")
Back to Journals » OncoTargets and Therapy » Volume 9
Next-generation EGFR/HER tyrosine kinase inhibitors for the treatment of patients with non-small-cell lung cancer harboring EGFR mutations: a review of the evidence
Authors Wang X, Goldstein D, Crowe P, Yang J
Received 17 May 2016
Accepted for publication 12 August 2016
Published 6 September 2016 Volume 2016:9 Pages 5461—5473
DOI https://doi.org/10.2147/OTT.S94745
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Xiaochun Wang,1,2 David Goldstein,3 Philip J Crowe,1,2 Jia-Lin Yang1,2
1Department of Surgery, 2Sarcoma and Nanooncology Group, Adult Cancer Program, Lowy Cancer Research Centre, 3Department of Medical Oncology, Prince of Wales Clinical School, Faculty of Medicine, University of New South Wales, Sydney, NSW, Australia
Abstract: Tyrosine kinase inhibitors (TKIs) against human epidermal growth factor receptor (EGFR/HER) family have been introduced into the clinic to treat cancers, particularly non-small-cell lung cancer (NSCLC). There have been three generations of the EGFR/HER-TKIs. First-generation EGFR/HER-TKIs, binding competitively and reversibly to the ATP-binding site of the EGFR TK domain, show a significant breakthrough treatment in selected NSCLC patients with activating EGFR mutations (actEGFRm) EGFRL858R and EGFRDel19, in terms of safety, efficacy, and quality of life. However, all those responders inevitably develop acquired resistance within 12 months, because of the EGFRT790M mutation, which prevents TKI binding to ATP-pocket of EGFR by steric hindrance. The second-generation EGFR/HER-TKIs were developed to prolong and maintain more potent response as well as overcome the resistance to the first-generation EGFR/HER-TKIs. They are different from the first-generation EGFR/HER-TKIs by covalently binding to the ATP-binding site, irreversibly blocking enzymatic activation, and targeting EGFR/HER family members, including EGFR, HER2, and HER4. Preclinically, these compounds inhibit the enzymatic activation for actEGFRm, EGFRT790M, and wtEGFR. The second-generation EGFR/HER-TKIs improve overall survival in cancer patients with actEGFRm in a modest way. However, they are not clinically active in overcoming EGFRT790M resistance, mainly because of dose-limiting toxicity due to simultaneous inhibition against wtEGFR. The third-generation EGFR/HER-TKIs selectively and irreversibly target EGFRT790M and actEGFRm while sparing wtEGFR. They yield promising efficacy in NSCLC patients with actEGFRm as well as EGFRT790M resistant to the first- and second-generation EGFR-TKIs. They also appear to have a lower incidence of toxicity due to the reduced inhibitory effect on wtEGFR. Currently, the first-generation EGFR/HER-TKIs gefitinib and erlotinib and second-generation EGFR/HER-TKI afatinib have been approved for use as the first-line treatment of metastatic NSCLC with actEGFRm. This review will summarize and evaluate a broad range of evidence of recent development of EGFR/HER-TKIs, with a focus on the second- and third-generation EGFR/HER-TKIs, in the treatment of patients with NSCLC harboring EGFR mutations.
Keywords: EGFR/HER, tyrosine kinase inhibitors, NSCLC, EGFR mutations, acquired resistance
Introduction
Lung cancer is currently the leading cause of cancer-related mortality worldwide, causing more than one-quarter of all cancer deaths (28% in males and 26% in females).1 As of 2016, it is estimated that 224,390 new cases of lung cancer will be diagnosed in the US and 158,080 deaths will be caused from lung cancer.2 Non-small-cell lung cancer (NSCLC), accounting for a high proportion (85%–90%) in lung cancer,3 is subdivided histologically into adenocarcinoma, squamous-cell carcinoma, large-cell carcinoma, and other types.4,5 In the last decade, the diagnosis and treatment of NSCLC has evolved dramatically from the traditional “one-size-fits-all” chemotherapeutic approach to new anticancer compounds molecularly targeting oncogenic driver mutations, due to the advances in cancer biology and technology. Various driver genomic alterations have been identified in oncogene-dependent NSCLC, especially two genes: the human epidermal growth factor receptor (EGFR/HER) and the anaplastic lymphoma kinase (ALK).6
The EGFR/HER family of receptor tyrosine kinases (TKs) has four members including EGFR (HER1, erbB-1), HER2 (erbB-2), HER3 (erbB-3), and HER4 (erbB-4), and their signaling pathways regulate cell growth, survival, adhesion, migration, and differentiation through three downstream pathways: RAS/RAF/mitogen-activated protein kinase, phosphoinositide 3-kinase/AKT, and Janus kinase/signal transducer and activator of transcription (JAK/STAT).7,8 Dysregulated signaling of HER family has been associated with the development of several malignancies including NSCLC.9 Many patients with NSCLC have somatic mutations of EGFR, first identified in 2004,10,11 which lead to aberrant constitutive signaling via EGFR/HER family and their downstream protein markers. The EGFR mutations, including activating and resistant mutations, mostly occur in exons 18 to 21 of the EGFR gene encoding the ATP-binding pocket of the intracellular TK domain. The activating EGFR mutations (actEGFRm) have been reported in ~10%–15% of Caucasian patients but in up to 60% of selected Asian populations with NSCLC (female, never/light smoker, and adenocarcinoma).12–14 The most frequent actEGFRm in NSCLC are in-frame deletions in exon 19 (EGFRDel19, ≈60%) and L858R point mutation in exon 21 (EGFRL858R, ≈30%).12,15,16 These oncogenic mutations interact and generate stabilization with ATP, intrinsically stimulate phosphorylation of tyrosine residues, and then result in the intracellular signal transduction activation in a ligand-independent manner.17,18 The NSCLC patients with actEGFRm become apparently dependent on EGFR activity to stimulate downstream signaling pathways to maintain the malignant phenotype (“oncogene addiction”).19,20 Therefore, blocking EGFR/HER family pathways with EGFR/HER TK inhibitors (TKIs) can suppress tumor cell proliferation and initiate apoptosis.
In the past decade, the EGFR/HER family has become a potential therapeutic target and has greatly changed the treatment paradigm for NSCLC patients, since the introduction of the first-generation EGFR/HER-TKIs gefitinib and erlotinib. Currently, these two agents and a second-generation EGFR/HER-TKI afatinib are approved for use in the first-line treatment of metastatic NSCLC with actEGFRm (EGFRDel19 and EGFRL858R), based on the outcomes of several clinical trials that demonstrate that these TKIs are superior to standard chemotherapy in terms of safety, efficacy, and quality of life.21–23 However, despite a good initial response, the development of acquired resistance in most of the patients limits the long-term efficacy of TKI therapy. Therefore, extensive investigations on better understanding of the mechanisms of resistance are being undertaken in order to robust the benefit of EGFR/HER-TKIs in NSCLC. New generations of EGFR/HER-TKIs have been developed to improve cancer treatment efficacy, overcome resistance, and reduce side effects. These EGFR/HER-TKIs are listed in Table 1. In this review, we will provide a broad overview of recent development of EGFR/HER-TKIs, with a focus on second- and third-generation EGFR/HER-TKIs, in the treatment of patients with NSCLC harboring EGFR mutations.
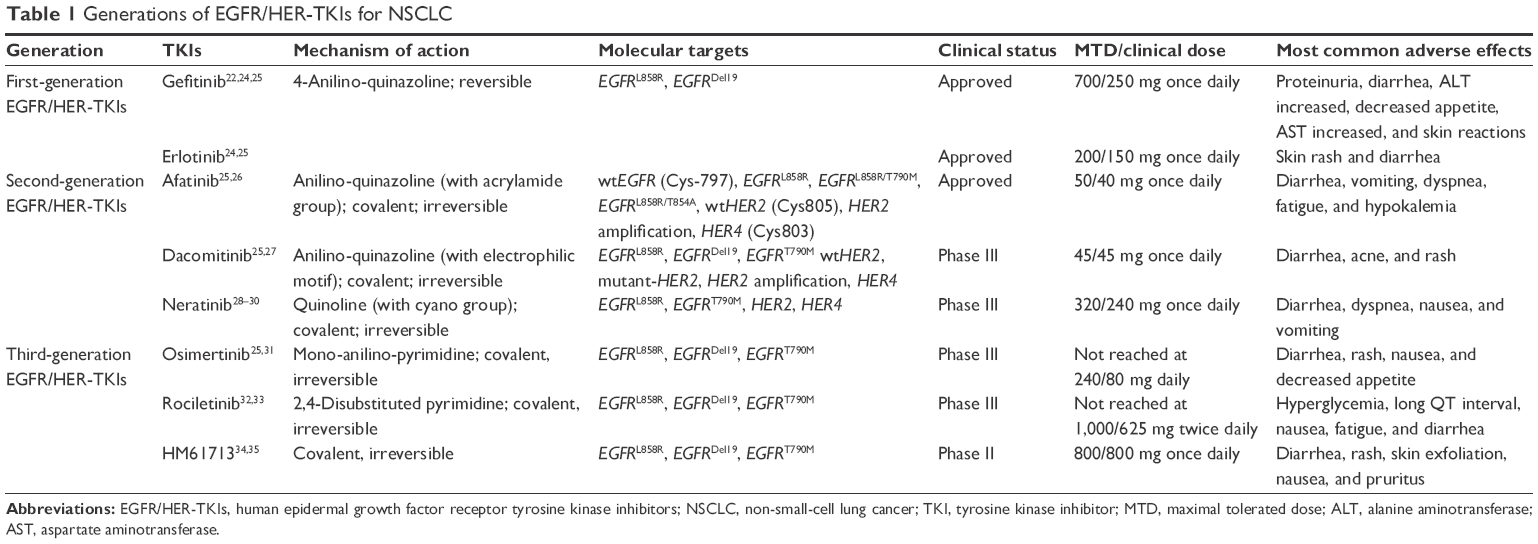 | Table 1 Generations of EGFR/HER-TKIs for NSCLC |
First-generation EGFR/HER-TKIs
Gefitinib (AstraZeneca plc, London, UK) and erlotinib (Astellas Pharma Inc., Tokyo, Japan) are the first-generation EGFR/HER-TKIs approved to use in the first-line setting for the treatment of advanced NSCLC patients with actEGFRm (EGFRDel19 and EGFRL858R).36,37 Both compounds (Figure 1) are orally active 4-anilino-quinzolines with antineoplastic activity and bind competitively and reversibly to the ATP-binding site of the TK domain of EGFR. This conformation of EGFR in this scenario prevents the autophosphorylation of the TK, blocks the activation of the EGFR signal transduction, inhibits tumor cell proliferation, and induces cell cycle arrest and apoptosis.38 Among the diverse (activating and resistant) mutations clustering around the catalytic cleft of EGFR TK domain, it has been demonstrated that the actEGFRm leads to increased affinity for EGFR/HER-TKIs, thus conferring more sensitivity to this treatment.13 Indeed, actEGFRm has been reported to bind 20-fold more tightly to TKIs than to the wild-type EGFR (wtEGFR).39
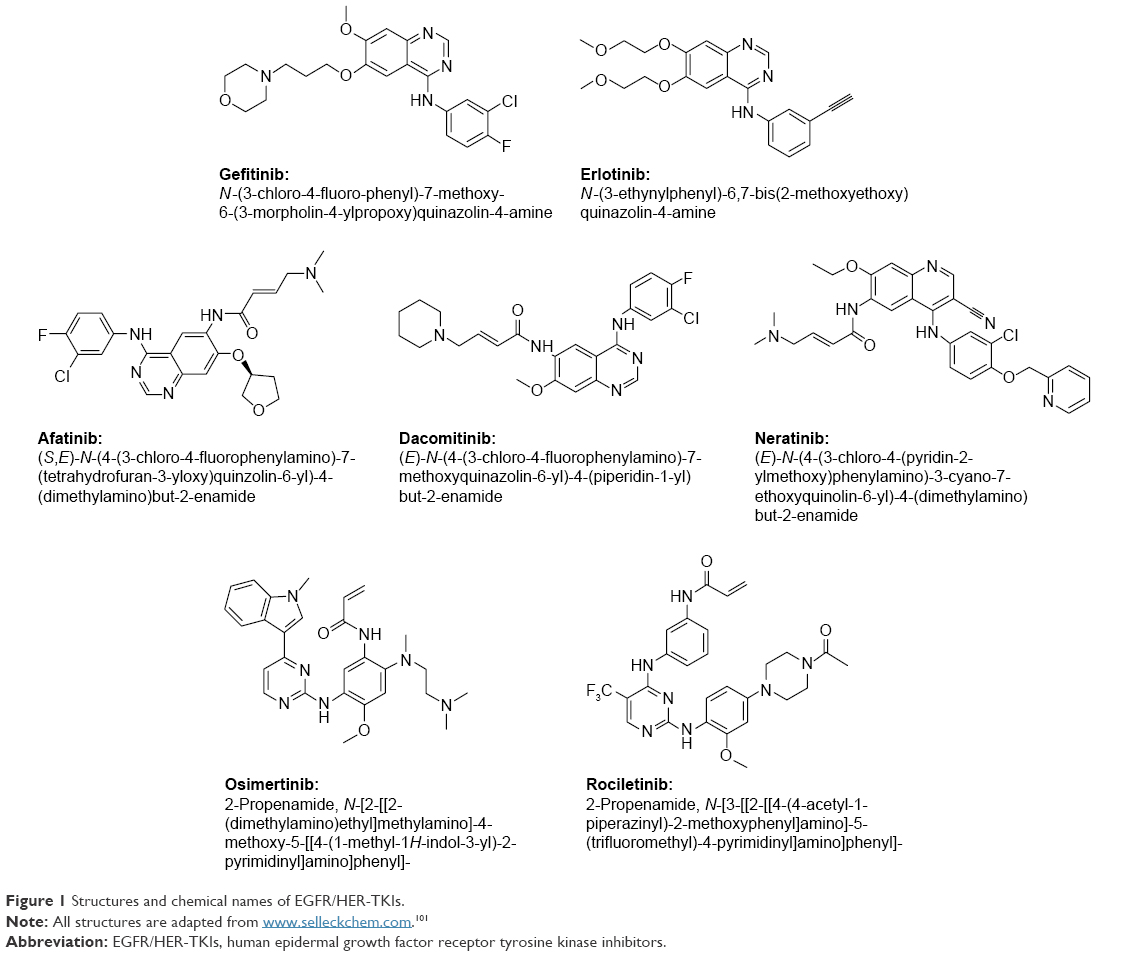 | Figure 1 Structures and chemical names of EGFR/HER-TKIs. |
Clinically, lung cancer patients with EGFRDel19 and EGFRL858R show a striking response to gefitinib and erlotinib treatment. Retrospective analysis of associations between EGFR gene mutations and EGFR/HER-TKIs sensitivity has shown that 70% of actEGFRm NSCLC patients are responsive to TKIs compared with 10% of wtEGFR patients.7,40 In the preselected subgroup of NSCLC patients with EGFRDel19 and EGFRL858R, the first-generation reversible EGFR/HER-TKIs gefitinib and erlotinib as a first-line treatment can dramatically affect patient outcomes, showing superiority to traditional platinum-based chemotherapy in terms of objective response rate (ORR), progression-free survival (PFS), and quality of life, and an acceptable toxicity profile.41–44 These studies resulted in their approval and widespread use for actEGFRm NSCLC patients.
On the other hand, resistant EGFR mutations (resEGFRm) either as primary or as secondary (acquired) events have also been reported, the most common being L747S and D761Y in exon 19, T790M and insertions in exon 20, and T854A in exon 21.17,45 The primary resistance (initially refractory to EGFR/HER-TKIs treatment) is seen in ~30% of NSCLC patients with actEGFRm, involving coexistent genetic alterations: resEGFRm, KRAS mutations, PTEN losses, PIK3CA mutations, BIM deletion, and 60% unknown factors.17,46–48 Additionally, although EGFR/HER-TKIs have great initial efficacy in 70% of patients with actEGFRm NSCLC, all those responders will inevitably develop acquired resistance (disease progression) within 1 year or 2 years.47 Approximately 50%–60% of patients with acquired resistance develop a secondary mutation in EGFR, most commonly the substitution of threonine at the “gatekeeper” amino acid 790 to methionine (T790M) occurring within exon 20, causing a bulky methionine side chain in TK domain.49,50 The EGFRT790M mutation results in the receptor becoming refractory to these reversible EGFR/HER-TKIs through a steric hindrance that prevents drugs binding to ATP-pocket and results in restored affinity to ATP.25,49,51,52 Preclinical modeling and analysis of tumor tissues obtained from patients after disease progression has also identified other less frequent mechanisms of acquired resistance, including bypass or alternative pathways (HER2 amplification, MET amplification, PIK3CA mutation, BRAF mutation, NF1 loss, and potentially FGFR signaling), histological/phenotypic transformation (small-cell lung cancer transformation or epithelial-to-mesenchymal transition), and unknown in 20%–30%.4,17,25,41,53–55 Understanding the biological basis responsible for the acquired resistance has therapeutic implications, and several strategies are currently under investigation. Based on the aforementioned mechanisms, several combinations with other therapies targeting bypass or alternative activating pathways have been explored in preclinical models or clinical trials. The potential candidate partners include MET-TKI tivanitinib,17 anti-MET antibody onartuzumab,56 MET/VEGFR-TKI TAS-115,57 anti-VEGF antibody bevacizumab,58,59 and STAT3 inhibitor S3I-201.60 Given the modest and nonoverlapping toxicities observed with EGFR/HER-TKIs compared with chemotherapy, a single Phase III trial demonstrated a significant improvement of intercalated chemotherapy and erlotinib in patients with advanced NSCLC harboring actEGFRm;61 however, four large Phase III studies failed to show superiority of combination treatment to standard platinum doublet chemotherapy in unselected chemotherapy-naive advanced NSCLC patients, making the value of this approach uncertain.62
Considering that EGFRT790M mutation represents the most frequent acquired resistance mechanism, targeting this mutation by irreversible next-generation (second and third generations) EGFR/HER-TKIs, therefore, represents an attractive strategy to overcome treatment resistance to first-generation EGFR/HER-TKIs.
Second-generation EGFR/HER-TKIs
The poor survival rate for NSCLC with coexpression of EGFR and HER263 suggests that HER family members should be simultaneously targeted for treatment. Aiming for a prolonged and more potent response and overcoming the resistance to first-generation EGFR/HER inhibitors, the second-generation EGFR/HER-TKIs,26 including afatinib (BIBW2992; Boehringer Ingelheim, Ingelheim am Rhein, Germany), dacomitinib (PF299804; Pfizer, Inc., New York, NY, USA), and neratinib (HKI272; Puma Biotechnology, Los Angeles, CA, USA), are designed to covalently bind to the ATP-binding site and irreversibly block enzymatic activation of EGFR and other HER family members including HER2 and HER4 (Table 1 and Figure 1). The second-generation irreversible EGFR/HER-TKIs were developed in part to inhibit the EGFRT790M mutation, in addition to the common actEGFRm.
Afatinib
In addition to gefitinib and erlotinib, a second-generation EGFR/HER-TKI afatinib is approved as a first-line treatment for the advanced NSCLC harboring actEGFRm in the US, Europe, Taiwan, Japan, and other countries.23 Afatinib is an ATP-competitive aniline-quinazoline derivate containing a reactive acrylamide group, covalently binding to EGFR, HER2, and HER4 and irreversibly inhibiting HER-family phosphorylation and signal transduction.26 The theoretical advantages and promising preclinical data indicated that afatinib irreversibly inhibited the enzymatic activation of wtEGFR, EGFRL858R, EGFRL858R/T790M, EGFRL858R/T854A, wtHER2, HER2 amplification, and wtHER4, as well as showed antitumor activities in both EGFR/HER-TKI-naive and resistant tumor cells and xenograft models.7,20
As a first-line treatment for advanced EGFR-mutated NSCLC, the Lux-Lung 2 single-arm Phase II trial64 showed that patients with common activating mutations (EGFRDel19 and EGFRL858R) were most sensitive to afatinib monotherapy with an ORR of 66%, median overall survival (OS) of 24 months, and median PFS of 14 months (Table 2), as well as indicated 40 mg/d was preferable for subsequent studies. Two larger Phase III clinical trials (Lux-Lung 3 and 6),65–67 comparing the second-generation TKI with the standard platinum-based chemotherapy, demonstrated that the first-line afatinib monotherapy for advanced actEGFRm lung adenocarcinoma had significantly prolonged median PFS and higher ORR as compared with up to six cycles of standard-of-care platinum-based (pemetrexed plus cisplatin or gemcitabine plus cisplatin) chemotherapy and improved quality of life (Table 2). The pooled analysis68,69 of both trials mentioned previously confirmed that afatinib used as the first-line treatment improved OS compared with chemotherapy for patients with EGFRDel19 (regardless of race/ethnicity), but no difference in unselective EGFR mutations and even in EGFRL858R subgroup. The improved OS has never been observed in the first-generation EGFR-TKIs, probably due to methodology differences in trial design. In April 2016, Lux-Lung 7 head-to-head international study,70 comparing the first- versus second-generation EGFR/HER-TKIs, reported that irreversible afatinib is superior to reversible gefitinib in treatment-naive patients with actEGFRm NSCLC in terms of efficacy and safety (Table 2). The results showed that afatinib achieved significant but clinically minor improved outcomes but did so with an improved safety profile.
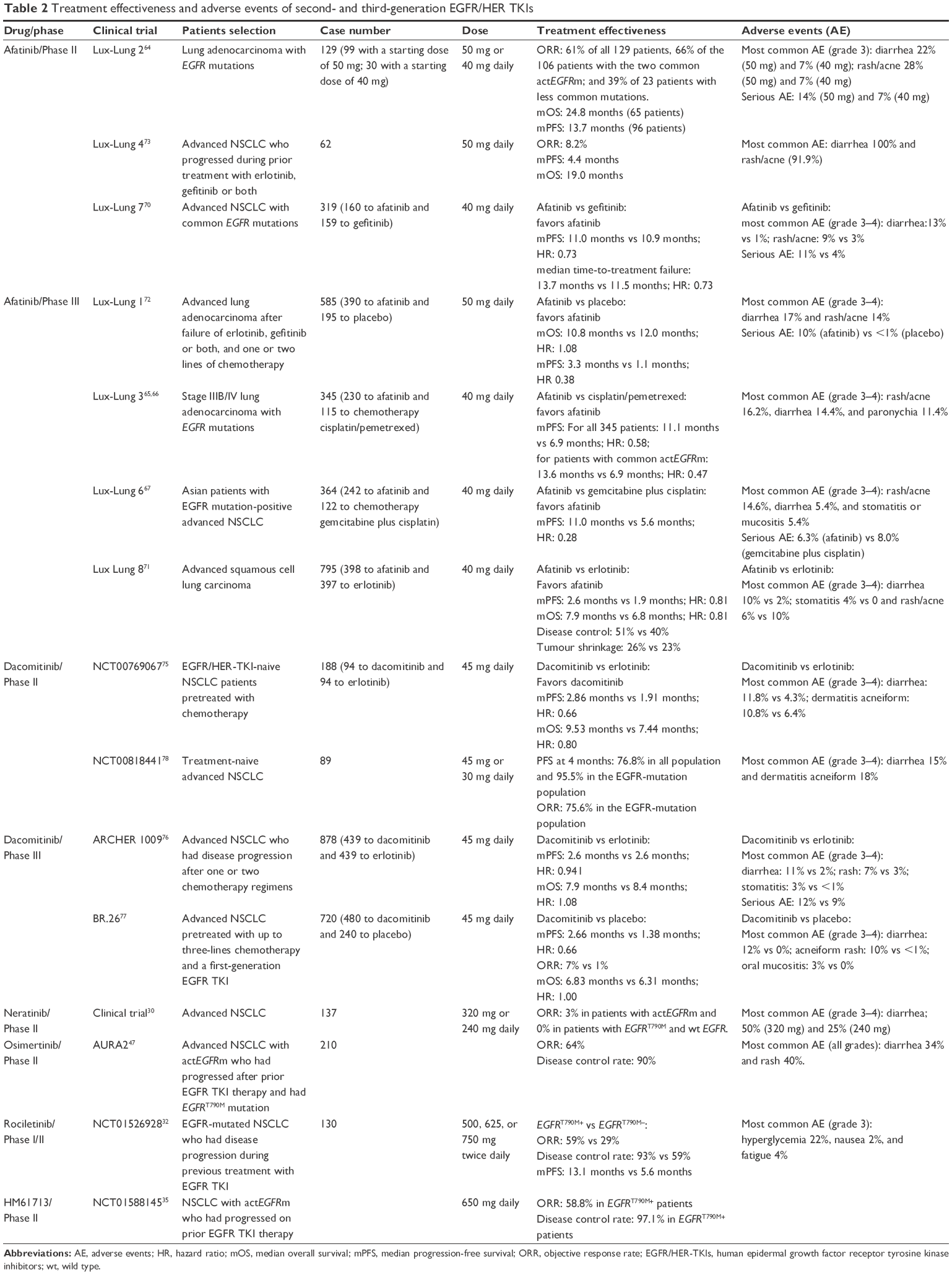 | Table 2 Treatment effectiveness and adverse events of second- and third-generation EGFR/HER TKIs |
Similarly, as a second-line treatment in patients with advanced squamous NSCLC who progressed on platinum-based chemotherapy, Lux-Lung 871 demonstrated that afatinib improved the PFS and OS in a modest way compared with erlotinib, but again with an improved safety profile (Table 2). Exploring the efficacy of afatinib as salvage therapy for advanced NSCLC patients, who were pretreated with one or two chemotherapy regimens and acquired resistance to first-generation EGFR/HER-TKIs, both single-arm Lux-lung 4 and two-arm Lux-lung 1 (Table 2) were carried out. The single-arm Lux-lung 4 showed an ORR of 8.2% and median PFS of 4.4 months in patients treated with afatinib at 50 mg/d, and the two-arm Lux-lung 1 observed a longer PFS and higher ORR along with improvements in lung cancer-related symptoms for patients treated with afatinib compared with placebo, despite failure to show the different median OS (primary endpoint).72,73 Both studies suggested that afatinib could overcome acquired resistance to gefitinib/erlotinib in some cases.
Dacomitinib
Dacomitinib (Figure 1) is an irreversible EGFR/HER-TKI against EGFR, HER2, and HER4 homodimers and heterodimers with a higher kinase inhibition than gefitinib/erlotinib, including actEGFRm (EGFRL858R and EGFRDel19), resEGFRm (EGFRT790M), wtHER2, HER2 amplification, and first-generation TKI-resistant HER2 mutations.27 Dacomitinib demonstrated activity in both gefitinib-sensitive and gefitinib-resistant NSCLC preclinical models.74
In relapsed/refractory setting, a Phase II study (NCT00769067) compared dacomitinib versus erlotinib as second- or third-line treatment in the EGFR/HER-TKI-naive NSCLC patients pretreated with chemotherapy.75 Significant results favored a small superiority for dacomitinib over erlotinib in PFS (Table 2). However, two recent Phase III studies with dacomitinib in this setting failed to achieve their primary objectives (Table 2). This demonstrates the critical importance of well-powered randomized trials to establish the true impact of new therapies. The ARCHER 1009 study, which compared dacomitinib with erlotinib as salvage therapy in patients with advanced NSCLC who had disease progression after one or two chemotherapy regimens did not show superiority for dacomitinib to erlotinib in an unselected population or in patients with wtKRAS.76 Another trial (BR.26), assessing dacomitinib versus placebo in patients pretreated with up to three-lines chemotherapy and a first-generation reversible EGFR/HER-TKI, showed similar OS regardless of EGFR mutation status, although improved OS was observed in wtKRAS and PFS that was significantly longer in dacomitinib group.77
In first-line setting, a Phase II study (NCT00818441) of dacomitinib in treatment-naive advanced NSCLC found an ORR of 73% and median PFS of 18.2 months in patients with actEGFRm (EGFRDel19 or EGFRL858R; Table 2).78 Based on these data, an ongoing randomized Phase III study (ARCHER 1050) has been designed to evaluate the efficacy of first-line dacomitinib versus gefitinib in locally advanced or metastatic NSCLC with actEGFRm.74 The results (estimated study completion in 2017) may demonstrate a more substantial role for dacomitinib in the earlier setting.
Neratinib
Neratinib (Figure 1) is an irreversible EGFR/HER-TKI targeting EGFR, HER2, and HER4, which was found to be more effective at suppressing cell proliferation than gefitinib in lung cancer cells with both actEGFRm and EGFRT790M in preclinical studies.28,79 However, due to the limitations of clinical dosing, neratinib had modest clinical efficacy in a Phase II study for TKI-resistant and TKI-naive patients, showing only 3% ORR in mutated-EGFR arm and no response in patients with EGFRT790M or wtEGFR (Table 2).30
In summary for the second-generation EGFR/HER-TKIs, in addition to inhibiting the two most common actEGFRm (EGFRL858R and EGFRDel19), these irreversible EGFR/HER-TKIs (afatinib, dacomitinib, and neratinib) demonstrated inhibitory activities in cells/tumors harboring EGFRT790M in preclinical models.80 However, they were most sensitive in patients with actEGFRm and were not clinically active in overcoming EGFRT790M resistance, perhaps because of dose-limiting toxicity (narrow therapeutic window, such as the crucial gastrointestinal [diarrhea and mucositis] and dermatologic toxicity) due to simultaneous inhibition against wtEGFR.66,67,72,73 Furthermore, the acquired resistance to these agents can develop due to EGFRT790M amplification.81,82
In view of the low probability of drug–drug interactions, multiple treatment strategies have been clinically used, including the combination of EGFR/HER-TKI with chemotherapy or other targeted therapy.20 The Phase III Lux-lung 5 study,83 evaluating the efficacy and safety of continued irreversible HER-family blockade with afatinib plus paclitaxel versus investigator’s choice of chemotherapy alone in patients with relapsed/refractory NSCLC following chemotherapy and acquired resistance to prior erlotinib/gefitinib and afatinib monotherapy, demonstrated that afatinib plus paclitaxel significantly improved PFS (5.6 months versus 2.8 months) and ORR (32.1% versus 13.2%) compared with single-agent chemotherapy. Another regimen that showed interesting clinical activity and a manageable safety profile is the combination of afatinib and cetuximab (anti-EGFR monoclonal antibody), which induced a PFS of 4.7 months and an ORR of 29% with median duration of response of 5.7 months in a small Phase IB trial for heavily pretreated patients with actEGFRm lung cancer and acquired resistance to erlotinib/gefitinib.84 This study indicated that the dual vertical blockade of EGFR (in the intracellular domain of the HER-family members with afatinib and the extracellular domain of EGFR with cetuximab) was effective regardless of the EGFRT790M status (ORR: 32% for EGFRT790M-positive and 25% for EGFRT790M-negative tumors, P=0.341). These combination studies on patients with acquired resistance to TKIs supported a focus upon rechallenging with EGFR/HER-TKI beyond disease progression in oncogene-addicted lung cancer, indicating that some tumor cells may remain dependent on HER signaling, due to either the type of acquired EGFR mutations, EGFR amplification, and/or HER2 amplification.84 However, current targeted therapeutic strategies for patients with EGFRT790M are limited, and no approved treatment options are available.
This has led to the development of third-generation EGFR-TKIs that are designed to more effectively and selectively target EGFRT790M and actEGFRm, while sparing the activity to wtEGFR.
Third-generation EGFR/HER-TKIs
Considering that the EGFRT790M mutation represents the most dominant acquired resistance to first- and second-generation EGFR/HER-TKI therapy, specific drugs to target this mutation are recently under clinical development and may bring a breakthrough for the treatment of NSCLC patients. The third-generation EGFR/HER-TKIs (Table 1 and Figure 1), such as osimertinib (AZD9291; AstraZeneca plc), rociletinib (CO-1686; Clovis Oncology Inc., Boulder, Colorado, USA), HM61713 (Hanmi Pharmaceutical, Songpa-gu, Seoul, Korea), EGF816 (Novartis International AG, Basel, Switzerland), and ASP8273 (Astellas Pharma Inc.), selectively and irreversibly target EGFRT790M and actEGFRm, and have the reduced affinity to wtEGFR.52,85 Clinically, they have yielded promising results of favorable benefit for the actEGFRm patients who had disease progression (especially with EGFRT790M mutation) following first-/second-generation-TKIs treatment, as well as showed very low inhibitory effect on wtEGFR, thus overcoming the toxicity limitation seen with earlier generation EGFR/HER-TKIs.86–89 Unlike previous generation inhibitors, most adverse effects, such as diarrhea, rash, and nausea, were mild (grades 1 and 2). There were no DLTs at any dose level, and maximal tolerated dose was not defined.
Osimertinib
Osimertinib, a mono-anillino-pyrimidine compound (Figure 1), is a double-mutant selective third-generation irreversible EGFR/HER-TKI.90 In cell culture and mouse models,25 it potently inhibited signaling pathways and cellular growth in cells/tumors with both actEGFRm (EGFRDel19 and EGFRL858R) and EGFRT790M, with 200-fold less activity in inhibiting wtEGFR, thus having the increased selectivity margin against wtEGFR.
As a second-line (or later) treatment, a global Phase I study (AURA)31 demonstrated that osimertinib was highly active in lung cancer patients with the EGFRT790M mutation who had disease progression during prior therapy with EGFR/HER-TKIs, showing a higher ORR (EGFRT790M positive: 61%; EGFRT790M negative: 21%) and longer median PFS (9.6 months versus 2.8 months, respectively) with low incidence of toxicity. AURA2, a global Phase II single-arm study47 for patients with actEGFRm who had progressed after prior EGFR/HER-TKI therapy and had EGFRT790M mutation, also showed antitumor efficacy of osimertinib suggesting it can overcome EGFRT790M-mediated acquired resistance. The ORR was 64%, disease control rate (DCR) was 90%, and PFS was not reached (Table 2). In November 2015, osimertinib was granted accelerated approval by FDA for the treatment of metastatic EGFRT790M-positive NSCLC who have progressed on or after EGFR/HER-TKI therapy.91,92 In the same population, a Phase III AURA3 trial is ongoing to compare osimertinib monotherapy with pemetrexed plus platinum chemotherapy.
In a first-line expansion cohort of AURA trial for treatment-naive advanced NSCLC with actEGFRm, osimertinib achieved promising anticancer activity with an ORR of 70%, DCR of 97%, and PFS at 3 months and 6 months of 93% and 87%, respectively.93 The ongoing Phase III FLAURA trial will assess the efficacy and safety of osimertinib versus gefitinib/erlotinib as the first-line treatment in patients with advanced NSCLC actEGFRm.
Rociletinib
Rociletinib, an oral 2,4-disubstituted pyrimidine covalent EGFR/HER-TKI (Figure 1), is a highly selective and irreversible inhibitor of both actEGFRm- and EGFRT790M-resistance mutation. In a preclinical study, rociletinib demonstrated a significant growth inhibitory effect toward EGFRT790M and actEGFRm cells/tumors with significantly less activity on wtEGFR in cell lines, xenograft, and transgenic mouse models.33
In a Phase I/II trial (TIGER X)32 for 130 patients with actEGFRm and acquired resistance to EGFR/HER-TKIs, rociletinib showed promising activity for EGFRT790M-positive patients, and to a lesser extent, to EGFRT790M-negative populations (ORR was 59% versus 29% for patients with EGFRT790M positive versus EGFRT790M negative, respectively, DCR was 93% versus 59%, and PFS was 13.1 months versus 5.6 months; Table 2). The modes of action against EGFRT790M-negative patients may include tumor heterogeneity, sensitivity of genotyping platform used, and activity against other resistant mechanisms.47 Rociletinib had infrequent EGFR-related toxicity (due to the low affinity to wtEGFR) but had a tendency to other concerning safety issues such as hyperglycemia and long QT interval, because of a rociletinib metabolite (M502), which inhibited insulin-like growth factor receptor-1.32 In May 2014, rociletinib was granted breakthrough therapy designation for NSCLC patients with EGFRT790M after progression on a prior TKI. This has allowed investigation in several TIGER trials in various treatment lines, such as the randomized Phase II/III TIGER 1 study, comparing rociletinib versus erlotinib as the first-line monotherapy for advanced actEGFRm NSCLC regardless of EGFRT790M status; the confirmatory Phase II single-arm TIGER 2 trial for advanced EGFR-mutated NSCLC progressed after previous EGFR/HER-TKI therapy and harboring EGFRT790M; the randomized Phase III TIGER 3 trial, evaluating rociletinib versus platinum doublet chemotherapy for second-line treatment for patients with actEGFRm and EGFRT790M after EGFR/HER-TKI failure.85,87,88,94
HM61713
HM61713 is another novel, oral mutant-selective inhibitor of actEGFRm and EGFRT790M, but not wtEGFR, which demonstrated good efficacy in animal models, especially those with concurrent actEGFRm and EGFRT790M mutations.34 An open-label Phase I/II trial (NCT01588145) in Korea with actEGFRm NSCLC patients who had progressed on prior EGFR/HER-TKI therapy demonstrated promising antitumor activity of HM61713 (especially with EGFRT790M mutation), showing an ORR of 21.7% and DCR of 67.5% in unselected population, an ORR of 29% and DCR of 75% in EGFRT790M-positive patients, and an ORR of 12% in EGFRT790M-negative group.88,95 The Phase II cohort (Table 2) showed an ORR of 58.8% and DCR of 97.1% in patients with centrally confirmed EGFRT790M who received HM61713 at a dose >650 mg/day, which also indicated that HM61713 had an encouraging clinical efficacy to overcome the EGFR/HER-TKI resistance.35 HM61713 caused mild side effects and can be controlled easily. A Phase II trial is currently enrolling patients with actEGFRm treated with first-line HM61713.
Several other third-generation mutant-EGFR specific TKIs are currently being investigated in NSCLC patients with actEGFRm and EGFRT790M, such as EGF816, ASP8273, AP26113, and poziotinib.96–98 In addition, the activity of third-generation EGFR/HER-TKIs has been further investigated in patients with central nervous system (CNS) metastases. The preliminary data99,100 indicated that the response rate of these TKIs (such as osimertinib and rociletinib) was not affected by the history of CNS disease, showing that the ORR among patients with versus without CNS metastases was 56% versus 64%, respectively, for the treatment with osimertinib, and 58% versus 45%, respectively, for the treatment with rociletinib. In summary, because of highly mutant-selective targeting, the third-generation EGFR/HER-TKIs are potent for the treatment of prior EGFR/HER-TKI-resistant patients harboring the EGFRT790M mutation, with probable lower EGFR treatment-related toxicity.
Although the efficacy of third-generation EGFR/HER-TKIs seems improved, responses are not durable, and disease progression still occurs. Possible mechanisms of acquired resistance have been described, including tertiary EGFR mutations (C797S, L844V, and L718Q), alternative/bypass signaling (increased RAS/RAF/ERK signaling by NRASE63K0 mutation, BRAFV600E mutation or increased MEK1 activity, HER2 amplification, MET amplification, and PIK3CAE545K mutation), and phenotypic alterations (epithelial–mesenchymal transition and small-cell lung cancer transformation).4,47 Due to the diversity of resistance mechanisms, various therapeutic regimens are under investigation in preclinical and clinical settings. The medical model of combination therapy may be the main trend to enhance their effectiveness. Clinical trials of combination therapy using third-generation EGFR/HER-TKIs are in process, combining with selumetinib (MEK inhibitor), savolitinib (AZD6094 and MET-TKI), necitumumab (anti-EGFR antibody), or navitoclax (inhibitor of Bcl-xl, Bcl-2, and Bcl-w).89
Conclusion
By virtue of the extensive investigation and better understanding of the EGFR/HER-family signaling pathway, NSCLC diagnosis and treatment has evolved dramatically from the traditional one-size-fits-all chemotherapeutic approach to the new personalized molecular target therapy. The application of the first-generation EGFR/HER-TKIs in selected NSCLC patients with actEGFRm (EGFRL858R and EGFRDel19) showed a significant superiority over the standard chemotherapy in terms of safety, efficacy, and quality of life. The second-generation EGFR/HER-TKIs similarly improved actEGFRm patients OS but failed to overcome the acquired EGFRT790M resistance. The third-generation EGFR/HER-TKIs selectively and irreversibly targeted EGFRT790M mutation and actEGFRm and were sparing to wtEGFR. They seem efficacious for TKI-resistant patients with EGFRT790M mutations. They have a lower incidence of toxicity due to the less inhibitory effect on wtEGFR. Currently, two first-generation EGFR/HER-TKIs gefitinib and erlotinib and one second-generation EGFR/HER-TKI afatinib have been used as the first-line treatment of metastatic NSCLC with actEGFRm (EGFRDel19 or EGFRL858R). Recently, the third-generation EGFR/HER TKIs osimertinib and rociletinib have been granted accelerated approval and breakthrough therapy designation, respectively, by the FDA for patients with metastatic EGFRT790M mutation-positive NSCLC. Their true place awaits definitive randomized trials.
Correct selection and use of these EGFR/HER-TKIs are mainly dependent upon identification of EGFR primary and secondary mutations. The pretreatment detection of EGFR mutations as predictive biomarkers maximizes the therapeutic index of personalized targeted therapy in lung cancer. Consequently, examining the genetic alterations is recommended for all new diagnosed NSCLC patients and when they experience disease progression, for better selection of the specific candidates for the targeted therapy. In addition, the rebiopsy will provide the genetic mechanisms for the development of acquired resistance and ultimately guide researchers to design the next generations of EGFR/HER-TKIs and strategies for cancer treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
American Cancer Society [webpage on the Internet]. Cancer Facts and Figures 2016. American Cancer Society. American Cancer Society: American Cancer Society. Available from: http://www.cancer.gov/types/lung/hp/non-small-cell-lung-treatment-pdq#cit/section_1.1. Accessed April 29, 2016. | ||
Reck M, Popat S, Reinmuth N, De Ruysscher D, Kerr KM, Peters S. Metastatic non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(suppl 3):iii27–iii39. | ||
Kanthala S, Pallerla S, Jois S. Current and future targeted therapies for non-small-cell lung cancers with aberrant EGF receptors. Future Oncol. 2015;11(5):865–878. | ||
Collins LG, Haines C, Perkel R, Enck RE. Lung cancer: diagnosis and management. Am Fam Physician. 2007;75(1):56–63. | ||
Shea M, Costa DB, Rangachari D. Management of advanced non-small cell lung cancers with known mutations or rearrangements: latest evidence and treatment approaches. Ther Adv Respir Dis. 2016;10(2):113–129. | ||
Wang X, Batty KM, Crowe PJ, Goldstein D, Yang JL. The potential of panHER inhibition in cancer. Front Oncol. 2015;5:2. | ||
Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–430. | ||
Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. | ||
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. | ||
Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. | ||
Rosell R, Moran T, Queralt C, et al; Spanish Lung Cancer Group. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361(10):958–967. | ||
Tartarone A, Lerose R. Clinical approaches to treat patients with non-small cell lung cancer and epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance. Ther Adv Respir Dis. 2015;9(5):242–250. | ||
Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol. 2009;10(5):432–433. | ||
Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod Pathol. 2008;21(suppl 2):S16–S22. | ||
Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015;4(1):36–54. | ||
Carrera S, Buque A, Azkona E, et al. Epidermal growth factor receptor tyrosine-kinase inhibitor treatment resistance in non-small cell lung cancer: biological basis and therapeutic strategies. Clin Transl Oncol. 2014;16(4):339–350. | ||
Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(suppl 1):S24–S31. | ||
Mok T, Lee K, Tang M, Leung L. Dacomitinib for the treatment of advanced or metastatic non-small-cell lung cancer. Future Oncol. 2014;10(5):813–822. | ||
Hirsh V. Next-generation covalent irreversible kinase inhibitors in NSCLC: focus on afatinib. BioDrugs. 2015;29(3):167–183. | ||
FDA.gov [webpage on the Internet]. FDA Approves Erlotinib; 2013. Available from: http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm352317.htm. Accessed April 29, 2016. | ||
FDA.gov [webpage on the Internet]. Gefitinib (Iressa). U.S. Food and Drug Administration; 2015. Available from: http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm454692.htm. Accessed April 29, 2016. | ||
FDA.gov [webpage on the Internet]. FDA Approves Afatinib. U.S. Food and Drug Administration; 2013. Available from: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm360574.htm. Accessed April 29, 2016. | ||
Roskoski R Jr. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun. 2004;319(1):1–11. | ||
Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. | ||
Solca F, Dahl G, Zoephel A, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343(2):342–350. | ||
Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67(24):11924–11932. | ||
Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64(11):3958–3965. | ||
Wong KK, Fracasso PM, Bukowski RM, et al. A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res. 2009;15(7):2552–2558. | ||
Sequist LV, Besse B, Lynch TJ, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(18):3076–3083. | ||
Janne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–1699. | ||
Sequist LV, Soria JC, Goldman JW, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;372(18):1700–1709. | ||
Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3(12):1404–1415. | ||
Lee K-O, Cha MY, Kim M, et al. Discovery of HM61713 as an orally available and mutant EGFR selective inhibitor. Cancer Res. 2014;74(19 suppl):LB–100. [Proceedings of the AACR Annual Meeting 2014]. | ||
Park K, Lee J-S, Lee KH, et al. Updated safety and efficacy results from phase I/II study of HM61713 in patients (pts) with EGFR mutation positive non-small cell lung cancer (NSCLC) who failed previous EGFR-tyrosine kinase inhibitor (TKI). J Clin Oncol. 2015;33:(sul;abstr8084). [Proceedings of the 2015 ASCO Annual Meeting 2015]. | ||
Khozin S, Blumenthal GM, Jiang X, et al. U.S. Food and Drug Administration approval summary: erlotinib for the first-line treatment of metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. Oncologist. 2014;19(7):774–779. | ||
Cohen MH, Williams GA, Sridhara R, Chen G, Pazdur R. FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets. Oncologist. 2003;8(4):303–306. | ||
Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315(3):971–979. | ||
Yun CH, Boggon TJ, Li Y, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11(3):217–227. | ||
Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007;98(12):1817–1824. | ||
Rooney C, Sethi T. Advances in molecular biology of lung disease: aiming for precision therapy in non-small cell lung cancer. Chest. 2015;148(4):1063–1072. | ||
Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. | ||
Rosell R, Carcereny E, Gervais R, et al; Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. | ||
Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. | ||
Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol. 2012;13(1):e23–e31. | ||
Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31(8):1070–1080. | ||
Tan CS, Cho BC, Soo RA. Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor-mutant non-small cell lung cancer. Lung Cancer. 2016;93:59–68. | ||
Lee JK, Shin JY, Kim S, et al. Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non-small-cell lung cancer harboring TKI-sensitive EGFR mutations: an exploratory study. Ann Oncol. 2013;24(8):2080–2087. | ||
Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. | ||
Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. | ||
Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res. 2010;70(3):868–874. | ||
Xu M, Xie Y, Ni S, Liu H. The latest therapeutic strategies after resistance to first generation epidermal growth factor receptor tyrosine kinase inhibitors (EGFR TKIs) in patients with non-small cell lung cancer (NSCLC). Ann Transl Med. 2015;3(7):96. | ||
Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2(10):922–933. | ||
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | ||
Stasi I, Cappuzzo F. Second generation tyrosine kinase inhibitors for the treatment of metastatic non-small-cell lung cancer. Transl Respir Med. 2014;2:2. | ||
Spigel DR, Ervin TJ, Ramlau RA, et al. Randomized phase II trial of onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31(32):4105–4114. | ||
Nakade J, Takeuchi S, Nakagawa T, et al. Triple inhibition of EGFR, Met, and VEGF suppresses regrowth of HGF-triggered, erlotinib-resistant lung cancer harboring an EGFR mutation. J Thorac Oncol. 2014;9(6):775–783. | ||
Yoshida K, Yamada Y. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harboring EGFR mutations (JO25567): an open-label, randomized, multicenter, phase II study. Transl Lung Cancer Res. 2015;4(3):217–219. | ||
Li H, Takayama K, Wang S, et al. Addition of bevacizumab enhances antitumor activity of erlotinib against non-small cell lung cancer xenografts depending on VEGF expression. Cancer Chemother Pharmacol. 2014;74(6):1297–1305. | ||
Wang X, Goldstein D, Crowe PJ, et al. Overcoming resistance of targeted EGFR monotherapy by inhibition of STAT3 escape pathway in soft tissue sarcoma. Oncotarget. 2016;7(16):21496–21509. | ||
Wu YL, Lee JS, Thongprasert S, et al. Intercalated combination of chemotherapy and erlotinib for patients with advanced stage non-small-cell lung cancer (FASTACT-2): a randomised, double-blind trial. Lancet Oncol. 2013;14(8):777–786. | ||
Asami K, Atagi S. Epidermal growth factor receptor tyrosine kinase inhibitors for non-small cell lung cancer. World J Clin Oncol. 2014;5(4):646–659. | ||
Brabender J, Danenberg KD, Metzger R, et al. Epidermal growth factor receptor and HER2-neu mRNA expression in non-small cell lung cancer is correlated with survival. Clin Cancer Res. 2001;7(7):1850–1855. | ||
Yang JC, Shih JY, Su WC, et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol. 2012;13(5):539–548. | ||
Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. | ||
Yang JC, Hirsh V, Schuler M, et al. Symptom control and quality of life in LUX-Lung 3: a phase III study of afatinib or cisplatin/pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3342–3350. | ||
Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213–222. | ||
Passaro A, Pochesci A, Spitaleri G, et al. Afatinib in first-line setting for NSCLC harbouring common EGFR mutations: new light after the preliminary results of LUX-Lung 7? J Thorac Dis. 2016;8(3):E217–E220. | ||
Yang JC, Wu YL, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16(2):141–151. | ||
Park K, Tan EH, O’Byrne K, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016;17(5):577–589. | ||
Soria JC, Felip E, Cobo M, et al; LUX-Lung 8 Investigators. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol. 2015;16(8):897–907. | ||
Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13(5):528–538. | ||
Katakami N, Atagi S, Goto K, et al. LUX-Lung 4: a phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol. 2013;31(27):3335–3341. | ||
Ou SH, Soo RA. Dacomitinib in lung cancer: a “lost generation” EGFR tyrosine-kinase inhibitor from a bygone era? Drug Des Devel Ther. 2015;9:5641–5653. | ||
Ramalingam SS, Blackhall F, Krzakowski M, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2012;30(27):3337–3344. | ||
Ramalingam SS, Janne PA, Mok T, et al. Dacomitinib versus erlotinib in patients with advanced-stage, previously treated non-small-cell lung cancer (ARCHER 1009): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(12):1369–1378. | ||
Ellis PM, Shepherd FA, Millward M, et al; NCI Naples Clinical Trials Unit. Dacomitinib compared with placebo in pretreated patients with advanced or metastatic non-small-cell lung cancer (NCIC CTG BR.26): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2014;15(12):1379–1388. | ||
Janne PA, Ou SH, Kim DW, et al. Dacomitinib as first-line treatment in patients with clinically or molecularly selected advanced non-small-cell lung cancer: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2014;15(13):1433–1441. | ||
Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102(21):7665–7670. | ||
Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–2075. | ||
Ercan D, Zejnullahu K, Yonesaka K, et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene. 2010;29(16):2346–2356. | ||
Kim Y, Ko J, Cui Z, et al. The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor. Mol Cancer Ther. 2012;11(3):784–791. | ||
Schuler M, Yang JC, Park K, et al; LUX-Lung 5 Investigators. Afatinib beyond progression in patients with non-small-cell lung cancer following chemotherapy, erlotinib/gefitinib and afatinib: phase III randomized LUX-Lung 5 trial. Ann Oncol. 2016;27(3):417–423. | ||
Janjigian YY, Smit EF, Groen HJ, et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4(9):1036–1045. | ||
Liao BC, Lin CC, Yang JC. Second and third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr Opin Oncol. 2015;27(2):94–101. | ||
Castellanos EH, Horn L. Generations of epidermal growth factor receptor tyrosine kinase inhibitors: perils and progress. Curr Treat Options Oncol. 2015;16(10):51. | ||
Yagishita S, Hamada A. Clinical pharmacology of EGFR/Met inhibitors in non-small cell lung cancer. Curr Drug Targets. 2014;15(14):1263–1272. | ||
Steuer CE, Khuri FR, Ramalingam SS. The next generation of epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of lung cancer. Cancer. 2015;121(8):E1–E6. | ||
Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9:34. | ||
Ward RA, Anderton MJ, Ashton S, et al. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J Med Chem. 2013;56(17):7025–7048. | ||
FDA.gov [webpage on the Internet]. Osimertinib. U.S. Food and Drug Administration; 2015. Available from: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm472565.htm. Accessed April 29, 2016. | ||
Fellner C. Pharmaceutical approval update. P T. 2016;41(1):26–59. | ||
Greig SL. Osimertinib: first global approval. Drugs. 2016;76(2):263–273. | ||
Chuang JC, Salahudeen AA, Wakelee HA. Rociletinib, a third generation EGFR tyrosine kinase inhibitor: current data and future directions. Expert Opin Pharmacother. 2016;17(7):989–993. | ||
Kim D-W, Lee DH, Kang JH, et al. Clinical activity and safety of HM61713, an EGFR-mutant selective inhibitor, in advanced non-small cell lung cancer (NSCLC) patients (pts) with EGFR mutations who had received EGFR tyrosine kinase inhibitors (TKIs). J Clin Oncol. 2014;32(5s):(sul:abstr8011). [Proceedings of the 2014 ASCO Annual Meeting 2014]. | ||
Camidge DR, Bazhenova L, Salgia R, et al. First-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: updated results. J Clin Oncol. 2013;31:(sul;abstra8031). [Proceedings of the 2013 ASCO Annual Meeting 2013]. | ||
Tan DS-W, Seto T, Leighl NB, et al. First-in-human phase I study of EGF816, a third generation, mutant-selective EGFR tyrosine kinase inhibitor, in advanced non-small cell lung cancer (NSCLC) harboring T790M. J Clin Oncol. 2015;33:(sul;abstr8013). [Proceedings of the 2015 ASCO Annual Meeting 2015]. | ||
Goto Y, Nokihara H, Murakami H, et al. ASP8273, a mutant-selective irreversible EGFR inhibitor in patients (pts) with NSCLC harboring EGFR activating mutations: preliminary results of first-in-human phase I study in Japan. J Clin Oncol. 2015;33:(sul;abstr8014). [Proceedings of the 2015 ASCO Annual Meeting 2015]. | ||
Camidge DR, Sequist LV, J-C Soria, et al. Activity of rociletinib in EGFR mutant NSCLC patients with a history of CNS involvement. In: Proceedings of the 16th World Conference on Lung Cancer 2015; Denver, Colorado, USA: Abstract #965. | ||
Mitsudomi T, Tsai C, Shepherd F, et al. AZD9291 in pre-treated T790M positive advanced NSCLC: AURA2 phase II study. In: Proceedings of the 16th World Conference on Lung Cancer 2015; Denver, Colorado, USA: Abstract #1406. | ||
Selleckchem.com [homepage on the Internet]. Houston: Selleck Chemicals; 2013. Available from: http://www.selleckchem.com/. Accessed August 30, 2016. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.