Back to Journals » OncoTargets and Therapy » Volume 9
Next-generation sequencing: advances and applications in cancer diagnosis
Authors Serratì S, De Summa S ![]() , Pilato B, Petriella D, Lacalamita R, Tommasi S
, Pilato B, Petriella D, Lacalamita R, Tommasi S ![]() , Pinto R
, Pinto R
Received 17 June 2016
Accepted for publication 28 September 2016
Published 2 December 2016 Volume 2016:9 Pages 7355—7365
DOI https://doi.org/10.2147/OTT.S99807
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Simona Serratì, Simona De Summa, Brunella Pilato, Daniela Petriella, Rosanna Lacalamita, Stefania Tommasi, Rosamaria Pinto
Molecular Genetics Laboratory, IRCCS Istituto Tumori Giovanni Paolo II, Bari, Italy
Abstract: Technological advances have led to the introduction of next-generation sequencing (NGS) platforms in cancer investigation. NGS allows massive parallel sequencing that affords maximal tumor genomic assessment. NGS approaches are different, and concern DNA and RNA analysis. DNA sequencing includes whole-genome, whole-exome, and targeted sequencing, which focuses on a selection of genes of interest for a specific disease. RNA sequencing facilitates the detection of alternative gene-spliced transcripts, posttranscriptional modifications, gene fusion, mutations/single-nucleotide polymorphisms, small and long noncoding RNAs, and changes in gene expression. Most applications are in the cancer research field, but lately NGS technology has been revolutionizing cancer molecular diagnostics, due to the many advantages it offers compared to traditional methods. There is greater knowledge on solid cancer diagnostics, and recent interest has been shown also in the field of hematologic cancer. In this review, we report the latest data on NGS diagnostic/predictive clinical applications in solid and hematologic cancers. Moreover, since the amount of NGS data produced is very large and their interpretation is very complex, we briefly discuss two bioinformatic aspects, variant-calling accuracy and copy-number variation detection, which are gaining a lot of importance in cancer-diagnostic assessment.
Keywords: hereditary breast cancer, melanoma, prostate cancer, thyroid cancer, lung cancer, colorectal cancer, hematologic cancer
Introduction
In recent years, next-generation sequencing (NGS) technologies have played an essential role in the understanding of the altered genetic pathways involved in human cancer. Compared to earlier genome-sequencing methods, numerous advantages characterize NGS. Primarily, this is a high-throughput method, as it allows massive parallel sequencing consisting of simultaneous sequencing of multiple targeted genomic regions in multiple samples in order to detect concomitant mutations in the same run. Another important advantage in routine tumor sequencing is the reduced turnaround time of analysis, which leads to reduced clinical reporting time. Moreover, an analysis in NGS requires very low input of DNA/RNA, in contrast to traditional sequencing methods. A variety of genomic aberrations with high accuracy and sensitivity can be screened simultaneously, such as single/multiple-nucleotide variants, small and large insertions and deletions, copy-number variations (CNVs), and fusion transcripts. The sensitivity of NGS is higher than Sanger sequencing (detection of 2%–10% versus 15%–25% allele frequency, respectively), and allows quantitative evaluation of the mutated allele.
NGS workflow is constituted by different steps, from nucleic acid extraction to variant annotation, as shown in Figure 1. There are currently three main companies offering NGS platforms: Roche, Illumina, and Life Technologies (Thermo Fisher Scientific, Waltham, MA, USA). Each of the available platforms uses different sequencing chemistry and methods for signal detection. Roche 454 platforms employ pyrosequencing, whereby a chemiluminescent signal indicates base incorporation and the intensity of the signal correlates with the number of bases incorporated through homopolymer reads.1 However, the NGS platforms most commonly used employ sequencing by synthesis, in which the DNA strand to be sequenced is used as a template, a complementary strand is synthesized, and consequently the sequence of the template strand is obtained. Illumina MiSeq and HiSeq sequencers use four distinct fluorescently labeled nucleotides and optical imaging to visualize the growing complementary strand. The error rate estimated for Illumina technology is <0.4%.2,3 Instead, Life Technologies uses a nonoptical approach and unlabeled nucleotides. Sequencing by synthesis is performed in microscopic wells interfaced with a semiconductor chip. The DNA is clonally amplified on microscopic beads. After incorporation of nucleotides one at a time, the protons released result in a change in pH, measured by the semiconductor chip. The error rate estimated for Ion Torrent technology is 1.8%–1.9%, mostly in the detection of homopolymer stretches.2,3
 | Figure 1 NGS workflow from nucleic acid extraction to variant annotation. |
NGS approaches are different, and concern tumoral DNA and RNA analysis. DNA sequencing includes whole-genome sequencing (WGS), whole-exome sequencing (WES), and targeted sequencing. WGS allows sequencing of the entire genome, requiring a large DNA sample. To detect clinical mutations accurately, 100- to 200-fold sequencing coverage may be needed, which is both time- and cost-prohibitive. Usually, a 30- to 60-fold sequencing, sufficient to identify structural rearrangements, is employed. WES focuses on the coding regions (exons) of a genome, typically ~2.5% of the human genome, to discover rare or common variants associated with a disorder or phenotype. WES reduces cost and time compared to WGS. The most common methods rely on hybridization by oligonucleotide probes to “capture” targeted DNA fragments, thereby enriching for exonic sequences. Targeted sequencing, focusing on a selection of genes of interest for a specific disease, could be more accurate and accessible in terms of time and cost for clinical applications for more laboratories.
RNA sequencing (RNA-Seq) facilitates the detection of alternative gene-spliced transcripts, posttranscriptional modifications, gene fusion, mutations/single-nucleotide polymorphisms (SNPs), and changes in gene expression. The extracted RNA is first enriched and reverse-transcribed into complementary DNA, which is then processed. Moreover, with the NGS approach, it is possible to investigate epigenetic alterations, such as promoter methylation, microRNAs, and the expression of other small RNAs, even if currently there are no relevant panels available to use in diagnostics. Life Technologies is engaging more in the setup of specific kits for the disease (Ion AmpliSeq Colon and Lung Panel version 2, BRCA1/2 Panel, AML Panel, and RNA Lung Fusion Panel) with respect to the Illumina approach, which is based on the development of a generic cancer-panel kit including information on the genes of several cancers (TruSeq Amplicon and TruSight Cancer).
Although NGS is extensively used for research purposes, its application in clinical practice has not been fully formalized with guidelines, due to the novelty of the approach. Despite this, NGS is beginning to be widely used for diagnostic requests. The Italian Society of Human Genetics has recently released early indications on this topic, summarizing criteria needed for new NGS-based molecular diagnosis.
This review includes the advances and initial clinical applications of NGS in solid and hematologic cancer diagnosis. Moreover, we briefly discuss two bioinformatic aspects that are gaining significant importance in cancer-diagnostic assessment: first, the accuracy and quality of variant calling, which is still an open question in terms of reducing the false-positive rate; and second, CNV detection, which is an essential analysis in the clinical setting.
NGS analysis for solid cancer diagnosis
Detection of critical cancer-gene alterations in solid-tumor samples better defines patient diagnosis and prognosis, and indicates what targeted therapies must be administered to improve the care of selected cancer patients in the personalized-medicine scenario. NGS studies on solid cancer, described here, offer a fundamental overview about how the cancer molecular approach is changing, evidencing advantages compared to traditional diagnostic methods.
Hereditary breast cancer
Hereditary breast cancers (HBCs) account for 5%–10% of all BCs, and in about 30% of cases are caused by BRCA1 and BRCA2 mutations. The BRCA1 and BRCA2 genes codify for tumor-suppressor proteins, essential for DNA repair and genomic stability. The presence of these mutations increases the lifetime risk of developing HBC, and so genetic counseling and a BRCA-gene test is recommended for BC patients with early onset or a significant family history.
Conventional DNA sequencing, such as direct Sanger sequencing, requires long analysis times and high costs, due to BRCA1 and BRCA2 gene lengths of 23 and 27 exons, respectively. Moreover, prescreening methods, such as denaturing high-performance liquid chromatography, have been suggested to speed up the molecular analysis.
Our lab experience and several recent papers have demonstrated how NGS methods are adequate to detect point mutations and indels in BRCA1/BRCA2 genes, revolutionizing this genetic analysis and reducing time and costs.4–6 This approach in fact is suitable in routine diagnostic workflow, since it is faster and more sensitive than denaturing high-performance liquid chromatography/Sanger sequencing methods. Data quality is assured by participation in international quality programs on BRCA1/BRCA2 testing with the NGS method (ie, the European Molecular Genetics Quality Network) that also allow obtainment of specific certification on correct results, sensitivity, specificity, and interpretation of variant calling.
Nowadays, other genes besides BRCA1/BRCA2 have been shown to confer high BC risk. NGS platforms allow the customization of gene panels, in order to give more chance to patients to determine their BC risk.7–9 Tung et al found that the frequency of mutations in non-BRCA1/BRCA2 genes was 4.3% in their 25-gene panel.7 Lin et al developed a sequencing panel containing 68 genes associated with cancer risk for patients with early onset or familial BC.8 They discovered alterations in RAD50, TP53, ATM, BRIP1, FANCI, MSH2, MUTYH, and RAD51C, which may be valuable in BC-risk assessment. Lhota et al performed NGS of 581 genes in 325 BC patients (negatively tested in previous BRCA1/BRCA2/PALB2 analyses), identifying 127 truncating variants.9
Despite several findings on HBC with NGS, a recent study demonstrated that with regard to the two most common platforms, neither the Illumina MiSeq sequencer with the supplied MiSeq Reporter software nor the Life Technologies Ion Torrent Personal Genome Machine (Ion PGM) with the supplied Torrent Suite software were completely suitable for clinical laboratory sequencing of BRCA1 or BRCA2.10 The inability of the MiSeq system is that it fails to detect insertions and deletions larger than nine base pairs. Similarly, the inability of the Ion PGM with Torrent Suite software was in detecting a ten-base pair insertion and 64-base pair deletion. However, the authors reported that an alternative alignment and variant-calling software, Quest Sequencing Analysis Pipeline (QSAP), was capable of detecting large deletions and insertions. With the combination of the MiSeq platform and QSAP alignment, they were able to design an assay with 100% sensitivity and specificity for BRCA1-and BRCA2-sequence variations. These results underline the strong impact of specific bioinformatic tools for alignment and variant calling depending on the application of interest, as we describe herein.
Melanoma
BRAF mutations play a key role in 40%–70% of malignant melanomas. According to the COSMIC database, 44% of the melanomas have BRAF mutations and 97.1% of these mutations are localized in codon 600 of the BRAF gene. Mutated BRAF can be inhibited by small-molecule kinase inhibitors, among which are vemurafenib (Roche), approved by the US Food and Drug Administration (FDA) in August 2011 for unresectable or metastatic melanoma, and dabrafenib. For these therapies, it is mandatory to detect BRAF alterations by gold-standard methods, such as Sanger sequencing and real-time polymerase chain reaction (PCR).
Ihle et al evaluated several parameters of different methods for BRAF-mutation analysis.11 They compared allele-specific PCR performed with the Cobas BRAFV600 test, pyrosequencing using the Therascreen BRAF Pyro kit, high-resolution melting analysis, immunohistochemistry, the NGS approach, and Sanger sequencing with regard to their sensitivity, specificity, costs, amount of work, feasibility, and limitations. They suggested the best method to be a combination of VE1-antibody staining and high-resolution melting for p.V600E-mutation analysis, associating the lowest detection limit with a fast method with 100% sensitivity. However, the authors reported the numerous NGS advantages for melanoma molecular diagnostics, supporting the future substitution of the current methods with an NGS approach.
However, there is a clinical need to analyze other genes, both in terms of finding other target-therapy types and understanding eventual resistance. Currently, validated diagnostic panels are not commercially available, and very few studies have been performed on the development of a custom-designed gene panel.12,13 van Engen-van Grunsven et al designed a panel containing hotspot alterations, such as BRAF exon 15, NRAS exons 2 and 3, HRAS exons 2 and 3, AKT1 exon 3, GNAQ exons 4 and 5, GNA11 exons 4 and 5, KIT exons 8, 9, 11, 13, and 14, and PDGFRA exons 12, 14, and 18.12 Our AmpliSeq custom panel includes eleven crucial full-length genes (BRAF, NRAS, PTEN, MITF, CDK4, MGMT, CTLA4, PIK3CA, MC1R, KIT, and RB1) involved in melanoma carcinogenesis and therapy-response pathways.13 We tested its clinical applicability on the NGS platform Ion PGM in order to individuate new or already known SNPs and mutations that could be related to different response duration to BRAF inhibitors. Our results showed higher sensitivity and specificity in detecting a wide range of genetic alterations compared to traditional sequencing methods. Moreover we identified alterations in CTLA4, MITF, PIK3CA, KIT, and MC1R related to BRAF-inhibitor response duration. This panel is now in validation, in order to use it routinely in diagnostic prognosis and therapy prediction.
Prostate cancer
Prostate cancer (PC) has become the leading cause of cancer death in many countries among males. The high tumor heterogeneity suggests that numerous genetic events are responsible for indolent and aggressive forms of the disease. Currently, there is no way to differentiate accurately between these two forms before treatment. Most men diagnosed with PC have clinically indolent disease that does not require immediate radical treatment, and overtreatment of these men could lead to worse quality of life. The clinical response to therapy varies widely from patient to patient, since some patients relapse shortly after treatment, whereas others remain disease-free for a long time before relapsing.
Recent advances in NGS technology have improved the understanding of PC biology and clinical variability. In particular, DNA-Seq, RNA-Seq, chromatin immunoprecipitation-Seq, and methyl-Seq experiments have better elucidated the major pathways affecting prostate tumorigenesis, which are the AR-signaling, PI3K–PTEN–Akt, and RTK–Ras–MAPK pathways.14
Two studies have evidenced the possibility for huge screening of PC patients in routine diagnosis.15,16 Manson-Bahr et al showed that DNA from cancer material dissected from transrectal ultrasound needle-core biopsy specimens can be analyzed.15 The authors observed a pattern of mutation consistent with those previously observed in PC surgical tissues, including TMPRSS2–ERG fusion and mutations in SPOP, TP53, ATM, and MEN1, while nonsense mutations were observed in the MAP2K5 and the NCOR2 genes. Iacono et al performed the first retrospective NGS study on 60 specimens: 30 high- and 30 intermediate-risk patients.16 They identified nonsynonymous variations and SNPs with an allelic frequency ≥10% in the TP53, CSFR1, KDR, KIT, PIK3CA, MET, and FGFR2 genes, evidencing their role in the progression and aggression of PC. However, at present the study of multiple genetic alterations in PC is not suggested for routine diagnostic purposes.
Thyroid cancer
Thyroid nodules, very frequent in the general population, are mostly benign, but an accurate identification of those nodules that could be a precursor of a cancer is needed. A common diagnostic approach that allows differential diagnosis between cancerous and benign nodules in most cases is represented by ultrasound-guided fine-needle aspiration (FNA) of the thyroid nodule followed by cytological examination. However, in approximately 25% of nodules, the diagnosis cannot be established by FNA cytology, since the limited diagnostic material available is not sufficient to perform a comprehensive molecular characterization by traditional techniques.
In the last few years, several studies have been performed on the possibility of improving thyroid cancer (TC) diagnosis by an NGS molecular test.17–19 In 2013, they developed the first custom gene panel, the ThyroSeq, which allowed the targeting of 284 mutational hotspots in 12 cancer genes. Sequencing was performed on 228 neoplastic and nonneoplastic thyroid samples, including 105 frozen, 72 formalin-fixed, and 51 FNA samples, representing all major types of TC.17 Using this approach, point mutations were detected in 30%–83% of specific types of TC and in only 6% of benign thyroid nodules.
In 2014, Nikiforov et al validated the performance of a new gene-mutation panel (ThyroSeq version 2) and a gene-fusion panel (ThyroSeq RNA) in a large series of thyroid nodules with follicular or oncocytic (Hürthle cell) neoplasms/suspicious for a follicular or oncocytic (Hürthle cell) neoplasm, demonstrating that it allowed accurate cancer-risk assessment in these nodules.18 In 2015, the same authors demonstrated the possibility to stratify patients with benign and malignant thyroid nodules diagnosed as atypia of undetermined significance/follicular lesion by cytology, with high sensitivity and specificity. The last custom panel developed by the authors (ThyroSeq version 2.1) included 14 genes analyzed for point mutations and 42 types of gene fusions occurring in TC.19
Recently, Simbolo et al investigated the diagnostic stratification of sporadic medullary TC by the use of the Ion AmpliSeq Hot Spot Cancer Panel version 2 (Life Technologies).20 Thirteen cases had a somatic RET mutation, and the authors showed that only ten were detected by both Sanger sequencing and NGS, while three were undetected by Sanger, revealing higher NGS sensitivity. In summary, these studies demonstrated that NGS offers the possibility of better classifying thyroid nodules. Moreover, this should improve patient management and allow clinicians to avoid diagnostic surgeries associated with significant costs and potential risks.
Lung cancer
Lung cancer (LC) is the leading cause of cancer-related death in developed countries, and is often diagnosed at an advanced stage. A comprehensive knowledge of predictive biomarkers has enabled the selection of LC patients for the use of tyrosine-kinase inhibitors (TKIs). In clinical practice, EGFR mutations must be evaluated to address patients for TKI treatment appropriately. Most (80%–90%) EGFR mutations are either small exon 19 deletions or the L858R mutation in exon 21, but other TKI-sensitive EGFR mutations can occur in exons 18–21. The mutation T790M in exon 20 needs to be investigated, because it is associated with first-generation TKI resistance but third-generation TKI sensitivity.21–24 Another marker of TKI resistance consists of ALK rearrangement. Indeed, to date, EGFR and ALK are the only actionable genes that have drugs approved by the FDA for LC treatment.
Formalin-fixed paraffin-embedded tissue is considered an optimal specimen for molecular analysis. The gold-standard technique to detect EGFR mutations for several years was Sanger sequencing, but recently other methods have been employed for molecular diagnostics (high resolution melting, restriction fragment-length polymorphism, mutant allele-specific PCR, peptide nucleic acid-mediated PCR, pyrosequencing, immunohistochemistry with specific EGFR antibodies, and the Scorpion Amplification Refractory Mutation System). Instead, to study ALK rearrangements, the gold standard is still immunohistochemistry or fluorescence in situ hybridization.
Several studies have indicated numerous changes due to the introduction of NGS into daily clinical practice for LC molecular diagnosis, reporting high sensitivity for detecting actionable alterations by the use of a gene panel on LC specimens.25–27 In fact, Lim et al recently reported that 58% of patients with wild type by standard testing for EGFR/KRAS/ALK showed alterations identified by NGS, thus giving these patients a therapeutic chance.28
However, tissue biopsies are not always available, because 60% of non-small-cell LCs (NSCLCs) are high-stage locally advanced and/or inoperable tumors that have already metastasized to distant sites when they are detected. The diagnosis of LC sometimes depends on metastatic lymph-node specimens obtained by FNA cytology. In these patients, cytology specimens are usually the only material available for histological typing and for molecular analysis. In these cases, the tumor-cell content may be very low, implying the need to use very sensitive methods. Scarpa et al demonstrated for the first time in 2013 the diagnostic relevance of the Ion AmpliSeq Colon and Lung Cancer Panel on lung adenocarcinoma cytological samples.29 The first version of this panel included 504 mutational hotspot regions in 22 cancer-related genes, and it was able to detect variants up to 1% of allelic frequency, which corresponds to 2% of cancer cells in a sample. An implementation of the Ion AmpliSeq Colon and Lung Cancer Panel was reported in a study in which seven different labs belonging to the OncoNetwork Consortium tested the NGS panel on the same samples.30 This final version of the panel was constituted of 1,825 selected mutational hotspots in 22 cancer-related genes.30 Recent studies have confirmed the sufficient and high quality of DNA from cytological LC samples for NGS molecular analysis.31,32
Neoplastic tissues remain the standard specimen for molecular analysis. However, the potential to obtain noninvasive sampling compared with tissue biopsy is very attractive. Blood collection is less invasive than tissue sampling, and can be used when tissue specimens are limited/not available or for critically ill patients. Moreover, it can allow for sampling at several time points to monitor the genetic evolution of the tumor and also to predict early treatment resistance or nonresponse.
Plasma DNA can also be used by NGS to detect cancer-related gene alterations useful in LC-treatment decisions, because plasma may reflect disease status compared with tumor biopsy.33–36 Moreover, during treatment, plasma analysis could reveal EGFR treatment-resistant mutations, indicating early clinical progression.37
In our lab, two NGS panels on Ion Torrent are in daily use in NSCLC patients: the Ion AmpliSeq Colon and Lung Cancer Panel version 2 and the Ion AmpliSeq RNA Fusion Lung Cancer Research Panel. We also participated in Thermo Fisher Scientific’s international validation program for the final version of this fusion panel. Routinely, NGS clinical analysis is performed on NSCLC formalin-fixed paraffin-embedded and cytological samples. A comparative study is ongoing on NGS application for tissue and plasma detection, obtaining encouraging results (manuscript in preparation). Moreover, the use of the Ion AmpliSeq Colon and Lung Cancer Panel is a fundamental step in our clinical analysis to characterize the EGFR deletion type, because specific in vitro diagnostic molecular tests on Rotor-Gene real-time PCR do not provide this information.
Colorectal cancer
EGFR, involved in cancer growth and survival, is targeted by several drugs in colorectal cancer (CRC) therapy. However, only a small subgroup of patients with metastatic CRC can benefit from anti-EGFR therapies (cetuximab or panitumumab), and thus prediction of patient responses is necessary to avoid side effects and to save costs. Ras proteins (HRas, KRas, and NRas) are important downstream effectors that transmit signals from EGFR to the intracellular signaling cascade. KRAS is considered a predictive biomarker for the efficacy of anti-EGFR therapy since KRAS-mutant CRC patients (codons 12 and 13 in exon 2) are resistant to treatment with EGFR inhibitors. However, approximately 40%–50% of patients harboring wild-type KRAS exon 2 do not benefit from these targeted agents, suggesting the potential involvement of other genetic alterations in pathways downstream of EGFR. In fact, a recent study suggested that additional mutations in KRAS and NRAS, as well as downstream mutations in BRAF or PIK3CA, may cause resistance to anti-EGFR treatment.38 Inter- and intratumoral genetic heterogeneity is another factor in predicting treatment failure and drug resistance in CRC therapies. The recently updated National Comprehensive Cancer Network guideline strongly recommends genotyping of tumor tissue (either primary tumor or metastasis) in all patients with metastatic CRC for RAS (exons 2–4 of KRAS and NRAS), and patients with any known KRAS or NRAS mutation should not be treated with cetuximab or panitumumab. To date, the gold standard for analysis of these genes is real-time PCR or pyrosequencing, methods that are time-consuming with low sensitivity.
In order to investigate CRC specimens with NGS in clinical practice, Tops et al developed a multigene panel, already used for LC investigation.30 This panel has also been employed by several other groups in CRC research, who have recommended it in clinics when compared to traditional methods.39–42 Another clinical application of NGS to CRC is represented by an interesting recent study in which a cutoff for mutational load can be identified via multigene tumor profiling to discriminate CRC patients in DNA-mismatch repair (MMR)-pathway proficiency and deficiency, since 15%–20% of CRC patients are deficient in one or more genes of MMR.43 This approach can be used for initial screening of Lynch syndrome. Moreover, the authors demonstrated the feasibility of analyzing MMR deficiency and RAS/BRAF mutations in CRC patients with the same panel, reducing time and costs of analysis.43
Lately, several custom gene panels have been developed with Illumina and Life Technologies to investigate many other crucial CRC genes.44–47 A multigene approach is in fact mandatory to obtain simultaneously a larger mutational spectrum, increasing the knowledge of CRC. Probably in the future, additional information emerging from these NGS studies will be useful for anti-EGFR therapy response duration or to develop other target therapies.
NGS and hematologic cancer
Hematological malignancies are grounded in genetic aberrations, in particular large mutations that are at the basis of the different phenotypes in the spectrum of hematologic cancers. NGS technologies have been applied to hematological disorders in a variety of contexts: guiding diagnosis (TCR gene rearrangement to establish T-cell clonality), subclassification (recurrent cytogenetic translocations in acute myeloid leukemia), prognosis (Philadelphia chromosome-positive in acute lymphoblastic leukemia), and minimal residual disease (MRD) testing (BCR–ABL transcripts in chronic myelogenous leukemia), often allowing the identification of novel mutations.48,49 The characterization of leukemias, lymphomas, and myelomas is continually evolving, and includes the precise identification of additional common mutations that may be of great prognostic value and clinical importance.
Multiple myeloma
Multiple myeloma (MM) is a malignancy of plasma cells. It is a multistep process, and an asymptomatic stage of monoclonal gammopathy of undetermined significance precedes virtually all cases of MM. This malignancy undergoes a multistep-transformation process. Its genetic landscape changes over time due to additional events, such as somatic mutations and epigenetic and chromosomal copy-number changes, driving its progression from monoclonal gammopathy of undetermined significance to symptomatic MM and ultimately to aggressive extramedullary disease in some patients.50
The first important event in plasma-cell transformation is represented by hyperdiploidy, observed in up to 55% of patients. The second is based on IGH translocations in 40%–50% of patients. Moreover, t(11;14) (dysregulation of the CCND1 gene, with its overexpression), t(4;14) (upregulation of FGFR3 and MMSET/WHSC1), and many other chromosomal rearrangements are present in the tumor plasma cells at the time of diagnosis. All these abnormalities have been known for a long time, because they are visible on the conventional karyotype. More recent data based on comparative genomic hybridization or SNP-array technologies have revealed other important chromosomal changes, especially homozygotic deletions.
With the development of NGS, the understanding of MM has been greatly improved in the past 5 years, confirming its wide heterogeneity at the molecular level, but also providing a clearer picture of the disease pathogenesis and progression. The quantitative nature of NGS data allows for higher resolution of the subclonal architecture of cancers. Nevertheless, initial reports of genomic evolution in MM using NGS were conducted on small cohorts, suggesting that MM shows a heterogeneous subclonal structure at diagnosis and only a few recurrent mutated genes of likely pathogenetic significance, including KRAS, NRAS, TP53, BRAF, and FAM46C.51,52
With NGS, Bolli et al confirmed subclonal KRAS, NRAS, and BRAF mutations in MM observed in about one-third of patients: acquisitions with crucial therapeutic implications in trials of Mek and BRaf inhibitors.53 Recently, Kortüm et al designed a 47-gene-targeting gene panel containing 39 genes known to be mutated in ≥3% of MM cases and eight genes in pathways therapeutically targeted in MM.53 Mutation analysis revealed KRAS as the most commonly mutated gene, followed by NRAS, TP53, DIS3, FAM46C, and SP140. They tracked clonal evolution and identified mutation acquisition and/or loss in FAM46C, FAT1, KRAS, NRAS, SPEN, PRDM1, NEB, and TP53, as well as two mutations in XBP1, a gene associated with bortezomib resistance.54
Lymphomas
In recent years, the development of NGS has also allowed the acquisition of important molecular information in a variety of lymphoid tumors, including Hodgkin’s lymphoma, diffuse large B-cell lymphoma, Burkitt’s lymphoma, chronic lymphocytic leukemia, follicular lymphoma, mantle-cell lymphoma, hairy-cell leukemia, and splenic marginal zone lymphoma. Although there have been many advances in this field, NGS panels are not yet available for clinical practice. The current modality to diagnose a hematological disease is based on fluorescence in situ hybridization, classic molecular biology, and radiographic studies. The latter in particular is associated with radiation exposure and limited specificity.
The new sequencing technologies, in addition to identifying somatic mutations involved in cancer progression (ie, mutations of BRAF, MYD88, and NOTCH2), have provided scientific evidence that might be useful for clinical treatment, as well as for the diagnosis and progression of these diseases.55 NGS aims to detect the tumor-specific clonotype and circulating tumor-specific sequence in the peripheral blood of patients with Hodgkin’s lymphoma. Quesada et al used this approach to identify lymphoma-specific immunoglobulin gene rearrangements in primary tumor samples at diagnosis or disease recurrence, as well as in follow-up. Moreover, the sequencing of B-cell lymphoma genomes has identified recurrent mutations, some of which have prognostic impact or serve as drug targets. Mutation of P53 predicts poor response to treatment and shortened overall survival across lymphoma entities, and mutations in NOTCH1 and SF3B1 have been shown to be independent predictors of poor outcome in chronic lymphocytic leukemia.56
Minimal residual disease
MRD is defined as the small number of cancer cells that persist in a patient during or after treatment, even though clinical and microscopic examinations confirm complete remission and the patient shows no signs or symptoms of disease. MRD detection and quantification are used for the evaluation of treatment efficiency, patient-risk stratification, and long-term outcome prediction in hematological malignancies.
Currently, flow cytometry is the most commonly used technique for the diagnosis and characterization of hematological malignancies and MRD. Although the method is widely used, a high level of expertise is required to interpret the data precisely when it comes to rare-event detection, such as MRD. The sensitivity for the detection of malignant cells varies according to the type of disorder, the panel of antibodies used, the number of cells analyzed, and the expertise of the laboratory. Furthermore, DNA and RNA tests usually lack the sensitivity required for MRD monitoring.
NGS approaches allow for searching not only for known mutations/translocations but also for all clonal gene mutations and rearrangements present in diagnostic samples to understand the possible evolution of MRD better. In a recent study, consensus primers and high-throughput sequencing were employed to amplify and sequence all rearranged IGH and TCR gene segments.57
Ladetto et al described a comparison between real-time quantitative PCR and LymphoSight NGS as methods for MRD detection using clonal IGH rearrangements.57 The primary results demonstrated that NGS enabled the detection of this molecular marker in a high proportion of cases, including a fraction in which standard PCR-based amplification failed. In addition, NGS showed a sensitivity comparable to that obtained by real-time quantitative PCR, allowing its use for detection of MRD.57
Unfortunately, NGS for this purpose is not yet routinely employed in clinical practice. NGS might overcome some disadvantages of PCR-based methods and avoid the need for patient-specific reagents. In addition, the NGS approach enables the analysis of genetic diversity and clonogenic heterogeneity which may contribute to our current understanding of disease biology and relapse kinetics.57,58 To date, only one CE (Conformité Européene)-marked in vitro diagnostic panel is commercially available: the LymphoTrack Dx assay (for Illumina MiSeq and Ion PGM) used for the identification of the DNA sequence, clonal prevalence, and V–J family identity for each gene rearrangement, as well as IGH assays, and the extent of IGHV somatic hypermutation.
Variant calling and copy-number variations
NGS provides large-scale data that continue to pose a major challenge.59 To call variants from NGS data, many aligners and variant callers have been developed and composed into diverse pipelines. A typical pipeline contains an aligner, which maps the sequencing reads to a reference genome, and a variant caller, which identifies variant sites and assigns a genotype to a subject. The performances of different aligners have been extensively studied, and great effort is still needed to correctly identify the best analysis pipeline.60,61
The Genome Analysis Toolkit (GATK; Broad Institute, Cambridge, MA, USA) is a powerful set of tools for NGS-data analysis. Recently, we focused on the optimization of GATK to call variants from data sets coming from an Ion Torrent targeted custom panel, including eleven genes involved in melanoma.13 In particular, the variant-filtration step was investigated. To this end, the variant quality-score recalibration (VQSR) step has been recently introduced. VQSR filtering uses annotation metrics (eg, quality by depth, mapping quality, strand bias) from a true variant, annotated in HapMap for instance, to generate an adaptive model. Such a model applied to the other variants allows calculation of the probability that a variant is true or false. Although this is a powerful method, it requires a large call set. Indeed, GATK’s best practices suggest not to apply VQSR in “small-scale experiments, such as targeted gene panels or exome studies with fewer than 30 exomes”. In these cases, hard filtering is the approach indicated by GATK. General rules are available, but appropriate filters have to be specifically set up for each study, considering also that GATK does not provide any technical documentation for Ion Torrent data. Therefore, starting from a comparison of results from GATK and proprietary Torrent Suite variant caller (TVC) analyses on the real data set, our aim was to determine a framework for GATK hard filtering in order to lower false-positive calls (Figure 2). We observed a high discrepancy between TVC and GATK, particularly for indels, suggesting that such type variants are difficult to detect with even the present bioinformatic tools. We then decided to simulate two data sets, each with a different coverage and each carrying alterations found in real data. Indeed, the importance of defining a “gold standard” data set to test variant calling methods is a very hot topic, and, recently “synthetic” matched tumor–normal samples have been created to compare performances of popular variant callers in the detection of “somatic” single nucleotide variants (SNVs).62 The first important result is that results are strictly correlated with coverage. We found that in a high-coverage data set, calling of SNVs led to a lower number of false positives than in a low-coverage data set. However, focusing on indels, the picture is more complex and the number of false-positive cases is high in both of the two data sets when looking at the variants suggested by GATK in the phase preceding the filtering of “good” variants. To be able to select opportune hard filters, we considered the most important parameters of quality indicated in the raw Variant Call Format file. We built up regression trees to be able to identify the best choice for hard filtering, in order to discriminate true and false calls better. We performed the analyses in SNV and indel subsets, both stratified by genotype, in high- and low-coverage data sets. Regression trees allowed us to set a series of filters for each type of alteration. Recently, Vanni et al used GATK to analyze sequencing data of the targeted AmpliSeq Colon and Lung Cancer Panel (Life Technologies).63 Methodologically, they filtered out variants with a Phred score of 5–30, marking them as low quality. Our results showed that such an approach might not be enough to have a high-quality GATK call set. In detail, we found that different parameters could be tuned depending on the type of mutations and genotypes suggested. The application of hard filtering was able to reduce the number of false positives. Sometimes, the loss of true variants could be high, in particular for indels, but it has to be noticed that the number of false variants was also high. Therefore, the application of hard filtering can help to drastically reduce such a high number of false positives, and we argue that increasing coverage should improve filtering results in terms of true variants not correctly discarded. We explored flanking regions of each type of alteration, in particular searching for recurrent homopolymeric strings, highlighting that they are partly responsible for false-positive calls. Hard filters were tested on a real independent cohort, which underwent sequencing by the same custom panel. We found almost 100% concordance regarding SNV calling (manuscript in preparation).
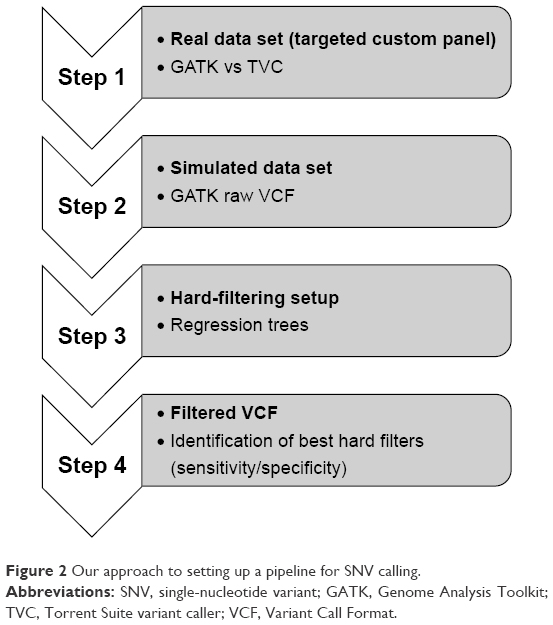 | Figure 2 Our approach to setting up a pipeline for SNV calling. |
Another NGS application is represented by CNV analysis. CNVs occur frequently during carcinogenesis, and thus the detection of these aberrations is essential for cancer-genome analysis to improve diagnosis and treatment. NGS-based CNV algorithms frequently manage WGS and WES data. A number of somatic CNV-detection programs for NGS data have been developed, each of them based on a different approach.64 However, with regard to targeted sequencing, the approach used in diagnostic settings, the bioinformatic challenge remains open. In essence, all pipelines for CNV detection in targeted-sequencing data work through the read-depth approach. In detail, they are based on the calculation of coverage of the amplicons and in the detection of outliers, subsequent to the opportune normalization step. Some algorithms require matched tumor–normal samples or a reference DNA, but recently an R package was introduced, Ioncopy, that does not need control samples.65–67 Different biases have to be considered in a read depth-based approach. PCR could lead to coverage distortion, because of nonuniform efficiency in amplification. An important issue deeply studied for CNV identification is guanine–cytosine bias, which affects read coverage. Moreover, another important bias regards the alignment step, because short reads might not be unambiguously mapped to the reference genome. In conclusion, even if a number of methods have been set up, validation is still needed in order to include them in a clinical setting.
Conclusion
Despite several critical points regarding mostly technology implementation and data interpretation, in this review, we have shown numerous benefits of an NGS approach (Figure 3). In fact, recent innovations in sequencing technologies, have allowed the obtainment of a wide spectrum of genomic alterations occurring within tumors.
 | Figure 3 Benefits obtained from the use of NGS methods in clinical molecular diagnostics. |
At present, the clinical utility and efficacy of comprehensive genomic profiling with the NGS are under evaluation, in order to introduce this technology in clinical guidelines for solid and hematologic cancer management. Initial results demonstrate that NGS might improve patient care, guiding them toward specific screening programs and targeted therapies with more accuracy and specificity than traditional sequencing methods, even if other many studies are needed.
Disclosure
The authors report no conflicts of interest in this work.
References
Li W, Zhao K, Kirberger M, Liao W, Yan Y. Next generation sequencing technologies in cancer diagnostics and therapeutics: a mini review. Cell Mol Biol (Noisy-le-grand). 2015;61(5):91–102. | ||
Quail MA, Smith M, Coupland P, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. | ||
Ross MG, Russ C, Costello M, et al. Characterizing and measuring bias in sequence data. Genome Biol. 2013;14(5):R51. | ||
D’Argenio V, Esposito MV, Telese A, et al. The molecular analysis of BRCA1 and BRCA2: next-generation sequencing supersedes conventional approaches. Clin Chim Acta. 2015;446:221–225. | ||
Tarabeux J, Zeitouni B, Moncoutier V, et al. Streamlined ion torrent PGM-based diagnostics: BRCA1 and BRCA2 genes as a model. Eur J Hum Genet. 2014;22(4):535–541. | ||
Pilato B, Pinto R, De Summa S, et al. BRCA1-2 diagnostic workflow from next-generation sequencing technologies to variant identification and final report. Genes Chromosomes Cancer. 2016;55(10):803–813. | ||
Tung N, Battelli C, Allen B, et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2015;121(1):25–33. | ||
Lin PH, Kuo WH, Huang AC, et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget. 2016;7(7):8310–8320. | ||
Lhota F, Zemankova P, Kleiblova P, et al. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2-negatively tested breast cancer patients. Clin Genet. 2016;90(4):324–333. | ||
Strom CM, Rivera S, Elzinga C, et al. Development and validation of a next-generation sequencing assay for BRCA1 and BRCA2 variants for the clinical laboratory. PLoS One. 2015;10(8):e0136419. | ||
Ihle MA, Fassunke J, König K, et al. Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer. 2014;14:13. | ||
van Engen-van Grunsven AC, Kusters-Vandevelde H, Groenen PJ, Blokx WA. Update on molecular pathology of cutaneous melanocytic lesions: what is new in diagnosis and molecular testing for treatment? Front Med (Lausanne). 2014;1:39. | ||
Pinto R, De Summa S, Strippoli S, et al. The next generation of metastatic melanoma: uncovering the genetic variants for anti-BRAF therapy response. Oncotarget. 2016;7(18):25135–25149. | ||
Chen J, Zhang D, Yan W, Yang D, Shen B. Translational bioinformatics for diagnostic and prognostic prediction of prostate cancer in the next-generation sequencing era. Biomed Res Int. 2013;2013:901578. | ||
Manson-Bahr D, Ball R, Gundem G, et al. Mutation detection in formalin-fixed prostate cancer biopsies taken at the time of diagnosis using next-generation DNA sequencing. J Clin Pathol. 2015;68(3):212–217. | ||
Iacono ML, Buttigliero C, Monica V, et al. Retrospective study testing next generation sequencing of selected cancer-associated genes in resected prostate cancer. Oncotarget. 2016;7(12):14394–14404. | ||
Nikiforova MN, Wald AI, Roy S, Durso MB, Nikiforov YE. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J Clin Endocrinol Metab. 2013;98(11):E1852–E1860. | ||
Nikiforov YE, Carty SE, Chiosea SI, et al. Highly accurate diagnosis of cancer in thyroid nodules with follicular neoplasm/suspicious for a follicular neoplasm cytology by ThyroSeq v2 next-generation sequencing assay. Cancer. 2014;120(23):3627–3634. | ||
Nikiforov YE, Carty SE, Chiosea SI, et al. Impact of the multi-gene ThyroSeq next-generation sequencing assay on cancer diagnosis in thyroid nodules with atypia of undetermined significance/follicular lesion of undetermined significance cytology. Thyroid. 2015;25(11):1217–1223. | ||
Simbolo M, Mian C, Barollo S, et al. High-throughput mutation profiling improves diagnostic stratification of sporadic medullary thyroid carcinomas. Virchows Arch. 2014;465(1):73–78. | ||
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | ||
Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. | ||
Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3(12):1404–1415. | ||
Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. | ||
Masago K, Fujita S, Muraki M, et al. Next-generation sequencing of tyrosine kinase inhibitor-resistant non-small-cell lung cancers in patients harboring epidermal growth factor-activating mutations. BMC Cancer. 2015;15:908. | ||
Rangachari D, VanderLaan PA, Le X, et al. Experience with targeted next generation sequencing for the care of lung cancer: insights into promises and limitations of genomic oncology in day-to-day practice. Cancer Treat Commun. 2015;4:174–181. | ||
Shao D, Lin Y, Liu J, et al. A targeted next-generation sequencing method for identifying clinically relevant mutation profiles in lung adenocarcinoma. Sci Rep. 2016;6:22338. | ||
Lim SM, Kim EY, Kim HR, et al. Genomic profiling of lung adenocarcinoma patients reveals therapeutic targets and confers clinical benefit when standard molecular testing is negative. Oncotarget. 2016;7(17):24172–24178. | ||
Scarpa A, Sikora K, Fassan M, et al. Molecular typing of lung adenocarcinoma on cytological samples using a multigene next generation sequencing panel. PLoS One. 2013;8(11):e80478. | ||
Tops BB, Normanno N, Kurth H, et al. Development of a semi-conductor sequencing-based panel for genotyping of colon and lung cancer by the Onconetwork consortium. BMC Cancer. 2015;15:26. | ||
Qiu T, Guo H, Zhao H, Wang L, Zhang Z. Next-generation sequencing for molecular diagnosis of lung adenocarcinoma specimens obtained by fine needle aspiration cytology. Sci Rep. 2015;5:11317. | ||
Treece AL, Montgomery ND, Patel NM, et al. FNA smears as a potential source of DNA for targeted next-generation sequencing of lung adenocarcinomas. Cancer Cytopathol. 2016;124(6):406–414. | ||
Uchida J, Kato K, Kukita Y, et al. Diagnostic accuracy of noninvasive genotyping of EGFR in lung cancer patients by deep sequencing of plasma cell-free DNA. Clin Chem. 2015;61(9):1191–1196. | ||
Schwaederle M, Husain H, Fanta PT, et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget. 2016;7(9):9707–9717. | ||
Que D, Xiao H, Zhao B, Zhang X, Wang Q, Wang G. EGFR mutation status in plasma and tumor tissues in non-small cell lung cancer serves as a predictor of response to EGFR-TKI treatment. Cancer Biol Ther. 2016;17(3):320–327. | ||
Xu S, Lou F, Wu Y, et al. Circulating tumor DNA identified by targeted sequencing in advanced-stage non-small cell lung cancer patients. Cancer Lett. 2016;370(2):324–331. | ||
Marchetti A, Palma JF, Felicioni L, et al. Early prediction of response to tyrosine kinase inhibitors by quantification of EGFR mutations in plasma of NSCLC patients. J Thorac Oncol. 2015;10(10):1437–1443. | ||
Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol. 2014;53(7):852–864. | ||
Gao J, Wu H, Wang L, et al. Validation of targeted next-generation sequencing for RAS mutation detection in FFPE colorectal cancer tissues: comparison with Sanger sequencing and ARMS-Scorpion real-time PCR. BMJ Open. 2016;6(1):e009532. | ||
Belardinilli F, Capalbo C, Buffone A, et al. Validation of the Ion Torrent PGM sequencing for the prospective routine molecular diagnostic of colorectal cancer. Clin Biochem. 2015;48(13–14):908–910. | ||
D’Haene N, Le Mercier M, De Neve N, et al. Clinical validation of targeted next generation sequencing for colon and lung cancers. PLoS One. 2015;10(9):e0138245. | ||
Haley L, Tseng LH, Zheng G, et al. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. Mod Pathol. 2015;28(10):1390–1399. | ||
Stadler ZK, Battaglin F, Middha S, et al. Reliable detection of mismatch repair deficiency in colorectal cancers using mutational load in next-generation sequencing panels. J Clin Oncol. 2016;34(18):2141–2147. | ||
Hsu HC, Thiam TK, Lu YJ, et al. Mutations of KRAS/NRAS/BRAF predict cetuximab resistance in metastatic colorectal cancer patients. Oncotarget. 2016;7(16):22257–22270. | ||
Malapelle U, Pisapia P, Sgariglia R, et al. Less frequently mutated genes in colorectal cancer: evidences from next-generation sequencing of 653 routine cases. J Clin Pathol. 2016;69(9):767–771. | ||
Jesinghaus M, Pfarr N, Endris V, et al. Genotyping of colorectal cancer for cancer precision medicine: results from the IPH Center for Molecular Pathology. Genes Chromosomes Cancer. 2016;55(6):505–521. | ||
Wang SR, Malik S, Tan IB, et al. Technical validation of a next-generation sequencing assay for detecting actionable mutations in patients with gastrointestinal cancer. J Mol Diagn. 2016;18(3):416–424. | ||
Hussaini M. Biomarkers in hematological malignancies: a review of molecular testing in hematopathology. Cancer Control. 2015;22(2):158–166. | ||
Black JS, Salto-Tellez M, Mills KI, Catherwood MA. The impact of next generation sequencing technologies on haematological research: a review. Pathogenesis. 2015;2(1–2):9–16. | ||
Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335–348. | ||
Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467–472. | ||
Walker BA, Wardell CP, Melchor L, et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia. 2014;28(2):384–390. | ||
Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. | ||
Kortüm KM, Langer C, Monge J, et al. Longitudinal analysis of 25 sequential sample-pairs using a custom multiple myeloma mutation sequencing panel (M3P). Ann Hematol. 2015;94(7):1205–1211. | ||
Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute: Working Group 1996 guidelines. Blood. 2008;111(12):5446–5456. | ||
Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44(1):47–52. | ||
Faham M, Zheng J, Moorhead M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120(26):5173–5180. | ||
Ladetto M, Bruggemann M, Monitillo L, et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia. 2014;28(6):1299–1307. | ||
Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12(6):443–451. | ||
Costa JL, Sousa S, Justino A, et al. Nonoptical massive parallel DNA sequencing of BRCA1 and BRCA2 genes in a diagnostic setting. Hum Mutat. 2013;34(4):629–635. | ||
Rothberg JM, Hinz W, Rearick TM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475(7356):348–352. | ||
Zook JM, Chapman B, Wang J, et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol. 2014;32(3):246–251. | ||
Vanni I, Coco S, Truini A, et al. Next-generation sequencing workflow for NSCLC critical samples using a targeted sequencing approach by Ion Torrent PGM platform. Int J Mol Sci. 2015;16(12):28765–28782. | ||
Liu B, Morrison CD, Johnson CS, et al. Computational methods for detecting copy number variations in cancer genome using next generation sequencing: principles and challenges. Oncotarget. 2013;4(11):1868–1881. | ||
Hoogstraat M, Hinrichs JW, Besselink NJ, et al. Simultaneous detection of clinically relevant mutations and amplifications for routine cancer pathology. J Mol Diagn. 2015;17(1):10–18. | ||
Grasso C, Butler T, Rhodes K, et al. Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. J Mol Diagn. 2015;17(1):53–63. | ||
Budczies J, Pfarr N, Stenzinger A, et al. Ioncopy: a novel method for calling copy number alterations in amplicon sequencing data including significance assessment. Oncotarget. 2016;7(11):13236–13247. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.