Back to Journals » International Journal of Nanomedicine » Volume 20
NIR-Programmed Trimodal Macrophage Nanovectors for Effective Anti-Tumor Therapy in Mice Model
Authors Xie Y, Shi Y, Li Z, Xu S, Chen Z, Ye X, Yan W
Received 25 May 2025
Accepted for publication 4 September 2025
Published 18 September 2025 Volume 2025:20 Pages 11295—11314
DOI https://doi.org/10.2147/IJN.S542647
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Farooq A. Shiekh
Yiwen Xie,1,* Yuwei Shi,2,* Zhihui Li,2,* Sumei Xu,1 Zhiyun Chen,2 Xuxia Ye,3 Wenxi Yan4
1Department of General Practice, The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang Provincial Hospital of Chinese Medicine), Hangzhou, Zhejiang, 310006, People’s Republic of China; 2Department of the Second Central Laboratory, The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang Provincial Hospital of Chinese Medicine), Hangzhou, Zhejiang, 310006, People’s Republic of China; 3School of Medical Technology and Information Engineering, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, 310053, People’s Republic of China; 4Department of Clinical Laboratory, The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang Provincial Hospital of Chinese Medicine), Hangzhou, Zhejiang, 310006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wenxi Yan, Department of Clinical Laboratory, The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang Provincial Hospital of Chinese Medicine), 54 Youdian Road, Hangzhou, Zhejiang, 310006, People’s Republic of China, Email [email protected]
Introduction: Chemotherapy remains the primary treatment modality for advanced and unresectable tumors; however, its antitumor efficacy is limited, and it has significant toxic effects on normal tissues.
Methods: To address these challenges, cell carrier- and nanomaterial-based strategies were employed in this study to engineer macrophages into functional “Trojan horses” loaded with ICG–CDDP mesoporous silicon nanoparticles, yielding an intelligent cell carrier chemotherapeutic drug delivery system with controlled light responsiveness and “on-demand” nanophotothermolysis capabilities.
Results: Our findings demonstrate that the intelligent macrophage drug delivery system actively homes to tumor sites and that indocyanine green (ICG) fluorescence can be used to visualize exogenous macrophages at the tumor site. In response irradiation of the delivery area with exogenous near-infrared (NIR) light, ICG generates a thermal effect, resulting in the lysis of macrophages and facilitating spatiotemporally controlled burst release of intracellular cisplatin, thereby precisely targeting tumor cells. Simultaneously, ICG can trigger immunogenic cell death (ICD) under NIR irradiation, transforming “cold tumors” into “hot tumors” and eliciting a prolonged antitumor immune response, thereby overcoming the limitations associated with chemotherapeutic drugs.
Conclusion: This collaborative trimodal strategy successfully represents a triple breakthrough in precise delivery, spatiotemporally controlled drug release, and immune activation. It is an innovative solution for the precise treatment of advanced tumors and has substantial potential for clinical translation.
Keywords: macrophage-mediated drug delivery, spatiotemporally controlled release, immunogenic cell death, ICD, trimodal therapy
Graphical Abstract:

Introduction
The incidence of malignant tumors is progressively increasing, posing a significant threat to human health.1,2 Chemotherapy continues to be the principal therapeutic approach for advanced tumors,3 yet its effectiveness is frequently undermined by associated toxic side effects.4 To address the limitations of conventional treatment approaches, our research focuses on the development of innovative therapeutic strategies, particularly for enhancing the localized delivery of chemotherapeutic agents to tumor sites. A critical aspect of this endeavor is identifying suitable drug carriers.5,6 Currently, a variety of nanocarriers are employed, and these carriers either passively accumulate at tumor sites by exploiting the enhanced permeability and retention effect of the tumor vasculature7,8 or actively target tumor tissues via modification of specific targeting molecules.5,9,10 Regrettably, these targeted carriers are susceptible to immune system attack,11,12 exhibit poor circulatory stability,13,14 and fail to provide accurate spatiotemporal regulation of the release of therapeutic compounds,15,16 thereby diminishing the efficacy of targeted therapies and potentially inducing severe systemic toxicity.17 Consequently, these strategies are suboptimal for effective treatment. To address these limitations, the investigation and development of various cellular carriers,18,19 particularly macrophages,20 have attracted significant attention.
As a principal cell type within the tumor microenvironment,21 macrophages exhibit inherent affinity for tumors.22,23 This tumor-directed chemotaxis enables macrophages to target and infiltrate tumor tissues effectively, adeptly navigating the complex physical and biological barriers encountered during drug delivery, akin to a “Trojan horse”.24,25 Consequently, macrophages hold considerable promise as drug carriers in antineoplastic therapy. However, the cytotoxic side effects of chemotherapeutic drugs on the carrier cells themselves remain a challenge.26,27
To optimize the use of macrophages and mitigate the cytotoxic effects of chemotherapeutic agents on carrier cells, we previously employed mesoporous silicon nanoparticles (MSNs) as intermediaries for drug delivery.28,29 Furthermore, we utilized electrostatic forces to coat the surface with negatively charged, cell-targeting hyaluronic acid (HA),30 thereby temporarily shielding against cytotoxic effects.31 By capitalizing on the inherent targeting capabilities of macrophages,32,33 we engineered a drug delivery system that facilitates the loading of secondary blocking agents onto macrophages after uptake, yielding a targeted chemotherapy modality for tumor treatment. This approach not only harnesses the active targeting abilities of macrophages within the tumor microenvironment but also reduces the cytotoxic effects of chemotherapeutic agents on these carrier cells. Despite the optimization of this targeted drug delivery system, we were unable to achieve precise spatiotemporal control of drug release. Consequently, our subsequent focus has been on developing a “switch” mechanism to enable the “burst release” of chemotherapeutic drugs. Notably, indocyanine green (ICG), which received FDA approval for clinical near-infrared fluorescence imaging of small organic molecules,34 demonstrates high thermal efficiency35 and favorable biocompatibility.36 By exploiting its fluorescence properties34 and photothermal conversion capabilities, we can employ ICG for the imaging and identification of metastatic lesions before irradiation with near-infrared (NIR) light, thereby inducing a thermal effect that lyses macrophages and facilitates the release of chemotherapeutic agents, enabling precise drug delivery. Thus, our objective is to achieve the simultaneous delivery of ICG and chemotherapeutic agents. By using cisplatin as a representative chemotherapeutic drug, we intend to leverage the interaction between the sulfonate group of ICG and platinum in cisplatin (CDDP) to coordinate the delivery of both ICG and CDDP.
In this study, we use macrophages as carriers. These cells are loaded with ICG-CDDP mesoporous silica nanoparticles, which function as Trojan horses to confer light-responsive capabilities and permit the controlled, “on-demand” release of chemotherapeutic drugs. When the intelligent macrophage drug delivery system actively targets the tumor site, exogenous macrophages can be detected at the tumor site by visualizing ICG fluorescence. The targeted area is subsequently irradiated with exogenous NIR light. In response to the NIR laser, ICG generates a thermal effect that lyses macrophages and facilitates the spatiotemporally controlled burst release of intracellular cisplatin, thereby enabling precise targeting of tumor cells.
Notably, reactive oxygen species (ROS) produced by ICG under NIR irradiation can induce endoplasmic reticulum stress in tumor cells, resulting in the translocation of calreticulin to the plasma membrane surface.37 As an “eat me” signal, calreticulin is recognized by antigen-presenting cells (APCs), particularly dendritic cells (DCs), and this promotes DC maturation and the phagocytosis of dead or dying tumor cells. This process leads to further recruitment and activation of cytotoxic T lymphocytes (CTLs) to the tumor site, where they release interferon-gamma (IFN-γ) and other cytokines, thereby reprogramming the tumor microenvironment to generate an “inflammatory infiltration” phenotype. Ultimately, this cascade induces immunogenic cell death (ICD),38,39 transforming “cold tumors” into “hot tumors”40 and eliciting a long-lasting antitumor immune response. This mechanism significantly compensates for the limitations of conventional chemotherapeutic drugs.
Materials and Methods
Indocyanine green (ICG) was purchased from Bidd Pharmaceutical Technology Co., Ltd. (Shanghai, China). Cisplatin (CDDP), 3-aminopropyltriethoxysilane (APTES), tetraethylorthosilicate (TEOS), and cetyltrimethylammonium bromide (CTAB) were obtained from Aladdin Chemical Co. (Shanghai, China). Hyaluronic acid (molecular weight 10 kDa) was obtained from Yuanye Biotechnology Co., Ltd. (Shanghai, China). Hoechst 33342 was acquired from Solarbio Science & Technology Co., Ltd. (Beijing, China). A Mouse Interferon Gamma (IFN-γ) ELISA Kit was acquired from ELK Biotechnology Co., Ltd. (Wuhan, China). Dihydroethidium (DHE) was obtained from MedChemExpress LLC (Monmouth Junction, NJ, USA). An anti-calreticulin polyclonal antibody was sourced from ProteinTech Group, Inc. (Wuhan, China).
Cell Culture and Animals
Murine colon carcinoma cells (CT26 cells) and mouse mononuclear macrophages (RAW 264.7 cells) were obtained from the National Collection of Authenticated Cell Cultures (NCACC) in Shanghai, China. CT26 cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium, while RAW 264.7 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM). Both media were supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics (streptomycin and penicillin).
Male BALB/c mice, aged 6 to 8 weeks, were procured from Slack Laboratory Animals LLC in Shanghai, China (Animal Use Permit Number: SYXK (Zhejiang) 2021-0012) and housed in a specific pathogen-free (SPF) environment. The experimental procedures commenced after a one-week acclimatization period. This study was conducted in compliance with the Laboratory Animal-Guideline for ethical review of animal welfare of the People’s Republic of China and received approval from the Animal Ethics and Welfare Committee of Zhejiang Chinese Medicine University (approval number IACUC-20220718-08).
Synthesis of Indocyanine Green-Cisplatin (ICG-CDDP) Complexes
CDDP (60 mg, 0.20 mM) was dissolved in 1 mL of deionized water (the resistivity of 18.2 MΩ·cm) and stirred at 800 rpm and 37 °C in the absence of light until fully dissolved, after which it was allowed to cool to room temperature. Silver nitrate (AgNO3) (66.2 mg, 0.39 mM) was subsequently carefully introduced to achieve a homogeneous mixture. This mixture was then heated and stirred at 800 rpm for 3 hours at 60 °C, followed by continuous stirring at 800 rpm at room temperature in the dark for an additional 48 hours. The resulting mixture was centrifuged at 12,000 × g for 15 minutes to separate the silver chloride (AgCl) precipitate formed during the reaction. The supernatant was subsequently filtered using a 0.22-µm Millipore Millex®-GP syringe filter (polyethersulfone, PES) for further use. ICG (131 mg, 0.2 mM) was dissolved in deionized water (2–5 mg/mL, the resistivity of 18.2 MΩ·cm) and added to the CDDP precursor solution at a 1:1 molar ratio. The final mixture was gently stirred at 800 rpm for 24 hours at room temperature in the dark to synthesize the ICG‒CDDP complex.
Synthesis of Amine-Group Modified Mesoporous Silica Nanoparticles (MSNs-NH2)
First, CTAB (1 g) and sodium hydroxide (NaOH, 0.28 g) were dissolved in 480 mL of deionized water and stirred at 1200 rpm for 1 h at 80 °C. TEOS (5 g) was subsequently added to the solution, and the mixture was stirred at 1200 rpm for an additional 2 hours at 80 °C. The resultant precipitate was isolated via centrifugation at 8000 rpm for 10 minutes and then thoroughly washed with methanol and distilled water; each step was repeated three times to eliminate impurities. The product was then subjected to vacuum freeze-drying. To introduce amino functionalities, the dried product (1 g) was redispersed in 75 mL of methanol, to which 2 mL of APTES was added. The mixture was stirred continuously at 1000 rpm for 24 hours at room temperature. The modified nanoparticles were again collected by centrifugation at 8000 rpm for 10 minutes, followed by three washes with methanol and distilled water. To eliminate the CTAB template, the product was extracted by stirring at 1000 rpm for 48 hours at 80 °C using a solution composed of 192 mL of methanol and 12 mL of 37% hydrochloric acid. Finally, the purified product was collected by centrifugation at 8000 rpm for 10 minutes, washed with methanol and distilled water three times each, and subsequently freeze-dried under vacuum for further use.25
Synthesis of Hyaluronic Acid-Coated, Amine-Group Modified Mesoporous Silica Nanoparticles Loaded with ICG-CDDP Complexes (ICG-CDDP@HMs)
ICG‒CDDP complexes (37.5 mg) were dissolved in 10 mL of deionized water, followed by the addition of amine-functionalized mesoporous silica nanoparticles (MSNs-NH2) (75 mg). The suspension was agitated at 1000 rpm for 12 hours at 4 °C in the dark to yield ICG-CDDP-loaded MSNs (ICG-CDDP@MSNs). Finally, hyaluronic acid (75 mg, in a 1:1 weight ratio) was added to the suspension, which was then stirred at 1000 rpm for an additional 2 hours in the dark, yielding hyaluronic acid-coated ICG-CDDP-loaded mesoporous silica nanoparticles (ICG-CDDP@HMs).
Characterization
The synthesized ICG-CDDP complexes were characterized using an Avance III 600 MHz Digital NMR Spectrometer (Bruker, Germany) and a liquid chromatography‒quadrupole time-of-flight mass spectrometer (LC/Q-TOF, Waters, SYNAPT G2-Si, USA; see Figure S1). The sizes of various nanoparticles, including MSNs-NH2, HMs, and ICG-CDDP@HMs, were determined using a particle size analyzer. The morphology and structure of these nanoparticles were examined via transmission electron microscopy (TEM, HITACHI H-7650, Japan). The particle size and zeta potential of various nanoparticles, including MSNs-NH2, HMs, and ICG-CDDP@HMs, was determined using dynamic light scattering (DLS; Nano-ZS 90; Malvern Instruments, UK). The ultraviolet–visible (UV–Vis) absorption of different synthetic materials, namely, MSNs-NH2, ICG, CDDP, ICG-CDDP, and ICG-CDDP@HMs, was measured using an ultraviolet–visible–near-infrared spectrophotometer (SHIMADZU, UV-3600Plus, Japan). The concentration of ICG-CDDP was determined using a standard curve, with the following equation: y = 0.0892055X - 0.0171386 (R² = 0.99839). The drug-loading rate (DL) was computed as (DL)=m(ICG-CDDP)/(m(ICG-CDDP) + m(HMs)) ×100%, resulting in a value of 19.64% (where m(ICG-CDDP) =18.33 mg and m(HMs) =75 mg). The specific surface area and pore size distribution were analyzed using the Brunauer–Emmett–Teller method (BET, 3FLEX, Micromeritics, USA).
Intracellular Uptake
Macrophages (RAW 264.7 cells) were incubated with 50 μg/mL ICG-CDDP@HMs for 8 hours, after which the medium was replaced with fresh DMEM containing 10% FBS to obtain macrophages loaded with ICG-CDDP@HMs (designated ICG-CDDP@HMs@Macs).
The uptake of ICG-CDDP@HMs by macrophages was assessed via confocal laser scanning microscopy. RAW 264.7 cells were seeded onto glass slides in 24-well plates (Corning®, tissue culture-treated) at a density of 2.5×105 cells per well. After a 24-h culture period in 0.5 mL of DMEM enriched with 10% FBS, the cells were subsequently exposed to 0.5 mL of newly prepared medium enriched with 10% FBS that contained either free ICG-CDDP or ICG-CDDP@HMs, both of which were administered at a concentration of 50 μg/mL. Following an incubation period of 8 hours, the cells were washed three times with 0.5 mL of cold PBS to eliminate any excess drugs or nanoparticles. Cells were harvested at 0 h, 24 h, and 48 h post-incubation, fixed with 4% paraformaldehyde for 15 minutes, and then incubated with 0.5 mL of Hoechst 33342 (10 μg/mL) in the dark for an additional 15 minutes at room temperature. Fluorescence images were acquired using a laser scanning confocal microscope (Zeiss LSM 880, Germany).
The fluorescence intensity of ICG in macrophages following the uptake of ICG-CDDP@HMs was assessed using a flow cytometry system. RAW 264.7 cells (5 × 105 cells per well) were seeded in 12-well plates (Corning®, tissue culture-treated). After 24 hours of culture in 1 mL of DMEM enriched with 10% FBS, the cells were exposed to 0.5 mL of fresh medium enriched with 10% FBS that included either free ICG-CDDP or ICG-CDDP@HMs at varying concentrations (2, 5, 10, 20, 50, 100, and 300 μg/mL). Following an 8-hour incubation period, the cells were washed three times with 1 mL of cold PBS to eliminate excess drugs and nanoparticles. The cells were subsequently harvested and resuspended in cold PBS containing 1% paraformaldehyde, and the fluorescence intensity of ICG within the macrophages was subsequently assessed utilizing a flow cytometry system (BD, FACS Canto, USA).
To evaluate the capacity of macrophages to retain ICG-CDDP@HMs, RAW 264.7 cells (5 × 105 cells per well) were seeded in 12-well plates (Corning®, tissue culture-treated). After 24 hours of culture in 1 mL of DMEM enriched with 10% FBS, the cells were subsequently exposed to 0.5 mL of fresh medium enriched with 10% FBS containing either free ICG-CDDP or ICG-CDDP@HMs at a concentration of 50 μg/mL. Following an 8-h incubation, the cells were washed three times with 1 mL of cold PBS to eliminate excess drugs and nanoparticles. The cells were subsequently transferred to 1 mL of fresh medium supplemented with 10% FBS and cultured for 24 or 48 hours. Following incubation, the cells were collected and resuspended in cold PBS containing 1% paraformaldehyde. The fluorescence intensity of ICG within the macrophages was subsequently assessed using a flow cytometry system (BD FACS Canto, USA).
In vitro Photothermal Imaging
The photothermal conversion efficiency of ICG-CDDP@HMs@Macs was evaluated in vitro using an infrared thermal imager (HIKMICRO, H10, China). Solutions of PBS, HMs (204 μg/mL), ICG (50 μg/mL), ICG-CDDP (50 μg/mL), and ICG-CDDP@HMs (50 μg/mL) were dispensed into Eppendorf tubes. Furthermore, macrophages (RAW 264.7 cells) were incubated with 50 μg/mL ICG-CDDP@HMs for 8 hours to yield ICG-CDDP@HMs@Macs. Following collection, the cells were resuspended in PBS and transferred to Eppendorf tubes at a density of 3×106 cells per tube. All the samples were exposed to irradiation with an NIR laser for 5 minutes after being centrifuged at 1000rpm for 5 minutes, with temperature readings taken every 30 seconds using an infrared thermal imager.
In vitro Drug Release Behavior
The in vitro release profile of ICG-CDDP from ICG-CDDP@HMs was systematically investigated. ICG-CDDP@HMs were suspended in 2 mL of phosphate-buffered saline (PBS) at pH levels of 7.4 and 5.0, or in PBS at pH levels of 5.0 containing hyaluronidase (HAase) at a concentration of 150 U/mL. The suspension was agitated at 200 rpm at a temperature of 37°C. At predetermined intervals, 2 mL of the release medium was withdrawn and replaced with an equivalent volume of fresh medium. The release was quantified using an ultraviolet–visible–near-infrared spectrophotometer (SHIMADZU, UV-3600Plus, Japan).
The relative amount of ICG-CDDP released from ICG-CDDP@HMs@Macs after NIR irradiation was determined by measuring the ICG fluorescence intensity. Specifically, RAW 264.7 cells (1×107 cells/well) were seeded in 10 cm Petri dishes (Corning®, tissue culture-treated) and incubated with 50 μg/mL ICG-CDDP@HMs for 8 hours to yield ICG-CDDP@HMs@Macs. The cells were subsequently washed twice with 2 mL of PBS to completely remove the residual ICG-CDDP@HMs and were subsequently collected by centrifugation at 1000 rpm for 5 min. The cells were irradiated with an NIR laser for 120 s (808 nm, 1.0 W/cm2) or not irradiated and then incubated at 37 °C in an incubator containing 5% CO2 for 0.5, 1, 2, or 4 h. Afterward, the cells and supernatant were separated by centrifugation at 1000 rpm for 5 min, after which the fluorescence intensity of ICG (excitation wavelength: 650 nm; emission wavelength: 700 nm) in the supernatant was measured using a multimode microplate reader (Synergy H1, BioTek, USA).
In vitro Cytotoxicity Analysis
The impact of NIR irradiation on the cytotoxicity of ICG-CDDP was assessed using a Cell Counting Kit-8 (CCK-8; GLPBIO, USA). CT26 cells were seeded in 96-well plates containing 200 μL of RPMI 1640 medium supplemented with 10% FBS at a density of 5×103 cells per well. The ICG-CDDP solution was subjected to NIR irradiation (808 nm, 1.0 W/cm2) for 5 minutes. After NIR irradiation, the CT26 cells were exposed to RPMI 1640 medium supplemented with 10% FBS and ICG-CDDP or ICG-CDDP at various concentrations (0.5 μg/mL, 10 μg/mL, 50 μg/mL, 100 μg/mL, 200 μg/mL, and 300 μg/mL) for 8 hours in a volume of 100 μL per well. Following a 48-hour incubation period in 200 μL of fresh medium supplemented with 10% FBS, cell viability was measured using the CCK-8 assay. Specifically, the medium in each well was replaced with 100 µL of fresh medium supplemented with 10% FBS and 10 µL of CCK-8 solution, and the cells were then incubated for 2 h at 37 °C. After being gently mixed on a shaker for 1 min, the absorbance was measured at 450 nm by a microplate reader (Gen5, BioTek, USA).
Additionally, the cytotoxic effects of ICG-CDDP@HMs on macrophages were evaluated using the CCK-8 assay. RAW 264.7 cells were seeded in 96-well plates at a density of 1×104 cells per well in 200 μL of DMEM supplemented with 10% FBS. The macrophages were then incubated in fresh DMEM supplemented with 10% FBS and either ICG-CDDP or ICG-CDDP@HMs at a concentration of 0.5 μg/mL, 2 μg/mL, 10 μg/mL, 50 μg/mL, 100 μg/mL, or 200 μg/mL for 8 hours in a volume of 100 μL per well. After a subsequent 48-hour incubation in 200 μL of fresh medium supplemented with 10% FBS, macrophage viability was assessed using the CCK-8 assay. Specifically, the medium in each well was replaced with 100 µL of fresh medium supplemented with 10% FBS and 10 µL of CCK-8 solution, and the cells were then incubated for 2 h at 37 °C. After being gently mixed on a shaker for 1 min, the absorbance was measured at 450 nm by a microplate reader (Gen5, BioTek, USA).
The cytotoxic effects of ICG-CDDP@HMs@Macs on CT26 cells following NIR irradiation were assessed using the CCK-8 assay. CT26 cells were seeded at a density of 5×103 cells per well in 96-well plates containing 200 μL of RPMI 1640 medium supplemented with 10% FBS. Concurrently, RAW 264.7 cells were seeded at a density of 3×106 cells per well in 6-well plates. After 24 hours of culture in 2 mL of DMEM supplemented with 10% FBS, the RAW 264.7 cells were treated with 1 mL of fresh medium supplemented with 10% FBS and either free ICG or ICG-CDDP@HMs at a concentration of 50 μg/mL. Following an 8-hour incubation period, the RAW 264.7 cells were collected in Eppendorf tubes and subjected to NIR irradiation (808 nm, 1.0 W/cm2) for 5 minutes. After a 30-minute culture period, RAW 264.7 cells at different densities (10 × 103, 50 × 103, and 100×103 cells per well) were added to the preseeded CT26 cells. After a 48-h culture period, the cells were washed three times with 0.2 mL of cold PBS, and the viability of the CT26 cells was evaluated using the CCK-8 assay. Specifically, the medium in each well was replaced with 100 µL of fresh medium supplemented with 10% FBS and 10 µL of CCK-8 solution, and the cells were then incubated for 2 h at 37 °C. After being gently mixed on a shaker for 1 min, the absorbance was measured at 450 nm by a microplate reader (Gen5, BioTek, USA).
Evaluation of in vitro Migration Ability
The migratory capacity of ICG-CDDP@HMs@Macs was investigated using Corning® 6.5 mm Transwell® plates with an 8.0-µm Pore Polycarbonate Membrane Insert in vitro. For this purpose, CT26 cells were seeded in 10 cm Petri dishes at a density of 6×106 cells per plate and cultured in 10 mL of RPMI 1640 medium supplemented with 10% fetal bovine serum. The culture medium was subsequently replaced with 8 mL of serum-free RPMI 1640 medium. After a 48-h culture period, all the culture media were collected and centrifuged at 1000 rpm to remove the sediment. Afterward, the CT26 cell culture supernatant and FBS-free RPMI 1640 medium were filtered through a 0.22-µm Millipore Millex®-GP syringe filter (polyethersulfone, PES) for further use. Either 0.6 mL of CT26 cell culture supernatant or FBS-free RPMI 1640 as a control was added to the lower chamber, while macrophages (RAW 264.7 cells) or macrophages loaded with ICG-CDDP@HMs (at an ICG-CDDP concentration of 50 μg/mL) were added to the upper chamber (3 × 104 cells each). Following a 48-h incubation period, the chambers were fixed with 600 µL of 4% paraformaldehyde fixative for 30 minutes. The chamber was subsequently washed once with PBS. Afterward, the macrophages on the Transwell membrane were stained with 600 µL of 0.1% crystal violet solution for 10 minutes at room temperature and visualized under a microscope after being washed three times with PBS. The number of migrated cells was quantified using ImageJ software.
Evaluation of in vivo Photothermal Imaging Using Mice Model
CT26 cells (5 × 105 cells per mouse) were subcutaneously injected into the right axilla of the mice to establish a subcutaneous CT26 colon cancer mouse model to assess the photothermal conversion efficiency of ICG-CDDP@HMs@Macs in vivo. Once the tumor volume reached 100 mm3, macrophages (RAW 264.7 cells, 3×106 cells per mouse) loaded with ICG-CDDP@HMs (at an ICG-CDDP concentration of 50 μg/mL) were administered to tumor-bearing mice via tail vein injection (n = 8 mice per group). The tumor region was subjected to irradiation with an NIR laser (808 nm, 1.5 W/cm2), and the local tumor temperature was monitored every minute using an infrared thermal imager (HIKMICRO, H10, China).
Evaluation of in vivo Tumor-Homing Ability Using Mice Model
CT26 cells (5 × 105 cells per mouse) were subcutaneously injected into the right axilla of mice to establish a subcutaneous CT26 colon cancer mouse model to evaluate the tumor-targeting ability of ICG-CDDP@HMs@Macs in vivo. Once the tumor volume reached 100 mm3, macrophages (Macs, HMs@Macs, ICG@HMs@Macs, and ICG-CDDP@HMs@Macs (ICG-CDDP concentration of 50 μg/mL); 3×106 cells per mouse), were labeled with the fluorescent probe 3,3′-dioctadecyloxacarbocyanine perchlorate (DIO; Beyotime, China) and administered to tumor-bearing mice via tail vein injection (n = 3 mice per group). The distribution and intensity of ICG fluorescence in the mice were monitored using via a small animal in vivo imaging system (IVIS® SPECTRUM, PerkinElmer, USA; excitation filter: 780 nm; emission filter:831; luminescent exposure: 15 sec) at 12, 24, and 48 hours post-injection, and the fluorescence intensity of DIO in mouse tumor tissue was evaluated through analysis of tissue sections at 48 hours post-injection. Tumor tissues were fixed with 4% paraformaldehyde and embedded in paraffin to prepare tumor sections (RM2245, Leica, Germany). After the nuclei were stained with DAPI, the samples were observed using a digital slide scanner (Pannoramic MID, 3DHISTECH, Hungary; DAPI, excitation wavelength, 330–380 nm; emission wavelength, 420 nm; DIO, excitation wavelength, 465–495 nm; emission wavelength, 515–555 nm; ICG, excitation wavelength, 510–560 nm; emission wavelength, 590 nm).
Evaluation of in vivo Antitumor Effects Using Mice Model
Additionally, CT26 cells (5 × 105 cells per mouse) were subcutaneously injected into the right axilla of mice to establish a subcutaneous CT26 colon cancer mouse model to evaluate the antitumor efficacy of ICG-CDDP@HMs@Macs in combination with NIR irradiation in vivo. Upon reaching a tumor volume of 100 mm3, macrophages (Macs, HMs@Macs, ICG@HMs@Macs, and ICG-CDDP@HMs@Macs; 3×106 cells per mouse, ICG-CDDP concentration of 50 μg/mL) were administered via the tail vein to tumor-bearing mice (n = 6 mice per group). The tumors were subsequently subjected to localized NIR laser irradiation (808 nm, 1.5 W/cm2) for 5 minutes at 24 hours and 48 hours post-injection. Tumor volume was assessed over a 14-day treatment period. After 14 days, the mice were euthanized, and blood samples were collected from the orbital sinus. Serum was obtained through centrifugation (3500 rpm, 5 minutes) for the analysis of IFN-γ. The tumor tissues were excised for tissue section analysis. Tumor tissues were fixed with 4% paraformaldehyde and embedded in paraffin to prepare tumor sections (RM2245, Leica, Germany) for hematoxylin and eosin (H&E) staining, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, dihydroethidium (DHE) staining and immunohistochemical staining. For immunohistochemical staining, the sections were blocked and incubated with a polyclonal primary antibody against calreticulin (Proteintech, China), followed by incubation with a horseradish peroxidase (HRP)-conjugated secondary antibody (zsbio, China); the nuclei were stained with hematoxylin. The samples were observed using microscope slide scanners (NanoZoomer S60, Hamamatsu, Japan).
In vivo Biosafety Evaluation Using Mice Model
CT26 cells (5 × 105 cells per mouse) were subcutaneously injected into the right axilla of the mice to establish a subcutaneous CT26 colon cancer mouse model to evaluate the biosafety of ICG-CDDP@HMs@Macs in combination with NIR irradiation in vivo. Once the tumor volume reached 100 mm3, macrophages (Macs, HMs@Macs, ICG@HMs@Macs, and ICG-CDDP@HMs@Macs; 3×106 cells per mouse, ICG-CDDP concentration of 50 μg/mL) were administered to tumor-bearing mice via tail vein injection, and tumor-bearing mice injected with PBS served as the control group (n = 3 mice per group). The tumors in the group treated with ICG-CDDP@HMs@Macs were subsequently subjected to local irradiation with an NIR laser (808 nm, 1.5 W/cm2) for 5 minutes 24 hours post-injection. Blood samples were collected from the orbital sinus of the mice at 3 days and 14 days post-injection, and serum was obtained via centrifugation (3500 rpm, 5 minutes) for the assessment of alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatine kinase (CK), creatine kinase MB form (CK-MB), creatinine (CREA), and blood urea nitrogen (BUN) levels using an automatic biochemical analyzer (HITACHI 3100, Japan). Additionally, tumor tissues were dissected, fixed with 4% paraformaldehyde, and embedded in paraffin to prepare tumor sections (RM2245, Leica, Germany) for hematoxylin and eosin (H&E) staining after the mice were sacrificed. The samples were observed using microscope slide scanners (NanoZoomer S60, Hamamatsu, Japan).
Statistical Analysis
The experimental results are reported as the means ± standard deviations (SDs). Statistical comparisons between two groups were conducted using Student’s t tests, and one-way ANOVA was employed for comparisons among multiple groups. The thresholds for statistical significance were as follows: *p < 0.05, **p < 0.01, and ***p < 0.001; N.S.: nonsignificance.
Results and Discussion
Firstly, hyaluronic acid-coated, amine-group modified mesoporous silica nanoparticles (HMs) were synthesized as previously described in the literature.25 ICG and CDDP molecules formed ICG‒CDDP complexes through coordinated reactions and were subsequently loaded into the aforementioned HMs, achieving a loading efficiency of 19.64%. Transmission electron microscopy (TEM) analysis revealed that the particle size of MSNs-NH2 was approximately 70 nm (Figures 1A and S2A) and increased to approximately 100 nm upon coating with hyaluronic acid (HA) (Figures 1B and S2B). Similarly, the particle size remained approximately 100 nm when the HMs were loaded with ICG-CDDP complexes (ICG-CDDP@HMs) (Figures 1C and S2C). MSNs-NH2 were coated with hyaluronic acid (HA) through electrostatic attraction between the amino groups of MSNs-NH2 and the carboxyl groups of hyaluronic acid. The zeta potential of MSNs-NH2 was 52 mV and −28 mV prior to and after coating with hyaluronic acid, respectively, indicating that the surface of the MSNs-NH2 was successfully coated with hyaluronic acid, whereas the zeta potential was −51 mV after ICG-CDDP was applied (Figure 1D). ICG-CDDP@HMs retained the characteristic absorption peak of ICG at 783 nm and exhibited a redshift in the absorbance spectrum, facilitating responsiveness to NIR irradiation (Figure 1E). The specific surface area of MSNs-NH2 was 234.0 m²/g, and the pores had a diameter of 1.2 nm to 7.0 nm (Figure 1F and G). Under varying conditions, the release dynamics of ICG-CDDP from ICG-CDDP@HMs were assessed. At pH 7.4 and 5.0, ICG-CDDP@HMs released only 10.3% and 23.5% of ICG-CDDP over a 72-hour period, demonstrating the stability of ICG-CDDP@HMs, which is advantageous for uptake by host macrophages. Moreover, in a cellular microenvironment at pH 5.0 with concurrent HAase treatment, the release of ICG-CDDP increased to 50.9% within 72 hours, indicating a significant enhancement in release rate and underscoring the efficacy of HA encapsulation (Figure 1H). Notably, the antitumor efficacy of ICG-CDDP remained unaffected by NIR irradiation (Figure 1I). The photothermal conversion efficiency of ICG (50 μg/mL) was demonstrated using a thermal imager, which revealed a rapid temperature increase to 83 °C under NIR irradiation (808 nm, 1.0 W/cm²) for 5 minutes. Similarly, (ICG-CDDP) (50 μg/mL) treatment resulted in a temperature of 76 °C under the same conditions. Upon the incorporation of HMs, the temperature profiles (Figure 1J) and representative infrared thermal images (Figure 1K) indicated that compared with those of free ICG and ICG-CDDP, the temperature of ICG-CDDP@HMs complexes was significantly elevated and stable under near-infrared (NIR) irradiation.
 |
Figure 1 Characterization and photothermal conversion capability of ICG-CDDP@HMs. (A) A representative TEM image of MSNs-NH2. Scale bar, 100 nm. (B) A representative TEM image of HMs. Scale bar, 100 nm. (C) A representative TEM image of ICG-CDDP@HMs. Scale bar, 100 nm. (D) Zeta potential of MSNs-NH2, HMs and ICG-CDDP@HMs. (E) Ultraviolet−visible absorbance spectra of HMs, CDDP, ICG, ICG-CDDP, and ICG-CDDP@HMs. (F) Adsorption/desorption isotherms of MSNs-NH2. (G) Pore diameter distributions of MSNs-NH2. (H) In vitro release profiles of ICG-CDDP from ICG-CDDP @HMs in PBS at pH 7.4 and 5.0, and in a pH 5.0 condition with HAase (150 U/mL). (I) The cytotoxicity of ICG-CDDP to CT26 cells under NIR laser irradiation (808 nm laser, 1.0 W/cm²). (J and K) Temperature profiles (J) and representative infrared thermal images (K) of solutions of HMs (204 μg/mL), ICG-CDDP@HMs (50 μg/mL ICG-CDDP), ICG-CDDP (50 μg/mL ICG-CDDP), and free ICG (50 μg/mL ICG) during NIR laser irradiation (808 nm laser, 1.0 W/cm²). Statistical significance is indicated as follows: N.S.: nonsignificance. “L” denotes NIR laser irradiation. |
By leveraging the potent phagocytic capabilities of macrophages, ICG-CDDP@HMs were bioengineered to form ICG-CDDP@HMs@Macs. The cytotoxic effects of ICG-CDDP on host macrophages were notably diminished following encapsulation by HMs, which is essential for the use of macrophages as drug carriers (Figure 2A). Compared with that in the free ICG-CDDP group, the fluorescence intensity of ICG within macrophages was significantly greater in the ICG-CDDP@HMs group at ICG-CDDP concentrations of 50 μg/mL and 100 μg/mL, increasing by 96% and 49%, respectively (Figure 2B), and remained markedly elevated after both 24 hours and 48 hours of retention (Figure 2C and D). Consistently, the results of laser confocal microscopy revealed that the red fluorescence intensity of the macrophages was greater in the ICG-CDDP@HMs group than in the ICG-CDDP group within 48 hours of retention (Figure 2D and E). These findings suggest that the HM-mediated loading of ICG-CDDP increases drug uptake by macrophages and prolongs drug retention, thereby facilitating subsequent responses to NIR irradiation at 808 nm.
 |
Figure 2 ICG-CDDP@HMs bioarmed with macrophages. (A) Assessment of the cytotoxicity of ICG-CDDP or ICG-CDDP@HMs in RAW 264.7 cells at a concentration of 50 μg/mL ICG-CDDP. (B) Quantification of the retained ICG fluorescence intensity in RAW 264.7 cells treated with ICG-CDDP@HMs or ICG-CDDP for 8 hours. (C) Quantification of the ICG fluorescence intensity at specified time points in RAW 264.7 cells treated with either ICG-CDDP@HMs or ICG-CDDP at a concentration of 50 μg/mL ICG-CDDP for 8 hours. (D) Schematic illustration of the experiments in (C and E). RAW 264.7 cells were incubated with ICG-CDDP@HMs at a concentration of 50 μg/mL for 8 hours, and then the medium was changed to facilitate the loading of macrophages with ICG-CDDP@HMs nanoparticles. The uptake of ICG-CDDP@HMs by macrophages was assessed using confocal laser scanning microscopy. The fluorescence intensity of intracellular ICG was quantified via flow cytometry and laser scanning confocal microscopy after 24 and 48 hours of continuous culture, respectively. (E) Confocal images illustrating ICG-CDDP uptake by and retention in RAW 264.7 cells. ICG is shown in red, and Hoechst is shown in blue. Scale bar, 10 µm. Statistical significance is indicated as follows: **p < 0.01, N.S.: nonsignificance. |
Subsequent evaluations were conducted to assess the photothermal conversion efficiency, drug release behavior, antitumor efficacy, and migratory capacity of ICG-CDDP@HMs@Macs within the tumor microenvironment in vitro. Upon exposure to NIR irradiation at 808 nm and 1.0 W/cm² for 5 minutes, the temperature profiles (Figure 3A) and representative infrared thermal images (Figure 3B) revealed a significant increase in the temperature of ICG-CDDP@HMs@Macs, reaching a peak of 122 °C, which was substantially higher than that observed in the ICG@HMs@Macs group. Next, whether ICG-CDDP could be released from ICG-CDDP@HMs@Macs irradiated with NIR light was determined. In response to NIR irradiation, approximately 100% of ICG-CDDP was rapidly released into the supernatant from ICG-CDDP@HMs@Macs, whereas approximately 14% release was observed from ICG-CDDP@HMs@Macs not subjected to NIR irradiation, representing a nearly 7-fold increase (Figure 3C). Furthermore, cytotoxicity assays revealed that ICG-CDDP@HMs@Macs had a concentration-dependent cytotoxic effect on CT26 tumor cells following 5 minutes of NIR irradiation (Figure 3D). A Transwell chamber assay (Figure 3E and F) revealed that ICG-CDDP@HMs@Macs migrated toward the tumor environment. This phenomenon may be attributed to the influence of chemokines such as CCL2,41,42 IL-6,43 and M-CSF,44 which are known to facilitate the migration of macrophages into tumor environment. Notably, ICG-CDDP@Macs markedly reduced the inherent migratory ability of macrophages, suggesting that the encapsulation of ICG-CDDP within HMs significantly mitigated the cytotoxic effects of chemotherapeutic drugs on host macrophages while enhancing their migratory capacity.
 |
Figure 3 ICG-CDDP@HMs@Macs retain enhanced photothermal conversion efficiency, exert enhanced therapeutic efficacy against tumors, and exhibit an increased capacity to migrate toward the tumor microenvironment in vitro. (A and B) Photothermal responses of ICG@HMs@Macs and ICG-CDDP@HMs@Macs under NIR irradiation. Temperature profiles (A) and representative infrared thermal images (B) of various cell-containing solutions following 5 minutes of NIR laser exposure (808 nm, 1.0 W/cm²). (C) The release profiles of ICG-CDDP from ICG-CDDP@HMs@Macs after incubation at 37 °C for 0.5 h, 1 h, 2 h, or 4 h following NIR irradiation (red line) or no (black line). (D) The cytotoxic effects on CT26 cells were evaluated following a 48-hour incubation with either macrophages (Macs or ICG-CDDP@HMs@Macs) or cell lysates of ICG@HMs@Macs or ICG-CDDP@HMs@Macs subjected to NIR irradiation (808 nm, 1.0 W/cm², 1 minute). Untreated CT26 cells served as the control group. (E and F) Representative images (E) and quantification (F) of macrophage (Macs, ICG-CDDP@Macs, HMs@Macs, and ICG-CDDP@HMs@Macs) migration toward either fresh medium or conditioned medium (CM) from CT26 colon carcinoma cells. Scale bar, 200 μm. The data are presented as the mean ± SD, with n = 3 per treatment group. Statistical significance is indicated as follows: **p < 0.01, N.S.: nonsignificance. “L” denotes NIR laser irradiation. |
Furthermore, to evaluate the tumor-homing ability of ICG-CDDP@HMs@Macs and respond to NIR irradiation in vivo, a subcutaneous CT26 colon cancer mouse model was established. Small animal in vivo imaging revealed that the ICG fluorescence intensity gradually increased in tumor tissues within 48 h after tail vein injection of ICG-CDDP@HMs@Macs (Figure 4A). Strong ICG fluorescence was observed in tumor tissues in both the ICG@HMs@Macs and the ICG-CDDP@HMs@Macs groups 48 h after administration (Figure 4B). We subsequently utilized NIR irradiation (808 nm, 1.0 W/cm2) to investigate the ability of ICG-CDDP@HMs@Macs to bind to tumor tissue in response to NIR light for photothermal conversion. The local tumor temperatures in the ICG@HMs@Macs and ICG-CDDP@HMs@Macs groups rapidly increased within 3 minutes of irradiation, followed by a sustained plateau, and the maximum temperature reached approximately 60 °C. Moreover, the maximum temperatures in the PBS, Macs, and HMs@Macs groups were similar at approximately 50 °C (Figure 4C and D). In comparison to the in vitro results, the temperature achieved in vivo is more moderate and safer, which supports the potential application of this strategy in living organisms. These results indicate that the macrophages still possessed tumor-homing ability after the phagocytosis of drug-bearing nanoparticles (ICG-CDDP@HMs) and subsequently exerted a photothermal effect in response to NIR irradiation in vivo.
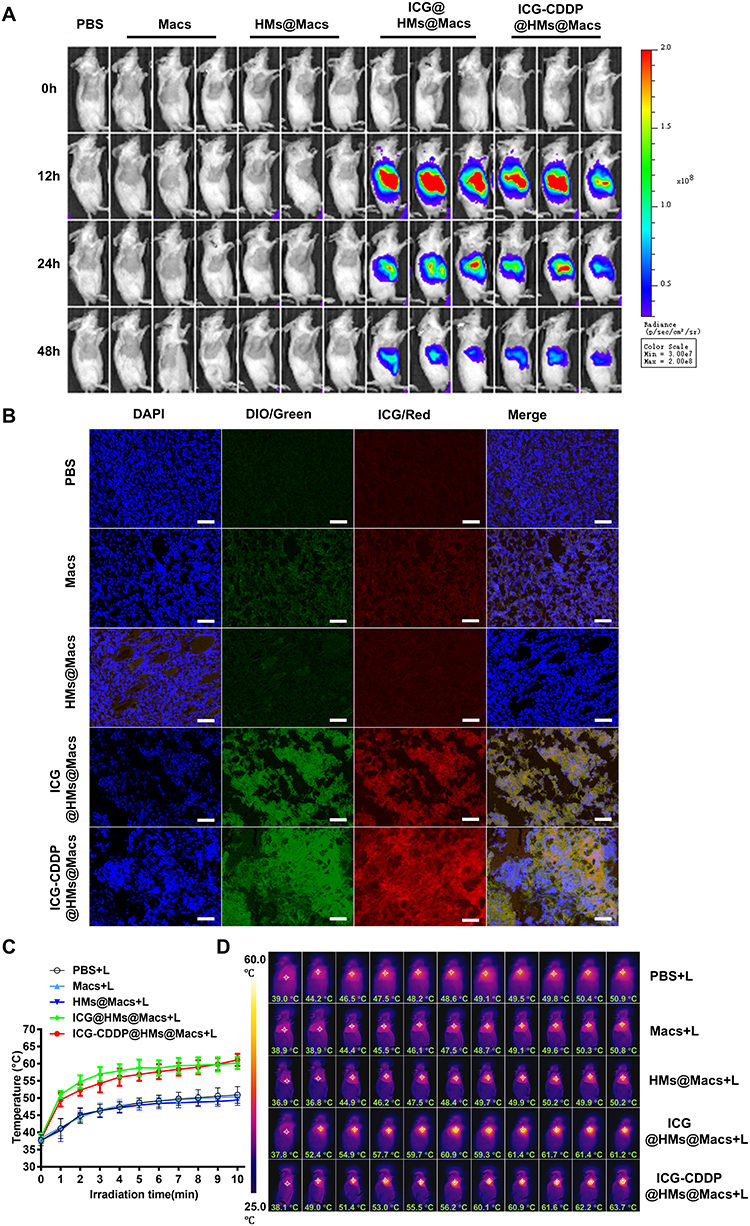 |
Figure 4 ICG-CDDP@HMs@Macs retain the ability to home to tumors and respond to NIR irradiation in vivo. (A) Real-time in vivo tracking of macrophages (Macs, ICG@HMs@Macs or ICG-CDDP@HMs@Macs) was conducted by monitoring ICG fluorescence at 12, 24, and 48 hours after intravenous injection into CT26 tumor-bearing mice. n = 3 per group. (B) Microscopy images of tumor tissues extracted from CT26 tumor-bearing mice 72 hours after intravenous administration of macrophages labeled with DiO (green) (Macs, ICG@HMs@Macs, or ICG-CDDP@HMs@Macs). The nuclei were counterstained with DAPI (blue). Scale bar, 50 μm. (C and D) Temperature profiles of tumors in mice 24 hours after intravenous injection of macrophages (Macs, ICG@HMs@Macs, or ICG-CDDP@HMs@Macs) during a 10-minute exposure to NIR laser irradiation (808 nm, 1.5 W/cm²). The data are shown as the means ± SDs. n = 8 per group. (D) Representative infrared thermal images corresponding to the data in (C). “L” denotes NIR laser irradiation. |
Next, we developed a subcutaneous mouse model of CT26 colon cancer to assess the anti-tumor efficacy of ICG-CDDP@HMs@Macs in conjunction with NIR irradiation. When the tumors reached a volume of 100 mm³, various drug formulations were administered via tail vein injection, and localized NIR irradiation was applied to the tumor site at 24 hours and 48 hours post-injection (Figure 5A). The PBS group served as the control group. Macs, HMs@Macs, and ICG@HMs@Macs exerted moderate antitumor effects, likely due to the intrinsic antitumor properties of macrophages.45,46 In contrast, the antitumor effect of ICG-CDDP@HMs@Macs was slightly enhanced, potentially because of the release of a small quantity of ICG-CDDP@HMs from the macrophages. Building on these observations, a significant improvement in antitumor efficacy was noted following the combined application of NIR irradiation. Remarkably, the antitumor activity of ICG-CDDP@HMs@Macs was enhanced after combined NIR irradiation (Figure 5B, and C).
Figure 5 Continued. Figure 5 ICG-CDDP@HMs@Macs combined with near-infrared irradiation exert excellent antitumor effects in vivo. (A) Schematic illustration of the in vivo therapeutic experiment. (B) The relative tumor volume of CT26 tumor-bearing mice over a 14-day period following the indicated treatment. The mice received intravenous injections of the designated agents on day 0, followed by NIR laser irradiation (1.5 W/cm², 5 minutes) on days 1 and 2 post-injection. (C) Photographs of tumors excised from mice in each treatment group at the conclusion of the study. (D) Serum IFN-γ levels in mice from each group at the end of the experimental period. (E and F) TUNEL, H&E, and immunohistochemical staining of tumor tissue sections obtained from mice at the end of the study. Scale bar, 250 µm. The data are presented as the means ± SDs. n = 6 animals per group. Statistical significance is indicated as follows: *p < 0.05, **p < 0.01. “L” denotes NIR laser irradiation.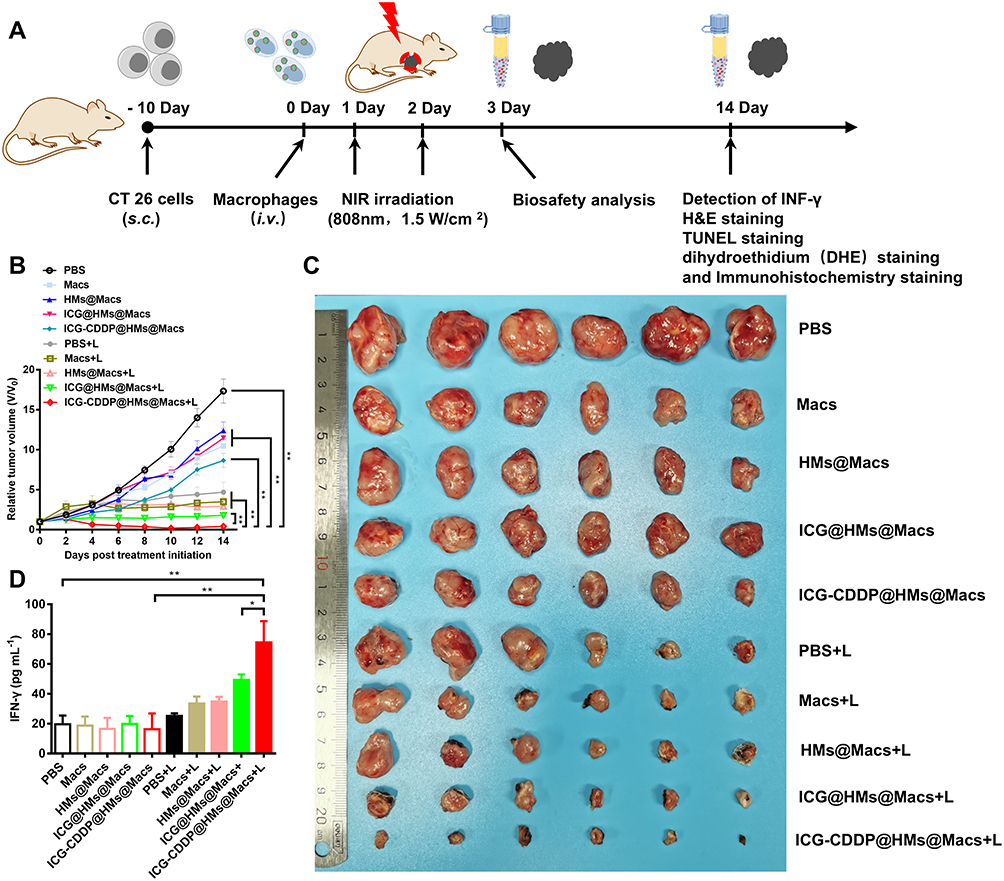

Subsequently, tumor tissues were collected for H&E and TUNEL staining. TUNEL staining revealed significantly more apoptotic cells in the ICG-CDDP@HMs@Macs group than in the other groups. Similarly, H&E staining revealed a marked reduction in tumor cell density in the ICG-CDDP@HMs@Macs group (Figure 5E). These findings demonstrate that ICG-CDDP@HMs@Macs, in conjunction with NIR irradiation, exerts a potent antitumor therapeutic effect.
Furthermore, the serum IFN-γ level in the ICG-CDDP@HMs@Macs group was significantly greater than that in the other groups (Figure 5D). The fluorescence staining and immunohistochemical staining results revealed significant increases in ROS (Figure S3) and calreticulin levels in the tumor tissues of the groups treated with ICG@HMs@Macs and ICG-CDDP@HMs@Macs plus NIR irradiation (Figure 5F). These findings suggest that the ability of ICG to respond to NIR irradiation is preserved when it is delivered by macrophages in ICG-CDDP@HMs, resulting in substantial ROS generation. This, in turn, facilitates calreticulin expression in tumor tissues, thereby inducing immunogenic cell death.
Finally, to evaluate the in vivo biosafety of ICG-CDDP@HMs@Macs in combination with NIR irradiation, we conducted a comprehensive assessment of serum biochemical indices and histological analysis of tumor-bearing mice. Serum biochemical parameters were measured on the 3rd and 14th days after treatment, and histological examination of tissues and organs was performed using H&E staining. Analysis of serum biochemical indices revealed no statistically significant differences in liver (ALT and AST), heart (CK and CK-MB), or kidney (CREA and BUN) parameters compared with those in the PBS control group (Tables 1 and 2). Furthermore, H&E staining revealed no discernible abnormalities in heart, liver, spleen, lung, or kidney tissues compared with those in the PBS group (Figure 6A and B). These findings collectively suggest that the combination of ICG-CDDP@HMs@Macs with NIR irradiation results in favorable biosafety profiles.
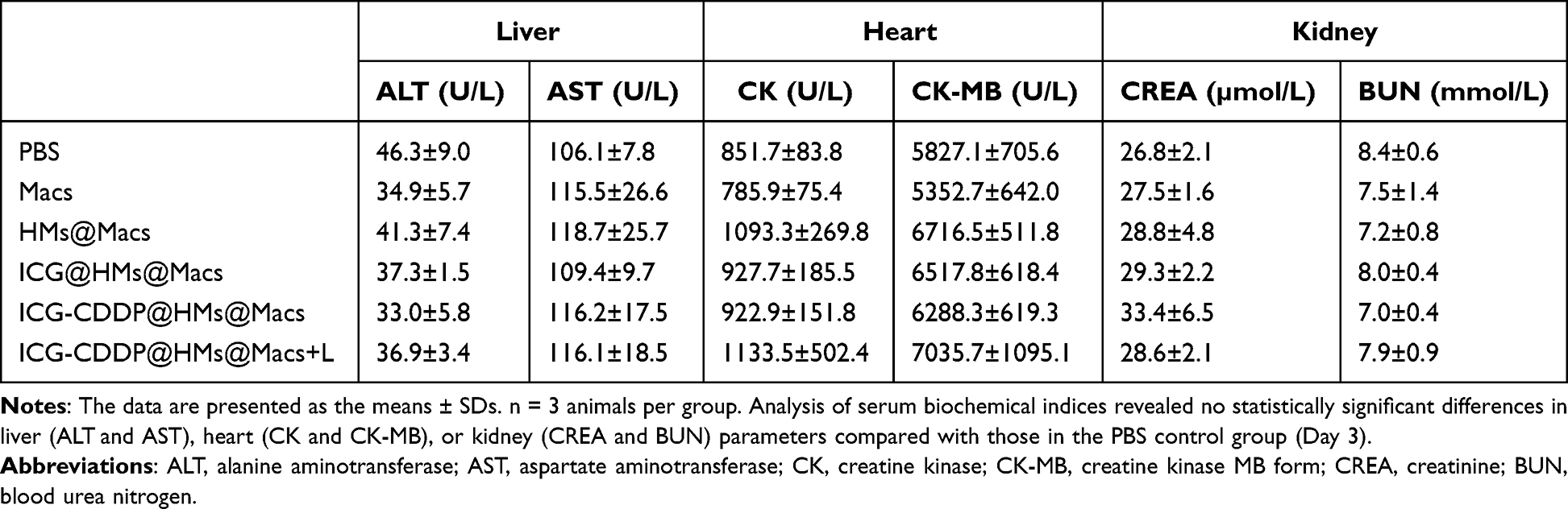 |
Table 1 Serum Was Collected from the Mice on Day 3 After Treatment for Biochemical Assays |
 |
Table 2 Serum Was Collected from the Mice on Day 14 After Treatment for Biochemical Assays |
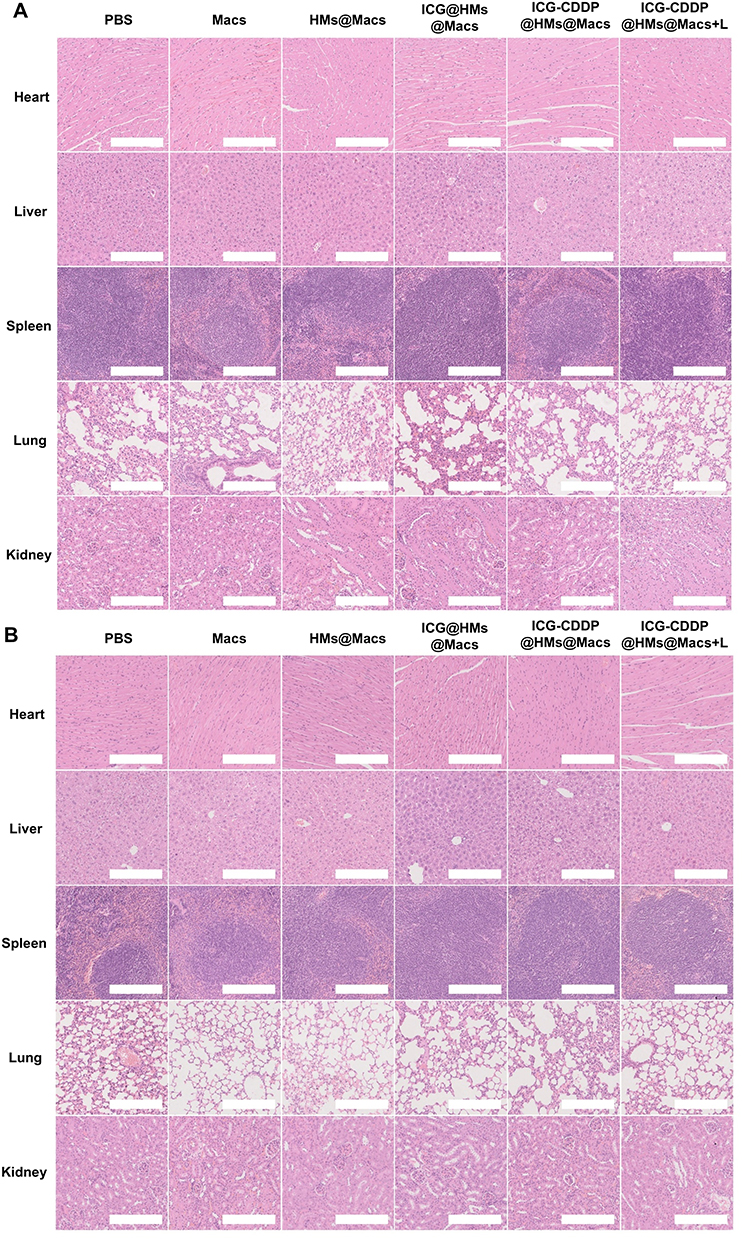 |
Figure 6 ICG-CDDP@HMs@Macs combined with NIR irradiation have good biosafety in vivo. (A and B) H&E-stained images of major organs, including the heart, liver, spleen, lung, and kidney, were obtained on day 3 (A) and day 14 (B) following the indicated treatment. Scale bar, 250 µm. |
Conclusion
In this study, we developed a precision tool for cancer treatment by employing autologous macrophages as drug carriers. These macrophages are capable of internalizing silicon spherical nanoparticles encapsulating ICG-CDDP complexes, thereby facilitating the simultaneous targeted delivery of photosensitizers and chemotherapeutic agents to tumor tissues. Upon exposure to local near-infrared (NIR) irradiation in vitro, the ICG within the macrophages generates a photothermal effect, destroying both the macrophages and tumor cells. This mechanism enables the controlled and sustained release of chemotherapeutic drugs within the tumor microenvironment, thereby exerting secondary cytotoxic effects on tumor cells. Furthermore, local photosensitizers within tumor tissues are activated by irradiation with NIR light to generate reactive oxygen species, which induce immunogenic cell death and ultimately have a lethal effect on tumor cells. The primary innovations of this research include three key aspects: (1) the novel integration of the inherent properties of macrophages with light-responsive nanocarriers to develop an advanced multifunctional drug delivery system; (2) the successful execution of a cascade reaction involving photothermal triggering, drug release, and immune activation; and (3) experimental evidence demonstrating that this approach significantly enhances the drug accumulation efficiency at tumor sites while mitigating systemic toxicity. These pivotal findings offer a promising new avenue for the advancement of precise anticancer therapies. We acknowledge the potential for further exploration in several key areas. First, concerning the experimental model, the current results were predominantly derived from the CT26 colon cancer model. It is imperative to assess the generalizability of these findings across various tumor models with different immune profiles, such as B16 melanoma and 4T1 breast cancer models, in future research. Second, mechanistic investigations are critically needed to further elucidate the dynamic alterations in immune cell subpopulations within the tumor microenvironment during treatment, as well as the underlying regulatory mechanisms. In summary, three significant advancements were made using the trimodal therapy developed in this study—precise delivery, spatiotemporally controlled drug release, and effective immune activation; thus, this therapy represents a novel and transformative strategy for clinical cancer treatment.
Acknowledgments
The authors are thankful to the Department of the Second Central Laboratory, The First Affiliated Hospital of Zhejiang Chinese Medical University, for providing technological support. This work was supported by the Zhejiang Provincial Natural Science Foundation of China under Grant No. LGF21H160019, Medical Science and Technology Project of Zhejiang Province under Grant No. 2025KY968, Zhejiang Traditional Chinese Medicine Administration under Grant Nos. 2024ZF012, 2025ZF007, 2025ZF035, Zhejiang Chinese Medical University under Grant No. 2022JKZKTS33.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by the Zhejiang Provincial Natural Science Foundation of China under Grant No. LGF21H160019, Medical Science and Technology Project of Zhejiang Province under Grant No. 2025KY968, Zhejiang Traditional Chinese Medicine Administration under Grant Nos. 2024ZF012, 2025ZF007, 2025ZF035, Zhejiang Chinese Medical University under Grant No. 2022JKZKTS33.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Paragomi P, Zhang Z, Abe SK, et al. Body mass index and risk of colorectal cancer incidence and mortality in Asia. JAMA Network Open. 2024;7:e2429494. doi:10.1001/jamanetworkopen.2024.29494
2. Thrift AP, Wenker TN, El-Serag HB. Global burden of gastric cancer: epidemiological trends, risk factors, screening and prevention. Nat Rev Clin Oncol. 2023;20:338–349. doi:10.1038/s41571-023-00747-0
3. Guo Y, Wang M, Zou Y, et al. Mechanisms of chemotherapeutic resistance and the application of targeted nanoparticles for enhanced chemotherapy in colorectal cancer. J Nanobiotechnol. 2022;20:371. doi:10.1186/s12951-022-01586-4
4. Xie P, Liu P. Formulation of DOX-dimer with bi-functionalized chitooligosaccharide for tumor-specific self-boosted drug release and synergistic chemo/chemodynamic therapy. Carbohydr Polym. 2023;320:121210. doi:10.1016/j.carbpol.2023.121210
5. Agwa MM, Elmotasem H, Moustafa RI, Abdelsattar AS, Mohy-Eldin MS, Fouda MMG. Advent in proteins, nucleic acids, and biological cell membranes functionalized nanocarriers to accomplish active or homologous tumor targeting for smart amalgamated chemotherapy/photo-therapy: a review. Int J Biol Macromol. 2023;253:127460. doi:10.1016/j.ijbiomac.2023.127460
6. Chen L, Hong W, Ren W, Xu T, Qian Z, He Z. Recent progress in targeted delivery vectors based on biomimetic nanoparticles. Ignal Transduct Target Ther. 2021;6:225. doi:10.1038/s41392-021-00631-2
7. Zhu M, Zhuang J, Li Z, et al. Machine-learning-assisted single-vessel analysis of nanoparticle permeability in tumour vasculatures. Nat Nanotechnol. 2023;18:657–666. doi:10.1038/s41565-023-01323-4
8. Tomita T, Kato M, Hiratsuka S. Regulation of vascular permeability in cancer metastasis. Cancer Sci. 2021;112:2966–2974. doi:10.1111/cas.14942
9. Ying N, Lin X, Xie M, Zeng D. Effect of surface ligand modification on the properties of anti-tumor nanocarrier. Colloids Surf B. 2022;220:112944. doi:10.1016/j.colsurfb.2022.112944
10. Ahmed SS, Baba MZ, Wahedi U, et al. Oral delivery of solid lipid nanoparticles surface decorated with hyaluronic acid and bovine serum albumin: a novel approach to treat colon cancer through active targeting. Int J Biol Macromol. 2024;279:135487. doi:10.1016/J.IJBIOMAC.2024.135487
11. Li A, Zhao Y, Li Y, Jiang L, Gu Y, Liu J. Cell-derived biomimetic nanocarriers for targeted cancer therapy: cell membranes and extracellular vesicles. Drug Delivery. 2021;28:1237–1255. doi:10.1080/10717544.2021.1938757
12. Ryu JH, Yoon HY, Sun IC, Kwon IC, Kim K. Tumor-targeting glycol chitosan nanoparticles for cancer heterogeneity. Adv Mater. 2020;32:e2002197. doi:10.1002/adma.202002197
13. Zhao F, Wang J, Zhang Y, et al. In vivo fate of targeted drug delivery carriers. Int J Nanomed. 2024;19:6895–6929. doi:10.2147/IJN.S465959
14. Edgar P-H, Alberto F-M. The reversed intra- and extracellular pH in tumors as a unified strategy to chemotherapeutic delivery using targeted nanocarriers. Acta Pharm Sin B. 2021;11:2243–2264. doi:10.1016/J.APSB.2021.01.012
15. Weide X, Jilong W, Qinghua L, et al. Cancer cell membrane-coated nanogels as a redox/pH dual-responsive drug carrier for tumor-targeted therapy. J Mat Chem B. 2021;9:8031–8037. doi:10.1039/D1TB00788B
16. Sharifi S, Caracciolo G, Mahmoudi M. Biomolecular corona affects controlled release of drug payloads from nanocarriers. Trends Pharmacol Sci. 2020;41:641–652. doi:10.1016/j.tips.2020.06.011
17. Jin Y, Wu Z, Wu C, et al. Size-adaptable and ligand (biotin)-sheddable nanocarriers equipped with avidin scavenging technology for deep tumor penetration and reduced toxicity. J Control Release. 2020;320:142–158. doi:10.1016/j.jconrel.2020.01.040
18. Bush LM, Healy CP, Javdan SB, Emmons JC, Deans TL. Biological cells as therapeutic delivery vehicles. Trends Pharmacol Sci. 2020;42:106–118. doi:10.1016/J.TIPS.2020.11.008
19. Combes F, Meyer E, Sanders NN. Immune cells as tumor drug delivery vehicles. J Control Release. 2020;327:70–87. doi:10.1016/j.jconrel.2020.07.043
20. Ning P, Du F, Wang H, et al. Genetically engineered macrophages as living cell drug carriers for targeted cancer therapy. J Control Release. 2024;367:697–707. doi:10.1016/J.JCONREL.2024.02.003
21. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019;30:36–50. doi:10.1016/j.cmet.2019.06.001
22. Qiu-Ping L, Yu-Ying C, Pei A, Khalid R, Xin L, Hong Z. Natural products targeting macrophages in tumor microenvironment are a source of potential antitumor agents. Phytomedicine. 2023;109:154612. doi:10.1016/J.PHYMED.2022.154612
23. Wang H, Wang X, Zhang X, Xu W. The promising role of tumor-associated macrophages in the treatment of cancer. Drug Resist Updat. 2024;73:101041. doi:10.1016/j.drup.2023.101041
24. Zeng X, Huabei H, Xiang X, et al. A near-infrared light-responsive extracellular vesicle as a “Trojan horse” for tumor deep penetration and imaging-guided therapy. Biomaterials. 2021;269:120647. doi:10.1016/J.BIOMATERIALS.2020.120647
25. Huang L, Yan W, Cai B, et al. Dual-engineered, “Trojanized” macrophages bio-modally eradicate tumors through biologically and photothermally deconstructing cancer cells in an on-demand, NIR-commanded, self-explosive manner. Biomaterials. 2020;250:120021. doi:10.1016/j.biomaterials.2020.120021
26. Timin AS, Litvak MM, Gorin DA, Atochina-Vasserman EN, Atochin DN, Sukhorukov GB. Cell-based drug delivery and use of nano-and microcarriers for cell functionalization. Adv Healthc Mater. 2018;7:2300107. doi:10.1002/adhm.201700818
27. Liu XL, Dong X, Yang SC, et al. Biomimetic liposomal nanoplatinum for targeted cancer chemophototherapy. Adv Sci. 2021;8:2003679. doi:10.1002/advs.202003679
28. Li T, Shi S, Goel S, et al. Recent advancements in mesoporous silica nanoparticles towards therapeutic applications for cancer. Acta Biomater. 2019;89:1–13. doi:10.1016/j.actbio.2019.02.031
29. Hang Y, Liu Y, Teng Z, Cao X, Zhu H. Mesoporous nanodrug delivery system: a powerful tool for a new paradigm of remodeling of the tumor microenvironment. J Nanobiotechnol. 2023;21:101. doi:10.1186/s12951-023-01841-2
30. Choi KY, Min KH, Yoon HY, et al. PEGylation of hyaluronic acid nanoparticles improves tumor targetability in vivo. Biomaterials. 2011;32:1880–1889. doi:10.1016/j.biomaterials.2010.11.010
31. Caon I, Bartolini B, Parnigoni A, et al. Revisiting the hallmarks of cancer: the role of hyaluronan. Semi Cancer Biol. 2020;62:9–19. doi:10.1016/j.semcancer.2019.07.007
32. Maoz A, Weiskopf K. Phagocytic cooperativity by tumour macrophages. Nat Biomed Eng. 2023;7:1057–1059. doi:10.1038/s41551-023-01088-0
33. Joffe AM, Bakalar MH, Fletcher DA. Macrophage phagocytosis assay with reconstituted target particles. Nat Protoc. 2020;15:2230–2246. doi:10.1038/s41596-020-0330-8
34. Zhou J, Fan X, Wu D, et al. Hot-band absorption of indocyanine green for advanced anti-stokes fluorescence bioimaging. Light. 2021;10:182. doi:10.1038/s41377-021-00627-1
35. Xue P, Yang R, Sun L, et al. Indocyanine green-conjugated magnetic prussian blue nanoparticles for synchronous photothermal/photodynamic tumor therapy. Nano-Micro Lett. 2018;10:74. doi:10.1007/s40820-018-0227-z
36. Zheng C, Zheng M, Gong P, et al. Indocyanine green-loaded biodegradable tumor targeting nanoprobes for in vitro and in vivo imaging. Biomaterials. 2012;33:5603–5609. doi:10.1016/j.biomaterials.2012.04.044
37. Kim DY, Pyo A, Yun M, et al. Imaging calreticulin for early detection of immunogenic cell death during anticancer treatment. J Nucl Med. 2021;62:956–960. doi:10.2967/jnumed.120.245290
38. Xiang Y, Chen L, Liu C, Yi X, Li L, Huang Y. Redirecting chemotherapeutics to the endoplasmic reticulum increases tumor immunogenicity and potentiates Anti-PD-L1 therapy. Small. 2022;18:e2104591. doi:10.1002/smll.202104591
39. Zhang Z, Pan Z, Li Q, Huang Q, Shi L, Liu Y. Rational design of ICD-inducing nanoparticles for cancer immunotherapy. Sci Adv. 2024;10:eadk0716. doi:10.1126/sciadv.adk0716
40. Khosravi GR, Mostafavi S, Bastan S, Ebrahimi N, Gharibvand RS, Eskandari N. Immunologic tumor microenvironment modulators for turning cold tumors hot. Cancer Commun. 2024;44:521–553. doi:10.1002/cac2.12539
41. O’Connor T, Heikenwalder M. CCL2 in the Tumor Microenvironment. Adv Exp Med Biol. 2021;1302:1–14. doi:10.1007/978-3-030-62658-7_1
42. Bess SN, Greening GJ, Rajaram N, Muldoon TJ. Macrophage-targeted anti-CCL2 immunotherapy enhances tumor sensitivity to 5-fluorouracil in a Balb/c-CT26 murine colon carcinoma model measured using diffuse reflectance spectroscopy. BMC immunol. 2022;23:20. doi:10.1186/s12865-022-00493-5
43. Alshamsan A. Induction of tolerogenic dendritic cells by IL-6-secreting CT26 colon carcinoma. Immunopharmacol Immunotoxicol. 2012;34:465–469. doi:10.3109/08923973.2011.625034
44. Popova A, Kzhyshkowska J, Nurgazieva D, Goerdt S, Gratchev A. Pro- and anti-inflammatory control of M-CSF-mediated macrophage differentiation. Immunobiology. 2011;216:164–172. doi:10.1016/j.imbio.2010.06.003
45. Li CX, Zhang Y, Dong X, et al. Artificially reprogrammed macrophages as tumor-tropic immunosuppression-resistant biologics to realize therapeutics production and immune activation. Adv Mater. 2019;31:e1807211. doi:10.1002/adma.201807211
46. Wei F, Liu H, Wang Y, Li Y, Han S. Engineering macrophages and their derivatives: a new hope for antitumor therapy. Biomed Pharmacother. 2024;177:116925. doi:10.1016/j.biopha.2024.116925
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.