")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
New Patient-Centric Approaches to the Management of Alpha-1 Antitrypsin Deficiency
Authors Chorostowska-Wynimko J , Barrecheguren M, Ferrarotti I , Greulich T , Sandhaus RA, Campos M
Received 14 October 2019
Accepted for publication 24 January 2020
Published 12 February 2020 Volume 2020:15 Pages 345—355
DOI https://doi.org/10.2147/COPD.S234646
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Joanna Chorostowska-Wynimko,1 Miriam Barrecheguren,2 Ilaria Ferrarotti,3 Timm Greulich,4 Robert A Sandhaus,5 Michael Campos6
1Department of Genetics and Clinical Immunology, National Institute of Tuberculosis and Lung Diseases, Warsaw, Poland; 2Department of Pneumology, University Hospital Vall d’Hebron, Barcelona, Spain; 3Department of Internal Medicine and Therapeutics, Pneumology Unit IRCCS San Matteo Hospital Foundation, University of Pavia, Pavia, Italy; 4Department of Medicine, Pulmonary and Critical Care Medicine, University Medical Centre Giessen and Marburg, Philipps-University, Member of the German Centre for Lung Research (DZL), Marburg, Germany; 5Division of Pulmonary, Critical Care and Sleep Medicine, National Jewish Health, Denver, CO, USA; 6Division of Pulmonary, Allergy, Critical Care and Sleep Medicine, University of Miami School of Medicine, Miami, FL, USA
Correspondence: Joanna Chorostowska-Wynimko
Department of Genetics and Clinical Immunology, National Institute of Tuberculosis and Lung Diseases, ul. Płocka 26, 01-138, Warsaw, Poland
Tel +48 22 43 12 158
Fax +48 22 43 12 358
Email [email protected]
Abstract: Alpha-1 antitrypsin deficiency (AATD) is a rare and underdiagnosed genetic predisposition for COPD and emphysema and other conditions, including liver disease. Although there have been improvements in terms of awareness of AATD and understanding of its treatment in recent years, current challenges center on optimizing detection and management of patients with AATD, and improving access to intravenous (IV) AAT therapy – the only available pharmacological intervention that can slow disease progression. However, as an orphan disease with geographically dispersed patients, international cooperation is essential to address these issues. To achieve this, new European initiatives in the form of the European Reference Network for Rare Lung Diseases (ERN-LUNG) and the European Alpha-1 Research Collaboration (EARCO) have been established. These organizations are striving to address the current challenges in AATD, and provide a new platform for future research efforts in AATD. The first objectives of ERN-LUNG are to establish a quality control program for European AATD laboratories and create a disease management program for AATD, following the success of such programs in the United States. The main purpose of EARCO is to create a pan-European registry, with the aim of understanding the natural history of the disease and supporting the development of new treatment modalities in AATD and access to AAT therapy. Going further, other patient-centric initiatives involve improving the convenience of intravenous AAT therapy infusions through extended-interval dosing and self-administration. The present review will discuss the implementation of these initiatives and their potential contribution to the optimization of patient care in AATD.
Keywords: Alpha-1 antitrypsin deficiency, registries, testing, self-administration, alternative dosing
Introduction
Alpha-1 antitrypsin deficiency (AATD) is a rare genetic disorder characterized by low levels or dysfunctional forms of an altered alpha-1 antitrypsin (AAT) protein, which predisposes affected individuals to develop pulmonary complications, including chronic obstructive pulmonary disease (COPD) and early-onset emphysema.1 Cigarette smoking is a major risk factor that accelerates the pulmonary complications associated with this condition. In addition, patients may also develop liver disease (chronic hepatitis, cirrhosis and hepatocellular carcinoma), which can present as early as childhood,2 and other less common disorders such as panniculitis3 and vasculitis.4 As diseases associated with AATD are progressive, early diagnosis is key in order to initiate proactive interventions to minimize pulmonary complications, such as smoking cessation and treatment with intravenous (IV) infusions of donor-derived purified human AAT to slow the progression of emphysema.5–7
Awareness of AATD as the underlying cause of COPD or emphysema is crucial as the condition has no unique clinical features. Patients often present with non-specific respiratory symptoms common to other conditions (ie, non-genetic COPD and asthma), such as wheezing, dyspnea, and shortness of breath.8 Signs and symptoms of fibrosis and cirrhosis caused by the misfolded AAT protein are also non-specific, and can manifest in the absence of, or alongside, respiratory symptoms.9 Therefore, only laboratory analyses can conclusively determine if a patient has AATD. Given the range of tests available to clinicians, efforts towards the standardization of laboratory diagnostics for AATD are under way and will be discussed later.10,11 Underdiagnosis, a continuing issue in the field with reports of delays of up to 6 years before a diagnosis of AATD is confirmed, is of particular significance as diagnostic delays have been associated with greater symptom burden at the time of eventual diagnosis.12 This may result from reluctance of the patient to seek medical attention, lack of health care practitioner awareness, and failure to conduct AATD testing when warranted.5,13–16 A recent study from the United Kingdom (UK) found that only ~2% of COPD patients diagnosed at <60 years had undergone any form of AATD testing.16 Because of this, efforts to increase disease awareness and testing rates are ongoing. For example, a program in Germany successfully increased diagnosis rates of AATD by combining an educational campaign with cost-free genetic testing.17
Another factor is the lack of uniform reimbursement for AAT augmentation therapy, the only available disease-modifying pharmacological intervention for AATD,6,10,18 which likely limits prioritization of AATD testing in countries where treatment is not available.19 Testing should be done regardless of the availability of specific treatment since preventative measures such as smoking cessation, evaluation of occupational exposures, and monitoring for other associated pathologies (eg, liver disease) may significantly modify the clinical course of affected individuals. Variation in reimbursement of AAT therapy is due to differences in levels of evidence required by health authorities.10 Although randomized controlled trials (RCTs) have demonstrated the efficacy of AAT therapy on the surrogate endpoint of lung density decline, as determined by computed tomography (CT),7 evidence of an effect on patient-reported outcomes (PROs), such as quality of life and exacerbation frequency/severity are still lacking. Studies designed to address the impact on PROs require sample sizes larger than those used in published RCTs and likely require longer duration of evaluation, which are both very difficult to accomplish in a rare disease. This is an area in which registries, which can recruit much larger numbers of patients and follow them over many years, can contribute valuable information. Observational data from registries have been used to overcome some of these obstacles in rare diseases such as AATD, by providing supplemental information on the efficacy and safety of interventions.20,21 Prior registries in AATD have been central to enhance understanding of the disease and identification of patients, and remain key to future efforts. The importance of registries to future research in AATD and supporting access to AAT therapy was highlighted by the 2017 European Respiratory Society (ERS) Statement.18 In this report, novel efforts to unify registries across countries were discussed.
The ERS statement also advocates that the care of patients with AATD should be managed through expert reference centers, including follow-up and regular monitoring/assessment of patients by a multidisciplinary team to ensure the best quality of care.18 In the United States (US), integrated care of AATD has been realized with the use of a nationwide disease self-management program, which has demonstrated a range of benefits to patients with AATD such as improved self-care (including uptake of smoking cessation and immunization), optimization of medication use, and improvements in PROs, including health-related quality of life and duration/frequency of exacerbations.22–24 In addition, this program may also reduce healthcare expenditure associated with the use of AAT therapy.24 Overall, these trends suggest a shift towards patient-centric care of AATD. In line with this, there is currently increasing interest in personalized use of AAT therapy, including the utilization of extended dosing intervals and treatment at home, these topics will also be discussed in this report.10,25
At the 2018 ERS International Congress in Paris, France, a symposium was held to discuss new European initiatives aiming to maximize healthcare provision for patients with AATD and emerging strategies aimed at optimizing the patient experience of AAT therapy provision. The present review will explore these pertinent topics in the field of AATD.
New European Initiatives in Rare Lung Diseases
Given that AATD patients and centers of clinical expertise are dispersed across Europe, increasing the connectivity of resources and knowledge across the European states is of utmost importance. To this end, new European initiatives will seek to establish an enhanced and integrated management of patients with AATD, and provide a platform for future research efforts (Figure 1). There are two arms to this new European AATD initiative – one is legislative-driven (via the European Commission), and the other will center on an ERS Clinical Research Collaboration – the European Alpha-1 Research Collaboration (EARCO; Figure 2).
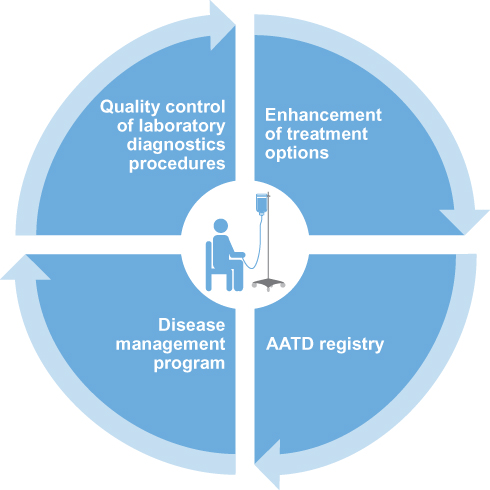 |
Figure 1 European strategies to improve patient care in AATD. Abbreviation: AATD, Alpha-1 antitrypsin deficiency. |
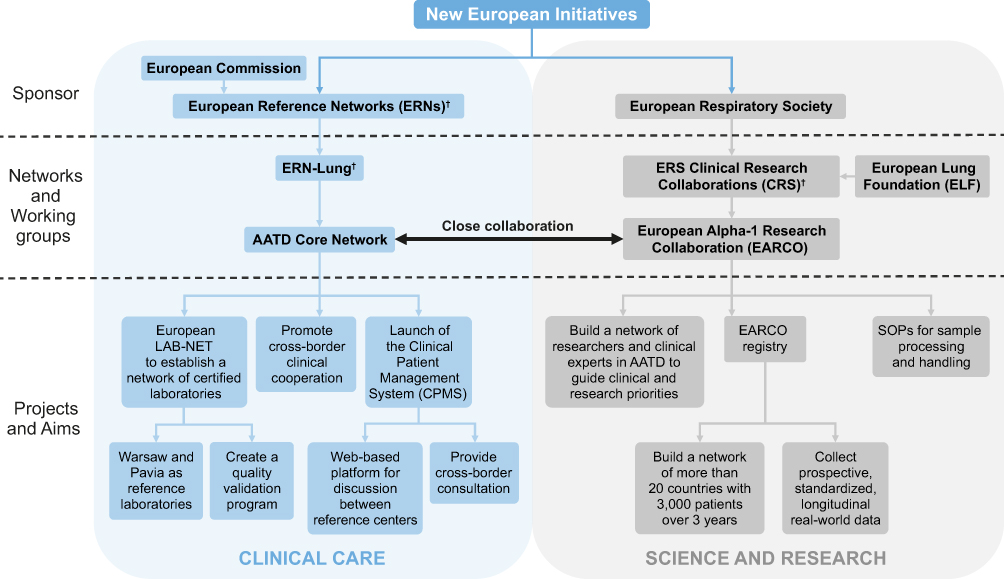 |
Figure 2 Structure of new European initiatives. †Includes various networks of complex or rare diseases, and/or respiratory diseases. Abbreviation: SOP, standard operating procedure. |
The legislative arm will seek to drive improvements in clinical care in AATD via the European Reference Network for Rare Lung Diseases (ERN-LUNG), a patient-focused, non-profit community committed to the prevention, diagnosis and treatment of rare respiratory diseases. The network consists of 60 centers in 12 countries, with nine core networks for conditions including AATD, cystic fibrosis, interstitial lung disease, idiopathic pulmonary fibrosis and pulmonary arterial hypertension.26 The ERN-LUNG AATD core network aims to ensure highly specialized healthcare for AATD patients in Europe. It currently incorporates six reference centers located in different countries across Europe: Netherlands (Leiden), Poland (Warsaw) and Italy (Pavia), as well as Spain (Barcelona), Germany (Homburg) and Ireland (Dublin), and will utilize a secure web-based platform designed to enable live discussions of clinical cases between reference centers. The AATD core network also aims to promote cross-border co-operation; to this end, resources of the network will be available to healthcare providers outside of ERNs for cross-border consultations/patients’ file assessment, and will work closely with EARCO. An operational checklist and patient care pathway has been established by ERN-LUNG for cross-border referral,27 and questions regarding compliance with the General Data Protection Regulation have recently been clarified by an EU Commission Implementing Decision, within which the requirements for written informed consent to enable patient participation with ERNs are outlined.28 The realization of cross-border care in AATD is a major step forward in terms of the access that patients will have to expert centers where equivalent experience is not available in their country. In addition, a current objective is to expand the number of contact point centers, with priority given to countries not currently involved, which will increase the access that non-specialist physicians will have to relevant expertise.
Furthermore, although evidence from the US in recent years has demonstrated the benefit of disease management programs in AATD, no such programs currently exist in Europe. Therefore, an additional objective of the AATD core network is to launch the Clinical Patient Management System (CPMS). Another immediate priority of the AATD core network is to improve the detection of AATD in Europe through quality improvement in AATD testing.
Standardization and Reliability of AATD Diagnostics
AATD is ultimately diagnosed through laboratory analyses, and since these tests are the basis of clinical decisions regarding potential treatment options for individual patients, including augmentation therapy and monitoring requirements, reliability of diagnostics is essential. However, in Europe, there is currently a lack of quality control procedures for AATD diagnostics.
Comprehensive testing for AATD includes quantification of AAT and C-reactive protein (CRP) serum levels, and qualitative assessment of phenotype by isoelectric focusing (IEF) and/or genetic testing, respectively.29 Determination of AAT serum levels is the first step in testing for AATD, and checking CRP is often used to determine if a patient has transiently elevated AAT levels (like CRP, AAT is an acute-phase reactant).30 The normal range for AAT serum levels as outlined by the American Thoracic Society (ATS) and ERS statement (2003), which remains a key resource for many laboratories, is 83–220 mg/dL by nephelometry.31 However, more recent data suggest a much narrower range (105–164 mg/dL),32 which may be due to the use of more accurate/modern assays and standards. This evolution in the accuracy of diagnostic techniques may not be uniformly reflected in equipment used across all European laboratories. Furthermore, specific cut-offs for AAT levels may be utilized in a pre-specified diagnostic algorithm to determine which samples require further qualitative testing, ie, reflex testing.33 Cut-off levels and algorithms may vary between laboratories.11
Qualitative testing results determine disease-risk and management requirements for patients. IEF is a useful technique available to most clinicians10 that can establish the presence of AAT variants by the isoelectric points of AAT isoforms, displayed by differences in banding patterns.11 The technique can detect a wide range of variants; however, many variants show similar banding patterns to the normal allele (M-like), and the technique cannot detect Null variants appearing in the heterozygous state. Furthermore, results of IEF ultimately require expert interpretation and the technique is therefore associated with high personnel costs.11 Targeted genetic testing by polymerase-chain reaction is a reliable technique used to screen for the more common deficiency alleles, ie, “Z” and “S”,10 and sometimes “F” and “I”. Being a cost-effective and high-throughput method, novel approaches are being developed to include several other mutations on initial testing; however, identification of more rare variants is limited by the availability of the corresponding primers.11,33 In cases such as these, sequencing of the SERPINA1 gene that encodes AAT is often employed and frequently discloses novel mutation variants. In this regard, the two countries to report high levels of novel variant detection are Italy and Portugal.34,35 Regional variation in the frequency of rare variants is expected, as a small-scale population study in Italy demonstrated that more isolated communities show the highest prevalence of rare variants.36 Variants such as these would have been missed without incorporation of sequencing into the testing workflow. One study found that among patients with no common deficiency alleles and low/low-normal AAT levels, 38% had additional variants only detectable by sequencing.37
Available sequencing methodologies can be broadly split between older methods, ie, Sanger sequencing, and newer, high-throughput methods, ie, next-generation sequencing (NGS). As NGS is becoming increasingly available, it is beginning to appear in testing algorithms for AATD.19,29,38 Despite the clear utility of sequencing in AATD diagnostics, it is a tool that requires careful validation when used in the clinical setting. The sequencing workflow can be broadly split into pre-analytical (preparation of the DNA template), analytical (the sequencing run itself), and post-analytical (data quality check and analysis) steps, with different standards applicable at each stage.39 The pre-analytical process focuses on the quality and amount of a sample – insufficient quantity of a blood sample and errors in storage, eg, introducing repeated freeze-thaw cycles of DNA/RNA samples, can affect the accuracy of results.39 During the analytical phase, polymerase errors can occur.39 For data analysis, there can be diversity in the file formats used (FASTQ being the most frequently used), and in how often the data are deposited in publicly accessible databases.39
As outlined above, there are numerous steps and parameters involved for a comprehensive and accurate diagnosis of AATD, and inaccuracies in results can be introduced at several stages, as different technologies and workflows are likely to vary between European centers. In particular, sequencing is not universally available to clinicians in Europe.10 Moreover, a publication by Miravitlles et al showed that there are distinct differences in the testing algorithms employed by three leading European AATD laboratories.11 In this work, several recommendations for best practice in Europe going forward were outlined, including, but not limited to, determining the most cost-effective approach to targeted detection, the preparation of laboratories’ own set of reference standards, and participation of laboratories in a quality control program. The latter of these recommendations is now possible with the launch of ERN-LUNG, as one of the aims of the ERN-LUNG AATD Core Group is the creation of a European quality validation program for AATD that will verify the accuracy of diagnostic procedures. This program will be implemented via a network of certified laboratories (European LAB-NET), with Warsaw and Pavia acting as the reference laboratories involved in assessing the quality of other laboratories within the network.
Although encouraging standardization in practice is a broad aim of the program, creation of a common European standard operating procedure for AATD diagnostics is a difficult proposition, owing to the diversity of available technologies and differences between healthcare systems. Nonetheless, access to a quality control program will add an additional verification step and help ensure that patients in Europe are being correctly diagnosed. A more basic issue that remains in the field of AATD is the clinical decision-making process by the non-specialist physicians that often deviate from guideline recommendations regarding testing for AATD. There is evidence to suggest that primary care physicians in particular have low awareness/knowledge of AATD, and do not always follow fundamental recommendations such as testing all newly diagnosed COPD patients for AATD.13 Underpinning this issue is continued low awareness of the disease, and this is an area that ERN-LUNG and EARCO will endeavor to address through raising the profile of AATD and by connecting specialist and non-specialist physicians.
EARCO and the EARCO Registry
The second pathway of the European initiatives, which will be implemented through EARCO, focuses on prioritizing international research and science in AATD. The collaboration aims to bring together a network of experienced and early career clinical researchers, experts in the field of AATD and industry sponsors to establish current and future research priorities, and to facilitate an increase in the number and quality of European clinical trials in AATD.40 In addition, as part of EARCO’s aim to include the patient’s voice at the center of its objectives and activities, the collaboration will work closely with patient advisors via the European Lung Foundation (Figure 3). The steering committee includes two AATD patients, and patients and patients’ representatives are welcome to the meetings and also participate in projects.
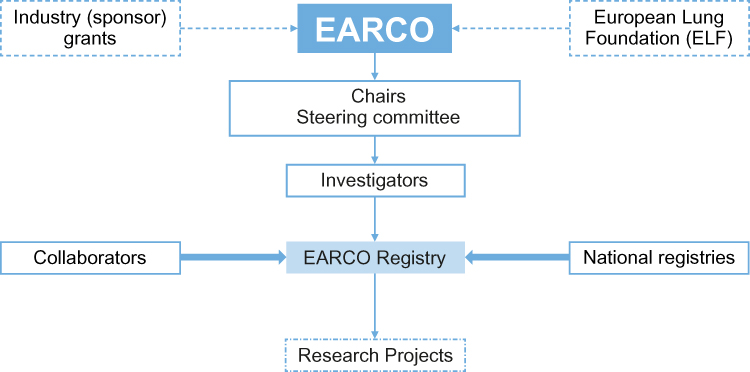 |
Figure 3 Structure of the European Alpha-1 Research Collaboration (EARCO). The center of the initiative is the EARCO registry, which works closely with national registries and all collaborators (investigators, physicians, clinicians) who will provide data and allow the registry to grow. |
The first project of EARCO is the foundation of a new pan-European AATD registry.40 Since the discovery of AATD, national and international registries have been critical to increasing knowledge and understanding of the disease. For example, one of the earliest AATD registries in Sweden provided crucial insight into the prevalence of AATD through neonatal screening,41 and has subsequently enabled assessment of health-status in these individuals through the first five decades of life.42 Furthermore, registries have been key to exploring the natural history of lung disease in AATD, particularly the effect of smoking and exposure to pollutants on spirometric decline, and the prognosis associated with index vs. non-index identification.43–46 In terms of therapeutic insight, registries in general can also provide longitudinal data to support the efficacy of treatments, which is particularly important in the field of orphan diseases where constructing sufficiently powered clinical trials can be difficult due to low patient numbers.20,47 In AATD, registries in both Europe and the US have been central to providing some of the first evidence for the efficacy of AAT therapy on clinical endpoints including lung function decline and mortality.21,48 More recently, incorporation of newer technology in the form of chest CT scans into routine registry data collection in the UK has provided crucial insight on the etiology of emphysema in AATD, and helped to support the validity of CT lung densitometry as a clinical endpoint for clinical trials in AATD.49,50
At a global level, the Alpha-1 International Registry (AIR) was formed in 1997 in response to the World Health Organization's memorandum on AATD.51 By the end of 2005, the registry had connected and pooled data from 21 countries across four continents.51 AIR has been central to raising awareness of AATD worldwide, for connecting a network of AATD experts globally, and in establishing a pool of patients for clinical trial recruitment. However, the registry has not been able to publish significant clinical data relating to the disease course in AATD and the influence of pharmacological interventions, which may be due to a lack of standardization in data collection. Therefore, one of the purposes of EARCO is to expand on the success of AIR by creating a prospective registry that includes a quality control system, with standardized data collection on a broad range of clinical parameters covering basic clinical chemistry, respiratory physiology, quality of life, sociodemographic factors and pharmacological intervention (Figure 4). In addition, of note is that EARCO will collect data on all aspects of AATD and its clinical manifestations, including clinical parameters related to liver disease, an under-appreciated aspect of AATD and significant contributor to the morbidity and mortality associated with the disease.52 It is hoped that comprehensive and quality-controlled data collection will facilitate improvements in future epidemiological research in AATD, and act as a valuable resource that will support claims regarding the efficacy of pharmacological interventions, particularly AAT therapy, and assist in the implementation of new clinical trials. Ultimately, the goal of the EARCO registry is to facilitate improvements in the clinical management of patients with AATD.
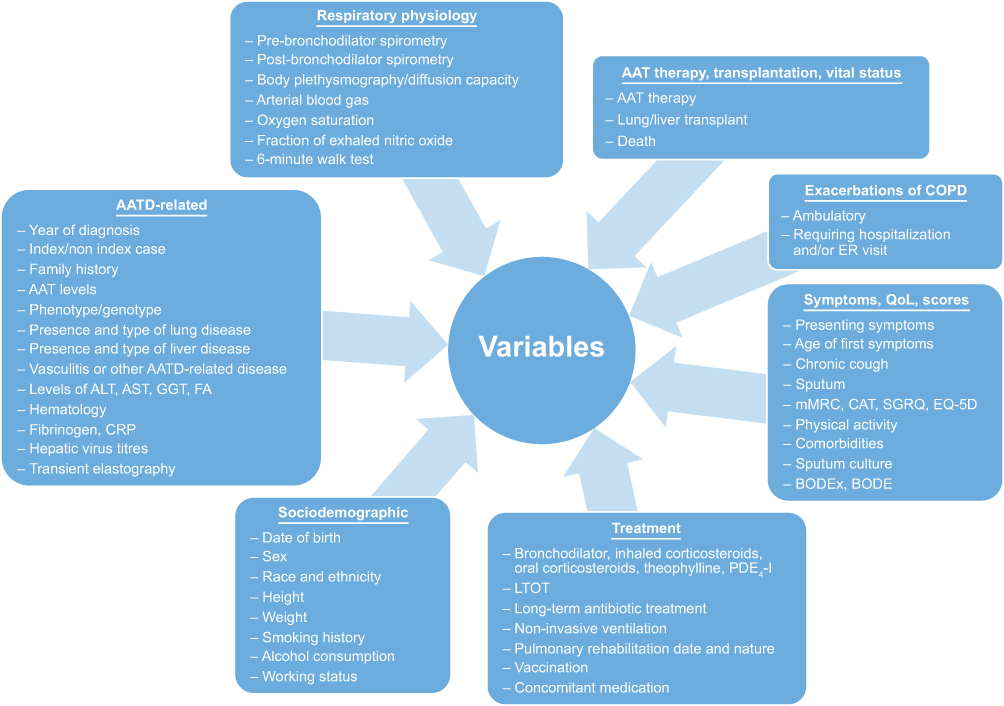 |
Figure 4 EARCO registry data collection. Abbreviations: AAT, Alpha-1 antitrypsin; AATD, Alpha-1 antitrypsin deficiency; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BODE, Body-mass index, Obstruction, Dyspnea, Exercise; CAT, COPD Assessment Test; COPD, chronic obstructive pulmonary disease; CRP, C-reactive protein; EQ-5D, EuroQol; ER, emergency room; FA, folic acids; GGT, gamma-glutamyltransferase; LTOT, long-term oxygen therapy; mMRC, modified Medical Research Council dyspnea scale; PDE4-I, phosphodiesterase-4-inhibtor; QoL, quality of life; SGRQ, St George’s Respiratory Questionnaire. |
Improving Convenience of AAT Therapy
One of the hurdles to improving the treatment experience of patients with AATD centers on the implementation of AAT therapy. AAT therapy is associated with the burden of IV administration, including frequent visits to the clinic, which can greatly reduce patients’ autonomy. Two strategies to combat the burden associated with IV administration are alternative dosing regimens and self-administration.
Alternative Dosing Regimens for AAT Therapy
The recommended and licensed dose for AAT therapy is 60 mg/kg body weight per week; thus, the combination of weekly travel to the clinic and frequent IV infusions can be inconvenient for patients and time consuming for clinicians. Therefore, extending the dosing interval by using larger doses, eg, 120 mg/kg body weight bi-weekly, has been highlighted as a method to improve convenience for patients. Although pharmacokinetic studies have shown trough levels to be slightly lower with bi-weekly than weekly dosing, bi-weekly dosing is broadly sufficient to maintain AAT plasma concentrations above 11 µM during the dosing interval.53–55 Furthermore, a recent survey of European AATD experts found that the majority of those surveyed would consider alternative dosing strategies such as bi-weekly dosing, particularly to cover vacations and for patients in employment.10 Additionally, in certain European countries such as Spain, France and Czech Republic, extended dosing intervals are already recommended and utilized.10,56,57
However, as the majority of the safety data related to AAT therapy are based on the standard 60 mg/kg dose, the safety profile of 120 mg doses has been largely unexplored. A recent post hoc analysis of the RAPID clinical trial program, the first clinical trials to demonstrate the clinical efficacy of AAT therapy,7,58 sought to address this issue. In the RAPID clinical trial program, a subgroup of patients received bi-weekly infusions at the discretion of the treating physician to cover periods where they were unable to attend the clinic, eg, during vacations.59 Overall, there were 933 bi-weekly infusions during the RAPID clinical trial program, and results showed similar rates, temporal distribution and severity of adverse events between weekly dosing with 60 mg/kg and bi-weekly dosing with 120 mg/kg AAT.59 Therefore, given the favorable pharmacokinetic and safety profiles of bi-weekly dosing, and the willingness of AATD experts to utilize alternative dosing strategies, there is an increasingly strong rationale for the implementation of extended-interval dosing when required. However, the standard dose remains the preferred choice of treatment as there are no specific efficacy data related to the 120 mg/kg bi-weekly dose and no data on the impact of differing trough levels between weekly and bi-weekly dosing on the efficacy of treatment. Nevertheless, it is certainly preferable for patients to receive bi-weekly doses than omitting doses due to vacations, or other situations where the patient is unable to attend the clinic on a weekly basis. Furthermore, if this treatment schedule is chosen, it has a similar safety profile to standard dosing.
Self-Administration of AAT Therapy
IV self-administration has been utilized in other therapy areas such as hereditary angioedema, and has demonstrated benefits in terms of reducing time off work, and reducing healthcare visits and episodes of hospitalization.60 Therefore, in a subset of patients with AATD, self-administration of AAT therapy has the potential to improve the convenience of weekly infusions. However, although self-administration has been shown to be feasible in AATD,55 there has been a lack of data concerning the frequency of self-administration utilization, and patients’ satisfaction with this therapeutic strategy. Therefore, AlphaNet, a US-based not-for-profit organization that provides customized patient care through the Alpha-1 Disease Management and Prevention (ADMAP) program,61 sought to evaluate the utilization of and patients’ satisfaction with self-administration.62 Of the 555 patients surveyed, 44 (7.9%) reported that they were actively self-administering their AAT therapy, and all patients self-infusing were either “very satisfied” (95.4%) or “satisfied” (4.6%) with their treatment overall. In addition, very few adverse events were reported related to self-administration. The most frequently cited reason for patients not choosing self-administration was that they were satisfied with their current regimens, which highlights that self-administration is not suitable for all patients (Figure 5). However, it is notable that many patients merely lacked the confidence to self-administer, suggesting that a further section of patients may benefit from self-administration with adequate support. A further interesting insight from the survey was that the primary source of information regarding self-administration was other patients with AATD (Figure 6), rather than healthcare professionals, which may indicate that educational programs are needed to raise awareness of self-administration among professionals involved in the care of AATD.
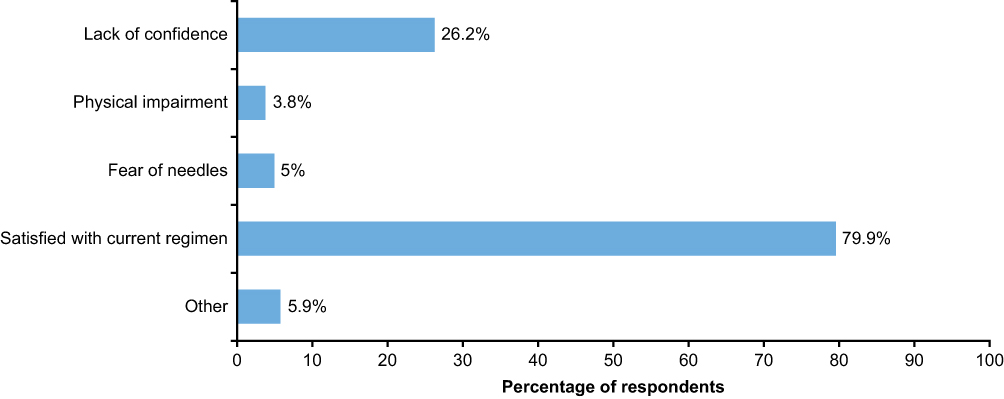 |
Figure 5 Reasons why patients chose not to self-administer. Abbreviation: AATD, Alpha-1 antitrypsin deficiency. |
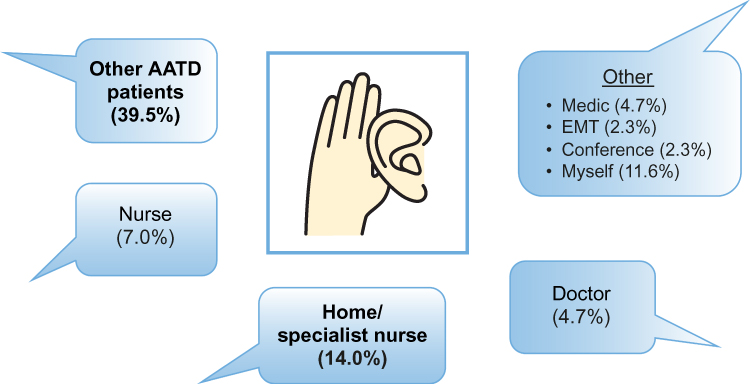 |
Figure 6 How patients heard about self-administration. Abbreviations: AATD, Alpha-1 antitrypsin deficiency; EMT, emergency medical technician. |
Overall, self-administration may be of great value to certain patients with AATD, eg, those in employment or those who would experience difficulty in traveling to the clinic,10 and is currently implemented successfully by a sub-section of patients in the US. Although at present there are no national self-administration programs in Europe, one of the AAT products on the market in Europe includes within its label the possibility to self-administer the therapy, in conjunction with adequate training and counselling.63 Furthermore, a recent expert survey found that physicians are generally supportive of utilizing self-administration in carefully selected patients.10 Therefore, there is potential for the roll-out of self-administration in Europe in the years to come, which will add a further dimension to the treatment options available to patients with AATD.
Conclusions
Although there have been great advances in the understanding of AATD and its treatment in recent years, AATD remains an underdiagnosed and undertreated condition, and future initiatives should focus on optimizing detection and patient management. In Europe, EARCO and ERN-LUNG will be central to future diagnostic and management efforts through enabling cooperation, communication and integration across countries. It is hoped that the EARCO registry will contribute to the development of new treatment modalities in AATD and support access to AAT therapy, with the ERN-LUNG AATD core network providing a basis for the implementation of a disease management program and improvements in the standardization and accuracy of AATD diagnosis. Furthermore, current patient-centric approaches to AAT therapy utilization, such as extended interval dosing and self-administration, will help alleviate the burden of weekly intravenous infusions, and may help to improve patients’ quality of life.
Abbreviations
AAT, Alpha-1 Antitrypsin; AATD, Alpha-1 Antitrypsin Deficiency; ADMAP, Alpha-1 Disease Management and Prevention; AIR, Alpha-1 International Registry; ATS, American Thoracic Society; COPD, Chronic obstructive pulmonary disease; CPMS, Clinical Patient Management System; CRP, C-reactive protein; CT, Computed tomography; EARCO, European Alpha-1 Research Collaboration; ERN-LUNG, European Reference Network for Rare Lung Diseases; ERS, European Respiratory Society; IEF, isoelectric focusing; IV, Intravenous; PRO, Patient-reported outcome.
Acknowledgment
Medical writing assistance was provided by Meridian HealthComms, funded by CSL Behring.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
Medical writing assistance was funded by CSL Behring; the funder had no role in the design of the paper.
Disclosure
Professor Joanna Chorostowska-Wynimko reports grants, personal fees, non-financial support from Grifols, Boehringer Ingelheim, and CSL Behring, personal fees, non-financial support from Novartis, and personal fees from GSK, outside the submitted work. Dr Miriam Barrecheguren reports personal fees from Grifols, Menarini, CSL Behring, GSK, Novartis, and Gebro Pharma, outside the submitted work. Dr Ilaria Ferrarotti reports personal fees from Grifols and grants from CSL Behring, outside the submitted work. Dr Timm Greulich reports personal fees from AstraZeneca, Berlin-Chemie, Boehringer-Ingelheim, Chiesi, CSL-Behring, Grifols, GSK, and Novartis, outside the submitted work. Professor Robert A Sandhaus reports participation in advisory boards for Grifols, CSL Behring, AstraZeneca, Mereo, and Inhibrx for which he receives reimbursement of travel expenses. Dr Michael Campos reports grants, non-financial support, and personal fees from CSL Behring, and grants and personal fees from Grifols, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Janciauskiene SM, Bals R, Koczulla R, Vogelmeier C, Kohnlein T, Welte T. The discovery of alpha1-antitrypsin and its role in health and disease. Respir Med. 2011;105(8):1129–1139.
2. Fairbanks KD, Tavill AS. Liver disease in alpha 1-antitrypsin deficiency: a review. Am J Gastroenterol. 2008;103(8):2136–2141. doi:10.1111/ajg.2008.103.issue-8
3. Gross B, Grebe M, Wencker M, Stoller JK, Bjursten LM, Janciauskiene S. New findings in PiZZ alpha1-antitrypsin deficiency-related panniculitis. Demonstration of skin polymers and high dosing requirements of intravenous augmentation therapy. Dermatology. 2009;218(4):370–375. doi:10.1159/000202982
4. Elzouki AN, Segelmark M, Wieslander J, Eriksson S. Strong link between the alpha 1-antitrypsin PiZ allele and Wegener’s granulomatosis. J Intern Med. 1994;236(5):543–548. doi:10.1111/j.1365-2796.1994.tb00842.x
5. Köhnlein T, Janciauskiene S, Welte T. Diagnostic delay and clinical modifiers in alpha-1 antitrypsin deficiency. Ther Adv Respir Dis. 2010;4(5):279–287. doi:10.1177/1753465810376407
6. Chorostowska-Wynimko J. Disease modification in emphysema related to alpha-1 antitrypsin deficiency. COPD. 2016;13(6):807–815. doi:10.1080/15412555.2016.1178224
7. McElvaney NG, Burdon J, Holmes M, et al. Long-term efficacy and safety of alpha1 proteinase inhibitor treatment for emphysema caused by severe alpha1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). Lancet Respir Med. 2017;5(1):51–60. doi:10.1016/S2213-2600(16)30430-1
8. Craig TJ. Suspecting and testing for alpha-1 antitrypsin deficiency-an allergist’s and/or immunologist’s perspective. J Allergy Clin Immunol Pract. 2015;3(4):506–511. doi:10.1016/j.jaip.2015.04.005
9. Strange C, Stoller JK, Sandhaus RA, Dickson R, Turino G. Results of a survey of patients with alpha-1 antitrypsin deficiency. Respiration. 2006;73(2):185–190. doi:10.1159/000088061
10. Horvath I, Canotilho M, Chlumsky J, et al. Diagnosis and management of alpha1-antitrypsin deficiency in Europe: an expert survey. ERJ Open Res. 2019;5(1). doi:10.1183/23120541.00171-2018
11. Miravitlles M, Herr C, Ferrarotti I, et al. Laboratory testing of individuals with severe alpha1-antitrypsin deficiency in three European centres. Eur Respir J. 2010;35(5):960–968. doi:10.1183/09031936.00069709
12. Tejwani V, Sanders C, Fye E, Nowacki A, Stoller J. Symptom and airflow correlates of delayed diagnosis in alpha-1 antitrypsin deficiency. Chest. 2017;152(4):A817. doi:10.1016/j.chest.2017.08.848
13. Esquinas C, Barrecheguren M, Sucena M, Rodriguez E, Fernandez S, Miravitlles M. Practice and knowledge about diagnosis and treatment of alpha-1 antitrypsin deficiency in Spain and Portugal. BMC Pulm Med. 2016;16(1):64. doi:10.1186/s12890-016-0222-4
14. Greulich T, Ottaviani S, Bals R, et al. Alpha1-antitrypsin deficiency - diagnostic testing and disease awareness in Germany and Italy. Respir Med. 2013;107(9):1400–1408. doi:10.1016/j.rmed.2013.04.023
15. Calle Rubio M, Soriano JB, Lopez-Campos JL, et al. Testing for alpha-1 antitrypsin in COPD in outpatient respiratory clinics in Spain: a multilevel, cross-sectional analysis of the EPOCONSUL study. PLoS One. 2018;13(6):e0198777. doi:10.1371/journal.pone.0198777
16. Soriano JB, Lucas SJ, Jones R, et al. Trends of testing for and diagnosis of alpha1-antitrypsin deficiency in the UK: more testing is needed. Eur Respir J. 2018;52(1). doi:10.1183/13993003.00360-2018
17. Greulich T, Nell C, Herr C, Vogelmeier C, Kotke V, Wiedmann S, et al. Results from a large targeted screening program for alpha-1-antitrypsin deficiency: 2003-2015. Orphanet J Rare Dis. 2016;11(1):75.
18. Miravitlles M, Dirksen A, Ferrarotti I, et al. European respiratory society statement: diagnosis and treatment of pulmonary disease in alpha1-antitrypsin deficiency. Eur Respir J. 2017;50:5. doi:10.1183/13993003.00610-2017
19. Greulich T, Vogelmeier CF. Alpha-1-antitrypsin deficiency: increasing awareness and improving diagnosis. Ther Adv Respir Dis. 2016;10(1):72–84. doi:10.1177/1753465815602162
20. Luisetti M, Balfour-Lynn IM, Johnson SR, et al. Perspectives for improving the evaluation and access of therapies for rare lung diseases in Europe. Respir Med. 2012;106(6):759–768. doi:10.1016/j.rmed.2012.02.016
21. Seersholm N, Wencker M, Banik N, et al. Does alpha1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study group. Eur Respir J. 1997;10(10):2260–2263. doi:10.1183/09031936.97.10102260
22. Perkins JT, Choate R, Mannino DM, Browning SR, Sandhaus RA. Benefits among patients with alpha-1 antitrypsin deficiency enrolled in a disease management and prevention program. Chronic Obstr Pulm Dis. 2016;4(1):56–64. doi:10.15326/jcopdf.4.1.2016.0161
23. Campos MA, Alazemi S, Zhang G, Wanner A, Sandhaus RA. Effects of a disease management program in individuals with alpha-1 antitrypsin deficiency. COPD. 2009;6(1):31–40. doi:10.1080/15412550802607410
24. Campos MA, Runken MC, Davis AM, Johnson MP, Stone GA, Buikema AR. Impact of a health management program on healthcare outcomes among patients on augmentation therapy for alpha 1-antitrypsin deficiency: an insurance claims analysis. Adv Ther. 2018;35(4):467–481. doi:10.1007/s12325-018-0690-4
25. Stockley RA, Miravitlles M, Vogelmeier C; Alpha One International R. Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis. 2013;8:149. doi:10.1186/1750-1172-8-149
26. Humbert M, Wagner TO. Rare respiratory diseases are ready for primetime: from rare disease day to the European reference networks. Eur Respir J. 2017;49:2. doi:10.1183/13993003.00085-2017
27. European Reference Networks. ERN-LUNG cross-border referral operational. Available from: https://ern-lung.eu/inhalt/wp-content/uploads/2019/07/Cross-border-referral-operational.pdf.
28. Commission implementing decision (EU) 2019/1269 of 26 July 2019 amending implementing decision 2014/287/EU setting out criteria for establishing and evaluating European reference networks and their members and for facilitating the exchange of information and expertise on establishing and evaluating such networks. C/2019/5470. Official Journal of the European Union. L 200, 29.7.2019; 35–43. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32019D1269&from=EN.
29. Kueppers F, Sanders C. State-of-the-art testing for alpha-1 antitrypsin deficiency. Allergy Asthma Proc. 2017;38(2):108–114. doi:10.2500/aap.2017.38.4031
30. Sanders CL, Ponte A, Kueppers F. The effects of inflammation on alpha 1 antitrypsin levels in a national screening cohort. COPD. 2018;15(1):10–16. doi:10.1080/15412555.2017.1401600
31. American Thoracic S; European Respiratory S. American Thoracic Society/European respiratory society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900.
32. Ferrarotti I, Thun GA, Zorzetto M, et al. Serum levels and genotype distribution of alpha1-antitrypsin in the general population. Thorax. 2012;67(8):669–674. doi:10.1136/thoraxjnl-2011-201321
33. Bornhorst JA, Procter M, Meadows C, Ashwood ER, Mao R. Evaluation of an integrative diagnostic algorithm for the identification of people at risk for alpha1-antitrypsin deficiency. Am J Clin Pathol. 2007;128(3):482–490. doi:10.1309/44J4KBCFQ8E9D1B8
34. Silva D, Oliveira MJ, Guimaraes M, Lima R, Gomes S, Seixas S. Alpha-1-antitrypsin (SERPINA1) mutation spectrum: three novel variants and haplotype characterization of rare deficiency alleles identified in Portugal. Respir Med. 2016;116:8–18. doi:10.1016/j.rmed.2016.05.002
35. Luisetti M, Ferrarotti I, Corda L, et al. Italian registry of patients with alpha-1 antitrypsin deficiency: general data and quality of life evaluation. COPD. 2015;12(Suppl 1):52–57. doi:10.3109/15412555.2015.1023393
36. Corda L, Medicina D, La Piana GE, et al. Population genetic screening for alpha1-antitrypsin deficiency in a high-prevalence area. Respiration. 2011;82(5):418–425. doi:10.1159/000325067
37. Graham RP, Dina MA, Howe SC, et al. SERPINA1 full-gene sequencing identifies rare mutations not detected in targeted mutation analysis. J Mol Diagn. 2015;17(6):689–694. doi:10.1016/j.jmoldx.2015.07.002
38. Kueppers F, Andrake MD, Xu Q, Dunbrack RL
39. Endrullat C, Glokler J, Franke P, Frohme M. Standardization and quality management in next-generation sequencing. Appl Transl Genom. 2016;10:2–9. doi:10.1016/j.atg.2016.06.001
40. Miravitlles M, Chorostowska-Wynimko J, Ferrarotti I, et al. The European Alpha-1 Research Collaboration (EARCO): a new ERS clinical research collaboration to promote research in alpha-1 antitrypsin deficiency. Eur Respir J. 2019;53(2):1900138. doi:10.1183/13993003.00138-2019
41. Laurell CB, Sveger T. Mass screening of newborn Swedish infants for alpha antitrypsin deficiency. Am J Hum Genet. 1975;27(2):213–217.
42. Mostafavi B, Piitulainen E, Tanash HA. Survival in the Swedish cohort with alpha-1-antitrypsin deficiency, up to the age of 43–45 years. Int J Chron Obstruct Pulmon Dis. 2019;14:525–530. doi:10.2147/COPD.S183205
43. Seersholm N, Kok-Jensen A, Dirksen A. Survival of patients with severe alpha 1-antitrypsin deficiency with special reference to non-index cases. Thorax. 1994;49(7):695–698. doi:10.1136/thx.49.7.695
44. Seersholm N, Kok-Jensen A. Clinical features and prognosis of life time non-smokers with severe alpha 1-antitrypsin deficiency. Thorax. 1998;53(4):265–268. doi:10.1136/thx.53.4.265
45. Piitulainen E, Tornling G, Eriksson S. Environmental correlates of impaired lung function in non-smokers with severe alpha 1-antitrypsin deficiency (PiZZ). Thorax. 1998;53(11):939–943. doi:10.1136/thx.53.11.939
46. Piitulainen E, Mostafavi B, Tanash HA. Health status and lung function in the Swedish alpha 1-antitrypsin deficient cohort, identified by neonatal screening, at the age of 37–40 years. Int J Chron Obstruct Pulmon Dis. 2017;12:495–500. doi:10.2147/COPD
47. Luisetti M, Campo I, Scabini R, et al. The problems of clinical trials and registries in rare diseases. Respir Med. 2010;104(Suppl 1):S42–S44. doi:10.1016/j.rmed.2010.03.016
48. The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med. 1998;158(1):49–59. doi:10.1164/ajrccm.158.1.9712017
49. Dawkins PA, Dowson LJ, Guest PJ, Stockley RA. Predictors of mortality in alpha1-antitrypsin deficiency. Thorax. 2003;58(12):1020–1026. doi:10.1136/thorax.58.12.1020
50. Green CE, Parr DG, Edgar RG, Stockley RA, Turner AM. Lung density associates with survival in alpha 1 antitrypsin deficient patients. Respir Med. 2016;112:81–87. doi:10.1016/j.rmed.2016.01.007
51. Stockley RA, Luisetti M, Miravitlles M, Piitulainen E, Fernandez P, Alpha One International Registry g. Ongoing research in Europe: Alpha one International Registry (AIR) objectives and development. Eur Respir J. 2007;29(3):582–586. doi:10.1183/09031936.00053606
52. Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Survival in severe alpha-1-antitrypsin deficiency (PiZZ). Respir Res. 2010;11:44. doi:10.1186/1465-9921-11-44
53. Barker AF, Iwata-Morgan I, Oveson L, Roussel R. Pharmacokinetic study of alpha1-antitrypsin infusion in alpha1-antitrypsin deficiency. Chest. 1997;112(3):607–613. doi:10.1378/chest.112.3.607
54. Soy D, de la Roza C, Lara B, Esquinas C, Torres A, Miravitlles M. Alpha-1-antitrypsin deficiency: optimal therapeutic regimen based on population pharmacokinetics. Thorax. 2006;61(12):1059–1064. doi:10.1136/thx.2005.057943
55. Piitulainen E, Bernspang E, Bjorkman S, Berntorp E. Tailored pharmacokinetic dosing allows self-administration and reduces the cost of IV augmentation therapy with human alpha(1)-antitrypsin. Eur J Clin Pharmacol. 2003;59(2):151–156. doi:10.1007/s00228-003-0589-z
56. Vidal R, Blanco I, Casas F, Jardi R, Miravitlles M. Committee on the national registry of individuals with alpha-1 antitrypsin D. [Guidelines for the diagnosis and management of alpha-1 antitrypsin deficiency]. Arch Bronconeumol. 2006;42(12):645–659. doi:10.1157/13095974
57. Gauvain C, Mornex JF, Pison C, et al. Health-related quality of life in patients with alpha-1 antitrypsin deficiency: the French experience. COPD. 2015;12(Suppl 1):46–51. doi:10.3109/15412555.2015.1022645
58. Chapman KR, Burdon JG, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991):360–368. doi:10.1016/S0140-6736(15)60860-1
59. Greulich T, Chlumsky J, Wencker M, et al. Safety of biweekly alpha1-antitrypsin treatment in the RAPID programme. Eur Respir J. 2018;52:5. doi:10.1183/13993003.00897-2018
60. Bygum A, Andersen KE, Mikkelsen CS. Self-administration of intravenous C1-inhibitor therapy for hereditary angioedema and associated quality of life benefits. Eur J Dermatol. 2009;19(2):147–151. doi:10.1684/ejd.2008.0603
61. AlphaNet. Available from: https://www.alphanet.org.
62. Sandhaus RA, Boyd BS. Alpha 1 antitrypsin therapy: a satisfaction survey of individuals self-administering. Am J Respir Crit Care Med. 2018;A46:A1758–A1758.
63. European Medicines Agency. Respreeza summary of product characteristics. Available from: https://www.ema.europa.eu/en/documents/product-information/respreeza-epar-product-information_en.pdf.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.