")
Back to Journals » Drug Design, Development and Therapy » Volume 16
New Series of VEGFR-2 Inhibitors and Apoptosis Enhancers: Design, Synthesis and Biological Evaluation
Authors Abdallah AE , Mabrouk RR, Elnagar MR , Farrag AM, Kalaba MH , Sharaf MH, El-Fakharany EM , Bakhotmah DA , Elkaeed EB , Al Ward MMS
Received 16 October 2021
Accepted for publication 28 January 2022
Published 3 March 2022 Volume 2022:16 Pages 587—607
DOI https://doi.org/10.2147/DDDT.S344750
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Abdallah E Abdallah,1 Reda R Mabrouk,1 Mohamed R Elnagar,2 Amel Mostafa Farrag,3 Mohamed H Kalaba,4 Mohamed H Sharaf,4 Esmail M El-Fakharany,5 Dina Abed Bakhotmah,6 Eslam B Elkaeed,7 Maged Mohammed Saleh Al Ward1
1Pharmaceutical Medicinal Chemistry & Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo, 11884, Egypt; 2Department of Pharmacology and Toxicology, Faculty of Pharmacy, Al-Azhar University, Cairo, 11884, Egypt; 3Pharmaceutical Chemistry Department, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo, Egypt; 4Botany and Microbiology Department, Faculty of Science, Al-Azhar University, Cairo, 11884, Egypt; 5Protein Research Department, Genetic Engineering and Biotechnology Research Institute, City of Scientific Research and Technological Applications, New Borg El Arab, Egypt; 6Chemistry Department, King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia; 7Department of Pharmaceutical Sciences, College of Pharmacy, AlMaarefa University, Riyadh, 13713, Saudi Arabia
Correspondence: Abdallah E Abdallah; Maged Mohammed Saleh Al Ward, Pharmaceutical Medicinal Chemistry & Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo, 11884, Egypt, Email [email protected]; [email protected]
Background: Cancer is still a major world health threat, causing a high rate of mortality. VEGFR-2 inhibitor anticancer agents are of great significance. However, they showed some serious side effects.
Purpose: To discover new effective and safer anticancer agents, a new series of piperazinylquinoxaline-based derivatives was designed and synthesized on the basis of the pharmacophoric features of VEGFR-2 inhibitor drugs.
Methods: The new candidates were evaluated against A549 lung cancer cells, HepG-2 hepatoma cells, Caco-2 colon cancer cells, MDA breast cancer cells, and VEGFR-2 kinase. Moreover, cell cycle kinetics and apoptosis rates were studied in HepG-2 cells treated with compound 11, which was the most promising candidate.
Results: The new derivatives revealed better antitumor results (IC50 from 6.48 to 38.58 μM) against the aforementioned cancer cell lines than sorafenib. Also, the new candidates showed VEGFR-2 inhibition with IC50 values ranging from 0.19 to 0.60 μM compared to 0.08 μM for sorafenib. Compound 11, meanwhile, showed IC50 values equal to 10.61, 9.52, 12.45, 11.52, and 0.19 μM against the cancer cell lines and VEGFR-2, respectively. Moreover, compound 11 raised the apoptosis rate in HepG-2 cells from 5% to 44% and caused 4, 2.3, and 3-fold increases in BAX/Bcl-2 ratio, caspase-3 level, and P53 expression, respectively, compared to control untreated cells. Finally, the new derivatives displayed the correct binding mode into VEGFR-2 kinase pocket, giving interactions with the essential residues.
Conclusion: This work suggests that compound 11 is a very significant anticancer candidate, and piperazinylquinoxaline is an important scaffold in the development of new potential effective and safer VEGFR-2 inhibitor agents.
Keywords: anticancer, apoptosis, molecular modeling, piperazinylquinoxaline, VEGFR-2 kinase, Western blot
Introduction
Cancer is still a significant health issue.1 It is a primary cause of death all over the world.2,3 Globally, huge efforts have been done to control cancer. However, the incidence and mortality of cancer are growing at a high rate.2 Worldwide, it is estimated that approximately 19.3 million new cancer cases emerged and about 10 million cancer deaths occurred in 2020.4 Lung cancer, colorectal cancer, liver cancer, and breast cancer are considered the main causes of cancer death.4 These facts clearly reveal that anti-cancer drug discovery is a rich field of research.
It was found that cancer cells express vascular endothelial growth factor-2 (VEGFR-2) at higher levels than normal cells. Relatively high levels of VEGFR-2 were detected in different cancer types, such as breast,5 non-small cell lung,6 colorectal,7 hepatocellular,8 and urothelial9 carcinomas. VEGFR-2 is crucial for angiogenesis10–12 in order to supply cancer cells with oxygen and nutrients, which are required for growth.13,14 Accordingly, VEGFR-2 is a significant target in order to develop selective and effective anticancer agents.15–19 Many VEGFR-2 inhibitors, such as sorafenib 1,20 regorafenib 2,21 sunitinib 3,22 pazopanib 4,23 vatalanib 5,24 and tivozanib 6,25 are currently FDA approved for treatment of different types of cancers.
According to binding properties, VEGFR-2 inhibitor drugs can be differentiated into two major categories.26 Type-I inhibitor drugs such as sunitinib were reported to bind effectively to the active conformation of the receptor. They interact with the region that is normally occupied by the adenine moiety of ATP.25,27 In contrast, type-II kinase inhibitor drugs, such as sorafenib, regorafenib, and tivozanib,25 were found to bind to a hydrophobic allosteric pocket which is specific to the inactive conformation (DFG-out) of VEGFR-2.28 Generally, type-II kinase inhibitors were reported to be more significant than type-I. This is because type-II kinase inhibitors show more specific kinase selectivity and slower off-rates.29
On the other hand, VEGFR-2 inhibitor drugs caused some side effects during clinical use.30 Hypertension was reported as a major side effect of sorafenib, regorafenib, and tivozanib.31–34 Meanwhile, quinoxaline-based compounds have been shown to inhibit VEGFR-2.35 Also, the piperazine moiety was incorporated into some potent anticancer compounds,36–38 some of which showed VEGFR-2 inhibiting effects.39 Moreover, both quinoxaline and piperazine derivatives have been shown to reduce hypertension,40–43 and substituted piperazinylquinoxalines have recently been proven to have an antihypertensive effect.44
In this study, in line with the reported four pharmacophoric features of type-II VEGFR-2 inhibitor agents25,45–48 (Figure 1), and depending on molecular hybridization technique,49,50 piperazinylquinoxaline scaffold was used to develop new VEGFR-2 inhibitor candidates as shown in Figure 1. The illustration shows that quinoxaline, which is a hetero aromatic moiety, was designed to occupy the ATP binding domain and show interactions with the key residues Cys919 and/or Glu917. At the same time, a piperazine moiety was incorporated to fill the space between the ATP binding region and the DFG domain. Thirdly, we constructed an amide group to interact by forming hydrogen bonds with the effective residues Glu885 and Asp1046. The amide group played such a role effectively in many potent VEGFR-2 inhibitors,47 eg AZD-2932 (see Figure 1). Finally, the terminal hydrophobic moiety was suggested to be substituted phenyl, unsubstituted phenyl, and benzyl. The substituents at the terminal phenyl were selected to be of different steric and electronic properties as well as of different polarity.
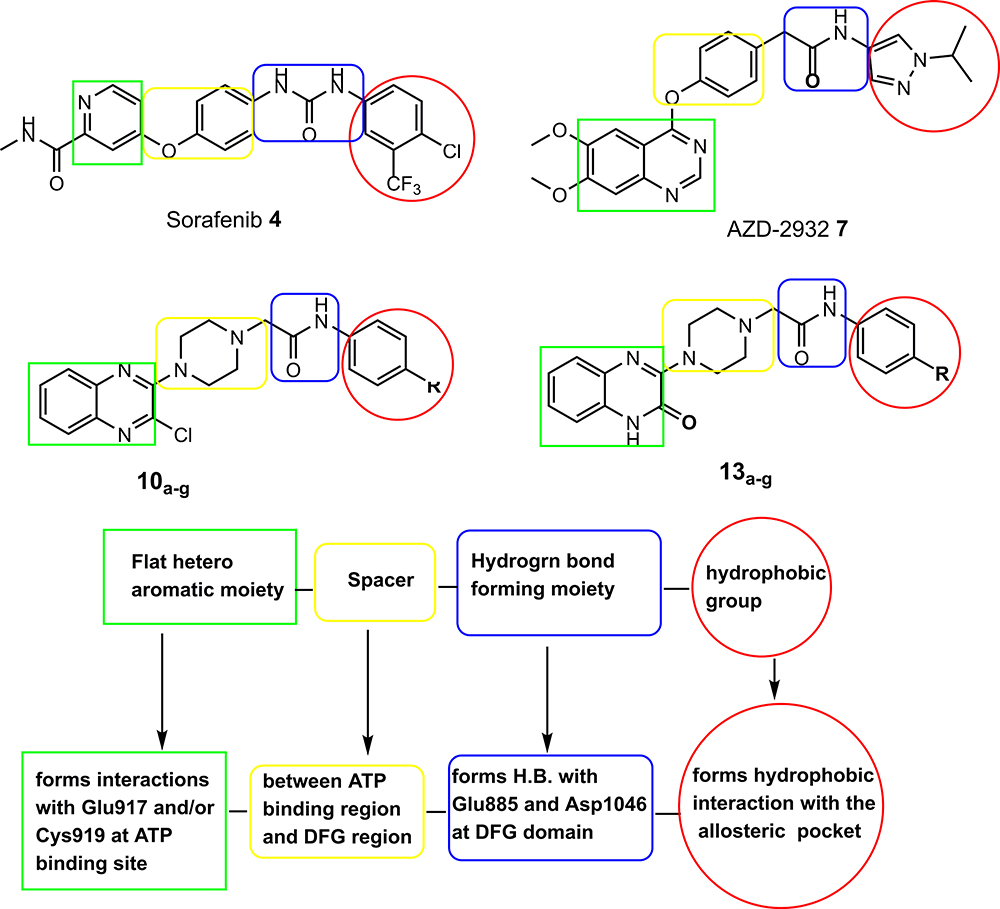 |
Figure 1 VEGFR-2 inhibitors (type II) pharmacophoric features as presented in sorafenib, AZD-2932, and the new derivatives and their corresponding binding regions as well. |
Materials and Methods
Chemistry
General Method for Synthesis of Final Compounds (10a-g) and 11
Equimolar amounts of 2-chloro-3-(piperazin-1-yl)quinoxaline (9), an appropriate intermediate (3a-g and 5) and NaHCO3 were refluxed in acetonitrile for 2h. The reaction mixture was cooled and then poured onto water with stirring. The obtained precipitate was collected by filtration, dried, washed with n-hexane and crystallized from ethanol.
4.1.1.1. 2-(4-(3-chloroquinoxalin-2-yl)piperazin-1-yl)-N-phenylacetamide (10a); white solid (yield 83.25%); m.p. = 208 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.78 (t, J = 4.4 Hz, 4H, 2CH2 piperazine), 3.25 (s, 2H, CH2CO), 3.58 (t, J = 4.4 Hz, 4H, 2CH2 piperazine), 7.07 (t, J = 7.4 Hz, 1H, Ar-H), 7.32 (dd, J = 8.1, 8.1 Hz, 2H, Ar-H), 7.65 (m, 3H, Ar-H), 7.75 (ddd, J = 8.3, 8.3, 1.3 Hz, 1H, Ar-H), 7.83 (dd, J = 8.3, 0.8 Hz, 1H, Ar-H), 7.89 (dd, J = 8.3, 0.9 Hz, 1H, Ar-H), 9.79 (s, 1H, NH) (Figure S5 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 49.11, 52.69, 61.99, 120.01, 123.94, 127.16, 127.48, 128.02, 129.13, 131.05, 138.00, 139.02, 139.94, 141.69, 152.76, 168.66 (Figure S6 in The supplementary materials).
4.1.1.2. 2-(4-(3-chloroquinoxalin-2-yl)piperazin-1-yl)-N-(4-methoxyphenyl)acetamide (10b); white solid (yield 84.32%); m.p. = 202 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.76 (t, J = 4.6 Hz, 4H, 2CH2 piperazine), 3.21 (s, 2H, CH2CO), 3.58 (t, J = 4.4 Hz, 4H, 2CH2 piperazine), 3.73 (s, 3H, CH3), 6.88 (d, J = 9.0 Hz, 2H, Ar-H), 7.55 (d, J = 9.0 Hz, 2H, Ar-H), 7.63 (ddd, J = 8.3, 8.3, 1.3 Hz, 1H, Ar-H), 7.75 (ddd, J = 8.3, 8.3, 1.3 Hz, 1H, Ar-H), 7.83 (dd, J = 8.2, 0.7 Hz, 1H, Ar-H), 7.89 (dd, J = 8.2, 0.8 Hz, 1H, Ar-H), 9.65 (s, 1H, NH) (Figure S7 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 49.11, 52.73, 55.64, 61.97, 114.23, 121.62, 127.17, 127.85, 128.02, 131.05, 132.21, 138.01, 139.96, 141.71, 152.79, 155.83, 168.13 (Figure S8 in The supplementary materials).
4.1.1.3. N-(4-chlorophenyl)-2-(4-(3-chloroquinoxalin-2-yl)piperazin-1-yl)acetamide (10c); White solid (yield 81.75%); m.p. = 232 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.77 (t, J = 4.5 Hz, 4H, 2CH2 piperazine), 3.25 (s, 2H, CH2CO), 3.58 (t, J = 4.5 Hz, 4H, 2CH2 piperazine), 7.36 (d, J = 8.9 Hz, 2H, Ar-H), 7.63 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H, Ar-H), 7.70 (d, J = 8.9 Hz, 2H, Ar-H), 7.75 (ddd, J = 8.2, 8.2, 1.3 Hz, 1H, Ar-H), 7.83 (dd, J = 8.2, 0.7 Hz, 1H, Ar-H), 7.89 (dd, J = 8.2, 0.8 Hz, 1H, Ar-H), 9.94 (s, 1H, NH) (Figure S9 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 49.05, 52.66, 61.92, 121.65, 127.15, 127.54, 127.82, 128.04, 129.01, 131.07, 137.98, 139.93, 141.68, 152.75, 168.94 (Figure S10 in The supplementary materials).
4.1.1.4. 2-(4-(3-chloroquinoxalin-2-yl)piperazin-1-yl)-N-(4-fluorophenyl)acetamide (10d); gray solid (yield 80.65%); m.p. = 195 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.76 (t, J = 4.4 Hz, 4H, 2CH2 piperazine), 3.24 (s, 2H, CH2CO), 3.58 (t, J = 4.3 Hz, 4H, 2CH2 piperazine), 7.16 (dd, J = 8.9 Hz, 2H, Ar-H), 7.64 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H, Ar-H), 7.68 (dd, J = 9.0, 5.0 Hz, 2H, Ar-H), 7.75 (ddd, J = 8.3, 8.3, 1.3 Hz, 1H, Ar-H), 7.83 (dd, J = 8.2, 0.6 Hz, 1H, Ar-H), 7.89 (dd, J = 8.2, 0.8 Hz, 1H, Ar-H), 9.86 (s, 1H, NH) (Figure S11 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 49.07, 52.69, 61.92, 115.55, 115.77, 121.88, 127.16, 127.84, 128.04, 131.07, 135.42, 137.99, 139.94, 141.70, 152.77, 168.65 (Figure S12 in The supplementary materials).
4.1.1.5. 2-(4-(3-chloroquinoxalin-2-yl)piperazin-1-yl)-N-(p-tolyl)acetamide (10e); white solid (yield 84.35%); m.p. = 215 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.26 (s, 3H, CH3), 2.76 (t, J = 4.5 Hz, 4H, 2CH2 piperazine), 3.22 (s, 2H, CH2CO), 3.58 (t, J = 4.3 Hz, 4H, 2CH2 piperazine), 7.11 (d, J = 8.3 Hz, 2H, Ar-H), 7.53 (d, J = 8.3 Hz, 2H, Ar-H), 7.64 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H, Ar-H), 7.75 (ddd, J = 8.2, 8.2, 1.2 Hz, 1H, Ar-H), 7.83 (d, J = 7.7 Hz, 1H, Ar-H), 7.89 (d, J = 7.6 Hz, 1H, Ar-H), 9.70 (s, 1H, NH) (Figure S13 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 20.89, 49.08, 52.68, 61.94, 120.05, 127.14, 127.82, 128.04, 129.51, 131.07, 132.93, 136.45, 137.97, 139.92, 141.69, 152.75, 168.47 (Figure S14 in The supplementary materials).
4.1.1.6. N-(4-acetylphenyl)-2-(4-(3-chloroquinoxalin-2-yl)piperazin-1-yl)acetamide (10f); white solid (yield 77.45%); m.p. = 210 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.54 (s, 3H, CH3), 2.78 (t, J = 4.4 Hz, 4H, 2CH2 piperazine), 3.30 (s, 2H, CH2CO), 3.58 (t, J = 4.4 Hz, 4H, 2CH2 piperazine), 7.63 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H, Ar-H), 7.75 (ddd, J = 8.3, 8.3, 1.2 Hz, 1H, Ar-H), 7.83 (3, 3H, Ar-H), 7.89 (dd, J = 8.2, 0.7 Hz, 1H, Ar-H), 7.93 (d, J = 8.7 Hz, 2H, Ar-H), 10.15 (s, 1H, NH) (Figure S15 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 26.90, 49.08, 52.64, 61.96, 119.20, 127.15, 127.83, 128.02, 129.85, 131.04, 132.35, 137.99, 139.93, 141.68, 143.39, 152.75, 169.37, 197.09 (Figure S16 in The supplementary materials).
4.1.1.7. 2-(4-(3-chloroquinoxalin-2-yl)piperazin-1-yl)-N-(4-nitrophenyl)acetamide (10g); yellowish solid (yield 75.35%); m.p. = 228 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.78 (t, J = 4.5 Hz, 4H, 2CH2 piperazine), 3.27 (s, 2H, CH2CO), 3.58 (t, J = 4.5 Hz, 4H, 2CH2 piperazine), 7.64 (ddd, J = 8.2, 7.2, 1.2 Hz, 1H, Ar-H), 7.75 (ddd, J = 8.3, 8.3, 1.3 Hz, 1H, Ar-H), 7.83 (dd, J = 8.3, 0.8 Hz, 1H, Ar-H), 7.89 (dd, J = 8.2. 0.8 Hz, 1H, Ar-H), 7.93 (d, J = 9.2 Hz, 2H, Ar-H), 8.23 (d, J = 9.2 Hz, 2H, Ar-H), 10.42 (s, 1H, NH) (Figure S17 and Figure S18 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 49.04, 52.60, 61.92, 119.65, 125.30, 127.14, 127.81, 128.02, 131.04, 137.97, 139.91, 141.66, 142.80, 145.23, 152.71, 169.83 (Figure S19 in The supplementary materials).
4.1.1.8. N-benzyl-2-(4-(3-chloroquinoxalin-2-yl)piperazin-1-yl)acetamide (11); white solid (yield 81.15%); m.p. = 205 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.69 (t, J = 4.6 Hz, 4H, 2CH2 piperazine), 3.10 (s, 2H, CH2CO), 3.54 (t, J = 4.5 Hz, 4H, 2CH2 piperazine), 4.33 (d, J = 6.2 Hz, 2H, CH2NH), 7.22–7.35 (m, 5H, Ar-H), 7.63 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H, Ar-H), 7.75 (ddd, J = 8.3, 8.3, 1.3 Hz, 1H, Ar-H), 7.82 (dd, J = 8.2, 0.8 Hz, 1H, Ar-H), 7.88 (dd, J = 8.2, 0.9 Hz, 1H, Ar-H), 8.39 (t, J = 6.2 Hz, 1H, NH) (Figure S20 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 42.36, 49.07, 52.85, 61.48, 127.15, 127.19, 127.63, 127.82, 128.04, 128.74, 131.05, 137.98, 139.93, 140.12, 141.69, 152.76, 169.82 (Figure S21 in The supplementary materials); Mass (m/z): 397.59 (M++2, 13.64%), 395.56 (M+, 34.12%), 388.96, 387.93, 386.12, 344.32, 314.20, 308.45, 398.22, 379.17, 377.97, 373.08, 365.60, 349.06, 286.33, 283.27, 274.38, 273.60, 265.28, 260.63, 246.46, 207.83, 201.13, 144.25, 105.18, 75.45, 69.43 (100%, base peak) (Figure S22 in The supplementary materials); Anal. Calcd. for C21H22ClN5O: C, 63.71; H, 5.60; N, 17.69. Found: C, 63.98; H, 5.72; N, 17.85%.
4.1.2. General method for synthesis of final compounds (13a-g) and 14: Equimolar amounts of 3-(piperazin-1-yl)quinoxalin-2(1H)-one (12), an appropriate intermediate (3a-g and 5) and NaHCO3 were heated under reflux in acetonitrile for 2h. The reaction mixture was then cooled, poured onto cold water and stirred for half an hour. The obtained precipitate was collected by filtration, dried, washed with n-hexane and crystallized from ethanol.
4.1.2.1. 2-(4-(3-oxo-3,4-dihydroquinoxalin-2-yl)piperazin-1-yl)-N-phenylacetamide (13a); white solid (yield 74.65%); m.p. = 243 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.66 (t, J = 4.8 Hz, 4H, 2CH2 piperazine), 3.19 (s, 2H, CH2CO), 3.96 (t, J = 4.8 Hz, 4H, 2CH2 piperazine), 7.06 (t, J = 7.4 Hz, 1H, Ar-H), 7.11–7.18 (m, 3H, Ar-H), 7.31 (dd, J = 8.2, 8.2 Hz, 2H, Ar-H), 7.38 (d, J = 7.4 Hz, 1H, Ar-H), 7.64 (d, J = 7.7 Hz, 2H, Ar-H), 9.80 (s, 1H, NHCO), 12.21 (s, 1H, NH quinoxaline) (Figure S23 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 46.52, 53.10, 62.05, 114.89, 120.03, 123.65, 123.96, 125.18, 125.56, 129.14, 129.79, 132.71, 138.99, 151.61, 152.53, 168.72 (Figure S24 in The supplementary materials).
4.1.2.2. N-(4-methoxyphenyl)-2-(4-(3-oxo-3,4-dihydroquinoxalin-2-yl)piperazin-1-yl)acetamide (13b); white solid (yield 73.23%); m.p. = 235 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.66 (s, 4H, 2CH2 piperazine), 3.12 (s, 2H, CH2CO), 3.72 (s, 3H, OCH3), 3.96 (s, 4H, 2CH2 piperazine), 6.88 (d, J = 8.6 Hz, 2H, Ar-H), 7.14–7.17 (m, 3H, Ar-H), 7.39 (d, J = 7.3 Hz, 1H, Ar-H), 7.54 (d, J = 8.6 Hz, 2H, Ar-H), 9.63 (s, 1H, NHCO), 12.13 (s, 1H, NH quinoxaline) (Figure S25 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 46.50, 53.11, 55.63, 61.97, 114.24, 114.84, 121.76, 123.72, 125.21, 125.56, 129.66, 132.03, 132.69, 151.58, 152.50, 155.89, 168.30 (Figure S26 in The supplementary materials).
4.1.2.3. N-(4-chlorophenyl)-2-(4-(3-oxo-3,4-dihydroquinoxalin-2-yl)piperazin-1-yl)acetamide (13c); white solid (yield 71.84%); m.p. = 264 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.65 (s, 4H, 2CH2 piperazine), 3.19 (s, 2H, CH2CO), 3.96 (s, 4H, 2CH2 piperazine), 7.12–7.17 (m, 3H, Ar-H), 7.36–740 (m, 3H, Ar-H), 7.68 (d, J = 8.8 Hz, 2H, Ar-H), 9.92 (s, 1H, NHCO), 12.12 (s, 1H, NH quinoxaline) (Figure S27 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 46.48, 53.10, 62.03, 114.83, 121.61, 123.64, 125.17, 125.57, 127.48, 129.00, 129.74, 132.70, 138.01, 151.59, 152.49, 168.88 (Figure S28 in The supplementary materials).
4.1.2.4. N-(4-fluorophenyl)-2-(4-(3-oxo-3,4-dihydroquinoxalin-2-yl)piperazin-1-yl)acetamide (13d); brown solid (yield 72.49%); m.p. = 227 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.66 (s, 4H, 2CH2 piperazine), 3.18 (s, 2H, CH2CO), 3.96 (s, 4H, 2CH2 piperazine), 7.13–7.17 (m, 5H, Ar-H), 7.39 (d, J = 7.4 Hz, 1H, Ar-H), 7.67 (dd, J = 8.8, 5.0 Hz, 2H, Ar-H), 9.84 (s, 1H, NHCO), 12.12 (s, 1H, NH quinoxaline) (Figure S29 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 46.48, 53.12, 62.00, 114.83, 115.54, 115.76, 121.93, 123.63, 125.16, 125.57, 129.47, 132.71, 135.42, 151.59, 152.49, 168.59 (Figure S30 in The supplementary materials).
4.1.2.5. 2-(4-(3-oxo-3,4-dihydroquinoxalin-2-yl)piperazin-1-yl)-N-(p-tolyl)acetamide (13e); white solid (yield 73.56%); m.p. = 248 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.26 (s, 3H, CH3), 2.66 (s, 4H, 2CH2 piperazine), 3.17 (s, 2H, CH2CO), 3.96 (s, 4H, 2CH2 piperazine), 7.11–7.17 (m, 5H, Ar-H), 7.39 (d, J = 7.4 Hz, 1H, Ar-H), 7.52 (d, J = 8.4 Hz, 2H, Ar-H), 9.68 (s, 1H, NHCO), 12.13 (s, 1H, NH quinoxaline) (Figure S31 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 20.91, 46.49, 53.13, 62.02, 114.83, 120.00, 123.61, 125.16, 125.57, 129.49, 129.77, 132.71, 132.82, 136.54, 151.59, 152.49, 168.31 (Figure S32 in The supplementary materials).
4.1.2.6. N-(4-acetylphenyl)-2-(4-(3-oxo-3,4-dihydroquinoxalin-2-yl)piperazin-1-yl)acetamide (13f); brownish solid (yield 68.50%); m.p. = 239 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.53 (s, 3H, CH3), 2.67 (s, 4H, 2CH2 piperazine), 3.24 (s, 2H, CH2CO), 3.96 (s, 4H, 2CH2 piperazine), 7.13–7.17 (m, 3H, Ar-H quinoxaline), 7.39 (d, J = 7.4 Hz, 1H, Ar-H), 7.80 (d, J = 8.7 Hz, 2H, Ar-H), 7.93 (d, J = 8.7 Hz, 2H, Ar-H), 10.12 (s, 1H, NHCO), 12.13 (s, 1H, NH quinoxaline) (Figure S33 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 26.89, 46.50, 53.05, 62.03, 114.84, 119.22, 123.69, 125.21, 125.57, 129.71, 129.86, 132.35, 132.69, 143.37, 151.59, 152.49, 169.40, 197.18 (Figure S34 in The supplementary materials).
4.1.2.7. N-(4-nitrophenyl)-2-(4-(3-oxo-3,4-dihydroquinoxalin-2-yl)piperazin-1-yl)acetamide (13g); yellowish solid (yield 65.30%); m.p. = 256 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.67 (s, 4H, 2CH2 piperazine), 3.27 (s, 2H, CH2CO), 3.96 (s, 4H, 2CH2 piperazine), 7.12–7.17 (m, 3H, Ar-H quinoxaline), 7.38 (d, J = 7.4 Hz, 1H, Ar-H), 7.92 (d, J = 9.1 Hz, 2H, Ar-H), 8.22 (d, J = 9.1 Hz, 2H, Ar-H), 10.42 (s, 1H, NHCO), 12.12 (s, 1H, NH quinoxaline) (Figure S35 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 44.37, 50.93, 59.95, 112.75, 117.52, 121.49, 123.04, 123.20, 123.45, 127.68, 130.60, 140.66, 143.18, 149.47, 150.40, 167.70 (Figure S36 in The supplementary materials).
4.1.2.8. N-benzyl-2-(4-(3-oxo-3,4-dihydroquinoxalin-2-yl)piperazin-1-yl)acetamide (14); white solid (yield 74.60%); m.p. = 230 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.59 (t, J = 4.5 Hz, 4H, 2CH2 piperazine), 3.04 (s, 2H, CH2CO), 3.93 (s, 4H, 2CH2 piperazine), 4.32 (d, J = 6.2 Hz, 2H, CH2NH), 7.12–7.17 (m, 3H, Ar-H quinoxaline), 7.22–7.35 (m, 5H, Ar-H), 7.38 (d, J = 7.3 Hz, 1H, Ar-H), 8.36 (s, 1H, NHCO), 12.12 (s, 1H, NH quinoxaline) (Figure S37 in The supplementary materials); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 42.35, 46.47, 53.24, 61.51, 114.83, 123.69, 125.20, 125.56, 127.20, 127.62, 128.74, 129.68, 132.69, 140.07, 151.56, 152.49, 169.88 (Figure S38 in The supplementary materials).
Biological Testing
In vitro Antitumor Assay
This assay was performed against four human cancer cell lines, namely, A549, HepG-2, Caco-2, and MDA. In line with the MTT protocol60 we carried out this colorimetric assay with quantitative measurement of the antitumor activity. The assay is primarily based on a biochemical reaction in which mitochondrial succinate dehydrogenase in viable cells converts the yellow tetrazolium bromide (MTT) to a purple formazan derivative. In general, the cells were cultured in RPMI-1640 medium with 10% fetal bovine serum. Penicillin (100 units/mL) and streptomycin (100µg/mL) were added in a 5% CO2 incubator at 37 °C. Seeding of the cells was in a 96-well plate at 37 C for 48 h under 5% CO2 and it was at a density of 1.0×104 cells/well. After the initial incubation, the cells were treated with the new derivatives at different concentrations and incubated for 24 h. Then we add 20 µL of MTT in a solution of 5mg/mL concentration and incubate for 4 h. To dissolve the obtained purple formazan, 100 µL of DMSO was added into each well. Measuring and recording the colorimetric assay were carried out by a plate reader (EXL 800, USA) at an absorbance of 570 nm. The percentage of relative cell viability was calculated.
VEGFR-2 (KDR) Kinase Assay
According to the manufacturer’s instructions, the VEGFR-2 Kinase Assay Kit (BPS-Bioscience, USA) was used for testing the VEGFR tyrosine kinase activity according to. In brief, the diluted VEGFR-2 enzyme or tested compound (at different dilution of 1000, 300, 100, 30, 10, 3, 1, and 0.30 nM) was added to the wells of a 96-well plate, and the reaction mixture was incubated at 30 °C for 30 min, then 25 mL of ADP-GloTM Reagent was added. The reaction plate was incubated at RT for 45 min, and the kinase detection reagent (Kinase-Glo MAX (Promega)) was added. A luminescence signal was detected and quantified by a BioTek Synergy 2 microplate reader for determination of IC50 of the tested compounds. A blank control was set up to which an equal volume of kinase assay buffer was added instead of the tested compounds. The activity for each protein kinase target was corrected by subtracting the blank control value.
Cell Cycle Analysis
This assay was conducted according to the reported method.61 In short, HepG-2 cells were seeded into six-well plates (2 × 105 cells per each well) and incubated for 24 h at 37 °C and 5% CO2. After that, they were treated with 9.52 µM of compound 11 dissolved in DMSO (1% v/v) for 48 h. After the cells were washed several times with cold phosphate buffered saline (PBS), fixed with ethyl alcohol (70%) and rinsed again with PBS, they were stained with the DNA fluorochrome propidium iodide and kept at 37 °C for 15 min in the dark. The cell distribution was analyzed using Epics XL-MCL™ Flow Cytometer (Beckman Coulter), and the data were analyzed using Flowing software (version 2.5.1, Turku Centre for Biotechnology, Turku, Finland).
Apoptosis Assay
Apoptosis detection was carried out using Annexin V fluorescein isothiocyanate (V-FITC)/PI kit according to the reported procedure.62 HepG-2 cells were seeded and incubated for 24 h, then treated with (9.52 µM) of compound 11 for 28 h. The cells were then collected, washed 3 times with PBS, fixed with ice-cold absolute ethanol (70%) and stained with Annexin-V-FITC/propidium iodide (PI) using a double staining kit for 20 min in the dark. Epics XL-MCL™ Flow Cytometer was used to evaluate the apoptosis.
Western Blot Analysis
In line with the manufacturer’s instructions,63 Western blot analysis was carried out for evaluation of the apoptotic effect of compound 11 (at its IC50) on the proteins; BAX, Bcl-2, caspase‐3 and p53 in HepG-2 cells. The cells were treated with compound 11 or DMSO (as control), lysed in cold lysis buffer (250 µL) containing Tris‐Base [10 mM], ethylenediaminetetraacetic acid [25 mM], ethylene glycol bis (2‐aminoethyl) 1% [v/v] NP‐40, tetraacetic acid [EGTA, 25 mM], NaCl [100 mM], 1% [v/v] Triton X‐100, and directly supplemented with 1:400 protease and phosphatase inhibitors mixture (Sigma) while pH was kept at 7.4. Additional lysis of the cancer cells was achieved by freezing for 1.5 hours at −80°C. The cells were then thawed, collected, sonicated, and centrifuged. The total protein concentration was measured using the Pierce 660 nm Protein Assay Reagent (Thermo Fisher Scientific, Rockford, IL), using bovine serum albumin (BSA) as a standard. An equal amount of protein (30 µg) was loaded into SDS‐polyacrylamide gel and separated by an electrophoresis unit (Cleaver Scientific Ltd, UK). After separation, the proteins were transferred onto polyvinylidene fluoride membranes (Bio‐Rad) for 35 min, blocked in 5% BSA in Tris-buffered saline containing 0.05% Tween, TBST. The membranes were then incubated with the primary antibody diluted in BSA (1:1000) at 4 °C overnight and washed 4 times with 1× TBST buffer. The secondary antibody (coupled to horseradish peroxidase) diluted in TBST buffer (1:2000) was added for 60 min at RT. The band signals were detected by Western Lightning enhanced chemiluminescence (ECL) reagents (Perkin Elmer, Waltham, MA), and captured by Chemi‐Doc imager (Bio‐Rad). The band intensities were calculated with normalization to the β-actin.63–65
Molecular Docking
Molecular operating environment (MOE) 2014 software was used to execute the molecular docking study. The crystal structure of VEGFR-2 (ID: 3VHE) was downloaded from PDB software. The downloaded protein is composed of one peptide chain, the co-crystallized ligand, and water molecules. The water molecules were eliminated, then protonation of 3D was done. The energy of the protein was minimized according to the default option (Forcefiled: MMFF94X, eps=r, cutoff] 8–10[). The pocket was obtained by isolating the receptor surface within 4.5 Å around the ligand atoms. Validation of the docking protocol was detected by redocking the ligand (pyrrolopyrimidine inhibitor). It was found that the RMSD of the redocked ligand was of 0.42 Å from the co-crystallized ligand. It was reported that RMSD of 1.5 Å or less is an indicator for validation of the docking procedure. The new derivatives were constructed at MOE, 3D protonation was done, and the energy of the new derivative was minimized based on the default option. The prepared 3D structures were added to a created database. Docking of the prepared database into the pocket was done according to the default protocol. MOE produced database results containing the scores of the best conformers in kcal/mol. 2D and 3D interactions between the conformers and the binding site of the enzyme were obtained.66–68
Statistical Analysis
The biological parts were investigated in triplicates (n = 3) and all data were expressed as Mean ± SEM. The significance of statistical analysis was assessed by the unpaired t-test to compare the mean of two independent groups using graph Pad Prism software 8, and differences were considered statistically significant at p-values <0.05.
Results and Discussion
Chemistry
Preparation of the intermediates, 3a-g and 5, was achieved by treatment of an appropriate amine with chloroacetyl chloride as illustrated in Scheme 1.
Synthesis of 2-chloro-3-(piperazin-1-yl)quinoxaline (9) was carried out according to the procedure which involves reaction of oxalic acid with o-phenylenediamine in 4N HCl to afford quinoxaline-2,3-dione which was then treated with POCl3 in presence of DMF to give 2,3-dichloroquinoxaline (8). Treatment of compound 8 with piperazine in absolute ethanol and in the presence of triethylamine gave compound 9 (Scheme 2).
Stirring of 2-chloro-3-(piperazin-1-yl)quinoxaline (9) in aqueous NaOH solution for 24 h at 50 °C resulted in 3-(piperazin-1-yl)quinoxalin-2(1H)-one (12) as presented in Scheme 3.
Finally, compounds 10a-g and 11 were obtained by treatment of compound 9 with 3a-g and 5, as shown in Scheme 2. While treatment of compound 12 with 3a-g and 5 afforded the final compounds: 13a-g and 14, respectively (Scheme 3).
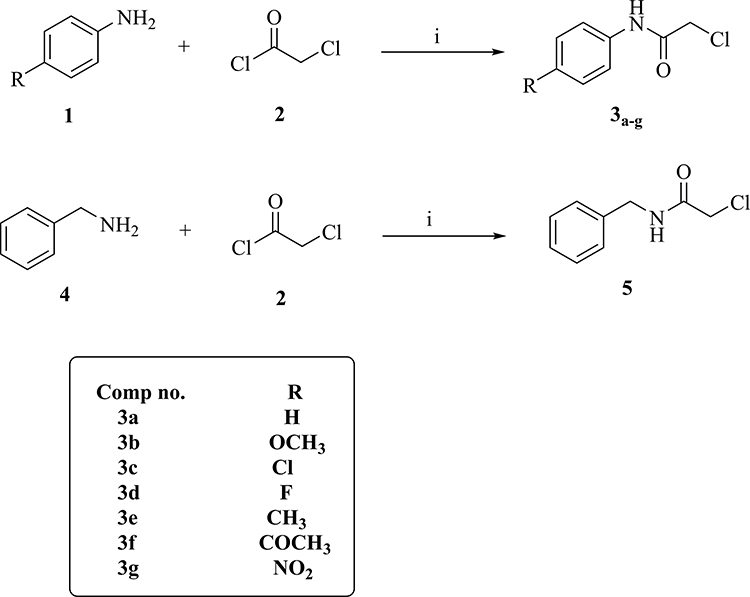 |
Scheme 1 General procedures for preparation of intermediates 3a-g and 5; conditions: (i) DMF, NaHCO3, ice bath, stirring, 1h. |
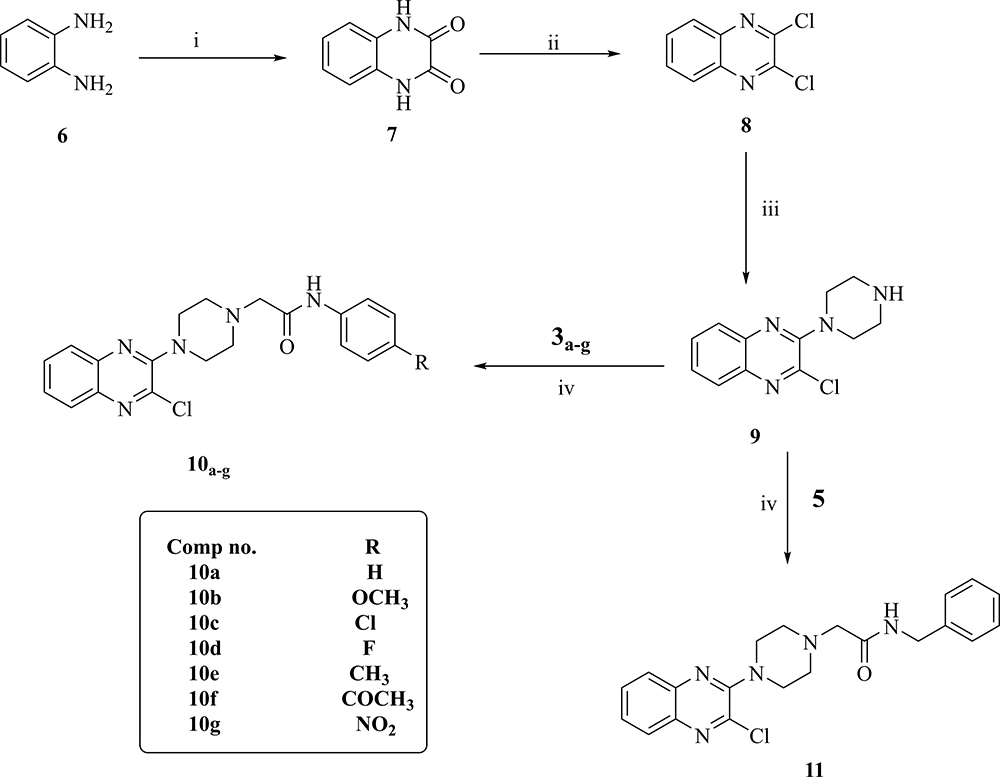 |
Scheme 2 General procedures for preparation of final compounds 10a-g and 11; Reagents and conditions: (i) oxalic acid, HCl, H2O, reflux. (ii) POCl3, DMF, reflux, 3h. (iii) piperazine, Et3N, absolute ethanol, ice bath. (iv) acetonitrile, NaHCO3, reflux, 2h. |
 |
Scheme 3 General procedures for synthesis of final compounds 13a-g and 14; Reagents and conditions: (i) NaOH, H2O, stirring, 50 °C. (ii) acetonitrile, NaHCO3, reflux, 2h. |
1H NMR charts of the new derivatives revealed that the 8 protons of the piperazine moiety appeared in two peaks; each was of 4 protons integration; the first peak was at δ value about 2.7 ppm, and the second one extended from 3.6 to 3.9 ppm. The peak of aromatic NH of acetanilide appeared at δ value ranging from 9.6 to 10.4 ppm. Its chemical shift was affected by the substituent at the benzene ring; it appeared shielded at 9.6 ppm by the electron donating effect of the methoxy group, but it was deshielded to appear at 10.4 ppm by the negative mesomeric effect of the nitro group. The aliphatic NH of compounds 11 and 14 were observed triplet at 8.4 ppm. Oxoquinoxaline derivatives showed a characteristic peak at about 12.12, ppm which is attributed to the NH of the quinoxalin-2-one nucleus.
13C NMR charts of the new derivatives showed peaks at about 169 ppm due to quinoxaline carbon attached to the piperazine moiety. They also demonstrated peaks at about 152 ppm due to CO of acetanilide. While the CO of quinoxalin-2-one nucleus appeared at about 151 ppm. The ketone CO of compounds 10f and 13f showed a characteristic peak at 197 ppm. The aromatic carbon attached to the methoxy group revealed a characteristic peak at about 155 ppm.
Biological Testing
In vitro Antitumor Assay
All the new molecules have been evaluated against 4 cancer cell lines: A549 lung cancer cells, HepG-2 hepatoma cells, Caco-2 colon cancer cells, and MDA breast cancer cells, which were obtained from American Type Culture Collection (ATCC, USA). Sorafenib was used as a standard anticancer agent.
The data presented in Table 1 indicate that the most sensitive cell line to the new candidates was A549. We can notice that all the new derivatives, except compound 14, were more potent than sorafenib, which showed an IC50 of 14.10 µM. The most potent candidate was 10b, with an IC50 of 6.48 µM; it demonstrated about twice the potency of sorafenib. Additionally, compounds 10g, 10a, 10c, 10f, 11, 10d, and 13b were far better than sorafenib, and they showed IC50 = 8.36, 9.03, 9.57, 10.31, 10.61, 10.90, and 10.97 µM, respectively. In general, 3-chloroquinoxaline derivatives displayed more potent results against A549 cells than 3-oxoquinoxaline compounds.
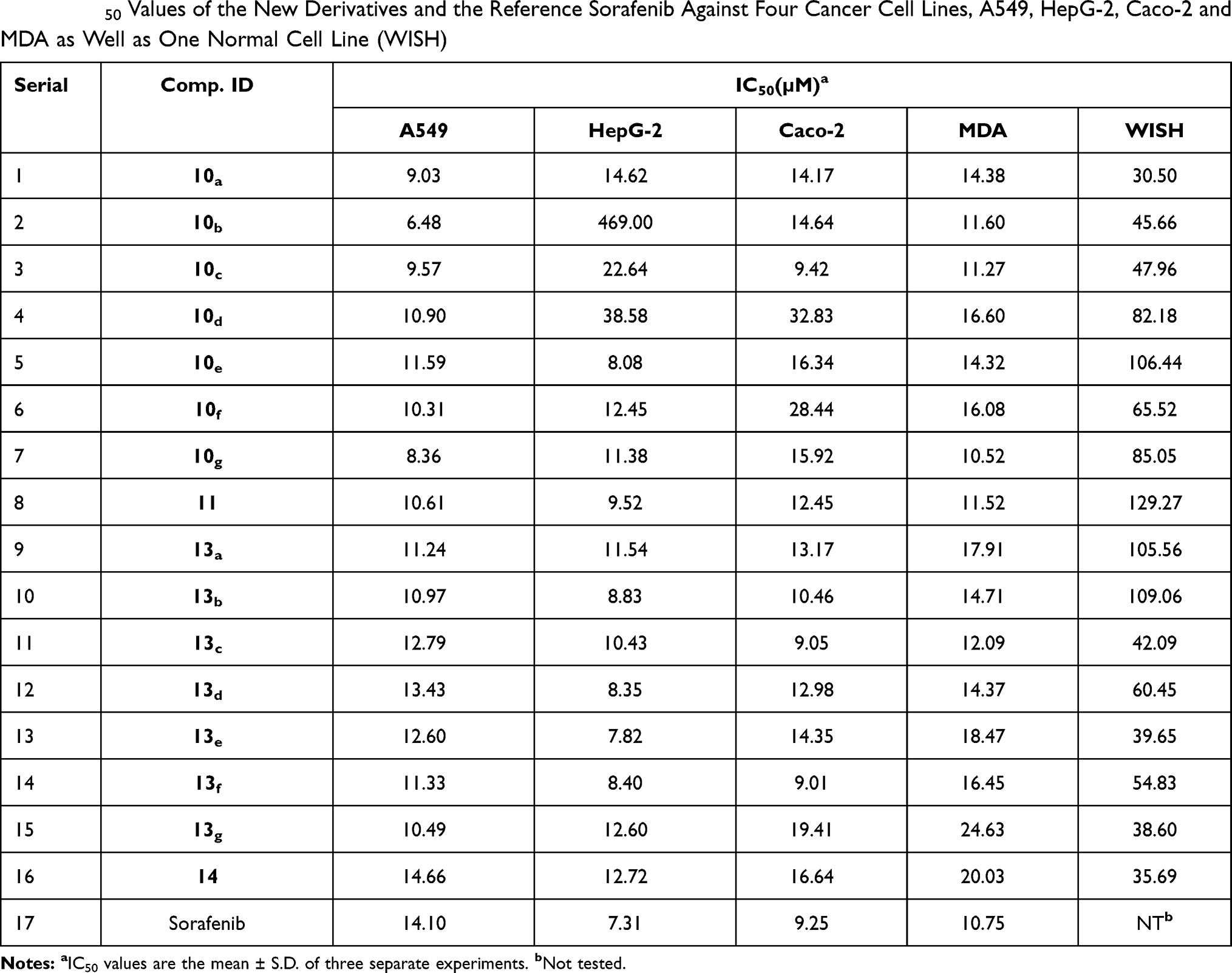 |
Table 1 IC50 Values of the New Derivatives and the Reference Sorafenib Against Four Cancer Cell Lines, A549, HepG-2, Caco-2 and MDA as Well as One Normal Cell Line (WISH) |
HepG-2 cell line came second in terms of sensitivity to the synthesized compounds. But in contrast to A549 cells, HepG-2 was more sensitive to 3-oxoquinoxaline derivatives than 3-chloroquinoxaline derivatives. Compounds 13e, 10e, 13b, 13d, and 13f were found to be the most promising candidates with IC50 = 7.82, 8.08, 8.35, 8.40, and 8.85 µM, respectively (Table 1). The results of these derivatives were weaker but close to sorafenib, which showed an IC50 of 7.31 µM. With the exception of compound 10b, the results of new compounds were considerable; IC50 ranged from 9.52 to 14.62 µM.
Regarding Caco-2 cells, we can notice that compounds 13f and 13c were more potent than sorafenib; they showed IC50 = 9.01 and 9.05 µM, respectively, compared to 9.25 µM for sorafenib. Compound 10c demonstrated an IC50 = 9.42 µM, which was comparable to that of sorafenib. Moreover, the data of compounds 13b, 11, 13d, and 13a were significant, exhibiting IC50 = 10.46, 12.45, 12.98, and 13.17 µM, respectively. Caco-2 was similar to HepG-2 in that 3-oxoquinoxaline derivatives showed better results than those of 3-chloroquinoxaline derivatives.
Finally, MDA sensitivity was higher to the effect of 3-chloroquinoxaline derivatives than 3-oxoquinoxaline derivatives. Compound 10g was found to be the most promising candidate, with an IC50 = 10.52 µM. Compared to sorafenib, which revealed an IC50 = 10.75 µM, 10g was slightly better. Furthermore, compounds 10c, 11, 10b, and 13c showed activities close to that of sorafenib; they demonstrated IC50 = 11.27, 11.52, 11.60, and 12.09 µM, respectively.
Initially, the cytotoxic effect of all the new derivatives was examined against the WISH normal cell line. The results given in Table 1 reveal good degrees of selectivity. It can be noticed that the results of compound 11 are of particular significance. It exhibited selectivity indices = 12.18, 13.57, 10.38, and 11.22 to A549, HepG-2, Caco-2, and MDA cell lines, respectively. The second most selective compound was 13b; its selectivity indices were 9.94, 12.35, 10.42, and 7.40 to the four cell lines, respectively. Compound 10e came in third place, with selectivity indices = 9.18, 13.17, 6.51, and 7.43 to the four cell lines, respectively. Furthermore, compounds 13a, 10g, 13f, 13d, 10f, 10c, 10d, 13e, and 10b showed good selectivity to one or more cancer cell lines.
Compounds 11, 13b, 10e, 13a, 10g, 13f, and 13d, which showed the most valuable results in cytotoxicity testing, have been selected for further evaluation against VEGFR-2 kinase.
VEGFR-2 (KDR) Kinase Assay
The most prominent seven candidates (11, 13b, 10e, 13a, 10g, 13f, and 13d) were evaluated in vitro against VEGFR-2 kinase. The results presented in Table 2 show that the new derivatives managed to inhibit VEGFR-2 kinase at sub-micromolar concentrations. The IC50 values of the new derivatives ranged from 0.192 to 0.602 µM. It is apparent from the data supplied that the most significant derivative was compound 11; it exhibited an IC50 of 0.192 µM compared to 0.082 µM for sorafenib (the positive control). Concentration-dependent kinase inhibition plots of compound 11 and sorafenib are presented in Figure 2 and Figure 3, respectively. The second most potent derivative was compound 10e, with an IC50 = 0.241 µM. Activity of the other candidates can be arranged in a descending order as follows: 13a (IC50 = 0.258 µM), 10g (IC50 = 0.332 µM), 13f (IC50 = 0.465 µM), 13b (IC50 = 0.471 µM), and 13d (IC50 = 0.602 µM).
 |
Table 2 VEGFR-2 IC50 of the Tested Derivatives Compared to Sorafenib |
 |
Table 3 Apoptosis and Necrosis Rates of HepG-2 Cells Treated with Compound 11 Compared to Control HepG-2 Cells |
 |
Table 4 The Impact of Compound 11 on Expression of the Tested Proteins in HepG-2 Cells Treated For 48 h |
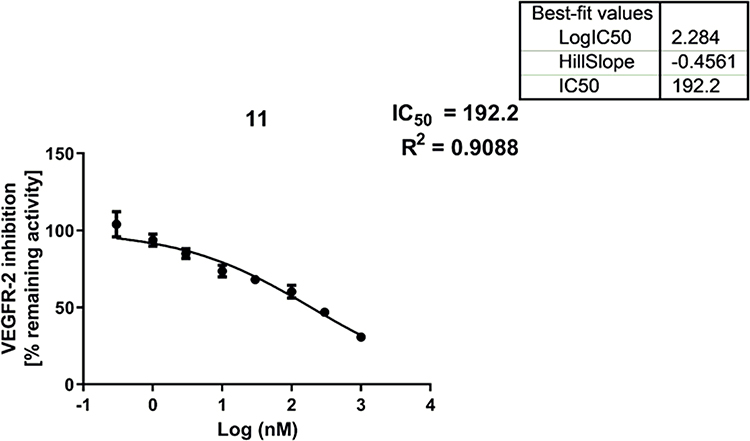 |
Figure 2 Concentration dependent kinase inhibition plot of compound 11. The percent VEGFR-2 inhibition was determined for compound 11 relative to control, and the data were fitted to the dose-response curve to obtain IC50 values. Error bars represent SEM of three replicates. |
 |
Figure 3 Concentration dependent kinase inhibition plot of sorafenib. The percent VEGFR-2 inhibition was determined for sorafenib relative to control, and the data were fitted to the dose-response curve to obtain IC50 values. Error bars represent SEM of three replicates. |
VEGFR-2 inhibitors have been shown to induce apoptosis.51 So, we evaluated cell cycle kinetics and apoptosis rates in HepG-2 treated with compound 11, which was the most potent VEGFR-2 inhibitor candidate. We aimed to examine reasons other than VEGFR-2 inhibition behind the significant antitumor results of the new derivatives.
Effect on Cell Cycle Kinetic
It is clear from Figure 4 that 9.52 μM of compound 11 displayed a strong impact on the different cell cycle phases of HepG-2 cells compared to control cells. We can notice that the most significant increase in cell accumulation rate was at S phase, from 20.14% to 46.10%. Also, cell accumulation in the G2/M phase was increased from 15.16% to 25.22%. On the other hand, there was a sharp decrease in cell accumulation percentage in phase G1 from 63.18% to 27.37%.
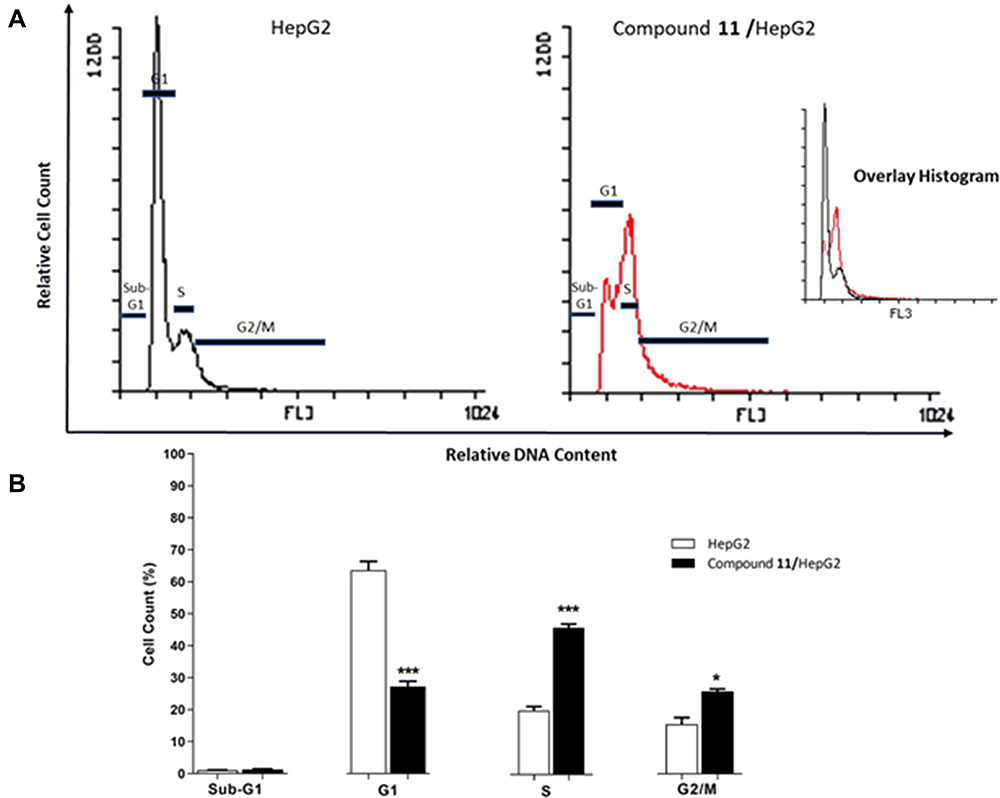 |
Figure 4 Flow cytometric analysis of HepG-2 cell cycle phases after 48 h treatment with 9.52 µM (IC50 value) of compound 11. After treatment with 11, HepG-2 cells were harvested, stained and then cell cycle phases were analyzed using Flowing software. (A) Illustrating histograms show the cell cycle distribution of both HepG-2 test cells and control. (B) Column diagram represents percentage of cells accumulation in the different phases of cell cycle. Results have been calculated as mean ± SEM of 3 experiments. *p < 0.05, ***p < 0.001 statistically indicate the significance of the obtained results compared to control (HepG-2). |
Annexin V-FITC Apoptosis Assay
It is apparent from the results illustrated in Table 3 and Figure 5 that there was a sharp increase in HepG-2 cells apoptosis due to the effect of 9.52 μM (IC50 value) of compound 11. As can be seen, the early apoptosis rate was increased exponentially by about 9-fold, from 4.75% to 43.60%. At the same time, late apoptosis was increased by 3-fold compared to control. On the other hand, the given information revealed a weak effect of compound 11 on HepG-2 necrosis. Additionally, there was a dramatic decrease in HepG-2 viable cells from 95.54% to 55.95% due to the effect of compound 11. Further evaluation was carried out to explore the mechanism of apoptosis induced by compound 11.
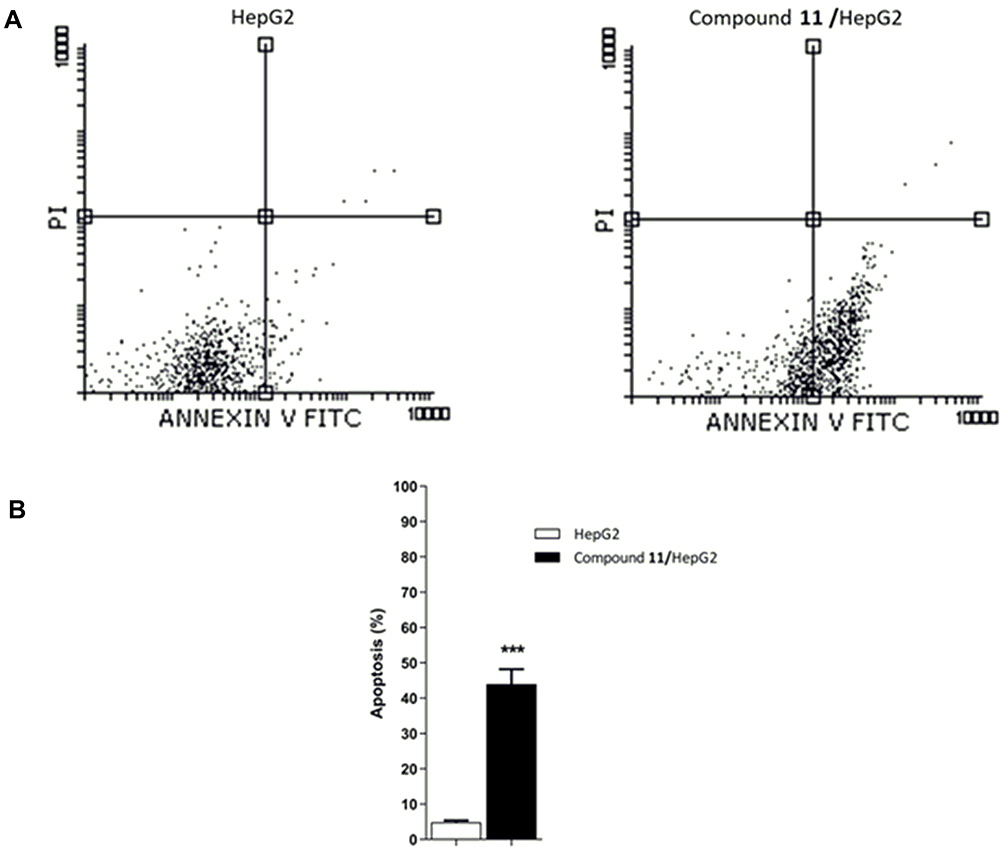 |
Figure 5 Apoptosis rate in HepG-2 cells treated with (9.52 µM) of compound 11 for 48 h in comparison to control HepG-2 cells. (A) The obtained flow cytometric figures for both test and control HepG-2 cells. (B) Data from quantification of apoptosis are presented as mean ± SEM of 3 experiments. ***p < 0.001 statistically indicate the significance of the obtained results in comparison to control (HepG-2). |
Effect on Levels of BAX and Bcl-2 Protein Expression
Western blot analysis provided data concerning the effect of 9.52 µM (IC50 value) of compound 11 on the expression of certain proteins in HepG-2 cells treated for 48h, as shown in Table 4 and Figure 6. It is clear from the results given that there was a double increase in BAX protein expression level compared to control HepG-2 cells. Meanwhile, Bcl-2 expression level decreased to half compared to control cells. Accordingly, a 4-fold rise in the BAX/Bcl-2 ratio was obtained. BAX was reported as a proapoptotic member of the Bcl-2 family. On the other hand, Bcl-2 is an antiapoptotic protein.52,53
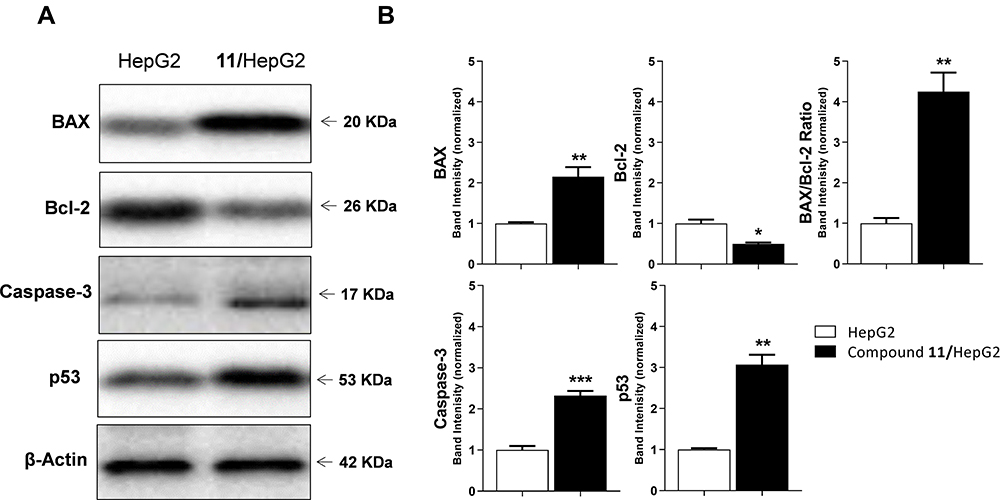 |
Figure 6 The immunoblotting of the expression of BAX, Bcl-2, Caspase-3, and p53 (Normalized to β-actin). (A) A given Western blot illustrations display the impact of 9.52 µM (IC50 value) of compound 11 on the tested proteins expression in HepG-2 cells treated for 48h. (B) The evaluated proteins quantity in the cell lysates. The reference HepG-2 cells were set to “1”, and all results from 3 experiments are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 indicate the importance of the obtained results statistically. |
Effect on Caspase-3 Level
Caspase-3 is known as the primary executioner caspase in apoptosis.54,55 Caspase-3 level was increased by 2.3-fold in HepG-2 cells treated with compound 11 (9.52 µM) compared to untreated cells, as presented in Figure 6 and Table 4.
Effect on P53 Level
Further evaluation of compound 11 (IC50 value) at HepG-2 showed an effective rise in the expression level of the tumor suppressor protein p53. A 3-fold increase in the expression level of p53 can be seen in Figure 6 and Table 4. P53 protein was reported to have many biological activities, such as apoptosis, DNA repair, and cell cycle arrest.56–59 It was also proven to inhibit tumor angiogenesis.59
The data obtained from Western blot analysis reveals that the apoptosis inducing effect of compound 11 is more likely to be attributed to its ability to increase the BAX/Bcl-2 ratio and levels of caspase-3 and p53 proteins.
Molecular Docking
We carried out docking studies for the new derivatives and sorafenib (reference of biology) in order to get insights into their binding and affinity to VEGFR-2. The protein (ID: 3VHE) was downloaded with its co-crystallized ligand (pyrrolopyrimidine inhibitor) from protein data bank (PDB) web site. The redocked ligand showed a root mean square deviation (RMSD) of 0.42 Å which means that the executed docking protocol was valid. Figure S1 in The supplementary data.pdf shows the overlay of the redocked and the co crystallized ligand. The supplied figure shows that the ligand displayed two hydrogen bonds with Glu885 and another one with Asp1046 via its urea moiety. One more hydrogen bond was formed between NH of the pyrrolopyrimidine nucleus and Cys919. Additionally, the planar aromatic moiety demonstrated a π cation interaction with Leu1035.
Sorafenib binding mode was found to be similar to that of the ligand as shown Figure S2 in The supplementary data.pdf; the urea moiety showed two hydrogen bonds (length: 1.87 Å and 1.97 Å) with Glu885 and another one (length: 1.77 Å) with Asp1046. Moreover, the phenoxy group showed a π cation interaction with the alkyl side chain of Val899.
With respect to the new derivatives, it was found that all compounds showed a good fit to the pocket with binding modes similar to that of the ligand. Figure 7 represents the binding mode of compound 11; as can be seen the amide group demonstrated two hydrogen bonds (length: 1.75 Å and 1.85 Å) with the essential amino acids Glu885 and Asp1046. Meanwhile, the quinoxaline moiety was oriented to occupy the ATP binding domain, showing a π cation interaction with the alkyl side chain of Val848. It was also found that the terminal benzylic group was directed to form hydrophobic interactions with the alkyl side chains of Ile888, Leu889, and Ile892 in the allosteric pocket. Figure 8 reveals that the binding patterns of compound 11 and the co-crystallized ligand are almost the same. These data clearly suggest that compound 11 is proposed to bind correctly and efficiently with VEGFR-2 kinase.
 |
Figure 7 Binding mode of compound 11. |
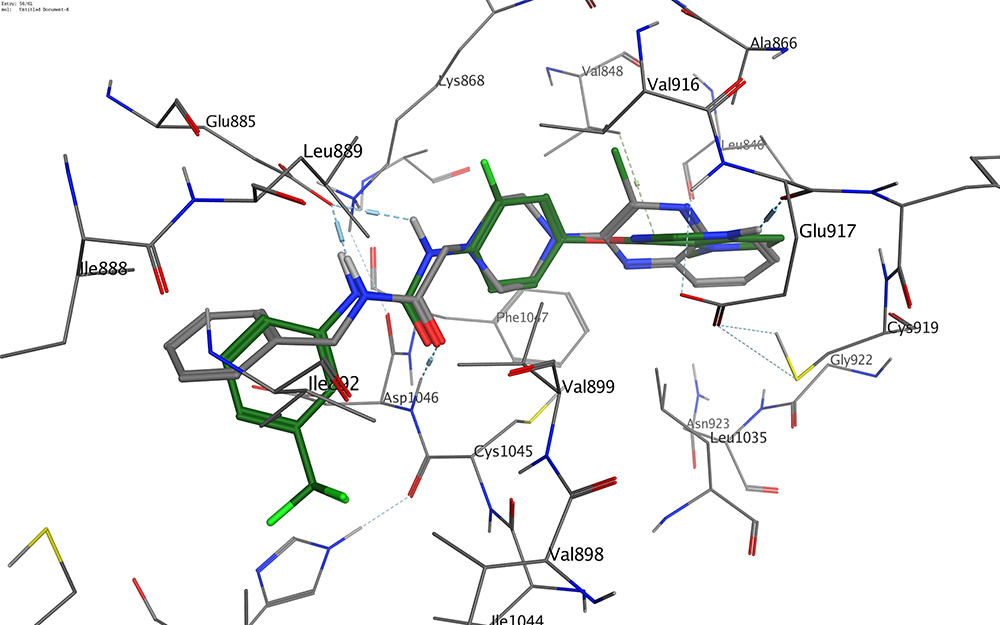 |
Figure 8 Overlay of compound 11 (gray colored) and the ligand (green colored). |
The binding pattern of compound 10e was also found to be similar to the ligand (Figure S3 in supplementary materials); we can see in the figure that the amide group showed a hydrogen bond with Glu885 and Asp1046. The methylene group of acetanilide demonstrated a π cation interaction with Glu885. The quinoxaline moiety revealed a π cation interaction with Val848. The terminal aromatic moiety demonstrated a π cation interaction with the alkyl side chain of Asp1046.
The binding mode of compound 13a is presented in Figure S4 in the supplementary materials. The amide group showed a hydrogen bond with Glu885 and another one with Asp1046. Moreover, the quinoxaline moiety displayed an essential hydrogen bond with Glu917 and a π cation interaction with Cys919. This π cation interaction improved the binding affinity of compound 13a.
Structure Activity Relationship
According to biological results obtained, relationships can be established between the structures and activities of the new derivatives as follows:
- Chloroquinoxaline derivatives are more potent antiproliferative agents against A549 and MDA than their corresponding oxoquinoxaline derivatives.
- Regarding HepG-2 and Caco-2, oxoquinoxaline derivatives showed better activity than chloroquinoxaline derivatives.
- The methoxy group at position 4 of the terminal phenyl ring revealed higher activity against A549 than other substituents at the same position.
- 4-methylphenyl as a terminal hydrophobic moiety was the most potent against HepG-2.
- 4-chlorophenyl as a terminal hydrophobic moiety was found to be the best against both Caco-2 and MDA.
- With regard to VEGFR-2 inhibition, the chlorine atom at position 2 of quinoxaline was superior to the oxygen at the same position.
- Non polar substituents at position-4 of the terminal phenyl showed better activity than polar substituents in terms of VEGFR-2 inhibition.
Conclusion
In accordance with the reported pharmacophoric features of VEGFR-2 inhibitor agents and on the basis of the antihypertensive property of the piperazinylquinoxaline moiety, we designed and synthesized new sixteen piperazinylquinoxaline derivatives as potential safer antitumor candidates. The obtained data showed that the new derivatives revealed considerable antitumor activities against four cancer cell lines: A549, HepG-2, Caco-2, and MDA, with considerable degrees of selectivity to cancer cells. Meanwhile, the studied candidates managed to inhibit VEGFR-2 kinase at sub-micromolar concentrations. Accordingly, we suggest that future work on the new derivatives by modification and further evaluation would be more likely to develop potent anticancer agents targeting VEGFR-2 kinase.
The most significant candidate to be considered for further work was compound 11, which displayed an encouraging profile of biological results and in silico data. It showed IC50 = 10.61, 9.52, 12.45, and 11.52 µM against A549, HepG-2, Caco-2, and MDA cell lines, respectively. While it showed IC50 of 129.27 µM against the WISH normal cell. Additionally, it demonstrated an IC50 = 0.192 µM against VEGFR-2 kinase. Moreover, HepG-2 treated with 9.52 µM of 11 revealed noticeable increases in BAX/Bcl-2 ratio, caspase-3 level, and P53 expression; these factors collectively account for the dramatic increase in apoptosis rate from 5% to about 44%.
Acknowledgment
The authors extend their appreciation to the Research Center at AlMaarefa University for funding this work under TUMA project agreement number (TUMA-2021-4).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Ma X, Yu H. Cancer issue: global burden of cancer. Yale J Biol Med. 2006;79(3–4):85.
2. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. El-Zahabi M. Design, molecular modeling and synthesis of new immunomodulatory agents for biological studies. Al-Azhar J Pharm Sci. 2021;64(2):1–20. doi:10.21608/ajps.2021.187700
4. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
5. Ghosh S, Sullivan CAW, Zerkowski MP, et al. High levels of vascular endothelial growth factor and its receptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome in breast cancer. Hum Pathol. 2008;39(12):1835–1843. doi:10.1016/j.humpath.2008.06.004
6. Seto T, Higashiyama M, Funai H, et al. Prognostic value of expression of vascular endothelial growth factor and its flt-1 and KDR receptors in stage I non-small-cell lung cancer. Lung Cancer. 2006;53(1):91–96. doi:10.1016/j.lungcan.2006.02.009
7. Duff SE, Jeziorska M, Rosa DD, et al. Vascular endothelial growth factors and receptors in colorectal cancer: implications for anti-angiogenic therapy. Eur J Cancer. 2006;42(1):112–117. doi:10.1016/j.ejca.2005.09.018
8. Chu JS, Ge FJ, Zhang B, et al. Expression and prognostic value of VEGFR-2, PDGFR-β, and c-Met in advanced hepatocellular carcinoma. J Exp Clin Cancer Res. 2013;32(1):1–8. doi:10.1186/1756-9966-32-16
9. Xia G, Kumar SR, Hawes D, et al. Expression and significance of vascular endothelial growth factor receptor 2 in bladder cancer. J Urol. 2006;175(4):1245–1252. doi:10.1016/S0022-5347(05)00736-6
10. Basagiannis D, Zografou S, Murphy C, et al. VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation through macropinocytosis. J Cell Sci. 2016;129(21):4091–4104. doi:10.1242/jcs.188219
11. Herkenne S, Paques C, Nivelles O, et al. The interaction of uPAR with VEGFR2 promotes VEGF-induced angiogenesis. Sci Signal. 2015;8(403):ra117–ra117. doi:10.1126/scisignal.aaa2403
12. Zhong M, Li N, Qiu X, et al. TIPE regulates VEGFR2 expression and promotes angiogenesis in colorectal cancer. Int J Biol Sci. 2020;16(2):272. doi:10.7150/ijbs.37906
13. Nishida N, Yano H, Nishida T, et al. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2(3):213. doi:10.2147/vhrm.2006.2.3.213
14. Yang Y, Sun M, Wang L, et al. HIFs, angiogenesis, and cancer. J Cell Biochem. 2013;114(5):967–974. doi:10.1002/jcb.24438
15. Xu WW, Li B, Lam AK, et al. Targeting VEGFR1-and VEGFR2-expressing non-tumor cells is essential for esophageal cancer therapy. Oncotarget. 2015;6(3):1790. doi:10.18632/oncotarget.2781
16. Jung K, Heishi T, Khan OF, et al. Ly6C lo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy. J Clin Invest. 2017;127(8):3039–3051. doi:10.1172/JCI93182
17. Pysz MA, Foygel K, Rosenberg J, et al. Antiangiogenic cancer therapy: monitoring with molecular US and a clinically translatable contrast agent (BR55). Radiology. 2010;256(2):519–527. doi:10.1148/radiol.10091858
18. Fontanella C, Ongaro E, Bolzonello S, et al. Clinical advances in the development of novel VEGFR2 inhibitors. Ann Trans Med. 2014;2(12):123.
19. Abdallah AE, Eissa SI, Al Ward MMS, et al. Design, synthesis and molecular modeling of new quinazolin-4 (3H)-one based VEGFR-2 kinase inhibitors for potential anticancer evaluation. Bioorg Chem. 2021;109:104695. doi:10.1016/j.bioorg.2021.104695
20. Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5(10):835–844. doi:10.1038/nrd2130
21. Strumberg D, Scheulen ME, Schultheis B, et al. Regorafenib (BAY 73-4506) in advanced colorectal cancer: a Phase I study. Br J Cancer. 2012;106(11):1722–1727. doi:10.1038/bjc.2012.153
22. Subbiah V, Meric-Bernstam F, Mills GB, et al. Next generation sequencing analysis of platinum refractory advanced germ cell tumor sensitive to Sunitinib (Sutent®) a VEGFR2/PDGFRβ/c-kit/FLT3/RET/CSF1R inhibitor in a Phase II trial. J Hematol Oncol. 2014;7(1):1–10. doi:10.1186/s13045-014-0052-x
23. Harris PA, Boloor A, Cheung M, et al. Discovery of 5-[[4-[(2, 3-dimethyl-2 H-indazol-6-yl) methylamino]-2-pyrimidinyl] amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem. 2008;51(15):4632–4640.
24. Kong LJ, Li H, Du Y-J, et al. Vatalanib, a tyrosine kinase inhibitor, decreases hepatic fibrosis and sinusoidal capillarization in CCl4-induced fibrotic mice. Mol Med Rep. 2017;15(5):2604–2610. doi:10.3892/mmr.2017.6325
25. Modi S, Kulkarni V. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: Medicinal Chemistry perspective. Med Drug Discov. 2019;2:100009. doi:10.1016/j.medidd.2019.100009
26. Zuccotto F, Ardini E, Casale E, et al. Through the “gatekeeper door”: exploiting the active kinase conformation. J Med Chem. 2010;53(7):2681–2694. doi:10.1021/jm901443h
27. Lintnerová L, García-Caballero M, Gregáň F, et al. A development of chimeric VEGFR2 TK inhibitor based on two ligand conformers from PDB: 1Y6A complex–Medicinal chemistry consequences of a TKs analysis. Eur J Med Chem. 2014;72:146–159. doi:10.1016/j.ejmech.2013.11.023
28. Blanc J, Geney R, Menet C. Type II kinase inhibitors: an opportunity in cancer for rational design. Anticancer Agents Med Chem. 2013;13(5):731–747.
29. Regan J, Pargellis CA, Cirillo PF, et al. The kinetics of binding to p38 MAP kinase by analogues of BIRB 796. Bioorg Med Chem Lett. 2003;13(18):3101–3104. doi:10.1016/S0960-894X(03)00656-5
30. Abdullaziz MA, Abdel-Mohsen HT, El Kerdawy AM, et al. Design, synthesis, molecular docking and cytotoxic evaluation of novel 2-furybenzimidazoles as VEGFR-2 inhibitors. Eur J Med Chem. 2017;136:315–329. doi:10.1016/j.ejmech.2017.04.068
31. Wu S, Chen JJ, Kudelka A, et al. Incidence and risk of hypertension with sorafenib in patients with cancer: a systematic review and meta-analysis. Lancet Oncol. 2008;9(2):117–123. doi:10.1016/S1470-2045(08)70003-2
32. Wang Z, Xu J, Nie W, et al. Risk of hypertension with regorafenib in cancer patients: a systematic review and meta-analysis. Eur J Clin Pharmacol. 2014;70(2):225–231. doi:10.1007/s00228-013-1598-1
33. Uschner FE, Schueller F, Nikolova I, et al. The multikinase inhibitor regorafenib decreases angiogenesis and improves portal hypertension. Oncotarget. 2018;9(90):36220. doi:10.18632/oncotarget.26333
34. Motzer RJ, Nosov D, Eisen T, et al. Tivozanib versus sorafenib as initial targeted therapy for patients with advanced renal cell carcinoma: results from a Phase III randomized, open-label, multicenter trial. J Clin Oncol. 2012;30(15):277S. doi:10.1200/jco.2012.30.15_suppl.4501
35. Shahin MI, Abou El Ella DA, Ismail NSM, et al. Design, synthesis and biological evaluation of type-II VEGFR-2 inhibitors based on quinoxaline scaffold. Bioorg Chem. 2014;56:16–26. doi:10.1016/j.bioorg.2014.05.010
36. Akhtar R, Zahoor AF, Rasul A, et al. Design, synthesis, in-silico study and anticancer potential of novel n-4-piperazinyl-ciprofloxacin-aniline hybrids. Pak J Pharm Sci. 2019;32(5):2215–2222.
37. Murty M, Ramalingeswara Rao B, Katiki MR, et al. Synthesis of piperazinyl benzothiazole/benzoxazole derivatives coupled with 1, 3, 4-oxadiazole-2-thiol: novel hybrid heterocycles as anticancer agents. Med Chem Res. 2013;22(10):4980–4991. doi:10.1007/s00044-013-0510-y
38. Rejmund M, Mrozek-Wilczkiewicz A, Malarz K, et al. Piperazinyl fragment improves anticancer activity of Triapine. PLoS One. 2018;13(4):e0188767. doi:10.1371/journal.pone.0188767
39. Abou-Seri SM, Eldehna WM, Ali MM, et al. 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: synthesis and in vitro biological evaluation. Eur J Med Chem. 2016;107:165–179. doi:10.1016/j.ejmech.2015.10.053
40. Alizadeh A, Moafi L. Simple access to spirooxadiazole compounds containing a quinoxaline moiety using a nitrile imine intermediate generated in situ. Heterocycl Commun. 2017;23(5):375–378. doi:10.1515/hc-2017-0084
41. Monge Vega A, Gil MJ, Basilio A, et al. 1-Hydrazinopyridazino [4, 5-b] quinoxaline, a new antihypertensive agent. Eur J Med Chem. 1986;21(3):251–254.
42. Xie S, Li X, Yu H, et al. Design, synthesis and biological evaluation of isochroman-4-one hybrids bearing piperazine moiety as antihypertensive agent candidates. Bioorg Med Chem. 2019;27(13):2764–2770. doi:10.1016/j.bmc.2019.05.004
43. Jianzhi S, Qizeng W, Bin L, et al. Piperazine ferulate exerts antihypertensive effect and improves endothelial function in vitro and in vivo via the activation of endothelial nitric oxide synthase. Cell Mol Biol. 2019;65(3):119–124. doi:10.14715/cmb/2019.65.3.18
44. Zheng C-B, Gao W-C, Pang -P-P, et al. Synthesis and vasorelaxant evaluation of novel 7-methoxyl-2, 3-disubstituted-quinoxaline derivatives. Bioorg Med Chem Lett. 2021;36:127785. doi:10.1016/j.bmcl.2021.127785
45. Lee K, Jeong K-W, Lee Y, et al. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur J Med Chem. 2010;45(11):5420–5427. doi:10.1016/j.ejmech.2010.09.002
46. Xie -Q-Q, Xie H-Z, Ren J-X, et al. Pharmacophore modeling studies of type I and type II kinase inhibitors of Tie2. J Mol Graph Model. 2009;27(6):751–758. doi:10.1016/j.jmgm.2008.11.008
47. Aziz MA, Serya RAT, Lasheen DS, et al. Discovery of potent VEGFR-2 inhibitors based on furopyrimidine and thienopyrimidne scaffolds as cancer targeting agents. Sci Rep. 2016;6(1):1–20. doi:10.1038/srep24460
48. Machado VA, Peixoto D, Costa R, et al. Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno [3, 2-b] pyridin-7-ylthio) phenyl] ureas: discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors. Bioorg Med Chem. 2015;23(19):6497–6509. doi:10.1016/j.bmc.2015.08.010
49. Viegas-Junior C, Danuello A, da Silva Bolzani V, et al. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr Med Chem. 2007;14(17):1829–1852. doi:10.2174/092986707781058805
50. Bosquesi PL, Melo TRF, Vizioli EO, et al. Anti-inflammatory drug design using a molecular hybridization approach. Pharmaceuticals. 2011;4(11):1450–1474. doi:10.3390/ph4111450
51. Harmey JH, Bouchier‐Hayes D. Vascular endothelial growth factor (VEGF), a survival factor for tumour cells: implications for anti‐angiogenic therapy. Bioessays. 2002;24(3):280–283. doi:10.1002/bies.10043
52. Bagci E, Vodovotz Y, Billiar TR, et al. Bistability in apoptosis: roles of bax, bcl-2, and mitochondrial permeability transition pores. Biophys J. 2006;90(5):1546–1559. doi:10.1529/biophysj.105.068122
53. Brady HJ, Gil-Gómez G. Molecules in focus Bax. The pro-apoptotic Bcl-2 family member, Bax. Int J Biochem Cell Biol. 1998;30(6):647–650. doi:10.1016/S1357-2725(98)00006-5
54. Slee EA, Adrain C, Martin SJ. Executioner caspase-3,-6, and-7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276(10):7320–7326. doi:10.1074/jbc.M008363200
55. Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326(1):1–16. doi:10.1042/bj3260001
56. Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6(3):a026104. doi:10.1101/cshperspect.a026104
57. Shaw PH. The role of p53 in cell cycle regulation. Pathol Res Pract. 1996;192(7):669–675. doi:10.1016/S0344-0338(96)80088-4
58. Pellegata NS, Antoniono RJ, Redpath JL, et al. DNA damage and p53-mediated cell cycle arrest: a reevaluation. Proc Natl Acad Sci. 1996;93(26):15209–15214. doi:10.1073/pnas.93.26.15209
59. Teodoro JG, Evans SK, Green MR. Inhibition of tumor angiogenesis by p53: a new role for The Guardian of the genome. J Mol Med. 2007;85(11):1175–1186. doi:10.1007/s00109-007-0221-2
60. Gerlier D, Thomasset N. Use of MTT colorimetric assay to measure cell activation. J Immunol Methods. 1986;94(1–2):57–63. doi:10.1016/0022-1759(86)90215-2
61. Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci. 2000;113(5):753–757. doi:10.1242/jcs.113.5.753
62. Lo KK-W, Lee TK-M, Lau JS-Y, et al. Luminescent biological probes derived from ruthenium (II) estradiol polypyridine complexes. Inorg Chem. 2008;47(1):200–208. doi:10.1021/ic701735q
63. Aborehab NM, Elnagar MR, Waly NE. Gallic acid potentiates the apoptotic effect of paclitaxel and carboplatin via overexpression of Bax and P53 on the MCF‐7 human breast cancer cell line. J Biochem Mol Toxicol. 2021;35(2):e22638. doi:10.1002/jbt.22638
64. Elnagar MR, Walls AB, Helal GK, et al. Functional characterization of α7 nicotinic acetylcholine and NMDA receptor signaling in SH-SY5Y neuroblastoma cells in an ERK phosphorylation assay. Eur J Pharmacol. 2018;826:106–113. doi:10.1016/j.ejphar.2018.02.047
65. Burnette WN. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981;112(2):195–203. doi:10.1016/0003-2697(81)90281-5
66. Abd-Elhamid AI, El-Gendi H, Abdallah AE, et al. Novel nanocombinations of l-tryptophan and l-cysteine: preparation, characterization, and their applications for antimicrobial and anticancer activities. Pharmaceutics. 2021;13(10):1595. doi:10.3390/pharmaceutics13101595
67. Dawoud N, El-fakharany E, El-gendi H, et al. Consolidated antimicrobial and anticancer activities through newly synthesized novel series of pyrazoles bearing indazolylthiazole moiety: characterization and molecular docking. Egypt J Chem. 2021;64(11):5–6. doi:10.21608/ejchem.2021.83623.4104
68. Abdallah AE, Alesawy MS, Eissa SI, et al. Design and synthesis of new 4-(2-nitrophenoxy)benzamide derivatives as potential antiviral agents: molecular modeling and in vitro antiviral screening. New J Chem. 2021;45(36):16557–16571. doi:10.1039/D1NJ02710G
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.