")
Back to Journals » The Application of Clinical Genetics » Volume 7
New insights into the genetic basis of infertility
Authors Venkatesh T, Suresh P, Tsutsumi R
Received 1 September 2014
Accepted for publication 25 October 2014
Published 1 December 2014 Volume 2014:7 Pages 235—243
DOI https://doi.org/10.2147/TACG.S40809
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Thejaswini Venkatesh,1 Padmanaban S Suresh,2 Rie Tsutsumi3
1Institute for Stem Cell Biology and Regenerative Medicine, National Centre for Biological Sciences, Bangalore, 2Centre for Biomedical Research, VIT University, Vellore, India; 3University of Tokushima, Institute of Health Bioscience, Department of Public Health and Nutrition, Tokushima, Japan
Abstract: Infertility is a disease of the reproductive system characterized by inability to achieve pregnancy after 12 or more months of regular unprotected sexual intercourse. A variety of factors, including ovulation defects, spermatogenic failure, parental age, obesity, and infections have been linked with infertility, in addition to specific karyotypes and genotypes. The study of genes associated with infertility in rodent models has expanded the field of translational genetics in identifying the underlying cause of human infertility problems. Many intriguing aspects of the molecular basis of infertility in humans remain poorly understood; however, application of genetic knowledge in this field looks promising. The growing literature on the genetics of human infertility disorders deserves attention and a critical concise summary is required. This paper provides information obtained from a systematic analysis of the literature related to current research into the genetics of infertility affecting both sexes.
Keywords: infertility, genetics, polycystic ovary syndrome, premature ovarian failure, spermatogenic failure, cystic fibrosis
Introduction
According to the World Health Organization, infertility is a disease of the reproductive system and is defined as the inability of sexually active couples taking no contraceptives to achieve pregnancy within 1 year. A systematic analysis of 277 health surveys of the prevalence of infertility (national, regional, and global) since 1990 suggests that 48.5 million couples were unable to have a child within 5 years in the year 2010.1 The issues of infertility are underappreciated in developing countries. Childlessness has negative psychological consequences and leads to social stigma. Nearly 70 million couples are infertile worldwide, and assisted reproductive technologies are expensive and particularly unaffordable for couples in developing countries.2
Reproduction is required for procreation of mammalian species and involves a complex coordination of several gene products.3 Human sexual development starts in the embryo and involves three processes, ie, sex determination, which is chromosomally established at fertilization, differentiation of the gonads into testes or ovaries, and differentiation of internal and external genitalia.4 Genital differentiation follows the male pattern in the presence of testicular hormones or the female pattern in their absence.4 In testicular development, the primordial cells are capable of undergoing proliferation before meiosis, and hence produce large numbers of sperm throughout the male life cycle. In ovarian development, the number of primordial follicles is finite. Thus, there is a subsequent loss of follicles during female development and puberty, and eventual follicle exhaustion leads to menopause. Moreover, reproduction physiology involves several paracrine, autocrine, and endocrine processes. All of these processes are tightly regulated by a plethora of genes (discussed in the successive sections), and discrepancies in any of these pathways can lead to infertility. The cause of infertility can be hormonal, immunological, infectious, or psychological.
Infertility is a complex disease, and hence an approach to identify a single causative gene is incomprehensive.5 Known genetic causes of infertility include chromosomal aberrations, single gene variants, and phenotypes with multifactorial inheritance.6 Genetic association studies are performed to identify and characterize natural variants or polymorphisms within genes that might be associated with the fertility phenotype.7 Karyotyping and fluorescence in situ hybridization analysis have been used to identify chromosomal aberrations. Unlike karyotyping, fluorescence in situ hybridization can be used on interphase cells and to analyze the sperm genetic component in cases of recurrent miscarriage.8,9 Sequencing methods are commonly used as tools to identify gene mutations. Further, animal models of reproductive disorders have been used to identify candidate genes for infertility.10 The molecular causes of infertility are not confined to genetic variations alone; it has been shown that spermatogonial histone retention, histone modifications, DNA methylation, and transcript levels play a role in male infertility.11 This review focuses on the genetic causes of male and female infertility, and outlines the research conducted in recent years in a few well studied infertility diseases.
Genetics of female infertility
Female infertility is defined as the inability to conceive naturally or to carry a pregnancy to full term.12 Female infertility is caused by genetic, hormonal, or environmental factors. In addition, pelvic inflammatory disease, uterine fibroids, age-related factors, tubal blockage, and hostile cervical mucus can cause infertility in females.12,13 Recent findings in the genetics of female infertility are discussed below.
Polycystic ovary syndrome
Polycystic ovary syndrome (PCOS) is a complex and heterogeneous endocrine condition that affects 5%–10% of women. PCOS is marked by hyperandrogenism, hyperinsulinemia, insulin resistance, and chronic anovulation.14,15 Pathological features include arrest of follicle growth at the small antral stage, minimal proliferation of granulosa cells, and hyperthecosis (hyperplasia of the theca interna of the ovary).16 The genetic basis of PCOS is not known, owing to the difficulties in determining the inheritability of PCOS. Several genetic variants have been reported to influence PCOS; however, the current literature dealing with the genetics of PCOS is inconsistent and inconclusive.17 Further, PCOS is influenced by obesity, and obesity itself has complex genetic associations. PCOS susceptibility genes are believed to be involved in sex hormone regulation, insulin sensitivity, and steroid biosynthesis.7 The genes that have been tested for association with PCOS include StAR (steroidogenic acute regulatory protein), CYP11A (CYP11A-cytochrome P450 side-chain cleavage enzyme), CYP17 (CYP17-cytochrome P450 17α- hydroxylase/17,20-desmolase), CYP19 (CYP19-cytochrome P450 aromatase), HSD17B1–3 (17-β hydroxysteroid dehydrogenase type 1–3), HSD3B1–2 (3-β hydroxysteroid dehydrogenase type I and type II), ACTR1 (activin receptor I), ACTR2A–B (activin receptor 2A and 2B), FS (follistatin), INHA (inhibin alpha), INHBA (inhibin β-A), INHC (inhibin C), SHBG (sex hormone binding globulin), LHCGR (luteinizing hormone/chorionic gonadotropin receptor), FSHR (follicle-stimulating hormone receptor), MADH4 (mothers against decapentaplegic homolog 4), AR (androgen receptor), MC4R (melanocortin 4 receptor), OB (leptin), OBR (leptin receptor), POMC (pro-opiomelanocortin), UCP2+3 (uncoupling proteins 2 + 3), IGF1 (insulin-like growth factor I), IGF1R (insulin-like growth factor receptor I), IGFBPI1+3 (insulin-like growth factor binding proteins 1 + 3), IR (insulin receptor), INSL (Leydig insulin-like protein), IRS1-2 (insulin receptor substrate I), and PPARG (peroxisome proliferator-activator receptor gene gamma).18 The polymorphisms associated with PCOS have been reviewed in detail.7 In this section, we aim to cover the latest reports of studies involving LHCGR, FSHR, VDR (vitamin D receptor), and insulin resistance in PCOS.
A genome-wide association study mapped PCOS-associated susceptibility loci to 2p16.3, 2p21, and 9q33.3 in a Chinese population.19 Among the three loci, LHCGR and FSHR on chromosome 2p16.3 were identified to have an association with PCOS in a European cohort.20 LHCGR is a receptor for two ligands, ie, luteinizing hormone and chorionic gonadotropin. In females, luteinizing hormone triggers the release of oocytes and is also required for maintaining the structure and function of the corpus luteum, while chorionic gonadotrophin stimulates luteal function, which maintains pregnancy. No somatic mutations were observed in LHCGR in a study involving 192 women with PCOS. However, promoter hypomethylation and reduced LHCGR transcript levels were observed.21 In another study, the G935A polymorphism within LHCGR was reported to be associated with PCOS.22
FSHR affects folliculogenesis and ovarian function. No somatic mutations were identified in the coding regions of FSHR in 124 Chinese Singaporean women.23 The known FSHR polymorphisms, A307T and N680S, have no association with PCOS.24 Several other studies have shown that the N680S polymorphism has no association with PCOS.25–27 Vitamin D is a steroid hormone that acts through the vitamin D receptor to maintain calcium homeostasis. Intriguingly, egg activation, oocyte maturation, follicular development, and mammalian embryo development are Ca2+-dependent.28 The VDR is a nuclear steroid hormone receptor involved in the regulation of gene transcription. Interestingly, VDR gene variants are associated with PCOS. For example, the CC genotype of rs731236 in exon 9 of VDR is associated with PCOS.29
PCOS is also characterized by insulin resistance. The elevated insulin levels facilitate secretion of androgens from the ovaries and adrenal glands, leading to hyperandrogenism. Elevated levels of androgens lead to menstrual disturbances and infertility.30 The role of the insulin pathway in the pathogenesis of PCOS is essential, and thus genetic variants of the insulin pathway associated with PCOS have been extensively reviewed.31
Widespread changes in transcription and transcriptional regulation in PCOS patients have also been found. First, DNA methylation regulates gene transcription. Forty genes have been shown to be differentially methylated in PCOS patients compared with the corresponding genes in normal individuals.32 Changes in methylation of EPHX1, LMNA, and GSK3A are associated with PCOS.32–34 MicroRNA (miRNA) profiles have also been shown to change in PCOS patients. Compared with their wild-type counterparts, rats with dihydrotestosterone-induced PCOS showed downregulation or upregulation of a total of 17 and 72 miRNAs, respectively, in the ovaries.15 This study showed that miR-132, which targets luteinizing hormone, was downregulated, and miRNAs such as miR-221/222 and miR-183, which regulate estrogen receptor-α signaling, were differentially expressed. Additionally, alterations in several miRNA expression patterns have been reported in the follicular fluid of PCOS-affected women.35
The genetic heterogeneity of PCOS suggests that the cellular mechanisms and signaling involved in the pathogenesis of the disease is complex. To conclude, although several genes have been associated with PCOS, there is no evidence to suggest that a unique gene or a dominant pathway is the sole causative factor.
XX gonadal dysgenesis
Sex determination in the mammalian embryo is marked by development of germ cells, migration of germ cells to the urogenital ridge, and development of either testes or ovaries from the bipotential gonad.36 These processes are controlled by complex molecular signaling; abnormalities in these signaling pathways can lead to gonadal (ovarian and testicular) dysgenesis. A well-known illustration of this type of dysgenesis is XX female gonadal dysgenesis (XX-GD). XX-GD is genetically heterogeneous, but phenotypically identified by the presence of gonadal streaks, lack of spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism.36 Ovarian insufficiency in this condition can range from lack of pubertal development to the onset of menopause before the age of 40 years. Mutations in FSHR, BMP15, NR5A1, EIF2B2, EIF2B5, HSD17B4, and HARS2 have been reported in XX-GD.36–39 NR5A1 (nuclear receptor subfamily 5, group A, member 1) is a nuclear receptor and transcriptional regulator of several genes involved in sexual development and reproduction, such as StAR, CYP17A1, CYP11A1, INHA, and CYP19A1 (Table 1). NR5A1-/- ovaries lack the corpora lutea. Mutations such as c.666delC, c.877G>A, and c.390delG within NR5A1 have been reported in 46,XX-GD patients.39 Another study has reported a pGlu201del mutation in PSMC3IP (proteasome 26S subunit, ATPase, 3-interacting protein) in a female with XX-GD. Mutations in PSMC3IP impair estrogen-driven transcription and hence lead to gonadal dysgenesis.36
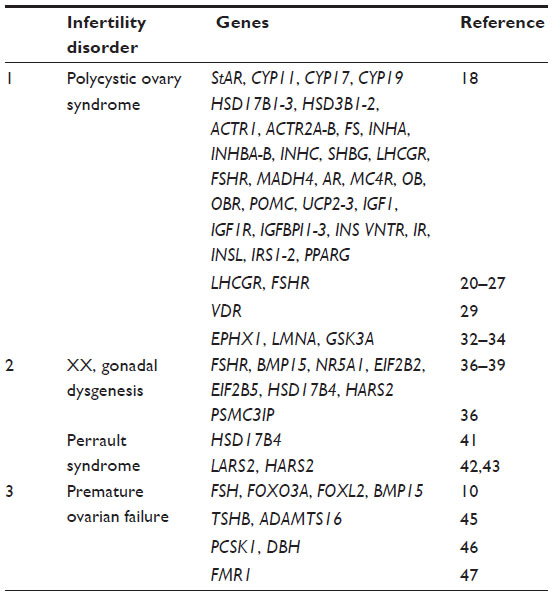 | Table 1 List of genes examined in female infertility as discussed in the review |
Intriguingly, XX-GD has been reported in both isolated and syndromic conditions. Ovarioleukodystrophy and Perrault syndrome are examples of syndromic cases of XX-GD. Perrault syndrome is characterized by 46,XX ovarian dysgenesis and sensorineural deafness in females. In a few instances, mental retardation, ataxia, and cerebellar hypoplasia have been reported in Perrault syndrome.40 Mutations such as c.650A>G (p.Y217C) and c.1704T>A (p.Y568X) in HSD17B4, which encodes 17 β-hydroxysteroid dehydrogenase type 4, also known as D-bifunctional protein, have been reported in a female with Perrault syndrome. HSD17B4/D-bifunctional protein is a multifunctional peroxisomal enzyme involved in fatty acid β-oxidation and steroid metabolism.41 In addition, two mitochondrial genes, LARS2 and HARS2, are linked to Perrault syndrome. LARS2, which encodes a mitochondrial leucyl-tRNA synthetase, shows homozygous c.1565C>A (p.Thr522Asn) in a consanguineous Palestinian family, and compound heterozygous c.1077delT and c.1886C>T (p.Thr629Met) mutations in a nonconsanguineous Slovenian family with Perrault syndrome.42 Compound mutations at two highly conserved amino acids, L200V and V368L, in mitochondrial histidyl-tRNA synthetase (HARS2) also cause this syndrome. These mutations implicate a role for the mitochondria in proper function of the ovaries.43
Premature ovarian failure
Premature ovarian failure (POF) is defined as the onset of menopause in women under the age of 40 years. The symptoms include amenorrhea due to hypoestrogenism, elevated gonadotrophin levels, and other menopause-related symptoms, such as hot flushes, night sweats, and vaginal dryness.10 POF is likely due to depletion of the follicles, which could be because of a decreased number of oocytes being formed during development or an increased rate of oocyte atresia during the reproductive lifespan.10 Paradoxically, some women affected by POF show the presence of follicles in their ovarian biopsies or ultrasound scans. Hence, our current understanding of the etiology of this disease is not clear, and it is likely that the disease is caused by several factors. POF can be influenced by environmental and genetic factors. Irreversible damage to the ovaries during radiation therapy, chemotherapy, or autoimmune disease conditions can cause POF. X-chromosome abnormalities and autosomal genetic defects can also cause POF.44
The genetic causes of POF have been reviewed in detail.10 Several genes have been studied in POF patients, and mutations in FSH, INHA, FOXO3A (forkhead box O3), FOXL2 (forkhead box L2), and BMP15 (bone morphogenetic protein 15) have been shown to cause POF (Table 1).10 It has been demonstrated that either of the two single nucleotide polymorphisms (rs7530810 and rs1321108) in the 5′-UTR (untranslated region) flanking region of TSHB (thyroid-stimulating hormone β) or the intronic single nucleotide polymorphism (rs13172105) within ADAMTS16 (ADAM metallopeptidase with thrombospondin type 1 motif, 16) increase the risk of POF in Korean women.45 Additionally, epistasis between the PCSK1 (proprotein convertase subtilisin/kexin type 1) and DBH (dopamine β-hydroxylase) genes is associated with POF.46 Deletions in Xq21.3–Xq27 have been observed in POF patients.44 The FMR1 (fragile X mental retardation 1) gene is located on Xq27, and expansion of the CGG repeats in its 5′-UTR is associated with POF.47 Since several genes have been associated with POF, next-generation sequencing with large sample sizes could be useful to identify highly associated genes that cause POF.10
Genetics of male infertility
Male infertility accounts for 50% of all infertility cases. Male infertility can be a multifactorial disorder. In 30% of cases, the cause of infertility is unknown.48 The genetic causes include chromosomal abnormalities, mitochondrial DNA mutations, and endocrine disorders of genetic origin. Chromosomal aberrations and single gene mutations account for 10%–15% of male infertility.49 The nongenetic causes include hypogonadism, testicular maldescent, structural abnormalities in the male genital tract, infection, impotence, chronic illness, medication, and immunological conditions.6,48
Leydig cell hypoplasia
In humans, the male genitals begin to develop around week 9 of gestation. Human chorionic gonadotropin (hCG) stimulates the fetal Leydig cells to secrete testosterone, which leads to the development of male genitals.50 At later stages of fetal development, the function of hCG is taken over by luteinizing hormone. Leydig cell hypoplasia is a rare autosomal recessive condition wherein the fetal Leydig cells are unresponsive to hCG. The features of Leydig cell hypoplasia include hypoplasia of the Leydig cells, complete feminization of the external genitals, and partial masculinization with micropenis.50 Leydig cell hypoplasia is caused by inactivating mutations in the LHCGR gene.51 LHCGR has eleven exons: the first ten exons code for the extracellular ligand binding domain, while exon 11 codes for the 7-transmembrane and intracellular domains of the receptor.50 Several LHCGR mutations have been reported to cause Leydig cell hypoplasia.52 A frame shift mutation (c.A589fs) in exon 11 of LHCGR has been reported in a Turkish child born of a consanguineous marriage.51 Additionally, a nonsense mutation (c.T1836G) in a 22-year-old Bedouin female is reported.50 The binding of luteinizing hormone to the LHCGR leads to activation of the mitogen-activated protein kinase (MAPK) cascade in primary cultures of postnatal Leydig cells.53 In agreement with crosstalk of luteinizing hormone and the MAPK pathway,53 it has been shown that the Leydig cell-specific deletion of MAPK 1/2 in mice leads to Leydig cell hypoplasia.54 Activation of the MAPK pathway leads to steroid synthesis.55
XY gonadal dysgenesis
Gonadal (testicular or ovarian) dysgenesis (GD) can be classified as complete or partial. Partial GD and complete GD in patients with a Y chromosome are discussed in this section. Partial GD with testicular dysgenesis is classified by 46,XY or 45,X karyotypes. The gonads in partial GD may be marked by the presence of few tubular structures or fibrous tissues, or may occur as streaks.56 In complete GD, patients have a completely female phenotype with no gonadal development.57 However, patients with complete GD have a higher risk for developing gonadoblastoma. Mutations in MAP3K1 (mitogen-activated protein kinase kinase kinase 1, E3 ubiquitin protein ligase) accompanied by normal SRY (sex determining region Y), SF1, and DHH (desert hedgehog) genes have been reported in 46,XY cases58 (Table 2). Mutations such as p.Glu121AlafsX25, p.Arg62Cys, and p.Ala154Thr in SF1 have been observed in Egyptian individuals with 46,XY disorders of sexual development.59 Recently, a research group has identified a p.N65D missense mutation in the SRY gene of a female with 46,XY gonadal dysgenesis.60 Another research group utilized the 1M array-comparative genomic hybridization (CGH) on the genomic DNA from 14 patients with 46,XY-GD to detect chromosomal aberrations.61 They reported duplication of the SUPT3H (suppressor of Ty 3 homolog [Saccharomyces cerevisiae]), PIP5K1B (phosphatidylinositol-4-phosphate 5-kinase, type I, beta), PRKACG (protein kinase, cAMP-dependent, catalytic, gamma), and FAM189A2 (family with sequence similarity 189, member A2) genes, and deletion of the C2ORF80 (chromosome 2 open reading frame 80) testicular gene. Additionally, haploinsufficiency of DMRT1 (doublesex and mab-3 related transcription factor 1) and mutations in MAMLD1 (mastermind-like domain containing 1) have been observed in 46,XY individuals.62,63 However, the detailed functional mechanisms for these genes are yet to be elucidated.
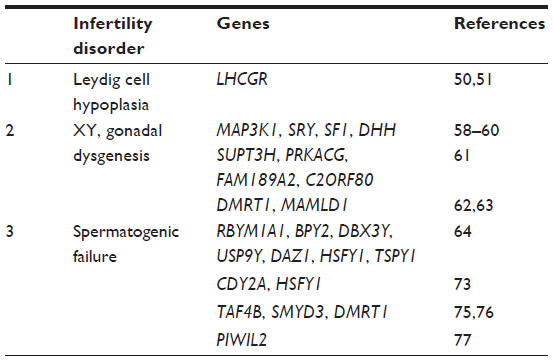 | Table 2 List of genes examined in male infertility as discussed in the review |
Spermatogenic failure
Spermatogenic failure is one the major causes of male infertility.64 Deficiency in spermatogenesis can lead to azoospermia, oligospermia, teratospermia, and asthenozoospermia. The cause of these spermatogenic failures can be genetic, acquired, or both.64 In 1976, the correlation between Y chromosome deletion and male infertility was identified.65 The sex-determining Y chromosome is the smallest human chromosome and consists of short (Yp) and long (Yq) arms. Within the Y chromosome, Yq microdeletions were detected in patients with spermatogenic failure. Three azoospermia factor (AZF) regions (AZFa, AZFb, and AZFc) have been defined on Yq11.66 Interestingly, deletions of AZF regions are thought to be caused by nonallelic homologous recombination. Deletions of ~792 kb and ~98 kb have been reported in the AZFa region.67–69 A ~3.5 Mb deletion of the AZFc region is recurrent, and termed the b2/b4 deletion.70 AZFa deletion leads to a complete absence of spermatozoa. Testicular biopsies from men with an AZFa deletion exhibit features of Sertoli cell-only syndrome.71 Sertoli cell-only syndrome, also referred to as germ cell aplasia, is a condition of the testes in which only Sertoli cells line the seminiferous tubules. AZFb deletion leads to arrest of spermatogenesis. Hence, intracytoplasmic sperm injections are not a viable treatment option for patients with either of these two deletions.71 Loss of AZFc results in azoospermia or oligospermia.72 The AZF-associated candidate genes, such as RBYM1A1 (RNA binding motif protein, Y-linked, family 1, member A1), BPY2 (basic charge, Y-linked, 2), DBX3Y (DEAD [Asp-Glu-Ala-Asp] box helicase 3, Y-linked), USP9Y (ubiquitin specific peptidase 9, Y-linked), DAZ1 (deleted in azoospermia 1), HSFY1 (heat shock transcription factor, Y-linked 1), and TSPY1 (testis specific protein, Y-linked 1) have been well studied. The genes tested for sperm defects have been reviewed and tabulated (Table 2).64
It has also been reported that CDY2A (chromodomain protein, Y-linked, 2A) and HSFY1 are highly expressed in men with obstructive azoospermia as compared with men with maturation arrest.73 Hence, these two Y chromosome-associated genes may have a role in sperm maturation. A study reported a unique t(Y; 1)(q12; q12) balanced reciprocal translocation with a loss of the heterochromatic region on chromosome 1 in an Indian male, which led to spermatogenesis arrest in meiosis I.74 Mutations in TAF4B (TAF4b RNA polymerase II, TATA box binding protein [TBP]-associated factor, 105kDa), SMYD3 (SET and MYND domain containing 3), and DMRT1 have also been reported in patients with azoospermia.75,76 Epigenetic changes are involved in various diseases, including male infertility. Using the Infinium HumanMethylation27 BeadChip, global genomic DNA methylation profiling was performed in three populations of human infertile male patients (conserved spermatogenesis, Sertoli cell-only syndrome, and spermatogenic failure due to germ cell maturation defects).77 The results of this study suggested that men with spermatogenic disorders show abnormal promoter methylation of the PIWIL2 (piwi-like RNA-mediated gene silencing 2) gene, causing defective production of piwi-interacting RNA and hypomethylation of the LINE-1 repetitive sequence.77
Genetics of infertility common to both sexes
Cystic fibrosis
Cystic fibrosis (CF) transmembrane conductance regulator (CFTR) is a cyclic adenosine monophosphate (cAMP)-activated Cl- and HCO3- ion conducting channel expressed in epithelial cells.78 Mutations in CFTR cause CF. Patients with CF suffer from systemic disorders in multiple organs. Infertility is also commonly observed in patients with CF.79 In males, CFTR mutations cause congenital bilateral absence of vas deferens or sperm abnormalities such as azoospermia, teratospermia, and oligospermia.80 Mutations such as F508del, R117H, and W1282X in CFTR have been reported in infertile males.81,82 FSH-induced cAMP production and phosphorylation of cAMP response element-binding protein is essential for spermatogenesis. It has been shown that defects in CFTR result in insufficient activation of FSH-induced signal transduction.80
Additionally, CFTR is involved in secretion of endometrial and oviduct HCO3-, which is necessary for sperm capacitation.83 CFTR is also expressed in the cervix, oviduct, ovary, and uterus, where it regulates fluid control in the female reproductive tract. CF is associated with menstrual irregularities, including amenorrhea, irregular cycles, and anovulation.84,85 CFTR may have fertility implications in non-CF patients. Ovarian hyperstimulation syndrome is an iatrogenic complication seen in individuals on fertility drugs and is characterized by massive cystic enlargement of the ovaries that can later result in ascites and pleural effusion.86 CFTR and aquaporin 1 have been implicated in ovarian hyperstimulation syndrome.86
Robertsonian translocations
Chromosomal abnormalities are also a cause of infertility. Translocations involving acrocentric chromosomes (13, 14, 15, 21, and 22) are defined as Robertsonian translocations.87 Translocations of chromosomes 13 and 14 constitute 75% of all Robertsonian translocations.87,88 Carriers of Robertsonian translocations are phenotypically normal; however, they exhibit reproductive dysfunction, such as oligospermia in males and miscarriages and infertility in females.89 To understand the wide range of phenotypes observed in patients with Robertsonian translocations, a study of eight boys with 45,XY, t(13;14) (q10;q10) and three girls with 45,XX, t(13;14) (q10;q10) karyotypes was conducted and reported hypogonadotrophic hypogonadism, precocious puberty, growth hormone deficiency, and short stature.89
Preimplantation genetic diagnosis has revealed a higher risk of producing aneuploid gametes in carriers of Robertsonian translocations.90 Aneuploidy in such cases occurs due to unusual pairing of derivative chromosomes with their structural normal homologues during meiosis. This phenomenon is called the interchromosomal effect. Aneuploid gametes can lead to miscarriage, reduced fertility, and also birth of children with congenital abnormalities.91 However, a few studies have shown that the interchromosomal effect is completely absent.92 Also, male carriers of balanced Robertsonian translocations show impaired gametogenesis and higher levels of nullisomic and disomic sperms.93 A study comparing 10,000 chromosomes from oocyte and embryonic samples of translocation carriers with 200,000 chromosomes from well matched normal patients attempted to determine if the interchromosomal effect truly exists.90 Alfarawati et al90 suggest that it does exist, but occurs in a narrow window of developmental stages. Aneuploidy was not seen in oocytes, but occurred during cleavage stages in the embryo. Further, no increase in aneuploidy was seen at blastocyst stages. Hence, the aneuploid embryos are rapidly eliminated. The data from this study could help future researchers achieve more successful assisted reproduction therapies.90
Conclusion
The genetics of infertility is a vast subject, and the genetic causes of several reproductive disorders are chromosomal, involve single genes, or are polygenic. In addition, there are several genetic syndromes that exhibit infertility. Despite extensive research, there are no well-defined genes that can be used for genetic testing of infertility conditions. Hence, there is a need for novel diagnostic technologies to identify both new and known infertility genes. Currently, several genetic association studies have been performed to identify genes for infertility. Increasing the sample size of these association studies may maximize the chance of identifying strong associations. Whole exome and whole genome sequencing can be considered, especially in idiopathic cases of infertility, although interpretation of high throughput sequencing data may itself be a challenge. In addition, genotype and phenotype correlation studies and microarray-based genome-wide studies from large numbers of patients could shed more light on the genetic causes of infertility disorders. Bisulfite sequencing, methylated DNA immunoprecipitation sequencing, and methylated DNA capture by affinity purification sequencing may aid future investigations. In the future, pursuing the most promising genetic variants, mutations, or polymorphisms may provide clinically relevant therapeutics for infertile individuals.
Acknowledgments
We acknowledge the management of VIT University for their support. We apologize to the many scientists whose work was not discussed due to space constraints, and in some cases, reviews have been cited at the expense of the original research.
Disclosure
The authors report no conflicts of interest in this work.
References
Mascarenhas MN, Flaxman SR, Boerma T, Vanderpoel S, Stevens GA. National, regional, and global trends in infertility prevalence since 1990: a systematic analysis of 277 health surveys. PLoS Med. 2012;9(12):e1001356. | |
Ombelet W, Cooke I, Dyer S, Serour G, Devroey P. Infertility and the provision of infertility medical services in developing countries. Hum Reprod Update. 2008;14(6):605–621. | |
Matzuk MM, Lamb DJ. The biology of infertility: research advances and clinical challenges. Nat Med. 2008;14(11):1197–1213. | |
dos Santos AP, Andrade JG, Piveta CS, et al. Screening of Y chromosome microdeletions in 46, XY partial gonadal dysgenesis and in patients with a 45, X/46, XY karyotype or its variants. BMC Med Genet. 2013;14:115. | |
Pouresmaeili F, Fazeli Z. Premature ovarian failure: a critical condition in the reproductive potential with various genetic causes. Int J Fertil Steril. 2014;8(1):1–12. | |
Shah K, Sivapalan G, Gibbons N, Tempest H, Griffin DK. The genetic basis of infertility. Reproduction. 2003;126(1):13–25. | |
Simoni M, Tempfer CB, Destenaves B, Fauser BC. Functional genetic polymorphisms and female reproductive disorders. Part I: Polycystic ovary syndrome and ovarian response. Hum Reprod Update. 2008;14(5):459–484. | |
Mühlmann M. Molecular cytogenetics in metaphase and interphase cells for cancer and genetic research, diagnosis and prognosis. Application in tissue sections and cell suspensions. Genet Mol Res. 2002;1(2):117–127. | |
Hwang K, Weedin JW, Lamb DJ. The use of fluorescent in situ hybridization in male infertility. Ther Adv Urol. 2010;2(4):157–169. | |
Shelling AN. Premature ovarian failure. Reproduction. 2010;140(5):633–641. | |
Jenkins TG, Carrell DT. The sperm epigenome and potential implications for the developing embryo. Reproduction. 2012;143(6):727–734. | |
Gaware VM, Parjane SK, Merekar Abhijit N, et al. Female infertility and its treatment by alternative medicine: a review. J Chem Pharm Res. 2009;1(1):148–162. | |
Olooto WE, Amballi AA, Banjo TA. A review of female infertility: important etiological factors and management. J Microbiol Biotech Res. 2012;2(3):379–385. | |
Walters KA, Allan CM, Handelsman DJ. Rodent models for human polycystic ovary syndrome. Biol Reprod. 2012;86(5):149, 1–12. | |
Hossain MM, Cao M, Wang Q, et al. Altered expression of miRNAs in a dihydrotestosterone-induced rat PCOS model. J Ovarian Res. 2013;6(1):36. | |
Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. 1997; 18(6):774–800. | |
Barber TM, Franks S. Genetics of polycystic ovary syndrome. Front Horm Res. 2013;40:28–39. | |
Jakubowski L. Genetic aspects of polycystic ovary syndrome. Endokrynol Pol. 2005;56:285–293. | |
Chen ZJ, Zhao H, He L, et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat Genet. 2011;43(1):55–59. | |
Mutharasan P, Galdones E, Peñalver Bernabé B, et al. Evidence for chromosome 2p16.3 polycystic ovary syndrome susceptibility locus in affected women of European ancestry. J Clin Endocrinol Metab. 2013;98(1):E185–E190. | |
Wang P, Zhao H, Li T, et al. Hypomethylation of the LH/choriogonadotropin receptor promoter region is a potential mechanism underlying susceptibility to polycystic ovary syndrome. Endocrinology. 2014;155(4):1445–1452. | |
Bassiouny YA, Rabie WA, Hassan AA, Darwish RK. Association of the luteinizing hormone/choriogonadotropin receptor gene polymorphism with polycystic ovary syndrome. Gynecol Endocrinol. 2014;30(6):428–430. | |
Tong Y, Liao WX, Roy AC, Ng SC. Absence of mutations in the coding regions of follicle-stimulating hormone receptor gene in Singapore Chinese women with premature ovarian failure and polycystic ovary syndrome. Horm Metab Res. 2001;33(4):221–226. | |
Unsal T, Konac E, Yesilkaya E, et al. Genetic polymorphisms of FSHR, CYP17, CYP1A1, CAPN10, INSR, SERPINE1 genes in adolescent girls with polycystic ovary syndrome. J Assist Reprod Genet. 2009;26(4):205–216. | |
Gu BH, Park JM, Baek KH. Genetic variations of follicle stimulating hormone receptor are associated with polycystic ovary syndrome. Int J Mol Med. 2010;26(1):107–112. | |
Fu L, Zhang Z, Zhang A, et al. Association study between FSHR Ala307Thr and Ser680Asn variants and polycystic ovary syndrome (PCOS) in Northern Chinese Han women. J Assist Reprod Genet. 2013;30(5):717–721. | |
Wu XQ, Xu SM, Liu JF, et al. Association between FSHR polymorphisms and polycystic ovary syndrome among Chinese women in north China. J Assist Reprod Genet. 2014;31(3):371–377. | |
Lee B, Palermo G, Machaca K. Downregulation of store-operated Ca2+ entry during mammalian meiosis is required for the egg-to-embryo transition. J Cell Sci. 2013;126 Pt 7:1672–1681. | |
Bagheri M, Abdi Rad I, Hosseini Jazani N, Nanbakhsh F. Vitamin D receptor TaqI gene variant in exon 9 and polycystic ovary syndrome risk. Int J Fertil Steril. 2013;7(2):116–121. | |
Stadtmauer LA, Wong BC, Oehninger S. Should patients with polycystic ovary syndrome be treated with metformin? Benefits of insulin sensitizing drugs in polycystic ovary syndrome-beyond ovulation induction. Hum Reprod. 2002;17(12):3016–3026. | |
Lakkakula BV, Thangavelu M, Godla UR. Genetic variants associated with insulin signaling and glucose homeostasis in the pathogenesis of insulin resistance in polycystic ovary syndrome: a systematic review. J Assist Reprod Genet. 2013;30(7):883–895. | |
Shen HR, Qiu LH, Zhang ZQ, Qin YY, Cao C, Di W. Genome-wide methylated DNA immunoprecipitation analysis of patients with polycystic ovary syndrome. PLoS One. 2013;8(5):e64801. | |
Sang Q, Li X, Wang H, et al. Quantitative methylation level of the EPHX1 promoter in peripheral blood DNA is associated with polycystic ovary syndrome. PLoS One. 2014;9(2):e88013. | |
Ting W, Yanyan Q, Jian H, Keqin H, Duan M. The relationship between insulin resistance and CpG island methylation of LMNA gene in polycystic ovary syndrome. Cell Biochem Biophys. 2013;67(3):1041–1047. | |
Roth LW, McCallie B, Alvero R, Schoolcraft WB, Minjarez D, Katz-Jaffe MG. Altered microRNA and gene expression in the follicular fluid of women with polycystic ovary syndrome. J Assist Reprod Genet. 2014;31(3):355–362. | |
Zangen D, Kaufman Y, Zeligson S, et al. XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes coactivation of estrogen-driven transcription. Am J Hum Genet. 2011;89(4):572–579. | |
Fogli A, Boespflug-Tanguy O. The large spectrum of eIF2B-related diseases. Biochem Soc Trans. 2006;34 Pt 1:22–29. | |
Ledig S, Röpke A, Haeusler G, Hinney B, Wieacker P. BMP15 mutations in XX gonadal dysgenesis and premature ovarian failure. Am J Obstet Gynecol. 2008;198(1):84. e1–e5. | |
Lourenço D, Brauner R, Lin L, et al. Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med. 2009;360(12):1200–1210. | |
Gottschalk ME, Coker SB, Fox LA. Neurologic anomalies of Perrault syndrome. Am J Med Genet. 1996;65(4):274–276. | |
Pierce SB, Walsh T, Chisholm KM, et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault syndrome. Am J Hum Genet. 2010;87(2):282–288. | |
Pierce SB, Gersak K, Michaelson-Cohen R, et al. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet. 2013;92(4):614–620. | |
Pierce SB, Chisholm KM, Lynch ED, et al. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci U S A. 2011;108(16):6543–6548. | |
Shelling AN. X chromosome defects and premature ovarian failure. Aust N Z J Med. 2000;30(1):5–7. | |
Pyun JA, Kim S, Cha DH, Kwack K. Epistasis between polymorphisms in TSHB and ADAMTS16 is associated with premature ovarian failure. Menopause. 2013;21(8):890–895. | |
Pyun JA, Kim S, Cha DH, Kwack K. Epistasis between polymorphisms in PCSK1 and DBH is associated with premature ovarian failure. Menopause. 2014;21(11):1249–1253. | |
Bodega B, Bione S, Dalprà L, et al. Influence of intermediate and uninterrupted FMR1 CGG expansions in premature ovarian failure manifestation. Hum Reprod. 2006;21(4):952–957. | |
Poongothai J, Gopenath TS, Manonayaki S. Genetics of human male infertility. Singapore Med J. 2009;50(4):336–347. | |
Ferlin A, Arredi B, Foresta C. Genetic causes of male infertility. Reprod Toxicol. 2006;22(2):133–141. | |
Salameh W, Choucair M, Guo TB, et al. Leydig cell hypoplasia due to inactivation of luteinizing hormone receptor by a novel homozygous nonsense truncation mutation in the seventh transmembrane domain. Mol Cell Endocrinol. 2005;229(1–2):57–64. | |
Richter-Unruh A, Korsch E, Hiort O, Holterhus PM, Themmen AP, Wudy SA. Novel insertion frameshift mutation of the LH receptor gene: problematic clinical distinction of Leydig cell hypoplasia from enzyme defects primarily affecting testosterone biosynthesis. Eur J Endocrinol. 2005;152(2):255–259. | |
Wu SM, Leschek EW, Rennert OM, Chan WY. Luteinizing hormone receptor mutations in disorders of sexual development and cancer. Front Biosci. 2000;5:D343–D352. | |
Shiraishi K, Ascoli M. Lutropin/choriogonadotropin (LH/CG) stimulate the proliferation of primary cultures of rat Leydig cells through a pathway that involves activation of the ERK1/2 cascade. Endocrinology. 2007;148(7):3214–3225. | |
Yamashita S, Tai P, Charron J, Ko C, Ascoli M. The Leydig cell MEK/ERK pathway is critical for maintaining a functional population of adult Leydig cells and for fertility. Mol Endocrinol. 2011;25(7):1211–1222. | |
Martinelle N, Holst M, Söder O, Svechnikov K. Extracellular signal-regulated kinases are involved in the acute activation of steroidogenesis in immature rat Leydig cells by human chorionic gonadotropin. Endocrinology. 2004;145(10):4629–4634. | |
Rohatgi M, Gupta DK, Menon PS, Verma IC, Mathur M. Mixed gonadal dysgenesis and dysgenetic male pseudohermaphroditism – a critical analysis. Indian J Pediatr. 1992;59(4):487–500. | |
McCann-Crosby B, Mansouri R, Dietrich JE, et al. State of the art review in gonadal dysgenesis: challenges in diagnosis and management. Int J Pediatr Endocrinol. 2014;2014(1):4. | |
Das DK, Rahate SG, Mehta BP, Gawde HM, Tamhankar PM. Mutation analysis of mitogen activated protein kinase 1 gene in Indian cases of 46, XY disorder of sex development. Indian J Hum Genet. 2013;19(4):437–442. | |
Tantawy S, Mazen I, Soliman H, et al. Analysis of the gene coding for steroidogenic factor 1 (SF1, NR5A1) in a cohort of 50 Egyptian patients with 46, XY disorders of sex development. Eur J Endocrinol. 2014;170(5):759–767. | |
Helszer Z, Dmochowska A, Szemraj J, et al. A novel mutation (c. 341A>G) in the SRY gene in a 46, XY female patient with gonadal dysgenesis. Gene. 2013;526(2):467–470. | |
Norling A, Lindén Hirschberg A, Iwarsson E, Persson B, Wedell A, Barbaro M. Novel candidate genes for 46, XY gonadal dysgenesis identified by a customized 1 M array-CGH platform. Eur J Med Genet. 2013;56(12):661–668. | |
Ledig S, Hiort O, Wünsch L, Wieacker P. Partial deletion of DMRT1 causes 46, XY ovotesticular disorder of sexual development. Eur J Endocrinol. 2012;167(1):119–124. | |
Kalfa N, Fukami M, Philibert P, et al. Screening of MAMLD1 mutations in 70 children with 46, XY DSD: identification and functional analysis of two new mutations. PLoS One. 2012;7(3):e32505. | |
Massart A, Lissens W, Tournaye H, Stouffs K. Genetic causes of spermatogenic failure. Asian J Androl. 2012;14(1):40–48. | |
Tiepolo L, Zuffardi O. Localization of factors controlling spermatogenesis in the nonfluorescent portion of the human Y chromosome long arm. Hum Genet. 1976;34(2):119–124. | |
Vogt PH, Edelmann A, Kirsch S, et al. Human Y chromosome azoospermia factors (AZF) mapped to different subregions in Yq11. Hum Mol Genet. 1996;5(7):933–943. | |
Blanco P, Shlumukova M, Sargent CA, Jobling MA, Affara N, Hurles ME. Divergent outcomes of intrachromosomal recombination on the human Y chromosome: male infertility and recurrent polymorphism. J Med Genet. 2000;37(10):752–758. | |
Sun C, Skaletsky H, Rozen S, et al. Deletion of azoospermia factor a (AZFa) region of human Y chromosome caused by recombination between HERV15 proviruses. Hum Mol Genet. 2000;9(15):2291–2296. | |
Ferlin A, Arredi B, Speltra E, et al. Molecular and clinical characterization of Y chromosome microdeletions in infertile men: a 10-year experience in Italy. J Clin Endocrinol Metab. 2007;92(3):762–770. | |
Kuroda-Kawaguchi T, Skaletsky H, Brown LG, et al. The AZFc region of the Y chromosome features massive palindromes and uniform recurrent deletions in infertile men. Nat Genet. 2001;29(3):279–286. | |
Krausz C, Quintana-Murci L, McElreavey K. Prognostic value of Y deletion analysis: what is the clinical prognostic value of Y chromosome microdeletion analysis? Hum Reprod. 2000;15(7):1431–1434. | |
Stouffs K, Lissens W, Tournaye H, Van Steirteghem A, Liebaers I. The choice and outcome of the fertility treatment of 38 couples in whom the male partner has a Yq microdeletion. Hum Reprod. 2005;20(7):1887–1896. | |
Stahl PJ, Mielnik AN, Barbieri CE, Schlegel PN, Paduch DA. Deletion or underexpression of the Y-chromosome genes CDY2 and HSFY is associated with maturation arrest in American men with nonobstructive azoospermia. Asian J Androl. 2012;14(5):676–682. | |
Pinho MJ, Neves R, Costa P, et al. Unique t(Y;1)(q12;q12) reciprocal translocation with loss of the heterochromatic region of chromosome 1 in a male with azoospermia due to meiotic arrest: a case report. Hum Reprod. 2005;20(3):689–696. | |
Lopes AM, Aston KI, Thompson E, et al. Human spermatogenic failure purges deleterious mutation load from the autosomes and both sex chromosomes, including the gene DMRT1. PLoS Genet. 2013;9(3):e1003349. | |
Ayhan Ö, Balkan M, Guven A, et al. Truncating mutations in TAF4B and ZMYND15 causing recessive azoospermia. J Med Genet. 2014;51(4):239–244. | |
Heyn H, Ferreira HJ, Bassas L, et al. Epigenetic disruption of the PIWI pathway in human spermatogenic disorders. PLoS One. 2012;7(10):e47892. | |
Quinton PM. [Too much salt, too little soda: cystic fibrosis]. Sheng Li Xue Bao. 2007;59:397–415. Chinese. | |
Welsh MJ, Fick RB. Cystic fibrosis. J Clin Invest. 1987;80(6):1523–1526. | |
Chen H, Ruan YC, Xu WM, Chen J, Chan HC. Regulation of male fertility by CFTR and implications in male infertility. Hum Reprod Update. 2012;18(6):703–713. | |
Stuppia L, Antonucci I, Binni F, et al. Screening of mutations in the CFTR gene in 1195 couples entering assisted reproduction technique programs. Eur J Hum Genet. 2005;13(8):959–964. | |
Schulz S, Jakubiczka S, Kropf S, Nickel I, Muschke P, Kleinstein J. Increased frequency of cystic fibrosis transmembrane conductance regulator gene mutations in infertile males. Fertil Steril. 2006;85(1):135–138. | |
Wang XF, Zhou CX, Shi QX, et al. Involvement of CFTR in uterine bicarbonate secretion and the fertilizing capacity of sperm. Nat Cell Biol. 2003;5(10):902–906. | |
Kent G, Oliver M, Foskett JK, et al. Phenotypic abnormalities in long-term surviving cystic fibrosis mice. Pediatr Res. 1996;40(2):233–241. | |
Ajonuma LC, Ng EH, Chow PH, et al. Increased cystic fibrosis transmembrane conductance regulator (CFTR) expression in the human hydrosalpinx. Hum Reprod. 2005;20(5):1228–1234. | |
Jin PY, Lu YC, Li L, Han QF. Co action of CFTR and AQP1 increases permeability of peritoneal epithelial cells on estrogen-induced ovarian hyper stimulation syndrome. BMC Cell Biol. 2012;13:23. | |
Gardner RJ, Sutherland GR. Chromosome Abnormalities and Genetic Counselling. 2nd ed. Oxford, UK: Oxford University Press; 1996. | |
Therman E, Susman B, Denniston C. The non-random participation of human acrocentric chromosomes in Robertsonian translocations. Ann Hum Genet. 1989;53 Pt 1:49–65. | |
Choi BH, Kim UH, Lee KS, Ko CW. Various endocrine disorders in children with t(13;14)(q10;q10) Robertsonian translocation. Ann Pediatr Endocrinol Metab. 2013;18 (3):111–115. | |
Alfarawati S, Fragouli E, Colls P, Wells D. Embryos of Robertsonian translocation carriers exhibit a mitotic interchromosomal effect that enhances genetic instability during early development. PLoS Genet. 2012;8(10):e1003025. | |
Scriven PN, Handyside AH, Ogilvie CM. Chromosome translocations: segregation modes and strategies for preimplantation genetic diagnosis. Prenat Diagn. 1998;18(13):1437–1449. | |
Munné S, Escudero T, Fischer J, et al. Negligible inter chromosomal effect in embryos of Robertsonian translocation carriers. Reprod Biomed Online. 2005;10(3):363–369. | |
Douet-Guilbert N, Bris MJ, Amice V, et al. Interchromosomal effect in sperm of males with translocations: report of 6 cases and review of the literature. Int J Androl. 2005;28(6):372–379. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.