ig (abatacept) therapy modulates T cell effector functions in autoantibody-positive rheumatoid arthritis patients. BMC Immunol. 2013;14:34. | |
Weinblatt M, Schiff M, Goldman A, et al. Selective co-stimulation modulation using abatacept in patients with active rheumatoid arthritis while receiving etanercept: a randomized clinical trial. Ann Rheum Dis. 2007;66:228–234. | |
Brennen FM, McLnnes IV. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–3545. | |
Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford). 2012; 51 Suppl 5:v3–v11. | |
Gernero P, Thompson E, Woodworth T, Smolen JS. Rapid and sustained improvement in bone and cartilage turnover markers with the anti-interleukin-6 receptor inhibitor tocilizumab plus methotrexate in rheumatoid arthritis patients with an inadequate response to methotrexate. Arthritis Rheum. 2010;62:33–43. | |
Dayer JM, Choy E. Therapeutic targets in rheumatoid arthritis: the interleukin-6 receptor. Rheumatology (Oxford). 2010;49:15–24. | |
Nunomura W, Hatakeyama M. [Binding of low density lipoprotein (LDL) to C-reactive protein (CRP): a possible binding through apolipoprotein B in LDL at phosphorylcholine-binding site of CRP]. Hokkaido Igaku Zasshi. 1990;65:474–480. Japanese. | |
Mattsson N, Magnussen CG, Rönnemaa T, et al. Metabolic syndrome and carotid intima-media thickness in young adults: roles of apolipoprotein B, apolipoprotein A-I, C-reactive protein, and secretory phospholipase A2: the cardiovascular risk in young Finns study. Arterioscler Thromb Vasc Biol. 2010;30:1861–1866. | |
Artl A, Marsche G, Lestavel S, Sattler W, Malle E. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler Thromb Vasc Biol. 2000;20:763–772. | |
Banka CL, Yuan T, de Beer MC, Kindy M, Curtiss LK, de Beer FC. Serum amyloid A (SAA): influence on HDL-mediated cellular cholesterol efflux. J Lipid Res. 1995;36:1058–1065. | |
Lee HY, Kim SD, Baek SH, et al. Serum amyloid A stimulates macrophage foam cell formation via lectin-like oxidized low-density lipoprotein receptor 1 upregulation. Biochem Biophys Res Commun. 2013; 433:18–23. | |
Patel AM, Moreland LW. Interleukin-6 inhibition for treatment of rheumatoid arthritis: a review of tocilizumab therapy. Drug Des Dev Ther. 2010;4:263–278. | |
Smolen JS, Beaulieu A, Rubbert-Roth A, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomized trial. Lancet. 2008;371:987–997. | |
Fleischmann RM, Halland AM, Brzosko M, et al. Tocilizumab inhibits structural joint damage and improves physical function in patients with rheumatoid arthritis and inadequate responses to methotrexate: LITHE study 2-year results. J Rheumatol. 2013;40(2):113–126. | |
Genovese MC, McKay JD, Nasonov EL, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to diseasemodifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum. 2008;58(10):2968–2980. | |
Genovese MC, McKay JD, Nasonov EL, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to diseasemodifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum. 2008;58(10):2968–2980. | |
Dougados M, Kissel K, Sheeran T, et al. Adding tocilizumab or switching to tocilizumab monotherapy in methotrexate inadequate responders: 24-week symptomatic and structural results of a 2-year randomised controlled strategy trial in rheumatoid arthritis (ACT-RAY). Ann Rheum Dis. 2013;72:43–50. | |
Jones G, Sebba A, Gu J, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69(1):88–96. | |
Salliot C, van der Heijde D. Long-term safety of methotrexate monotherapy in patients with rheumatoid arthritis: a systematic literature research. Ann Rheum Dis. 2009;68:1100–1104. | |
Hansel B, Bruckert E. [Lipid profile and cardiovascular risk in patients with rheumatoid arthritis: effect of the disease and of drug therapy]. Ann Endocrinol (Paris). 2010;71:257–263. French. | |
Oliveira S. Patient with a 9-year history of rheumatoid arthritis and hepatitis B co-infection who responds favorably to tocilizumab monotherapy after progression or intolerance to several disease-modifying anti-rheumatic drugs. Adv Ther. 2012;29 Special Issue:12–13. | |
Rose-John S. IL-6 Trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. 2012;8:1237–1247. | |
Nowell MA, Williams AS, Carty SA, et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol. 2009;182:613–622. | |
Barkhausen T, Tschernig T, Rosenstiel P, et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit Care Med. 2011;39:1407–1413. | |
Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. | |
Tanaka Y, Yamaoka K. JAK inhibitor tofacitinib for treating rheumatoid arthritis: from basic to clinical. Mod Rheumatol. 2013;23:415–424. | |
van der Heijde D, Tanaka Y, Fleischmann R, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013;65(3):559–570. | |
Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Eng J Med. 2012;367(6):495–507. | |
van Vollenhoven RF, Fleischmann R, Cohen S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Eng J Med. 2012;367(6):508–519. | |
Kremer J, Li ZG, Hall S, et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159(4):253–261. | |
Bresnihan B, Alvaro-Gracia JM, Cobby M, et al. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41:2196–2204. | |
Cohen S, Hurd E, Cush J, et al. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:614–624. | |
Cohen SB, Moreland LW, Cush JJ, et al. A multicentre, double blind, randomised, placebo controlled trial of anakinra (Kineret), a recombinant interleukin 1 receptor antagonist, in patients with rheumatoid arthritis treated with background methotrexate. Ann Rheum Dis. 2004;63:1062–1068. | |
Thaler K, Chandiramani DV, Hansen RA, Gerald G. Efficacy and safety of anakinra for the treatment of rheumatoid arthritis: an update of the Oregon Drug Effectiveness Review Project. Biologics. 2009;3: 485–498. | |
Reichert JM. Which are the antibodies to watch in 2013? MAbs. 2013;5: 1–4. | |
Tak PP, Mease PJ, Genovese MC, et al. Safety and efficacy of ocrelizumab in patients with rheumatoid arthritis and an inadequate response to at least one tumor necrosis factor inhibitor: results of a forty-eight-week randomized, double-blind, placebo-controlled, parallel-group phase III trial. Arthritis Rheum. 2012;64:360–370. | |
Rigby W, Tony HP, Oelke K, et al. Safety and efficacy of ocrelizumab in patients with rheumatoid arthritis and an inadequate response to methotrexate – results of a forty-eight-week randomized, double-blind, placebo-controlled, parallel-group Phase III trial. Arthritis Rheum. 2012;64:350–359. | |
Aletaha D, Alasti F, Smolen JS. Rituximab dissociates the tight link between disease activity and joint damage in rheumatoid arthritis patients. Ann Rheum Dis. 2013;72:7–12. | |
Smolen JS, Martinez Avila JC, Aletaha D. Tocilizumab inhibits progression of joint damage in rheumatoid arthritis irrespective of its anti-inflammatory effects: dissociation of the link between inflammation and destruction. Ann Rheum Dis. 2012;71:687–693. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.