Back to Journals » Drug Design, Development and Therapy » Volume 10
Non-imidazole-based histamine H3 receptor antagonists with anticonvulsant activity in different seizure models in male adult rats
Authors Sadek B ![]() , Saad A, Latacz G, Kuder K, Olejarz A, Karcz T, Stark H
, Saad A, Latacz G, Kuder K, Olejarz A, Karcz T, Stark H ![]() , Kieć-Kononowicz K
, Kieć-Kononowicz K ![]()
Received 1 July 2016
Accepted for publication 26 August 2016
Published 25 November 2016 Volume 2016:10 Pages 3879—3898
DOI https://doi.org/10.2147/DDDT.S116192
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jianbo Sun
Bassem Sadek,1 Ali Saad,1 Gniewomir Latacz,2 Kamil Kuder,2 Agnieszka Olejarz,2 Tadeusz Karcz,2 Holger Stark,3 Katarzyna Kieć-Kononowicz2
1Department of Pharmacology and Therapeutics, College of Medicine and Health Sciences, United Arab Emirates University, Al Ain, United Arab Emirates; 2Department of Technology and Biotechnology of Drugs, Faculty of Pharmacy, Jagiellonian University Medical College, Kraków, Poland; 3Department of Pharmaceutical and Medicinal Chemistry, Institute of Pharmaceutical and Medicinal Chemistry, Heinrich Heine University, Düsseldorf, Germany
Abstract: A series of twelve novel non-imidazole-based ligands (3–14) was developed and evaluated for its in vitro binding properties at the human histamine H3 receptor (hH3R). The novel ligands were investigated for their in vivo protective effects in different seizure models in male adult rats. Among the H3R ligands (3–14) tested, ligand 14 showed significant and dose-dependent reduction in the duration of tonic hind limb extension in maximal electroshock (MES)-induced seizure model subsequent to acute systemic administration (5, 10, and 20 mg/kg, intraperitoneally), whereas ligands 4, 6, and 7 without appreciable protection in MES model were most promising in pentylenetetrazole (PTZ) model. Moreover, the protective effect observed for ligand 14 in MES model was lower than that observed for the reference drug phenytoin and was entirely abrogated when rats were co-administered with the brain-penetrant H1R antagonist pyrilamine (PYR) but not the brain-penetrant H2R antagonist zolantidine (ZOL), demonstrating that histaminergic neurotransmission by activation of postsynaptically located H1Rs seems to be involved in the protective action. On the contrary, PYR and ZOL failed to abrogate the full protection provided by 4 in PTZ model and the moderate protective effect by 14 in strychnine (STR) model. Moreover, the experimental and in silico estimation of properties such as metabolism was performed for five selected test compounds. Also, lipophilicity using planar reversed-phase thin-layer chromatography method was included for better understanding of the molecular properties of the tested compounds. Additionally, the absorption, distribution, metabolism, and elimination and toxicity parameters were evaluated for the most promising compounds 2, 4, 6, 7, and 14 utilizing in vitro methods. These interesting results highlight the potential of H3R ligands as new antiepileptic drugs or as adjuvants to available epilepsy medications.
Keywords: histamine H3 receptors, antagonists, anticonvulsant, R-(α)-methyl-histamine, pyrilamine, zolantidine
Introduction
Epilepsy is a group of disorders characterized in common by high predisposition to recurrent seizures.1 Etiology can be due to genetic, congenital, and metabolic factors or secondary to stroke, trauma, tumors, drugs, alcohol, or medications.1 Mechanistically, these causes alter intrinsic (eg, pumps, ion channels, transporters) activity, modify synaptic transmission, or induce “re-wiring” of neurons, resulting in reduced seizure threshold, ie, epileptogenesis.1 Currently existing antiepileptic drugs (AEDs) could not help all epileptic patients as practically 30% of them are not responding to current therapy. In addition, as long life medication is necessary, the search for new and more effective AEDs with enhanced safety profile is a laudable therapeutic goal. Up to date, previous research studies showed that brain histamine and seizure pathophysiology are strongly associated.2,3 Hence, histamine itself is considered as an endogenous anticonvulsant,2 whereas histamine H1 receptor (H1R) antagonists have, in earlier studies, been found to induce convulsions.4,5 In addition, in previous preclinical studies, inhibition of N-methyltransferase, the histamine-metabolizing enzyme in the central nervous system (CNS), by metoprine was found to increase brain histamine content and to reduce seizure susceptibility.6–9 Conversely, inhibition of histidine decarboxylase (HDC), the histamine-synthesizing enzyme in the CNS, by alpha-fluoromethylhistidine has been found to diminish histamine synthesis and consequently to increase seizure activity in experimental animal models.10 Similarly, HDC knock-out mice were more prone to seizures as compared to wild-type mice.11 Interestingly, in a recent study, lamotrigine-resistant kindled model of epilepsy in mice exhibited reduced level of brain histamine, suggesting a role of this neurotransmitter in resistance development to the currently available AEDs.12 Clinically, children with febrile seizures had lower cerebrospinal fluid histamine than those with fever but no convulsions, suggesting the antiseizure effect of histamine.13,14 In addition, it has been found that H1R antagonists increased the incidence of seizures in children.4 Moreover, abnormal levels of H1Rs around the epileptic focus in complex partial seizures were noticed in human positron emission tomography studies.15 Furthermore, research during the last 2 years has provided more information concerning anticonvulsant activities of histamine H3R (hH3R) antagonists/inverse agonists investigated in a variety of animal epilepsy models.16 Accordingly, both imidazole17 and non-imidazole hH3R18–20 ligands provided some protective effects in the maximal electroshock (MES)-induced seizure in Wistar rats. Among a number of active H3R antagonists/inverse agonists achieved in both academia and industry, so far only one H3R antagonist, namely, pitolisant (1) (PIT; Bioprojet Biotech; Figure 1), successfully completed Phase III of clinical trials, with positive outcomes in narcolepsy and daytime excessive sleepiness in patients with Parkinson’s disease, and is under evaluation as an orphan drug (Wakix®; pitolisant hydrochloride) by Bioprojet Pharma.21–23 Also, a clinical utility of PIT was similarly tested in early Phase II studies involving human photosensitivity model.24 Accordingly, PIT tested in doses of 20, 40, and 60 mg/kg significantly provided a suppressive effect in 64% of investigated patients.24 Moreover, in search for potent hH3R antagonists/inverse agonists, our research group recently succeeded in developing a novel series of H3R antagonists/inverse agonists belonging to the non-imidazole class and of high antagonist affinities for human hH3Rs.19,25 Encouraged by the aforementioned results in experimental and clinical studies, we decided to investigate selected H3R antagonists in MES-, pentylenetetrazole (PTZ)-, and strychnine (STR)-induced seizure models in rats.19,25 The results observed showed that few of these antagonists provided promising anticonvulsant activity, among which was the H3R antagonist DL-77 (2) that significantly and dose-dependently (5, 10, and 15 mg/kg, intraperitoneally [i.p.]) reduced MES-induced seizure duration with protective action comparable to that of the reference drug phenytoin (PHT) (Figure 1).19 Encouraged by these results in experimental epilepsy models, and as a continuation of our research, this study investigated N-alkyl-substituted (homo)piperidine ether derivatives (3–14) (Scheme 1). As lead structures for the introduced modifications, the drug candidate PIT (1, Wakix) and DL-77 (2) have been chosen. Accordingly, the structural modifications included the introduction of different cycloalkylamine moieties in the basic part of target ligands, ascending alkyl chain length (five to six methylene groups), and variable position of methyl at the basic piperidine ring (3-CH3 or 4-CH3). The general structures of final ligands 3–14 are presented in Scheme 1. In addition, in vitro pharmacological evaluation of ligands 3–14 included hH3R affinity in a binding assay (human embryonic kidney (HEK)-293 cells) and in cAMP accumulation assay at HEK-293 cells. Moreover, the novel series (3–14) has been examined for its anticonvulsant activity in MES-, PTZ-, and STR-induced seizure models in adult rats. Furthermore and as absorption, distribution, metabolism, and elimination (ADME) are very important properties of ligands, in vitro studies are often applied at the earlier stages of drug discovery. Therefore, the metabolic stability and safety tests of five ligands with the most promising anticonvulsive effect was evaluated. Furthermore, lipophilicity using planar reversed-phase thin-layer chromatography (RP-TLC) method was also included for better understanding of the molecular properties of the compounds.
 | Figure 1 Chemical structures, in vitro affinities, and in vivo potencies of previously described H3R ligands PIT (1, Wakix®)22 and DL-77 (2).19,30,72 |
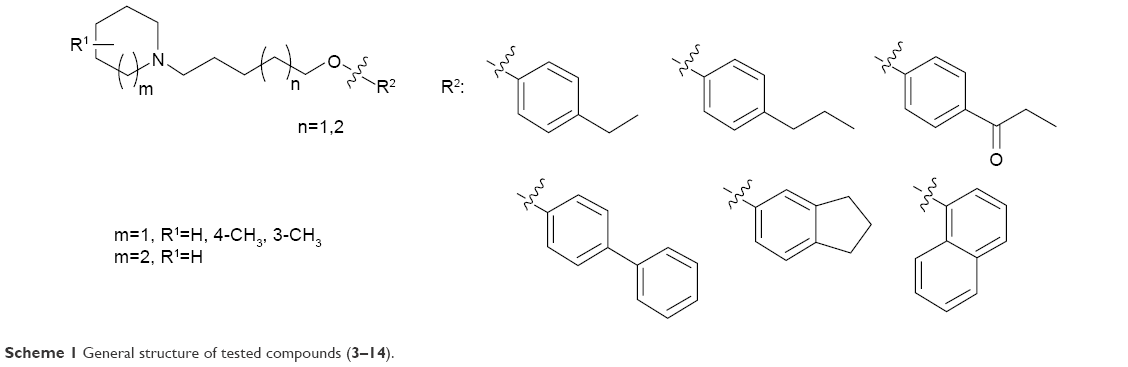 | Scheme 1 General structure of tested compounds (3–14). |
Materials and methods
In vitro pharmacology
Antagonist binding to human hH3R
The displacement binding assay was carried out as described by Kottke et al.26 In summary, frozen crude membrane preparations of HEK-293 cells stably expressing the full-length recombinant hH3R were thawed, homogenized, incubated for 90 min at 25°C, and shook at 250 rpm with [3H]Nα-methylhistamine ([3H]Nα-MeHA; 2 nM), and seven different concentrations of the test compounds, namely, 0.01 mM, 0.1, 1, 10, and 100 nM, and 1 and 10 μM, in a final assay volume of 200 μL per well were used. As described in previous publications,17–19,25,27,28 the antagonist affinity for human hH3Rs was tested utilizing radioligand binding assays and HEK-293 cell membrane preparations. The competition binding experiments were conducted on membranes (20–25 μg/well in a final volume of 0.2 mL binding buffer) that were incubated with [3H]Nα-MeHA (2 nM; 85 Ci/mmol) and a variety of concentrations range of the respective test ligand. For each compound, assays were conducted at least in triplicates with seven appropriate concentrations in the range of 0.01 nM to 10 μM of the respective test compound, and the produced non-specific binding was measured in the presence of unlabeled PIT (10 μM). Accordingly, the maximal binding concentration (Bmax) was determined to be 0.89 pmol/mg and the Kd value of [3H]Nα-MeHA was found to be 2.98 nmol/L (pKd =8.53±0.01) in saturation binding experiments. Moreover, filtration through GF/B filters pretreated with 0.3% m/v polyethyleneimine using an Inotech cell harvester was utilized to separate the bound radioligand from free radioligand. Accordingly, unbound radioligand was removed from the mixture by three washing cycles with 0.3 mL/well of ice-cold 50 mM Tris–HCl buffer, pH 7.4, containing 120 mM NaCl. The filters were saturated in a sample bag with 9 mL scintillator liquid (Betaplate Scint; PerkinElmer, Germany) and counted using a PerkinElmer MicroBeta Trilux scintillation counter. Consequently, the analysis of competition binding data was conducted by the software GraphPad Prism 3.02 using non-linear least squares fit. Ki values were calculated from the IC50 values according to the Cheng–Prusoff equation.29
In vivo pharmacology
Animals
Inbred male Wistar rats (Central Animal Facility of the UAE University) of body weight 180–220 g were used for this study. The animals were retained in an air-conditioned animal facility room with controlled temperature (24°C±2°C) and humidity (55%±15%) under a 12-h light/dark cycle. The animals were allowed free access to food and water. The experiments of this study were carried out between 9 and 12 h, and all procedures were performed according to the guidelines of the European Communities Council Directive of November 24, 1986 (86/609/EEC) and were previously approved for epilepsy study by the College of Medicine and Health Sciences, United Arab Emirates University (Institutional Animal Ethics Committee, approval number; A9-14).
Drugs
H1R antagonist pyrilamine (PYR, 10 mg/kg, i.p.), H2R antagonist (zolantidine [ZOL], 10 mg/kg, i.p.), PTZ (60 mg/kg, i.p.), STR (3.5 mg/kg, i.p.), PHT, and valproic acid (VPA) were purchased from Sigma-Aldrich (St Louis, MO, USA). The H3R ligands: 1-(5-(4-propylphenoxy)pentyl)piperidine hydrogen oxalate (3) – unpublished; 3-methyl-1-(5-(4-propylphenoxy)pentyl)piperidine hydrogen oxalate (4) – unpublished; 4-methyl-1-(5-(4-propylphenoxy)pentyl)piperidine hydrogen oxalate (5) – unpublished; 1-(5-(biphenyl-4-yloxy)pentyl)-3-methylpiperidine hydrogen oxalate (6) – unpublished; 1-(5-(4-propylphenoxy)pentyl)azepane hydrogen oxalate (7) – unpublished; 1-(5-(2,3-dihydro-1H-inden-5-yloxy)pentyl)azepane hydrogen oxalate (8) – unpublished; 1-(6-(4-ethylphenoxy)hexyl)-3-methylpiperidine hydrogen oxalate (9) – unpublished; 3-methyl-1-(6-(4-propylphenoxy)hexyl)piperidine hydrogen oxalate (10) – unpublished; 1-(4-(6-(3-methylpiperidin-1-yl)hexyloxy)phenyl)propan-1-one hydrogen oxalate (11) – unpublished; 1-(6-(biphenyl-4-yloxy)hexyl)-3-methylpiperidine hydrogen oxalate (12) – unpublished; 3-methyl-1-(6-(naphthalen-1-yloxy)hexyl)piperidine hydrogen oxalate (13) – unpublished; 1-(6-(naphthalen-1-yloxy)hexyl)azepane hydrogen oxalate (14) – unpublished; PIT (1); and DL 77 (2) were designed and synthesized in the Department of Technology and Biotechnology of Drugs (Kraków, Poland) according to previously described procedures30,31 (Figure 1; Table 1). Liquid chromatography–mass spectrometry was conducted on a system that involves a Waters Acquity UPLC, attached to a Waters TQD mass spectrometer. The retention times (tR) are provided in minutes, and RP-TLC data were acquired with Merck Silica gel 60 RP-18 F254S glass plates by means of planar chromatographic CHROMDES chambers. The solutions of test compounds were applied using Hamilton 25 μL syringes. The compounds were detected by means of the staining of TLC plates using iodine vapors. Isotonic saline solutions of test compounds 3–14, PIT, PHT, VPA, PYR, and ZOL were administered i.p. at a volume of 1 mL/kg for the in vivo studies. Doses of all test compounds (5, 10, and 20 mg/kg, i.p.) were expressed in terms of the free base. For each test compound, a group of seven rats was used to carry out the anticonvulsant study.
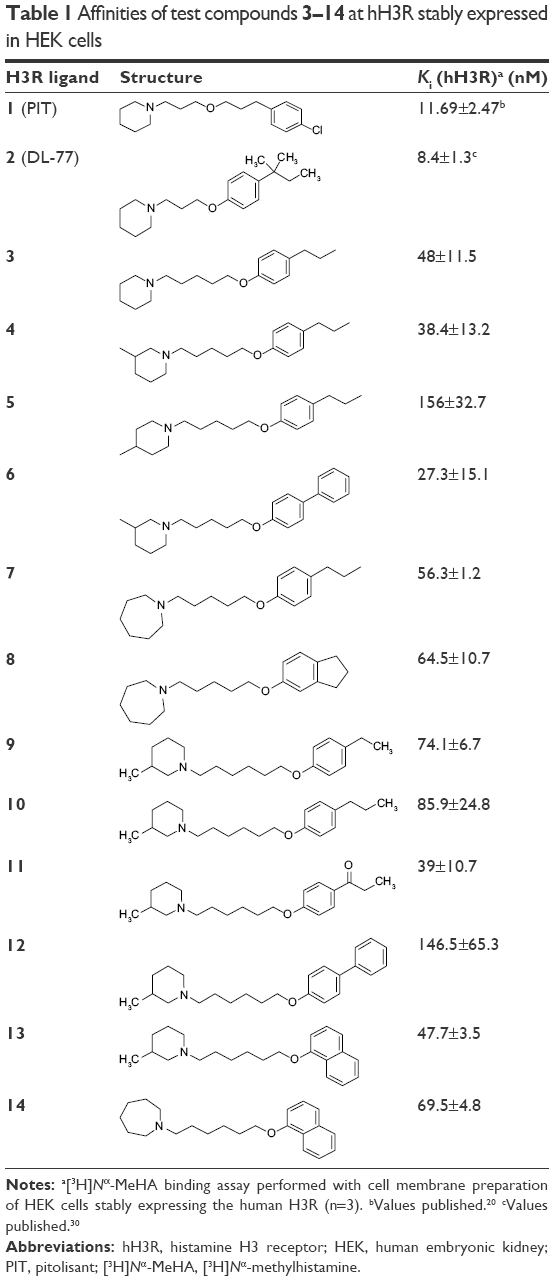 | Table 1 Affinities of test compounds 3–14 at hH3R stably expressed in HEK cells |
MES-induced seizure
As previously described,17–19,25,27,28 the convulsions were induced in rats utilizing a 50-Hz alternating current with 120 mA of intensity. The current was applied through ear electrodes for a duration of 1 s, and the protection against the spread of MES-induced convulsion was defined as the abolition of the tonic hind limb extension (THLE) component of the convulsion.17,18,25,32 The animals were divided into groups of seven rats. In the positive control group, rats were injected with PHT at a dose of 10 mg/kg, this being the minimal dose of PHT that protected animals against the spread of MES-induced seizures without mortality in rats. Animals in the experimental groups were administered test ligands at doses of 5, 10, or 20 mg/kg, i.p., 30 min before the MES challenge. In a further experiment, the most effective dose of ligand 14 was selected for further analysis. In two separate groups of seven rats, the selected dose of 14 was co-administered with PYR (10 mg/kg, i.p.) or ZOL (10 mg/kg, i.p.) 5 min apart and before challenge to the MES test.
Chemically induced seizures
In the current project and according to previously used experimental procedures, two chemical agents, namely, PTZ and STR, were utilized to induce convulsions.17–19,25,27,28 First, different doses of these proconvulsant compounds have been examined (in six to eight rats per dose) to define the minimum dose that induces convulsions in all treated animals.19,33–37 This minimal proconvulsant dose was then applied to screen potential anticonvulsant activities of the test compounds 3–14. Consequently, PTZ (60 mg/kg, i.p.) or STR (3.5 mg/kg, i.p.) was administered to all experimental test groups (six to eight rats per group), ie, saline and treated rats. Saline, VPA (100 mg/kg, i.p.),19 PIT (10 mg/kg, i.p.), or tested ligands 3–14 (10 mg/kg, i.p.) were injected 30 min prior to PTZ (60 mg/kg, i.p.) or STR (3.5 mg/kg, i.p.) administration, and rats were immediately observed for 30 min (experiment period) for any convulsive signs. The convulsion signs were observed, and graded scores have been used to assess the severity of convulsions according to the following scale: score 0= no seizures, score 1= eye or facial twitches, score 2= convulsive waves across the body, score 3= myo-clonic jerks or rearing, score 4= turn over on to one side position, and score 5= turn over on to back position, generalized tonic–clonic seizures, or die during the experiment period. The animals were divided into groups of seven rats. In the positive control group, rats were injected with VPA at a dose of 100 mg/kg, this being the minimal dose of VPA that protected animals against PTZ-induced seizures without mortality in rats. Animals in the experimental groups were administered test compounds at doses of 5, 10, or 20 mg/kg, i.p., 30 min before the PTZ or STR treatment. In a further experiment, the most protective dose of test ligand 4 was selected for further analysis. In two separate groups (seven animals each), the selected dose of 4 was co-administered with PYR (10 mg/kg, i.p.) or ZOL (10 mg/kg, i.p.) 5 min apart and prior to PTZ treatment – or in case of ligand 14 STR treatment. Notably, the current treatment used in the MES study was the one that produced convulsions in 100% of rats without mortality. Likewise, the dose of PTZ and STR used in this study formed convulsions in 100% of animals without mortality.
Statistical analysis
For statistical comparisons, the software package SPSS 20.0 (IBM Middle East, Dubai, UAE) was used. All data were expressed as the mean ± standard error of the mean. The effects of test compounds on epilepsy were analyzed using a mixed repeated-measures two-way analysis of variance (ANOVA) with Treatment (vehicle or test compound) and Dose (test compound) as the between-subjects factor. The effect of selected test compound in combination with PYR and ZOL on anticonvulsant effect was analyzed using one-way ANOVA with Treatment as the between-subject factor. In case of a significant main effect, post hoc comparisons were performed with Bonferroni’s test. The criterion for statistical significance was set at a P-value of <0.05.
Lipophilicity studies
Lipophilicity, by means of RM0 values, was assessed by means of RP-TLC planar method, and a methanol/acetic acid/water (with constant, 10% acid concentration) solvent mixture was used as a mobile phase. The organic solvent concentration varied from 85% to 55%, by 5% for each step. The compositions of the mobile phase and concentrations were selected experimentally. For each concentration, 10 μL of ligands methanol solution in 1 mg/mL concentration was used in the test, and the saturation of planar chromatographic chambers was achieved with suitable mixture for 45 min, followed by 15 min saturation together with the plate. Evaluation of glass plates was carried out at a distance of 90 mm, the glass plates were dried, and the resulting spots were envisioned utilizing iodine fumes. For each of the test ligands and considering the base of Rf values, the RM values were determined. RM0 values were read from RM/methanol concentration charts, after extrapolation to zero methanol concentration using in-house script.
Selected ADME-Tox parameter studies
Metabolic stability
Commercial, pooled, human (adult male and female) liver microsomes (HLMs) were purchased from Sigma-Aldrich. The biotransformations were conducted using 1 mg/mL of HLMs in 200 μL of reaction buffer containing 0.1 M Tris–HCl (pH 7.4) and the respective test compound (H3R ligand) with a final volume of 50 μM. The reaction mixture was preincubated at 37°C for 5 min, and then, the reaction was commenced by adding 50 μL of NADPH Regeneration System (Promega, Madison, WI, USA). The reaction was ended after 120 min by the addition of cold methanol (200 μL). The mixture was next centrifuged at 14,000 rpm for 15 min, and the ultra-performance liquid chromatography–mass spectrometry (UPLC/MS) analysis of the supernatant was performed. Mass spectra were recorded on UPLC/MS system that consisted of a Waters Acquity UPLC (Waters, Milford, CT, USA), joined to a Waters TQD mass spectrometer (electrospray ionization mode ESI-tandem quadrupole). The in silico investigation was achieved by MetaSite 4.1.1 provided by Molecular Discovery Ltd, and the probability sites with the highest metabolism were analyzed during this study by liver computational model.
Influence on recombinant human cytochromes P450 (CYPs) 3A4 and 2D6
The luminescent CYP3A4 P450-Glo™ and CYP2D6 P450-Glo™ assays and protocols were provided by Promega.38 The reference drugs ketoconazole (KE) and quinidine (QD) were obtained from Sigma-Aldrich. The enzymatic reactions were accomplished in white polystyrene, flat-bottom Nunc™ MicroWell™ 96-Well Microplates (Thermo Fisher Scientific, Waltham, MA, USA). The luminescence signal was measured by using a microplate reader in luminescence mode (EnSpire; PerkinElmer, Waltham, MA, USA). The IC50 value of KE was determined and calculated as reported previously.39 The IC50 value of the reference drug QD was determined according to the manufacturer’s recommendations at the final concentrations of 0.1–100 nM. The final concentrations of test ligands were comparable for both CYP3A4 and CYP2D6 assays and were in the range of 0.025–25 μM.
Safety profile
Antiproliferative activity
Human embryonic kidney HEK-293 cell line (ATCC CRL1573) was kindly provided by Prof Dr Christa Müller (Pharmaceutical Institute, Pharmaceutical Chemistry I, University of Bonn). As described previously, the cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco) with 10% fetal bovine serum, 100 mg/mL streptomycin, and 100 U/mL penicillin at 37°C in an atmosphere containing 5% of CO2.40 The HEK-293 cells were seeded in 96-well plates at a concentration of 1×104 cells/well in 200 μL culture medium and cultured for 24 h to reach 60% confluence. Next, the 25 mM stock solutions of H3R ligands in dimethyl sulfoxide (DMSO) were diluted into fresh growth medium and added into the microplates at the final concentrations of 0.1–250 μM (DMSO concentration did not exceed 1%). After 48 h of incubation, 20 μL of EZ4U labeling mixture (Biomedica) was added to each well, and the cells were incubated under the same conditions for 5 h. The absorbance of the samples was measured using a microplate reader (PerkinElmer) at 492 nm. The IC50 value of the reference drug doxorubicin (DX) was determined and calculated as reported previously.40 GraphPad Prism™ software (version 5.01, San Diego, CA, USA) was used to calculate IC50 value of H3R ligands.
Genotoxic activity (Ames test)
Ames microplate fluctuation protocol (MPF) assay was performed as described previously with Salmonella typhimurium strain TA98, enabling the detection of frameshift mutations.41 Bacterial strain as well as exposure and indicator medium were purchased from Xenometrix AG (Allschwil, Switzerland). The mutagenic potential of test ligands was assessed by incubation of bacteria, incapable of producing histidine, with particular concentration of compound for 90 min in exposure medium, containing limited amount of histidine. The manifestation of reversion events to histidine prototrophy was detected as a growth of bacteria in the indicator medium without histidine after 48 h of incubation in room temperature, and the growth of bacteria in 384-well plates was projected by a color change in the medium from violet to yellow upon addition of pH indicator dye. The respective test compound was classified as mutagenic depending on the ratio of positive wells and the solvent control baseline (FIB, >2-fold). FIB was defined as the mean number of positive wells in the negative control sample, increased by one standard deviation. The reference mutagen (0.5 μM) nonyl-4-hydroxyquinoline-N-oxide (NQNO) was used as a positive control in performed experiments.
Results
Pharmacology
In vitro antagonist affinities at hH1R, hH3R, and hH4Rs
The novel compounds were first tested for their antagonist H3R affinity obtained by [3H]Nα-MeHA binding assay on HEK-293 cell membrane preparation stably expressing hH3R. The observed antagonist affinities for hH3Rs are summarized in Table 1.
In vivo seizure models
Protective activity of H3R ligands 3–14 in MES-induced seizure model
The protective activities of acute systemic administration of H3R ligands 3–14 on MES-induced seizures in rats were investigated (Figure 2A). The results showed that pretreatment with PHT, PIT, and H3R ligands 3–14 (10 mg/kg, i.p.) provided a significant protective action against MES-induced convulsions as confirmed by applying one-way ANOVA with [F(14,90) =29.13; P<0.001]. Among the H3R ligands tested, compounds 13 and 14 at a dose of 10 mg/kg significantly exhibited the most promising protective effects against MES-induced convulsion in rats when compared with the saline-treated group with [F(1,12) =73.13 and F(1,12) =76.68, respectively, all P<0.001] (Figure 2A). Moreover, the results show that ligands 13 and 14 provided comparable protection compared to PIT with [F(1,12) =2.21 and F(1,12) =3.43, respectively, all P>0.05], whereas the provided protection observed was significantly lower than that of the reference drug PHT with [F(1,12) =26.65 and F(1,12) =20.19, respectively, all P<0.001].
 | Figure 2 Protective effect of acute systemic injection of H3R ligands 3–14 on MES-induced convulsions in rats. |
Dose-dependent protective activity of H3R ligand 14 in MES-induced seizure model
The results show that animals pretreated with 5 mg/kg of H3R ligand 14 were significantly protected against convulsions when compared with the saline-treated group [F(1,12) =9.82, P<0.005]. In addition, substantial intensifications of protective activities were observed upon acute systemic injection with 10 and 20 mg/kg of H3R ligand 14 when compared with 5 mg/kg of the same compound with [F(1,12) =7.52, P<0.05] and [F(1,12) =8.25, P<0.001], respectively (Figure 2B).
Effect of PYR and ZOL pretreatment on the protective activity provided by H3R ligand 14 on MES-induced seizures
The reversal of protection provided by H3R ligand 14 was tested by co-administration with CNS-penetrant hH1R antagonist PYR (10 mg/kg, i.p.) or the CNS-penetrant ZOL (10 mg/kg, i.p.) 30 min before MES challenge with [F(1,12) =0.22; P=0.645, for the comparison of saline–saline vs 14+PYR] and [F(1,12) =0.88; P=0.3678, for the comparison of saline–14 vs 14+ZOL], respectively (Figure 2B). Moreover, PYR and ZOL when administered alone did not affect MES-induced seizures with (P=0.883 saline–saline vs saline–PYR) and (P=0.759 saline–saline vs saline–ZOL), respectively (Figure 2B).
Protective activities of H3R ligands 3–14 pretreatment on PTZ-induced convulsions
The effects of H3R ligands 3–14 (10 mg/kg, i.p.) and PIT were assessed and compared with the protective effect of the reference AED VPA on PTZ-induced seizures in rats (Figure 3A). The results showed that ligands 4, 6, and 7 delivered significant protective activities when compared to the saline-treated group after 10, 20, and 30 min of observation [all P<0.05] (Figure 3A). However, PIT did not show protective activity in the PTZ model after 10, 20, and 30 min [F(1,12) =0.03, 0.01, and 0.50, respectively, all NS]. Notably, VPA (100 mg/kg) provided significant protection when compared with saline-treated group after 10, 20, and 30 min with [F(1,12) =101.4, 44.74, and 16.8; all P<0.05].
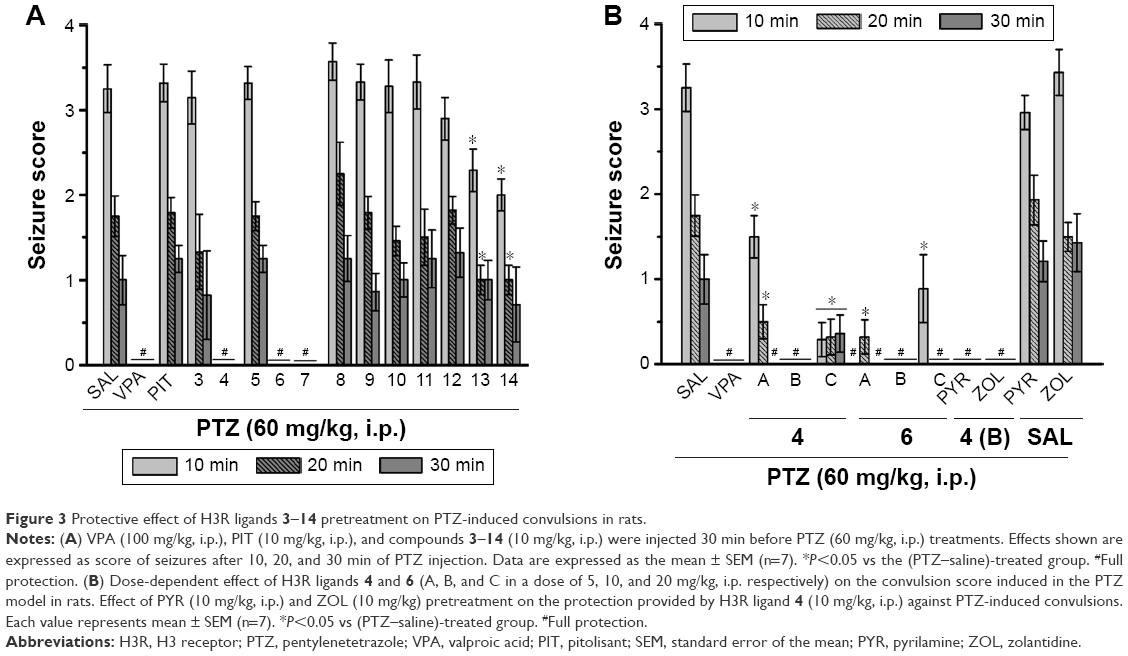 | Figure 3 Protective effect of H3R ligands 3–14 pretreatment on PTZ-induced convulsions in rats. |
Dose-dependent protective activity of H3R ligands 4 and 6 on PTZ-induced convulsions
One-way ANOVA showed that pretreatment with reference AED VPA and ligands 4 and 6 (5, 10, and 20 mg/kg, i.p.) exerted a significant protective action against PTZ-induced convulsions with [F(7,48) =25.63; P<0.001], [F(7,48) =13.47; P<0.001], and [F(7,48) =8.10; P<0.001] after 10, 20, and 30 min of observation, respectively (Figure 3B). In addition, the results showed that significant and full protection was provided at 10, 20, and 30 min after acute systemic administration with 10 mg/kg of ligands 4 and 6 (P<0.001) (Figure 3B). Moreover, the results showed that PYR and ZOL when administered alone failed to reverse the protection provided by H3R ligand 4 (10 mg/kg, i.p.) (Figure 3B).
Protective activities of H3R ligands 3–14 pretreatment on STR-induced convulsions
In STR-induced seizure model, only H3R ligand 2 provided significant protective activity when compared to the saline-treated group after 10, 20, and 30 min of observation [P<0.05] (Figure 4A). Similar to the reference drug VPA, ligands 13 and 14 exhibited significant protective effect as compared with the saline-treated group after 10, 20, and 30 min [all P<0.001] (Figure 4A). Moreover, PIT (10 mg/kg, i.p.) did not provide protective action after 10, 20, and 30 min [F(1,12) =0, 3.32, and 3.50; all NS], respectively. However, the reference AED VPA (300 mg/kg, i.p.) provided significant protection when compared with saline-treated group after 10, 20, and 30 min with [F(1,12) =41.82; P<0.05], [F(1,12) =144.15; P<0.05], and [F(1,12) =81.82; P<0.05], respectively.
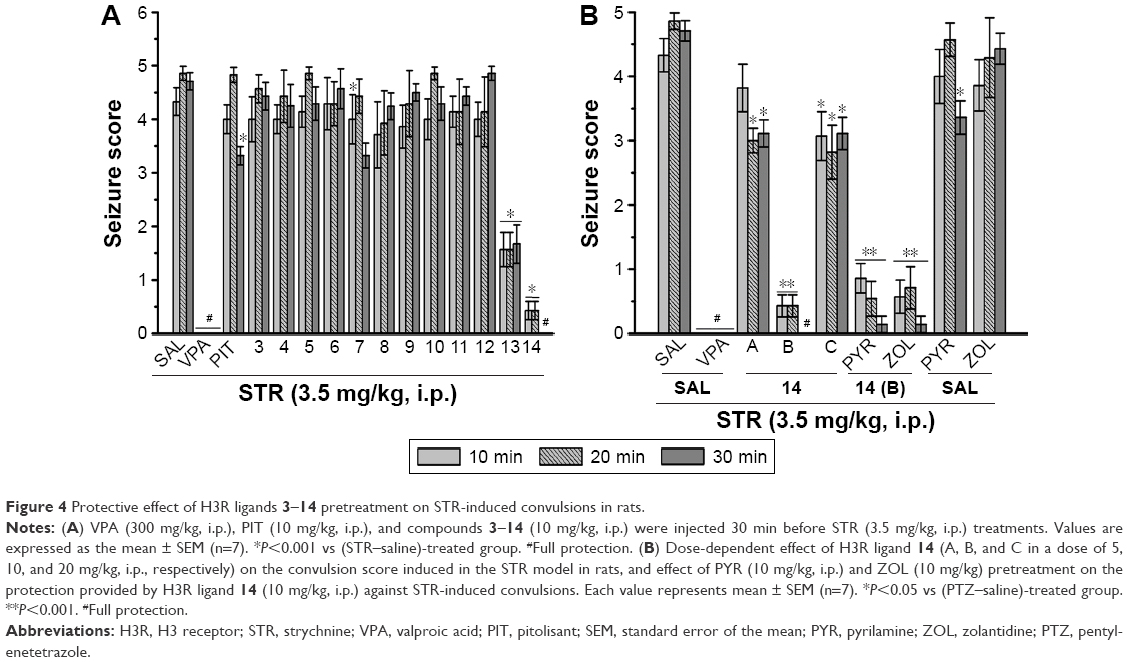 | Figure 4 Protective effect of H3R ligands 3–14 pretreatment on STR-induced convulsions in rats. |
Dose-dependent protective activity of H3R ligand 14 on STR-induced convulsions
The results show that acute systemic administration with reference AED VPA and ligand 14 (5, 10, and 20 mg/kg, i.p.) exerted a significant protective effect against STR-induced convulsions as confirmed with one-way ANOVA with [F(4,30) =33.27; P<0.001], [F(4,30) =57.46; P<0.001], and [F(4,30) =80.07; P<0.001] for 10, 20, and 30 min observations, respectively (Figure 4B). In addition, the results revealed that significant and full protective activity was obtained at 30 min after administration with only 10 mg/kg of 14 (P<0.001). However, lower dose (5 mg/kg, i.p.) and higher dose (20 mg/kg, i.p.) of 14 showed merely moderate protection against STR-induced convulsions when compared at 10, 20, and 30 min to the dose of 10 mg/kg of the same compound [all P<0.05]. Similar to the results observed in the PTZ model, PYR and ZOL failed, also, to reverse the protection provided by H3R ligand 14 (10 mg/kg, i.p.) in STR-induced convulsion model (Figure 4B).
Discussion
In vitro hH3R antagonist affinity
All compounds in their hydrogen oxalate forms were evaluated in radioligand displacement binding assays for affinity at human recombinant hH3R (hH3R) stably expressed in human embryonic kidney (HEK-293) cells as described by Kottke et al.26 The radioligand [3H]Nα-MeHA was used as a competitor. Pharmacological results are assembled in Table 1. All tested compounds showed affinities for hH3R in nanomolar concentration range (Ki: 27–156 nM). For the tested group of novel H3R ligands, among four different cycloalkylamine moieties used (piperidine, 3-methylpiperidine, 4-methylpiperidine, homopiperidine), it is obvious that 3-methylpiperidine is the most preferable basic moiety for hH3R binding pocket. Interestingly, 3-methylpiperidine derivative 6 with hH3R Ki values of 27.3±15 with pentyl alkyl spacer showed higher antagonist affinity in comparison to their corresponding derivatives with hexyl alkyl chain (eg, 10 and 12 with hH3R Ki values of 85.9±24.8 and 146.5±65.3 nM, respectively) (Table 1). On the other hand, for 4-phenylsubstituted compound 6, having the alkyl chain of pentyl resulted in the increment of hH3R antagonist affinity (6 vs 12; hH3R Ki =27.3±15.1 vs Ki =146±653 nM, respectively), whereas in the 4-substituted propylphenyl derivatives a replacement of 3-methylpiperidine with homopiperidine resulted in high decrement of the affinity (eg, 4 vs 7; hH3R Ki =38.4±13.2 nM vs hH3R Ki =56.3±1.2 nM, respectively) (Table 1). Noticeably, elongation of alkyl spacer from propyl in the lead compound 2 (PIT) to pentyl (eg, in compound 3) or hexyl (eg, in compound 10) resulted in a dramatic decrease of antagonist affinity for hH3Rs (2 vs 3 vs 10; hH3R Ki =8.4±1.3 nM vs hH3R Ki =48±11.5 nM vs hH3R Ki =85.9±24.8 nM, respectively). Compound 6 (hH3R Ki =27.3±15.1 nM) showed in fact the highest affinity toward hH3R in the current tested group of compounds. However, compound 12, being direct structural homolog of 6, showed radical loss of affinity (6 vs 12; hH3R Ki =27.3±15.1 nM vs 146.5±65.3 nM, respectively), demonstrating clearly that the length of alkyl spacer and consequently the position of ether moiety play a crucial role in receptor–ligand interactions for this series of derivatives. In summary, compounds incorporating a pentyl alkyl spacer with 3-methylpiperidine and a phenyl substituent in the para-position of the phenyl ring seem to confer higher affinity than those of 4-substituted methylpiperidine derivative (being 6 with hH3R Ki value of 27.3±15.1 nM). In conclusion and among the examples discussed so far in the present small series, phenyl substitution in the 4-position of phenyl moiety appears to be the most advantageous position to provide the respective ligand with high affinity for the binding pocket of hH3R.
In vivo anticonvulsant activity
In the MES model and as compared with standard H3R antagonist PIT (10 mg/kg, i.p.), the saline-treated group, and the H3R antagonist 14-treated group at a dose of 5 mg/kg, the results showed that H3R antagonists 13 and 14 provided the most promising protection after systemic injection with 10 and 20 mg/kg, i.p. (Figure 2A). Consequently, the results indicated a dose-dependent relationship of the observed protection and the presence of a ceiling for H3R antagonist 14 in MES-induced convulsions. Particularly, the protection exhibited by H3R antagonist 14 (10 or 20 mg/kg, i.p.) was similar to that observed in the group pretreated with the reference AED PHT (10 mg/kg, i.p.) (Figure 2A). The latter results are in agreement with recent studies that described the dose-dependent anticonvulsant effect of H3R antagonists in MES-induced seizure models and, also, for PIT in a photosensitivity seizure model in adult patients.17–19,24,25 A further experiment in this study indicated that the protection observed for H3R ligand 14 was abrogated when rats were co-administered with the CNS-penetrant hH1R antagonist PYR (10 mg/kg, i.p.) but not with the CNS-penetrant hH2R antagonist ZOL (10 mg/kg, i.p.) in the MES model (Figure 2B). Interestingly, PYR (10 mg/kg) and ZOL (10 mg/kg) administered alone did not show either a protective or an epileptogenic effect in rats challenged by the MES-induced convulsion. These findings propose that the protection of H3R antagonist 14 in the MES-induced convulsion is facilitated, at least in part, through H3R blockade and interactions of released histamine with postsynaptically located hH1Rs. The latter observation is in agreement with the previously observed protective effects for H3R antagonists.6,17,19,25 Hereafter, H3Rs are auto-receptors located on presynaptic histaminergic terminal neurons and inhibit the synthesis and the release of histamine.42 Consequently, antagonizing these H3Rs by compound 14 would result in an increased neuronal release of brain histamine that provides the protective action observed in the MES model. Particularly, comparable effects of imidazole- and non-imidazole-based H3R ligands have been described previously to be reversed either by H3R agonists or by CNS-penetrant H1R antagonists, but not by CNS-penetrant H2R antagonists, representing an interaction of the H3R antagonism-released histamine with H1Rs on postsynaptic neurons.6,17,19,25,43 However, a functional in vivo bioassay, eg, drinking bioassay, is essentially required to comprehend the in vivo H3R antagonist potency of the described novel ligands, eg, ligand 14 to further confirm their capability to exert an in vivo antagonist effect through interaction with H3Rs. On the other hand and while VPA (100 mg/kg, i.p.) provided full protection in the PTZ-induced convulsion model, acute systemic administration of H3R antagonists 13 and 14 (10 mg/kg, i.p.) offered moderate protective activity (Figure 3A). Notably, PIT used as the standard H3R antagonist (10 mg/kg, i.p.) did not exhibit any protective effect in chemically induced convulsions. Moreover, H3R ligands 4 (10 mg/kg, i.p.), 6 (10 mg/kg, i.p.), and 7 without any considerable protection in the MES model provided full protective activity in the PTZ-induced convulsion model. Furthermore, lower doses (5 mg/kg, i.p.) or higher doses (20 mg/kg, i.p.) of both ligands (4 and 6) failed to demonstrate a dose-dependent protection in the PTZ model (Figure 3A). In an additional experiment, the protective effect of H3R ligand 4 (10 mg/kg, i.p.) was not reversed when rats were pretreated with PYR (10 mg/kg, i.p.) or ZOL (10 mg/kg, i.p.) before PTZ challenge (Figure 3B). These findings suggest that the protective effects observed for H3R ligand 4 in the PTZ model are not mediated through histaminergic neurotransmission. The latter finding could, also, be due to differences in the trigger or the mechanism of seizures and the type of seizures each model represents. Accordingly, MES is a model of generalized tonic–clonic seizures,44 while PTZ (60 mg/kg, i.p.) induces generalized myoclonic and/or tonic–clonic convulsions.45 Moreover, electrically induced seizures result from stimulation of all neuronal pathways where the insult only lasts for short time (1 s). However, in chemically induced convulsions, the trigger remains until the elimination of the proconvulsant, eg, PTZ. Furthermore, chemical agents have in previous studies been found to affect only some neuronal pathways, eg, PTZ provides blockage of t-butylbicyclophosphorothionate site at GABAA receptors and reduces GABA neurotransmission.44–46 Notably, previous studies have, also, shown that other neurotransmitters such as GABA could be the additional player(s) in the mechanism of the protection provided in the PTZ-induced animal convulsion model. It can, also, be hypothesized that H3R antagonists (eg, 4 and 6) could also block histamine H3 heteroreceptors that control the release of other neurotransmitters including GABA.47,48 Interestingly in a previous study, the H3R antagonist clobenpropit was found to increase GABA release and to protect from NMDA-induced excitotoxicity in rat cultured cortical neurons.49
In a further experiment and among tested ligands 3–14, the results observed showed that acute systemic administration of H3R antagonists 13 and 14 (10 mg/kg, i.p.) offered some appreciable protection in the STR-induced convulsion model, whereas the standard AED VPA (300 mg/kg, i.p.) exhibited full protection following 10–30 min time observation (Figure 4A). Moreover, the lower dose (5 mg/kg, i.p.) and higher dose (20 mg/kg, i.p.) of H3R antagonist 14 failed to provide a dose-dependent protection against STR model (Figure 4B). Furthermore and in similarity to the results observed in the PTZ model, additional experiment indicated that the protective effect of H3R antagonist 14 against STR-induced convulsion was not reversed when rats were pretreated with 10 mg/kg, i.p., of the CNS-penetrant hH1R antagonist PYR or with the CNS-penetrant hH2R antagonist ZOL (10 mg/kg, i.p.) 30 min before STR challenge (Figure 4B). These findings further comprehend our results that the protective effects observed for H3R antagonist 14 in the PTZ-induced seizure model, considered also a chemically induced convulsion model, are not mediated through histaminergic neurotransmission. Moreover, it is well recognized that STR functions as a competitive antagonist of the inhibitory amino acid glycine. As a result, the failure of H3R ligand 14 to provide a dose-dependent protection against STR-induced convulsion advocates little or no effect on the glycine receptors, since the mechanism underlying STR-induced convulsions is thought to be credited to its blocking effect on glycine receptors in the brain and in the spinal cord.35 Notably, the discrepancy in the anticonvulsant effects observed for the described ligands of varied in vitro antagonist affinities could also be clarified by the different constructs and predictive validities of the MES, PTZ, and STR models. Hereafter, the end point in the MES test is THLE, which is considered to be a predictive model for generalized tonic–clonic seizures, whereas the PTZ or STR test is used as a predictor of anticonvulsant ligand activity against non-convulsive (absence or myoclonic) seizures. It is, also, noteworthy to mention that most marketed AEDs were not effective in all convulsion models conducted during preclinical drug development. For instance, carbamazepine, oxcarbazepine, and PHT were found to be – and are still – highly effective in the MES-induced convulsion model; however, they lack any protective effects against convulsions induced by PTZ, STR, or picrotoxin.50,51 On the contrary, ethosuximide and tiagabine, which provide high protection in chemically induced convulsion models, are inactive in the MES-induced convulsion model when used at non-toxic doses.51 These different preclinical activities were translated into the clinical utility of PHT, carbamazepine, and oxcarbazepine but not ethosuximide or tiagabine in generalized tonic–clonic convulsions.
Lipophilicity evaluation by planar RP-TLC
At the early stage of preclinical research, there is an influential aim to investigate physicochemical properties of novel compounds. Therefore, the lipophilicity of promising hH3R ligands was experimentally assessed (expressed by RM0 values) applying planar RP-TLC method.52 As a mobile phase, ternary solvent mixture – ethanol/acetic acid/water – was used, with constant, 10% acetic acid concentration and varying methanol concentration in the range of 50%–85% (5% by each step). Tested series of H3R ligands exhibited RM0 values in the range of 1.43–1.91 as depicted for the observed lipophilicity studies data in Table 2. As it was anticipated, lipophilic character of the respective derivative increases with the elongation of the alkyl chain. In addition, values resulted in the similar range for structural analogs, eg, ligands 4 and 10 as well as 13 and 14, respectively. Comparable dependence was observed for 3- and 4-methylpiperidine structural isomeric analogs, eg, ligands 4 and 5, and to some extent, lower values were observed for homopiperidine derivatives in comparison with their respective 3(4)-methylpiperidine analogs (Table 2). However, association between lipophilicity and hH3R affinity was not observed for the tested series as well as for lipophilicity and anticonvulsant activity. With the aim of comparing the theoretical partition coefficient parameters (clogP) with practical ones, calculations using various computer programs, Chem3D, Marvin, and QikProp for Schrödinger, were also implemented as described previously,53–55 since such obtained theoretical values appear to be more representative, considering the reality that all the test H3R ligands are present in their salt forms as hydrogenoxalates. While most comparable, the ratio (theoretical-to-practical) values were obtained using Marvin software, the best correlation was obtained using QikProp by Schrödinger (R=0.783), (R2=0.613).
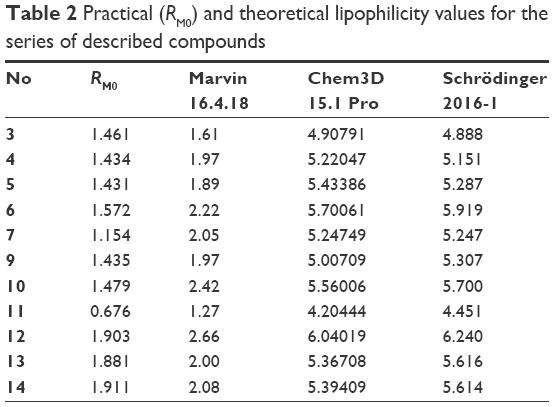 | Table 2 Practical (RM0) and theoretical lipophilicity values for the series of described compounds |
Selected ADME-toxicity (Tox) parameter study
In the modern drug discovery and development process, it is required to evaluate not only the pharmacological properties but also the ADME-Tox parameters together as early as possible. The chosen most active H3R ligands described in this study were screened to identify beneficial and adverse ADME-Tox properties using in vitro methods imitating in vivo conditions and based on the eukaryotic and prokaryotic cell culture growths, HLMs, and bioluminescent enzymatic assays.
Metabolic stability
The metabolic stability of ligands 2, 4, 6, 7, and 14 was studied first in silico by the computational procedure MetaSite.56 The most probably predicted site of metabolism (blue circle marked; Figure 5) was different depending on the respective structure investigated in this study. However, the piperidine and homopiperidine moieties were shown for all compounds as the sites with the very high probability of metabolic transformation (the darker red color of the marked functional group indicates its higher probability to be involved in the metabolism pathway; Figure 5), whereas according to in vitro data, among tested H3R ligands, the homopiperidine moiety of only compound 14 was involved in the reaction of biotransformation (Figures 5–7). In this study, the HLMs were used to determine in vitro the metabolic stability of H3R ligands. The UPLC analyses of the reaction mixtures after 120 min of 2, 4, 6, 7, and 14 incubation with HLMs are shown in Figure 6. Moreover, the number and molecular masses of the respective H3R ligands’ metabolites, identified by MS analysis, are included in Table 3. The exact ion fragment analysis of the investigated compounds and their main metabolites (M1s) allowed to determine the primary metabolism pathways of the respective H3R ligands. As shown in Figure 7A–E, the M1s of compounds 2, 4, 6, and 7 were obtained similarly by hydroxylation at the 4-substituted phenyl moieties. However, for compound 14, a different metabolism pathway was determined. According to the precise analysis of the fragment ions produced by 14 and M1, the main reaction of biotransformation of 14 includes two possibilities: double hydroxylation at the homopiperidine moiety and the degradation of homopiperidine moiety followed by oxidation (Figure 7E). The second option would be in accordance with our data obtained previously for 1 (PIT),57 where the degradation of the piperidine moiety of compound 1 followed by oxidation was observed as a main reaction of biotransformation.
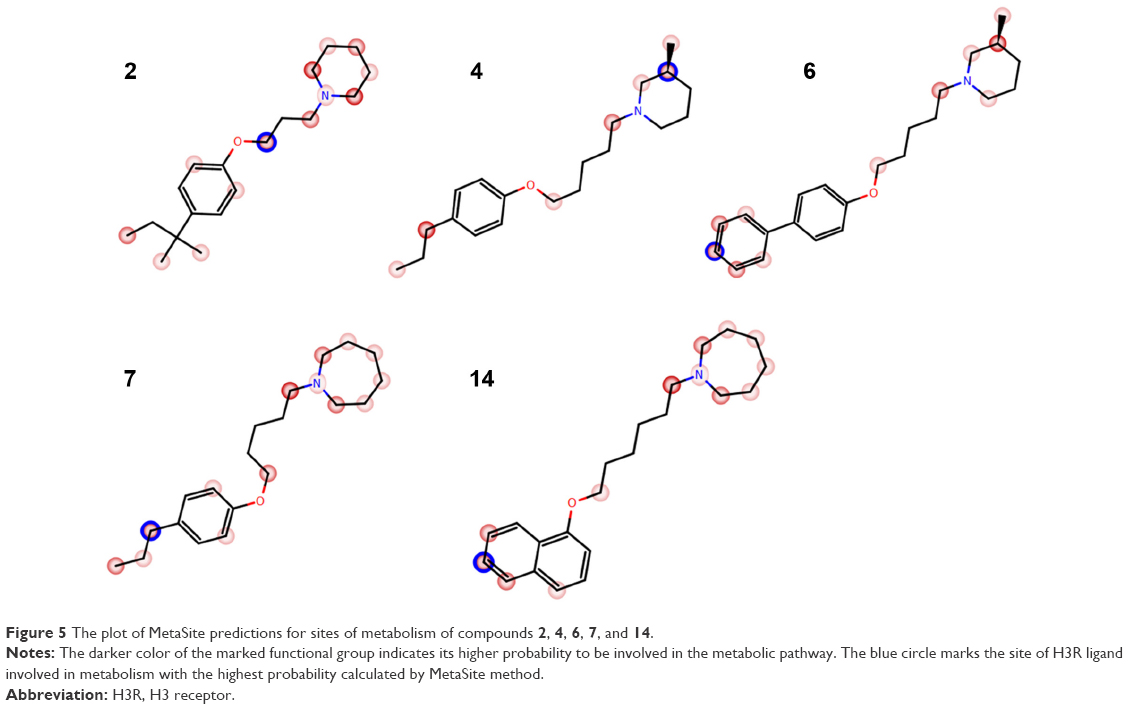 | Figure 5 The plot of MetaSite predictions for sites of metabolism of compounds 2, 4, 6, 7, and 14. |
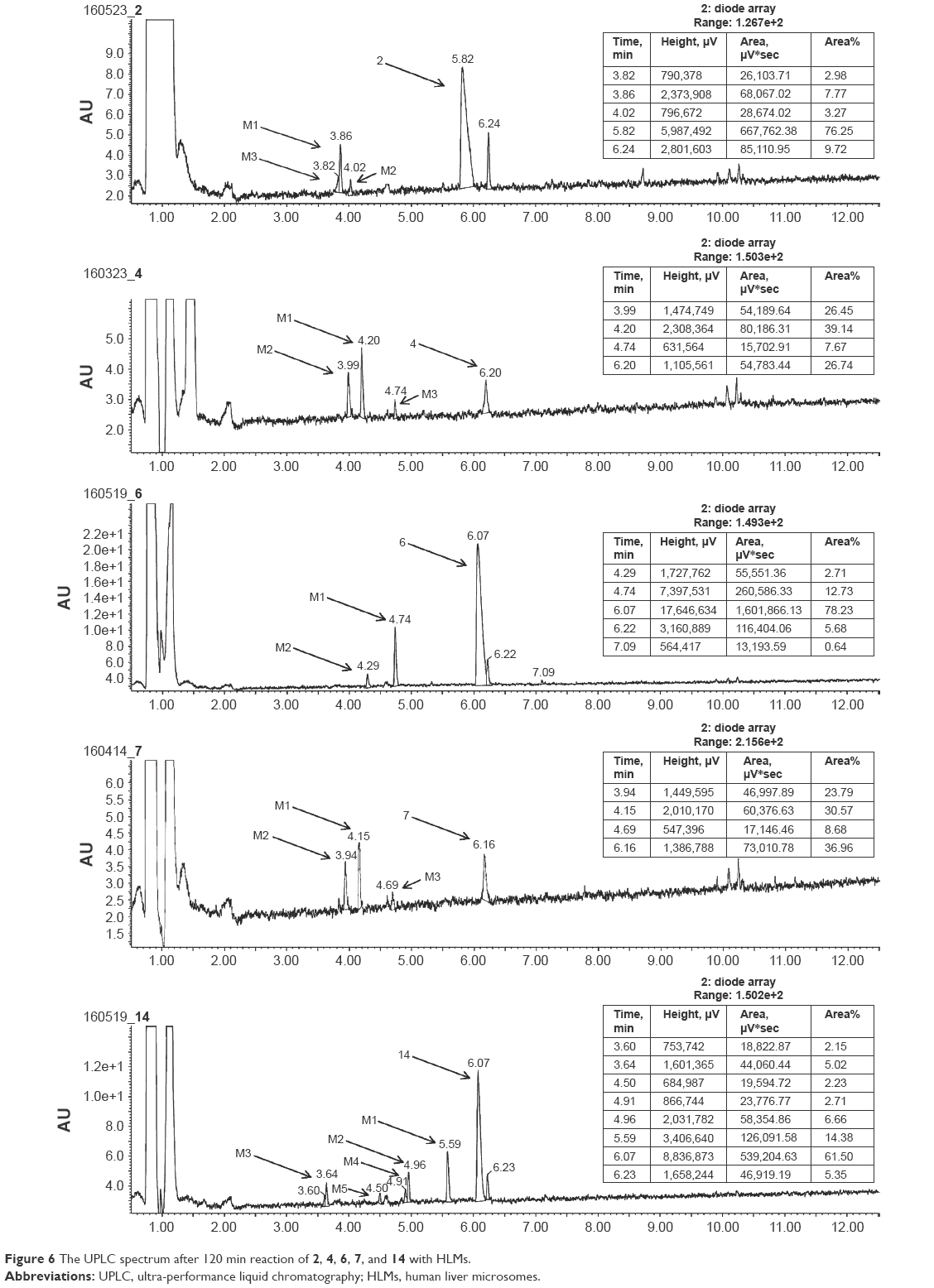 | Figure 6 The UPLC spectrum after 120 min reaction of 2, 4, 6, 7, and 14 with HLMs. |
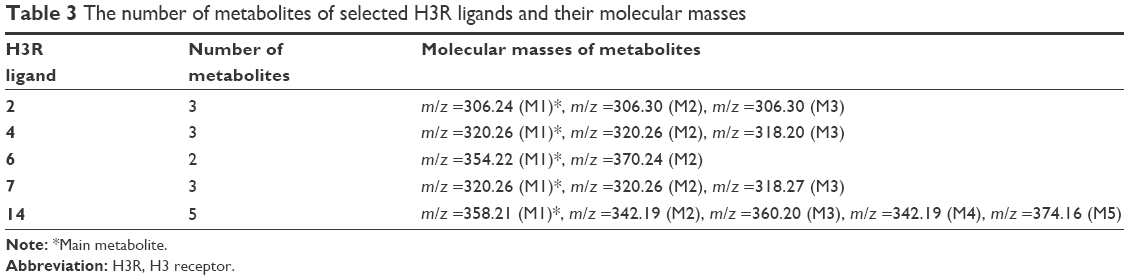 | Table 3 The number of metabolites of selected H3R ligands and their molecular masses |
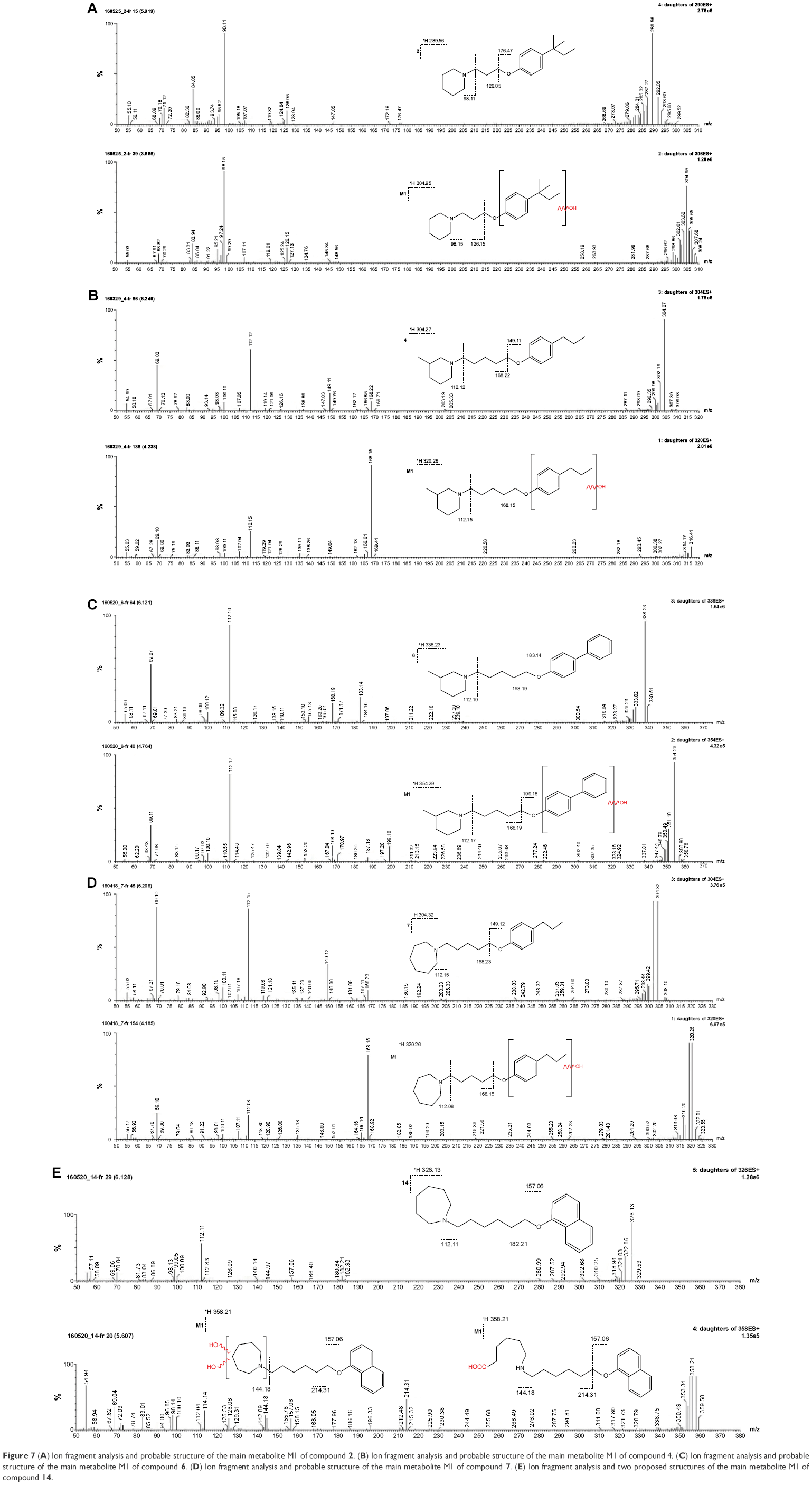 | Figure 7 (A) Ion fragment analysis and probable structure of the main metabolite M1 of compound 2. (B) Ion fragment analysis and probable structure of the main metabolite M1 of compound 4. (C) Ion fragment analysis and probable structure of the main metabolite M1 of compound 6. (D) Ion fragment analysis and probable structure of the main metabolite M1 of compound 7. (E) Ion fragment analysis and two proposed structures of the main metabolite M1 of compound 14. |
Influence on recombinant human CYP3A4 and 2D6 cytochromes activity
CYPs 3A4 and 2D6 are responsible for the metabolism of ~40%–50% of all marketed drugs; therefore, their potential inhibition may be a source of dangerous drug–drug interaction (DDI).58 To predict potential DDI, ligands 2, 4, 6, 7, and 14 were examined to determine their influence on CYPs 3A4 and 2D6 activity. For CYP 2D6, an additional study was performed for 1 (not included in Kuder et al57). To this purpose, we used the luminescence CYP3A4 and CYP2D6 P450-Glo™ assays based on the conversion of the beetle D-luciferin derivative into D-luciferin by recombinant human CYPs 3A4 or 2D6 isoenzymes.38 As reference compounds, the following strong CYP inhibitors were used: CYP3A4 inhibitor KE and CYP2D6 inhibitor QD. The CYP3A4 assay showed either very weak inhibition for ligands 2, 4, and 14 or very weak induction of CYP3A4 cytochrome for ligands 6 and 7 (Figure 8A). Similar to H3R ligands 2, 4, and 14, a very weak inhibition of CYP3A4 was also observed for ligand 1, which was described previously.57 Interestingly, almost all examined H3R ligands showed strong effect on 2D6 cytochrome at the high concentrations of ≥2.5 μM. Surprisingly, no effect on CYP2D6 for the lead structure 2, with the shorter, aliphatic linker was shown (Figure 8B).
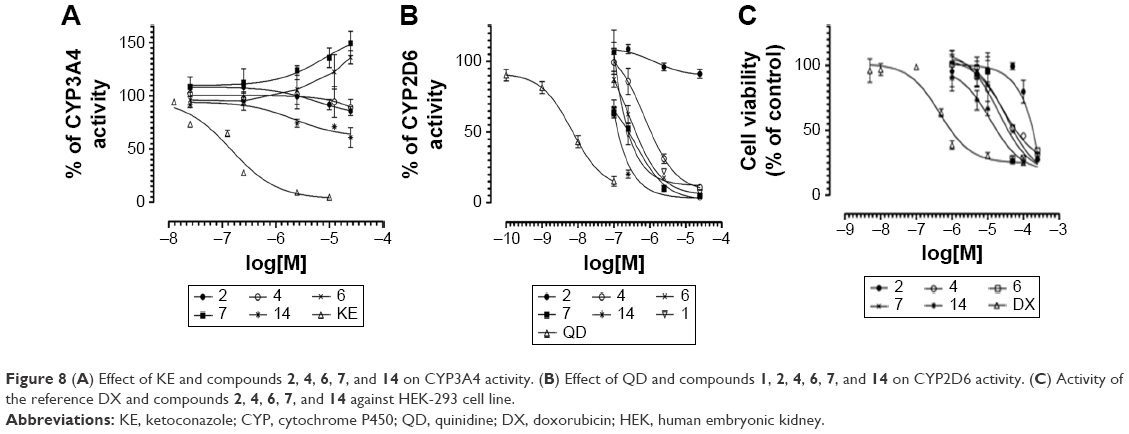 | Figure 8 (A) Effect of KE and compounds 2, 4, 6, 7, and 14 on CYP3A4 activity. (B) Effect of QD and compounds 1, 2, 4, 6, 7, and 14 on CYP2D6 activity. (C) Activity of the reference DX and compounds 2, 4, 6, 7, and 14 against HEK-293 cell line. |
Safety profile
The preliminary evaluation of safety profile for the H3R ligands 2, 4, 6, 7, and 14 was performed by the formazan dye-based EZ4U assay, which determines the influence of the examined compounds on the proliferation of eukaryotic cell lines and by Ames MPF assay to evaluate the risk of genotoxic effect. To determine the antiproliferative effect, the human embryonic cell line (HEK-293) was incubated in the presence of H3R ligands for 48 h. The antiproliferative drug DX was used as a reference. All tested H3R ligands, excluded 2, showed weak antiproliferative effect on examined cell line in IC50 range from 11.82 μM (14) to 33.26 μM (7). However, for compound 2, the antiproliferative effect was observed only at the highest concentration of 250 μM (Figure 8C). Furthermore, S. typhimurium strain TA98 was used for the evaluation of potential genotoxicity of promising H3R ligands. Strain TA98 is incapable of producing histidine, but mutagenic events may lead to reverse mutation that allows reverted bacteria to grow in histidine-deficient media. The criteria used to evaluate Ames MPF test result in a number of revertants after the exposure to tested chemicals. The increase of revertants is calculated relative to solvent control baseline (FIB), and the threshold value of FIB equal to 2.0 is considered as a cutoff parameter for the identification of potential mutagens. In this experiment, the H3R ligands 2, 4, 6, 7, and 14 were tested in two concentrations, 1 and 10 μM, and NQNO at 0.5 μM was used as a positive control. As depicted in Figure 9, none of the tested H3R ligands showed the genotoxic effect in Ames MPF test.
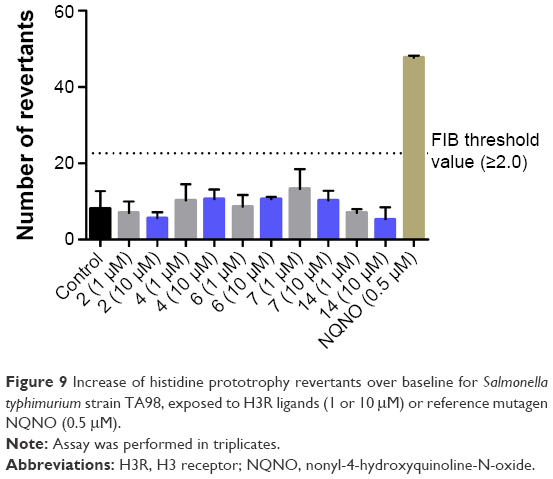 | Figure 9 Increase of histidine prototrophy revertants over baseline for Salmonella typhimurium strain TA98, exposed to H3R ligands (1 or 10 μM) or reference mutagen NQNO (0.5 μM). |
Conclusion
In the current series of H3R ligands, compounds with promising nanomolar affinity were obtained. While elongation of alkyl spacer length in the examined group of phenoxyalkylamine derivatives (as compared to DL-77 [2]) significantly influences the antagonist affinities of the investigated compounds for hH3R, the highest affinities were observed for 3-methylpiperidine derivative 6 (hH3R Ki values of 27.3±15.1 nM) with five carbons alkyl chain. Likewise, the character of the respective cyclic amine had an enormous effect on the affinity of the evaluated H3R ligands with 3-methylpiperidine derivatives being the most potent for this group. The results of the in vivo anticonvulsive testing revealed that compounds 4 and 14 showed promising protective effects subsequent to acute systemic administration in the STR-, PTZ-, and MES-induced seizure models, respectively. Notably, the protective effect found for ligand 14 in the MES-induced seizure model was entirely abolished when rats were pretreated with the CNS-penetrant H1R antagonist PYR, however, not with the centrally acting H2R antagonist ZOL, demonstrating that H1Rs histaminergic pathways seem to be involved in the provided anticonvulsant activity observed for H3R ligand 14. On the contrary, PYR and ZOL failed to abrogate the protective effects provided by 4 in the PTZ- and STR-induced seizure models, respectively. Moreover, the selected, most promising compounds show satisfying, selected ADME-Tox parameters. The metabolic stability and main metabolic pathways were evaluated. No genotoxic and weak cytotoxic effect in comparison to the reference drug DX was shown. Moreover, very weak influence on CYP3A4 and strong influence on CYP2D6, but only in the highest concentrations, were observed. Unlike the H3R ligands with long aliphatic chain, for the lead structure 2, no antiproliferative and no mutagenic effects, as well as no influence on examined CYPs, were observed.
Acknowledgments
Bassem Sadek was supported by intermural funds from the College of Medicine and Health Sciences and the Office of Graduate Studies and Research, UAE University. The authors acknowledge the partial support of National Science Center granted on the basis of decision numbers DEC-2011/02/A/NZ4/00031 and K/ZDS/004689. Support was kindly provided by the EU COST Actions CM1207 and CA15135 (HS and KK) as well by DFG INST 208/664-1 FUGG (HS).
Disclosure
The authors report no conflicts of interest in this work.
References
Fisher RS. Commentary: consciousness of epilepsy. Epilepsia. 2014;55(8):1153. | ||
Kamei C. Involvement of central histamine in amygdaloid kindled seizures in rats. Behav Brain Res. 2001;124(2):243–250. | ||
Kamei C, Ishizawa K, Kakinoki H, Fukunaga M. Histaminergic mechanisms in amygdaloid-kindled seizures in rats. Epilepsy Res. 1998;30(3):187–194. | ||
Miyata I, Saegusa H, Sakurai M. Seizure-modifying potential of histamine H1 antagonists: a clinical observation. Pediatr Int. 2011;53(5):706–708. | ||
Ago J, Ishikawa T, Matsumoto N, Ashequr Rahman M, Kamei C. Mechanism of imipramine-induced seizures in amygdala-kindled rats. Epilepsy Res. 2006;72(1):1–9. | ||
Kakinoki H, Ishizawa K, Fukunaga M, Fujii Y, Kamei C. The effects of histamine H3-receptor antagonists on amygdaloid kindled seizures in rats. Brain Res Bull. 1998;46(5):461–465. | ||
Yawata I, Tanaka K, Nakagawa Y, Watanabe Y, Murashima YL, Nakano K. Role of histaminergic neurons in development of epileptic seizures in EL mice. Brain Res Mol Brain Res. 2004;132(1):13–17. | ||
Onodera K, Yamatodani A, Watanabe T. Effects of alpha-fluoromethylhistidine on locomotor activity, brain histamine and catecholamine contents in rats. Methods Find Exp Clin Pharmacol. 1992;14(2):97–105. | ||
Tuomisto L, Tacke U. Is histamine an anticonvulsive inhibitory transmitter? Neuropharmacology. 1986;25(8):955–958. | ||
Zhang LS, Chen Z, Huang YW, Hu WW, Wei EQ, Yanai K. Effects of endogenous histamine on seizure development of pentylenetetrazole-induced kindling in rats. Pharmacology. 2003;69(1):27–32. | ||
Hirai T, Okuma C, Harada C, et al. Development of amygdaloid kindling in histidine decarboxylase-deficient and histamine H1 receptor-deficient mice. Epilepsia. 2004;45(4):309–313. | ||
Singh E, Pillai KK, Mehndiratta M. Characterization of a lamotrigine-resistant kindled model of epilepsy in mice: evaluation of drug resistance mechanisms. Basic Clin Pharmacol Toxicol. 2014;115(5):373–378. | ||
Kiviranta T, Tuomisto L, Airaksinen EM. Histamine in cerebrospinal fluid of children with febrile convulsions. Epilepsia. 1995;36(3):276–280. | ||
Takano T, Sakaue Y, Sokoda T, et al. Seizure susceptibility due to antihistamines in febrile seizures. Pediatr Neurol. 2010;42(4): 277–279. | ||
Tuomisto L, Lozeva V, Valjakka A, Lecklin A. Modifying effects of histamine on circadian rhythms and neuronal excitability. Behav Brain Res. 2001;124(2):129–135. | ||
Sadek B, Saad A, Sadeq A, Jalal F, Stark H. Histamine H3 receptor as a potential target for cognitive symptoms in neuropsychiatric diseases. Behav Brain Res. 2016;312:415–430. | ||
Sadek B, Shehab S, Wiecek M, et al. Anticonvulsant properties of histamine H3 receptor ligands belonging to N-substituted carbamates of imidazopropanol. Bioorg Med Chem Lett. 2013;23(17):4886–4891. | ||
Sadek B, Schwed JS, Subramanian D, et al. Non-imidazole histamine H3 receptor ligands incorporating antiepileptic moieties. Eur J Med Chem. 2014;77:269–279. | ||
Sadek B, Saad A, Subramanian D, Shafiullah M, Lazewska D, Kiec-Kononowiczc K. Anticonvulsant and procognitive properties of the non-imidazole histamine H3 receptor antagonist DL77 in male adult rats. Neuropharmacology. 2016;106:46–55. | ||
Sadek B, Stark H. Cherry-picked ligands at histamine receptor subtypes. Neuropharmacology. 2016;106:56–73. | ||
Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475–482. | ||
Schwartz JC. The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br J Pharmacol. 2011;163(4):713–721. | ||
Kuhne S, Wijtmans M, Lim HD, Leurs R, de Esch IJ. Several down, a few to go: histamine H3 receptor ligands making the final push towards the market? Expert Opin Investig Drugs. 2011;20(12):1629–1648. | ||
Kasteleijn-Nolst Trenite D, Parain D, Genton P, Masnou P, Schwartz JC, Hirsch E. Efficacy of the histamine 3 receptor (H3R) antagonist pitolisant (formerly known as tiprolisant; BF2.649) in epilepsy: dose-dependent effects in the human photosensitivity model. Epilepsy Behav. 2013;28(1):66–70. | ||
Sadek B, Kuder K, Subramanian D, et al. Anticonvulsive effect of nonimidazole histamine H3 receptor antagonists. Behav Pharmacol. 2014;25(3):245–252. | ||
Kottke T, Sander K, Weizel L, Schneider EH, Seifert R, Stark H. Receptor-specific functional efficacies of alkyl imidazoles as dual histamine H3/H4 receptor ligands. Eur J Pharmacol. 2011;654(3):200–208. | ||
Sander K, Kottke T, Weizel L, Stark H. Kojic acid derivatives as histamine H(3) receptor ligands. Chem Pharm Bull (Tokyo). 2010;58(10):1353–1361. | ||
Sadek B, Schreeb A, Schwed JS, Weizel L, Stark H. Drug-likeness approach of 2-aminopyrimidines as histamine H3 receptor ligands. Drug Des Devel Ther. 2014;8:1499–1513. | ||
Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. | ||
Lazewska D, Ligneau X, Schwartz JC, Schunack W, Stark H, Kiec-Kononowicz K. Ether derivatives of 3-piperidinopropan-1-ol as non-imidazole histamine H3 receptor antagonists. Bioorg Med Chem. 2006;14(10):3522–3529. | ||
Meier G, Apelt J, Reichert U, et al. Influence of imidazole replacement in different structural classes of histamine H(3)-receptor antagonists. Eur J Pharm Sci. 2001;13(3):249–259. | ||
Sadek B, Khanian SS, Ashoor A, et al. Effects of antihistamines on the function of human alpha7-nicotinic acetylcholine receptors. Eur J Pharmacol. 2014;746:308–316. | ||
Loscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure. 2011;20(5):359–368. | ||
Branco Cdos S, Scola G, Rodrigues AD, et al. Anticonvulsant, neuroprotective and behavioral effects of organic and conventional yerba mate (Ilex paraguariensis St. Hil.) on pentylenetetrazol-induced seizures in Wistar rats. Brain Res Bull. 2012;92:60–68. | ||
Sowemimo AA, Adio O, Fageyinbo S. Anticonvulsant activity of the methanolic extract of Justicia extensa T. Anders. J Ethnopharmacol. 2011;138(3):697–699. | ||
Sancheti J, Shaikh MF, Chaudhari R, et al. Characterization of anticonvulsant and antiepileptogenic potential of thymol in various experimental models. Naunyn Schmiedebergs Arch Pharmacol. 2013;387(1):59–66. | ||
Coppola G, Arcieri S, D’Aniello A, et al. Levetiracetam in submaximal subcutaneous pentylentetrazol-induced seizures in rats. Seizure. 2010;19(5):296–299. | ||
Cali JJ, Ma D, Sobol M, et al. Luminogenic cytochrome P450 assays. Expert Opin Drug Metab Toxicol. 2006;2(4):629–645. | ||
Lazewska D, Wiecek M, Ner J, et al. Aryl-1,3,5-triazine derivatives as histamine H4 receptor ligands. Eur J Med Chem. 2014;83:534–546. | ||
Grosicki M, Latacz G, Szopa A, Cukier A, Kiec-Kononowicz K. The study of cellular cytotoxicity of argireline – an anti-aging peptide. Acta Biochim Pol. 2014;61(1):29–32. | ||
Kaminska K, Ziemba J, Ner J, et al. (2-Arylethenyl)-1,3,5-triazin-2-amines as a novel histamine H4 receptor ligands. Eur J Med Chem. 2015;103:238–251. | ||
Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302:832–837. | ||
Bhowmik M, Khanam R, Vohora D. Histamine H3 receptor antagonists in relation to epilepsy and neurodegeneration: a systemic consideration of recent progress and perspectives. Br J Pharmacol. 2012;167(7):1398–1414. | ||
Loscher W, Fassbender CP, Nolting B. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. II. Maximal electroshock seizure models. Epilepsy Res. 1991;8(2):79–94. | ||
Loscher W, Honack D, Fassbender CP, Nolting B. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. III. Pentylenetetrazole seizure models. Epilepsy Res. 1991;8(3):171–189. | ||
Corda MG, Orlandi M, Lecca D, Carboni G, Frau V, Giorgi O. Pentylenetetrazol-induced kindling in rats: effect of GABA function inhibitors. Pharmacol Biochem Behav. 1991;40(2):329–333. | ||
Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Prog Neurobiol. 2001;63(6):637–672. | ||
Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88(3):1183–1241. | ||
Dai H, Fu Q, Shen Y, et al. The histamine H3 receptor antagonist clobenpropit enhances GABA release to protect against NMDA-induced excitotoxicity through the cAMP/protein kinase A pathway in cultured cortical neurons. Eur J Pharmacol. 2007;563(1–3):117–123. | ||
Swinyard EA, Sofia RD, Kupferberg HJ. Comparative anticonvulsant activity and neurotoxicity of felbamate and four prototype antiepileptic drugs in mice and rats. Epilepsia. 1986;27(1):27–34. | ||
White HS. Comparative anticonvulsant and mechanistic profile of the established and newer antiepileptic drugs. Epilepsia. 1999;40(suppl 5):S2–S10. | ||
Sherma J. Planar chromatography. Anal Chem. 2002;74(12):2653–2662. | ||
Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. | ||
Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–6196. | ||
Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–1759. | ||
Cruciani G, Carosati E, De Boeck B, et al. MetaSite: understanding metabolism in human cytochromes from the perspective of the chemist. J Med Chem. 2005;48(22):6970–6979. | ||
Kuder K, Lazewska D, Latacz G, et al. Chlorophenoxy aminoalkyl derivatives as histamine H(3)R ligands and antiseizure agents. Bioorg Med Chem. 2016;24(2):53–72. | ||
Nassar AF. Drug Metabolism Handbook: Concepts and Applications. Hoboken, NJ: John Wiley & Sons; 2009. | ||
Garbarg M, Arrang JM, Rouleau A, et al. S-[2-(4-imidazolyl)ethyl]isothiourea, a highly specific and potent histamine H3 receptor agonist. J Pharmacol Exp Ther. 1992;263(1):304–310. | ||
Garbarg M, Barbin G, Bischoff S, Pollard H, Schwartz JC. Dual localization of histamine in an ascending neuronal pathway and in non-neuronal cells evidenced by lesions in the lateral hypothalamic area. Brain Res. 1976;106(2):333–348. | ||
Garbarg M, Pollard H, Trung Tuong MD, Schwartz JC, Gros C. Sensitive radioimmunoassays for histamine and tele-methylhistamine in the brain. J Neurochem. 1989;53(6):1724–1730. | ||
Ligneau X, Lin J, Vanni-Mercier G, et al. Neurochemical and behavioral effects of ciproxifan, a potent histamine H3-receptor antagonist. J Pharmacol Exp Ther. 1998;287(2):658–666. | ||
Ligneau X, Garbarg M, Vizuete ML, et al. [125I]iodoproxyfan, a new antagonist to label and visualize cerebral histamine H3 receptors. J Pharmacol Exp Ther. 1994;271(1):452–459. | ||
Ligneau X, Morisset S, Tardivel-Lacombe J, et al. Distinct pharmacology of rat and human histamine H(3) receptors: role of two amino acids in the third transmembrane domain. Br J Pharmacol. 2000;131(7):1247–1250. | ||
Amon M, Ligneau X, Camelin JC, Berrebi-Bertrand I, Schwartz JC, Stark H. Highly potent fluorescence-tagged nonimidazole histamine H3 receptor ligands. ChemMedChem. 2007;2(5):708–716. | ||
Isensee K, Amon M, Garlapati A, et al. Fluorinated non-imidazole histamine H3 receptor antagonists. Bioorg Med Chem Lett. 2009;19(8):2172–2175. | ||
Tomasch M, Schwed JS, Paulke A, Stark H. Bodilisant-a novel fluorescent, highly affine histamine h3 receptor ligand. ACS Med Chem Lett. 2013;4(2):269–273. | ||
Schibli R, Schubiger PA. Current use and future potential of organometallic radiopharmaceuticals. Eur J Nucl Med Mol Imaging. 2002;29(11):1529–1542. | ||
Schlotter K, Boeckler F, Hubner H, Gmeiner P. Fancy bioisosteres: metallocene-derived G-protein-coupled receptor ligands with subnanomolar binding affinity and novel selectivity profiles. J Med Chem. 2005;48(11):3696–3699. | ||
van Staveren DR, Metzler-Nolte N. Bioorganometallic chemistry of ferrocene. Chem Rev. 2004;104(12):5931–5985. | ||
Sander K, Kottke T, Stark H. Histamine H3 receptor antagonists go to clinics. Biol Pharm Bull. 2008;31(12):2163–2181. | ||
Bahi A, Sadek B, Nurulain SM, Więcekc M, Kieć-Kononowicz K. The histamine H3 receptor antagonist DL77 reduces voluntary alcohol intake and ethanol-induced conditioned place preference in mice. Physiol Behav. 2015;151:189–197. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.