Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 7
New developments in the treatment of osteoarthritis – focus on biologic agents
Authors Torrero J, Martínez C
Received 19 February 2015
Accepted for publication 7 May 2015
Published 22 July 2015 Volume 2015:7 Pages 33—43
DOI https://doi.org/10.2147/OARRR.S50058
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Chuan-Ju Liu
Jose Ignacio Torrero,1 Carlos Martínez2
1BioTrauma Centre, Escaldes, Principality of Andorra; 2University of Illinois Hospital and Health Sciences System, Chicago, IL, USA
Abstract: Osteoarthritis (OA) is one of the most common diseases around the world. Medical, social, and financial consequences oblige clinicians, surgeons, and researchers to focus on finding the best treatment option, to eradicate and stop this degenerative joint disease, in order to avoid surgical options which in many instances are over-indicated. Noninvasive treatments, such as anti-inflammatory drugs, physiotherapy, orthotic devices, dietary supplements, have demonstrated lack of effectiveness. The possibility to perform intra-articular injections with hyaluronic acid, corticosteroids, or the newest but criticized treatment based on platelet-rich plasma (PRP) has changed the management of OA disease. The use of PRP has led to many differences in treatment since there is a lack of consensus about protocols, indications, number of doses, cost-effectiveness, and duration of the treatment. Many publications have suggested efficacy in tendon injuries, but when PRP has been indicated to treat cartilage injuries, things are more inconsistent. Some authors have reported their experience treating OA with PRP, and it seems that, if well indicated, it is an option as a supplementary therapy. Therefore, we need to understand that OA is a mechanical disease which not only produces changes in radiographs, but also affects the quality of life. Pathogenesis of OA has been well explained, providing us new knowledge and future possibilities to improve the clinical approach. From basic science to surgery, there is a great field we all need to contribute to, because the general population is aging and total joint replacements should not be the only solution for OA. So herein is an actual review of the developments for treating OA with biologics, intended to be useful for the population inside orthopedics who could be called bio-orthopedists, since OA is a molecular homeostasis disbalance between catabolism and anabolism triggered by mechanical stress.
Keywords: plasma, platelets, interleukines, receptors, orthokine
Introduction
As the world population ages, osteoarthritis (OA) becomes a disease with physical and economic consequences for both patients and communities. Actual treatments provide, at best, symptomatic pain and inflammation relief, and include analgesics, nonsteroidal anti-inflammatory drugs, topical anti-inflammatory creams, intra-articular injections, dietary supplements, acupuncture, physiotherapy, and orthotic devices, all last-resort options to avoid surgery. In fact, patients desire treatment options that avoid aggressive treatments like surgery. Therefore, the recent social emphasis on physical fitness in our environment has resulted in an increase of patients in the age range of 30–50s suffering injuries to hyaline cartilage due to repetitive movements and the increase of stress on cartilage. Finally, surgeons are accustomed to treating patients who have undergone a knee arthroscopy procedure precipitated by a meniscal tear, for example, and observing during the surgical procedure a degradation of weight areas inside the joint. As a result, the clinical follow-up will not be as good as the patient or surgeon would expect.
Furthermore, aggressive treatments cannot explain improvement based on the unbalanced metabolism of OA.1 Structural and symptomatic stages and comorbidities, as well as individualized factors such as sex, age, and genetics, can affect decisions regarding follow-up treatment.2–5
Definition of OA: pathogenesis and etiology
OA is a complex disease involving cartilage degradation leading finally to bone-on-bone contact with loss of articular balance and strength and episodes of inflammation. Knee, hip, ankle, and foot joints are the most frequent to suffer OA because of their relevance as weight-bearing joints.
The appearance of cartilage is normally white and smooth with a tensile and compressive resistance which can differ between hyaline and meniscal types. Hyaline and meniscal cartilages have a high percentage in water; their cells are named chondrocytes and fibrochondrocytes respectively and are located inside typical lagoons, and an extracellular matrix (ECM) which is composed of type II collagen for hyaline cartilage and type I for fibrocartilage and joint capsule, the latter with a deposition in layers for a better distribution in radial stress. Collagen fibers form a scaffold with hyaluronic acid (HA) polymers to which aggrecan molecules are fixed through a linker protein. HA has a glycoprotein, lubricin, which is responsible for lubrication. Recent studies support the promise of using lubricin as a therapy for OA.6 Aggrecans are formed by a core protein which is attached to chondroitin and keratin sulfate molecules (glycosaminoglycans), negative in their electrical properties to enhance water retention for ECM. Other proteins include noncollagenous, aggrecans, leucine-rich repeated, structural, regulatory, and others (Table 1, Figures 1 and 2).7
 | Table 1 PGs and proteins of the cartilage extracellular matrix |
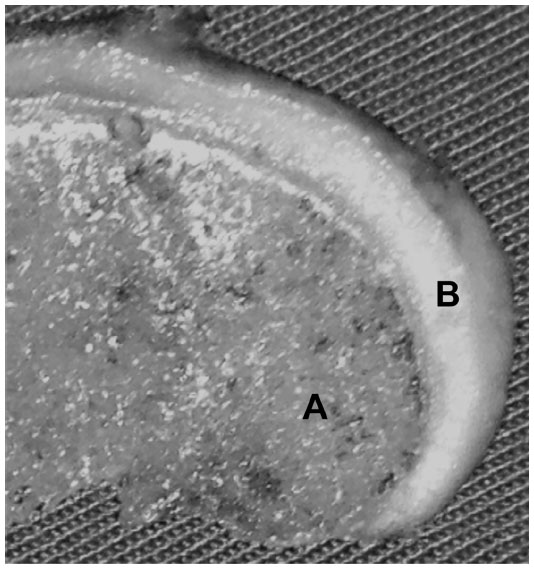 | Figure 1 Normal appearance of the hyaline cartilage and the subchondral bone in a fragment of the femoral condyle. |
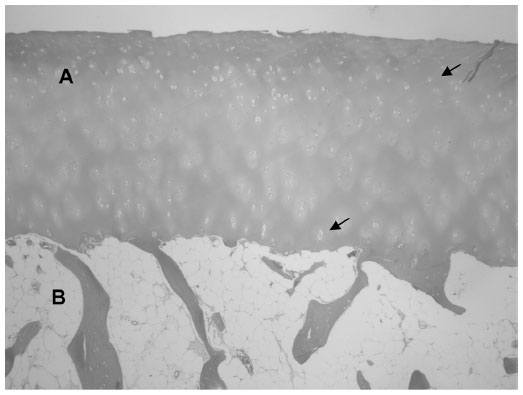 | Figure 2 Normal histological appearance of the hyaline cartilage (A) and the subchondral bone (B). |
All these properties and elements can be altered because hyaline cartilage is avascular, alymphatic, and aneural and is subjected to repetitive mechanical stress. A lack of blood supply compels the cartilage to acquire nutrition directly from synovial fluid which supplies all the nutrients and oxygen to the chondrocytes, enabling normal metabolism in a relative hypoxic medium due to gene expression of Sox9 and hypoxia inducible factor 1 alpha (HIF-1α).6 Synovial fluid, being produced by blood vessels from the synovial membrane, may contain macrophages, fibroblasts, adipocytes, and leukocytes, all directly influencing the pathogenesis of OA.8–10 Subchondral bone also has an important influence during the process of OA because osteoclasts demineralize bone through the action of acid proteases, like cathepsin K. Remodeling action is a turnover between resorption and bone apposition, governed by the bone morphogenetic protein (BMP) 2 and transforming growth factor (TGF)-β, which also plays a role in the pathogenesis of OA.6 In fact, OA can be explained as a compound of inflammatory cytokines and degradation products from the ECM, as a consequence of a biomechanical stress or micro-impact repetitive injury. The final result is an alteration of the subchondral bone as well, pointing the normal homeostasis balance toward a cellular catabolism, including apoptosis and aberrant expression of inflammatory genes. This process occurs in chondrocytes, synovium, and other joint tissues.1 Although we are dealing with a diverse etiology with different factors in place, the end result is a phenotypic image of an OA joint where enzymes and proinflammatory and anti-inflammatory cytokines are involved.3–5
The main enzymes involved in ECM breakdown are metzincin proteolytic enzymes like matrix metalloproteinases (MMPs), MMP-13 being the most involved in cleaving type II collagen and aggrecans and MMP-7 as an early predictor of OA, as much as 10 years ahead of the disease.6,11 The pathways of MMPs are sequentially activated by an amplifying cascade from fragmented proteins required for the joint homeostasis. These fragments are known to feedback and stimulate further matrix destruction.12 MMP is counterbalanced by the tissue inhibitors of MMPs (TIMPs), whose synthesis is low in OA cartilage.
Recently there has been published the importance of joint tissues such as synovium and cartilage within the innate immune inflammatory network implicated in OA progression.13–15 Aggrecan breakdown is caused by enzymes known as a disintegrin and metalloproteinase with thrombospondin motif (ADAMTS). There have been reported 23 MMPs, 18 ADAMTS, and four TIMPs.16–19
Joint inflammation is the result of proteolytic enzymes and cytokines. Cytokines can be divided into two major groups: inflammatory and anti-inflammatory.20 Inflammatory cytokines are responsible to the greatest extent for the loss of metabolic homeostasis of tissues that are inside joints (like cartilage), by promoting catabolic and destructive processes as well as other inflammatory compounds and enzymes.
The greatest inflammatory cytokines are TNFα, interleukins (IL-1β, IL-6, IL-15, IL-17, and IL-18), lymphokines (granulocyte-macrophage colony-stimulating factor [GM-CSF], interferon-gamma [IFN-γ]), and chemokines (IL-8).6 IL-1β induces inflammatory reactions. It is synthesized in the joint by chondrocytes, osteoblasts, cells forming the synovial membrane, and mononuclear cells that were previously present in the joint or infiltrated its structure during the inflammatory response.10,21 Patients affected with OA have been shown with elevated levels of IL-1β, which interacts with its cellular receptor IL-1R1.21–24 The expression of IL-1R1 receptor is increased in patients with OA on the surface of chondrocytes and fibroblast-like synoviocytes compared to treatment groups.25,26 IL-1β affects cartilage destruction by inhibition of collagen and aggrecan levels and stimulates the release of MMP-1, -3, and -13 as well as IL-6, IL-8, monocyte chemoattractant protein (MCP-1), and regulated on activation normal T-cell expressed and secreted (RANTES). TNFα is secreted by the same cells in the joint that synthesize IL-1β.21–24 TNFα works by blocking the synthesis of proteoglycan components, binding proteins, and type II collagen from chondrocytes.27 Cytokines modulate the expression of MMP and ADAMTS as well as alter the cellular mechanisms of catabolic molecules such as inducible nitric oxide synthase (iNOS), soluble phospholipase A2, cyclooxygenase-2 (COX-2), and membrane prostaglandin E synthase leading to prostaglandin E2 (PGE2) expression. IL-1β and TNFα result in upregulation of PGE2, which has been targeted as an anti-inflammatory strategy, and can only lead to gastrointestinal irritation and cardiac damage. IL-1β and TNFα also stimulate the production of reactive oxygen species which are free radicals like hydrogen peroxide, hypochlorous acid, hydroxyl radical, superoxide, and chloramines, that are highly destructive for living cells unless contained or neutralized by molecules which downregulate OA (antioxidants). Therefore, IL-1β and TNFα are also related to the promotion of chondrocyte hypertrophy and several transcription pathways like c-Jun N terminal kinase, p38 mitogen-activated kinase, and nuclear factor kappa B (NF-κB).6 IL-6 strongly activates the immune system, usually as a response to IL-1β and TNFα, mainly implemented by chondrocytes, osteoblasts, fibroblasts, macrophages, and adipocytes.28,29 The effect of IL-6 is not different from other cytokines and, in synergy with them, causes a decrease in the production of type II collagen and increases the production of MMP.30,31 IL-15 is increased in the synovial fluid in the early stages of OA.32 Increased IL-15 level in the serum correlates with both the sensation of pain and the severity of lesions in the X-ray image. It has also been noted that the presence of IL-15 can stimulate the secretion of certain MMPs.33 IL-17 is stimulated by CD4+ T-cells and mast cells which infiltrate the synovial membrane and the entire joint through blood vessels during the process of OA.34 IL-17 has been shown to inhibit the synthesis of proteoglycans by chondrocytes and promotes the production of enzymes of the MMP group.35 Furthermore, IL-17 influences the secretion of IL-1β, TNFα, IL-6, nitric oxide (NO) free radicals, and PGE2,36,37 which inhibits collagen and proteoglycan formation, as well as the production of the natural antagonist to IL-1β. The IL-1 receptor antagonist (IL-1Ra) has been shown to positively affect joint inflammation and OA with marginal clinical improvement.38 IL-18 is determined by chondrocytes, osteoblasts, fibroblasts, and macrophages.39,40 IL-18 affects chondrocytes by inducing regulation of its own receptor, IL-18 R alpha, on the surface and stimulates the synthesis of MMP-1, MMP-3, and MMP-13. In addition, there is an inhibition of synthesis of proteoglycan aggrecan and type II collagen; moreover, chondrocytes exhibit morphological changes typical of cells entering apoptosis.41,42 IL-8 is a cytokine that is chemoattractive, causing leukocytes to migrate toward areas in which they eventually concentrate. Nevertheless, the action of anti-inflammatory cytokines mainly involves the synthesis of inflammatory cytokines, particularly IL-1β and TNFα.43 IL-4 production is determined by T-cells (Th2) that infiltrate the synovium of the joint by blood vessels.44 IL-4 is associated with a strong chondroprotective effect, inhibiting the degradation of proteoglycans by inhibiting the secretions of MMP,45 as well as exhibiting properties of apoptosis inhibition of both chondrocytes and fibroblasts. Additionally, there is a decrease in IL-1β and TNFα production and a decrease in inflammatory mediators such as PGE2 and COX-2.45,46 IL-10 is structurally related to interferons. Chondrocytes express both IL-10 and the receptor IL-10R.47 It has been proven that IL-10 is involved in stimulating the synthesis of type II collagen and aggrecans and decreases TNFα and IL-1β plus the inhibitions of MMPs, thanks to the stimulations of IL-1β antagonist (IL-1Ra) and TIMP-1 as well as growth factors.48,49 IL-10 reduces the effect of TNFα and the number of TNFα receptors in synovial fibroblasts sampled from patients with OA.46 It is not known what increases the secretion of IL-10, but some authors argue that it is due to the inductive effect of the increased intra-articular hydrostatic fluid pressure on cells.50,51 IL-13 has a similar structure to IL-4. Its effects are inhibition of IL-1β, TNFα, and MMP-3 with a simultaneous increase in the level of IL-1Ra at the same time that it inhibits the synthesis of COX-2.52
Micro-impacts inside the joint are the most prevalent etiologic factor to induce OA. A mechanical injury can induce the release of reactive oxygen species that stimulate chondrocyte death and the production of stress kinases that upregulate MMP-13, ADAMTS-5, TNFα, and transcription factors such as ETS and HIF 2α.53,54
Integrins from cartilage cell surface play an important role as they interact with the ECM to maintain the cartilage homeostasis and interaction between ECM and integrins, indicating growth and differentiation of the cartilage. During OA, abnormal activation of integrin expression alters cell/ECM signaling and modifies the gene expression of the chondrocytes to a synthesis of cytokines with a destructive effect over the regulatory factors. IL-1β and TNFα, apart from being pain inducers, activate the enzymatic degradation of the cartilage matrix by MMP-13, with no inhibition signals being produced. OA chondrocytes express IL-1β by itself that can induce the expression of MMP-13 and predominantly the aggrecanases ADAMTS-4.55,56 At the same time, the chondrocyte expresses TNFα that upregulates the ADAMTS-4 considered to be the major aggrecan degradation enzymes.13 ADAMTS-5 is produced constitutively and is not under the regulation of IL-1β or TNFα.56,57 It is also known that IL-1β and TNFα can upregulate COX-2 and increase the synthesis of prostaglandins and consequentially the increase of NO2, reducing the cell survival.58,59
IL-1β and TNFα can promote the synthesis of proinflammatory cytokines such as IL-6, leukemia inhibitor factor, IL-17, IL-18, and IL-8.13 All of these cytokines work in the same direction and synergize in the promotion of cartilage catabolism as they also inhibit in vitro the expression of COL2A1 by chondrocytes.60–62 Therefore, other anabolic activity, by promoting COL2A1 transcription, has been shown by cytokine-induced synthesis of PGE2.61,63 IL-1β stimulates early growth response factor 1 (EGR-1) that is a cytokine that induces and works as a transcriptor factor inhibiting the COL2A1.64 All of these cytokines are subject to regulation by NF-κB pathways,65 which are abnormally activated in osteoarthritic chondrocytes due to the activation of the integrins and by the ECM products such as fibronectin fragments.66–68 IL-1β and TNFα also induce the expression of toll-like receptor (TLR)-2 and TLR-4, which are promoters of chronic inflammation and tissue remodeling to endogenous ligands.68,69 Low-molecular-weight hyaluronan as small-sized HA molecules might have a proinflammatory effect in animal models.70 High mobility group box chromosomal protein 1 (HMGB-1) which are increased in OA joints are ligands of TLR2. This TLR interaction with these two ligands promotes the MyD88 signaling pathway that will stimulate NF-κB-dependent genes like MMP-13.71 MMP-13 is upregulated by growth arrest and DNA damage-inducible GADD45β and ELF3, an ETS transcription factor family. However, GADD45β, ELF3, and MMP-13 are direct targets of canonical NF-κB signaling pathways.72 Oxidative stress produced by impact and genotoxicity can increase the levels of NF-κB and induce GADD45β. GADD45β contributes to alter the matrix homeostasis by suppressing COL2A1 promoter activity, but also protects from TNFα-induced apoptosis.73 HIF 2α can be activated by proinflammatory cytokines and other atypical stress-related events such as avascular, hypoxic environment. HIF2 impacts the production of MMP-13 by increasing its production.74,75 Discoidin domain receptor 2 (DDR2) and integrins are also involved in the response to mechanical stress that upregulates MMP-13 and other proteinases.76–78 However, physiological loading on cartilage may be chondroprotective by stopping the canonical NF-κB cascade, since cytokines that react to IKB kinase complex (IKKB) are inhibited by physiological loading, decreasing the amount of NF-κB transcripted.79 Retinoid receptor ligands and nuclear orphan receptor (NURRI) attenuate the binding of AP-1 and ETS transcription factors,80 so inhibiting the MMP-13, but also from engagement of integrins with fibronectin or collagen fragments that activate the focal adhesion kinase (FAK) which, at the end of the pathway, stimulates AP-1 and ETS transcription factor to attach to the MMP-13 gene.78,81
ELF3, a transcription factor from the family of the ETS, is expressed in inflammatory states induced by IL-1β and TNFα.82 This increase in ELF3 during inflammation disrupts the fine balance between the different ETS, resulting in a downregulation of COL2A1.83 ADAMTS-5 expression is downregulated by miR140 gene in normal cartilage, but miR140 is blocked by IL-1β.84 IL-1β binds to the receptor IL-1R1 (TLR) and recruits the protein MyD88,85 thereby activating the transcription factor NF-κB.86
Platelet-rich plasma
Platelet therapy is not new. More than 20 years ago, several authors focused on the possibility of enhancing the healing process by applying growth factors in tissues with poor vascular supply. Primary treatments have been reported in orofacial damages in order to promote tissue formation.87 Platelet concentrate has been proved to facilitate stem cell-mediated chondrogenesis in animals with artificially induced OA.88 Tendon, ligament, and disease cartilage including OA have all been reported to be successfully treated in humans with platelet therapy.89–96
There are four categories of platelet-rich plasma (PRP): pure platelet-rich plasma (P-PRP), leukocyte- and platelet-rich plasma (L-PRP), pure platelet-rich fibrin (P-PRF), and leukocyte- and platelet-rich fibrin (L-PRF).97 PRP allows the possibility of repairing the damage of joint cartilage, prevents degeneration, reduces pain, and accelerates joint functionality. PRP induces the proliferation of several cell types, inhibits the release of IL-1 from macrophages and reduces the proliferation of the macrophages themselves by limiting the initial inflammatory process, and stimulates quiescent stem cells in order to differentiate them into the type of the damaged tissue. Recent research shows that platelets have the possibility of reducing pain, probably due to the proteases which activate the receptor 4 in nociceptive neurones having anti-nociceptive properties. Further, activated PRP in chondrocytes reduces the transactivating activity of NF-κB, an important target of inflammation, and decreases COX-2 and CXCR4 gene expression. Hepatocyte growth factor (HGF) and TNFα disrupt NF-κB transactivating activity that happens when activated PRP is injected, resulting in an anti-inflammatory effect at a genetic level. Therefore, activated PRP showed a chemotaxis reduction in U937-monocytic cells by chemokine transactivation and CXCR4-receptor inhibition. In this manner, a focal inflammatory reduction in cartilage is granted.98
Although we are not able to prevent some of the causes of OA genetic factors, such as aging and trauma, we can try to alter the course of the disease at the level of cellular communication through a better understanding of the sensitive dependencies in the network of cytokines. All of this is achieved by counteracting the balance toward an anabolic joint homeostasis. A blood derivative with a higher platelet concentration compared with the whole blood before concentration is defined as a PRP. There are several actual devices and manufacturers to obtain PRP, and, depending on the method, we will have different platelet concentration as well as the presence or absence of white blood cells (WBCs). PRP which contains WBCs is defined as L-PRP, which has a high concentration of platelets with growth factors and plasma containing anti-inflammatory proteins and anabolic growth factors, plus WBCs that produce and mediate the production of anti-inflammatory cytokines. Even though proinflammatory cytokines are found, these are of lower concentration than the anti-inflammatory cytokines. PRP from OA patients contained high concentrations of anti-inflammatory proteins including IL-1Ra, sIL-1RII, sTNF-RI, and sTNF-RII.99 WBCs can augment the levels of TGF-β and may decrease the risk of infection following intra-articular injection, the primary reason to defend the presence of WBCs in the preparations of PRP.100–107 Finally, red blood cells seem to have no effect in their presence on PRP preparation, only to give the red color characteristic in some closed devices.
Numerous growth factors are contained within the platelet α-granules. The most well known include HGF, vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and TGF-β. Mitogenic activity to mesoderm-derived cells such as fibroblasts, vascular muscle cells, glial cells, and chondrocytes is the primary effect of PDGF. However, angiogenesis and chemotaxis for fibroblast and collagen production are the primary specific activities of PDGF. This is a key factor, because it has been shown in vivo that, when injected into a skeletally immature rat knee, it stimulates wound healing and matrix synthesis in the growth plate chondrocytes with no adverse effects observed in the cartilage or synovial membrane. The TGF-β superfamily are structurally related and only active as homo- or heterodimers linked together with a single disulfide bond. The latest study performed in vitro demonstrates the capacity of cartilage-derived morphogenic protein (CDMP)-1 (also known as GDF-5 [growth differentiation factor-5]) and CDMP-2 to stimulate cartilage matrix synthesis. On the other hand, TGF-β1 increases chondrocyte activity and lowers the catabolic activity of IL-1β. In vitro TGF-β1 starts chondrogenesis of the synovial lining and mesenchymal stem cells (MSCSs) from bone marrow. Other studies in rabbits showed promising effects following TGF-β1-stimulated cartilage repair. Fibroblast growth factor (FGF) has positive effects on cartilage repair; FGF-2 (also known as basic FGF [bFGF]) is found in relative abundance in the pericellular matrix of cartilage. During physiologic loading, FGF-2 binds itself to cell surface receptors and anabolic pathways activate, diminishing aggrecanase activity. However, proteoglycan concentration is not altered. VEGF is the major regulator of vasculogenesis and angiogenesis, playing an important role in tissue regeneration. Connective tissue growth factor (CTGF) is one of the newest growth factors described recently by Kubota et al,108 as reported in a work by Civinni et al;106 platelets adhere to CTGF at injured tissue wound sites, where it is overexpressed along with the platelet coagulation process. Nonactivated platelets contain considerable amounts of CTGF, which are released by activated PRP, stimulating angiogenetic activity and cartilage regeneration as shown in their experiments.106,108
So with all these cytokines being delivered to the precise place, L-PRP induces the proliferation of several cell types and inhibits IL-1β from macrophages. Furthermore, L-PRP diminishes the transactivity of NF-κB in chondrocytes, a critical regulator of the inflammatory process, due to HGF that downregulates the NF-κB.109 L-PRP also stops another inflammatory pathway by diminishing COX-2 and CXCR4 gene expression (no receptor, no chemotaxis). On the other hand, PRP increases the production of HGF, IL-4, and TGF-β that inhibit inflammation. MMPs are inversely related to increased concentrations of L-PRP due to the high concentration of TIMPs shown in L-PRP.110 In addition, L-PRP enhances the expression of HA synthases HAS-2 over HAS-3, increasing the number of the large molecules of HA.110,111 Anti-inflammatory activity from platelets have been found, since they were shown to stop the activity of the NF-κB pathway.111 The equilibrium between catabolism and anabolism could be corrected by controlling the biochemical factors that inhibit MMPs (IL-4, IL-10, IL-13, TIMPs, insulin-like growth factor [IGF], and TGF-β) and the biochemical elements that increase their synthesis (IL-1b, TNFα, IL-8, IL-6, IL-17, IL-18, leukemia inhibitory factor, and NO).112
Actually, it should be desirable to have an option to avoid aggressive surgical treatments, focused on reducing pain, increasing joint function, and of course altering the natural progression of OA. Due to the good results in tendon injuries treated with PRP injections, the same option has been available to treat cartilage injuries, with initial better results when compared to HA injections. Currently, PRP is an alternative for treating mild-to-moderate levels of chondromalacia if etiologic factors have been eliminated.
Role of HA
HA has always been the focus of attention since it is the primary component of synovial fluid. Synovial fluid is the main lubricant for the articular surface, reducing surface stress extension of the load zone, and is the transport of chondronutritive substances from the synovia. Decreased molecular weight, decreased elasticity, and a reduced concentration of HA in synovial fluid from an arthritic joint have been noted.113
Over the past 2 decades, numerous studies have been performed with an overall conclusion that HA is a common treatment to prevent or inhibit OA progression in two ways, by viscosupplementation and by visco-induction. Viscosupplementation is characterized by recovering the mechanical and viscoelastic properties of synovial fluid, and visco-induction is characterized by stimulating endogenous production of HA by synoviocytes and chondrocytes. HA treatment is offered for advanced knee OA before surgical intervention. HA can be used as an alternative treatment to nonsteroidal anti-inflammatory drugs and cortisone-based compounds when they are contraindicated, not tolerated, or ineffective. The most common approach is one injection per week for 3–5 weeks. However, the therapeutic strategy is determined by the molecular weight of the drug.113–115
Quality evidence has been advised when HA is compared to PRP in treating OA; the results of a clinical cohort study by Kon et al supported that the use of PRP in young patients with low or moderate levels of OA had better results when compared to HA treatment.116 Randomized aleatory studies comparing HA with PRP in OA have demonstrated that results were superior in patients treated with PRP suffering low or moderate degrees of OA.117,118
Autologous protein solution
The main inflammatory cytokines related to the progression of OA are IL-1β and TNFα,119 both of which can induce cartilage degradation and pain. Although not yet proved, antagonists of IL-1β or TNFα, such as recombinant IL-1Ra or the soluble receptor for TNFα (sTNF-R), have been proposed as OA therapies. Maybe inhibiting inflammatory signaling is the key for future strategies in treating OA.120
Another blood technique to consider is Orthokine, which has the advantage of anti-inflammatory cytokines with better results when compared to steroids or placebo injections. These results, however, are not conclusive.120 An autologous protein solution (APS) prepared from PRP has been developed to enhance the concentration of anabolic and anti-inflammatory cytokines.120 This APS is a compound of WBCs which contain anti-inflammatory proteins, platelets with growth factors, and concentrated plasma with anti-inflammatory proteins and anabolic growth factors.99,121 Treatments with APS have demonstrated in preclinical cell cultures a protective effect and anti-inflammatory target for cartilage injuries. Initial benefits in animal models should support further evaluations of APS in human OA therapies. Future research is focused on positive results to treat cartilage injuries with PRP and the presence of WBCs, given their relationship with the production of anti-inflammatory cytokines. L-PRP reduces the activation of NF-κB, a primary mediator of the inflammatory process in cultured articular chondrocytes challenged with TNFα. Therefore, it is suggested that autologous products containing WBCs may play a role in modulating inflammation and should be further explored as a potential treatment for OA.99
Discussion
Although there are several points that must be elucidated about the use of platelet therapies, like indications, contraindications, number of doses, time of good follow-up, cost-effectiveness, preparation, and legal regulations which can differ from one country to other, a number of manuscripts are being published about this technique. As always, we know long-term studies, randomized, controlled, double-blind, and, if possible, multi-centric, will be necessary in order to try to explain the initial results that have been published, but still cannot prove any conclusion definitively. PRP therapy should be indicated with careful reflection. Based on the clinical and scientific evidence as well as basic science, we could suggest it as a supplement, not as a primary treatment. Tendinopathy, understanding it as an histopathologic entity, could be one of the main diseases to be treated with PRP if standard treatment has failed. Usually it should be applied in those patients with a clinical situation which causes an important disruption in quality of life (measured with the several scores available for every anatomical area). Multiple authors have argued PRP as a valid option to treat tennis elbow, based on its physiopathology.122 Indications could be transferred to other anatomical areas such as the patellar, Achilles, plantar, and flexor–pronator elbow tendons as the most frequent to be treated.
When cartilage is analyzed, conclusions are not so objective, perhaps because if we want to understand when PRP should be indicated, physiopathology of OA is mandatory to keep in mind, even though it is more complicated to understand. However, OA is a degenerative and progressive process that affects not only the cartilage but also subchondral bone. There are a lot of biomolecules implicated, but the main cause is a biomechanical overpressure or micro-impact injury. OA should be recognized as a disease to be treated individually, always evaluating possible mechanical factors. Limb length discrepancies, valgus or varus misalignment, instabilities, and posttraumatic sequelae should be compensated for or, if possible, corrected. Secondary OA should not be treated with PRP as a primary option. Therefore, OA should be approached under the point of view of the clinical status and if possible macroscopical appearance (eg, Outerbridge degrees), because there is not always a corelation to the radiological images. A degree with a soft fibrillation or partial lack of thickness of the cartilage is not the same situation compared to a degree with an exposure of the subchondral bone; the possibilities of a biological response without the existence of hyaline cartilage is quite impossible. For example, when a soft fibrillation or a partial lack of thickness of the cartilage is seen in an arthroscopy, there is often not a good correlation to the magnetic resonance imaging and with the clinical status. Therefore, if we are thinking about the possibility to treat the injury with a biologic product, the response of the tissue will not be the same if a total lack of cartilage is present. It is necessary to have live tissue in order to stimulate with biologics. The most recent results concerning the use of PRP in OA suggests that it should be indicated as a supplement, not as a primary treatment; that there are better results in young patients with low or moderate degrees of OA in whom any undesirable mechanical factors have already been corrected. It seems that only one injection should be considered, because the literature cannot provide us any explanation to argue performing two, three, or more injections from the baseline. Future possibilities, such as blocking the receptors of IL with antagonists, will demonstrate whether or not OA is an entity that should be treated as soon as possible in order to avoid more aggressive treatments. The general population is aging around the world, and all the orthopedic societies are aware of the over-indications of total joint replacements and their consequences.
Conclusion
Herein, we have presented an opinion and actual review of the physiopathology and approach of OA with biological treatments already available. There is not any evaluation of the various devices, but only a scientific point of view and our personal experience. We have always judged that the etiologic treatment should be the perfect treatment, PRP being only a supplement that can help the patient in several conditions, but is not a panacea. We would like to thank the investigators who continuously contribute to the understanding of disease from the perspective of molecular medicine. Perhaps a new era or subspecialty is evolving within orthopedics.
Acknowledgments
The authors thank Mr Mark A Waldron, Director of Synodical Placement and Colloquy, Concordia University Chicago, River Forest, IL, USA, for his collaboration on the revision of this manuscript.
Disclosure
Dr Torrero is a professional consultant of Biomet Biologics®, and has not received any research funding from Biomet®. The authors report no other conflicts of interest in this work.
References
Goldring MB, Otero M, Plumb DA, et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur Cell Mater. 2011;21:202–220. | |
Heinegård D, Saxne T. The role of the cartilage matrix in osteoarthritis. Nat Rev Rheumatol. 2011;7(1):50–56. | |
Goldring MB, Berenbaum F. The regulation of chondrocyte function by proinflammatory mediators: prostaglandins and nitric oxide. Clin Orthop Relat Res. 2004;(427 Suppl):S37–S46. | |
Kobayashi M, Squires GR, Mousa A, et al. Role of interleukin-1 and tumor necrosis factor alpha in matrix degradation of human osteoarthritic cartilage. Arthritis Rheum. 2005;52(1):128–135. | |
Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010; 6(11):625–635. | |
Ortolano G, Wenz B. A review of the pathogenesis of osteoarthritis and the use of intra-articular platelet therapy for joint disease in animals and humans. Bone Tissue Regen Insights. 2014;5:1–13. | |
Roughley PJ. Articular cartilage and changes in arthritis: noncollagenous proteins and proteoglycans in the extracellular matrix of cartilage. Arthritis Res. 2001;3:342–347. | |
Zhao J, Zhang P, Qin L, Pan XH. Hypoxia is essential for bone-tendon junction healing: the molecular biological evidence. Int Orthop. 2011;35(6):925–928. | |
Gibson JS, Milner PI,White R, Fairfax TP, Wilkins RJ. Oxygen and reactive oxygen species in articular cartilage: modulators of ionic homeostasis. Pflugers Arch. 2008;455(4):563–573. | |
de Lange-Brokaar BJ, Ioan-Facsinay A, van Osch GJ, et al. Synovial inflammation, immune cells and their cytokines in osteoarthritis: a review. Osteoarthritis Cartilage. 2012;20(12):1484–1499. | |
Knäuper V, López-Otin C, Smith B, Knight G, Murphy G. Biochemical characterization of human collagenase-3. J Biol Chem. 1996;271(3):1544–1550. | |
Ayhan E, Kesmezacar H, Akgun I. Intraarticular injections (corticosteroid, hyaluronic acid, platelet rich plasma) for the knee osteoarthritis. World J Orthop. 2014;5(3):351–361. | |
Goldring MB, Otero M. Inflammation in osteoarthritis. Curr Opin Rheumatol. 2011;23(5):471–478. | |
Liu-Bryan R. Synovium and the innate inflammatory network in osteoarthritis progression. Curr Rheumatol Rep. 2013;15(5):323. | |
Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51(2):249–257. | |
Webster NL, Crowe SM. Matrix metalloproteinases, their production by monocytes and macrophages and their potential role in HIV-related diseases. J Leukoc Biol. 2006;80(5):1052–1066. | |
Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2011;41(2):271–290. | |
Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803(1):55–71. | |
Jones GC, Riley GP. ADAMTS proteinases: a multi-domain, multi-functional family with roles in extracellular matrix turnover and arthritis. Arthritis Res Ther. 2005;7(4):160–169. | |
Goldring SR, Goldring MB. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res. 2004;(427 Suppl):S27–S36. | |
Farahat MN, Yanni G, Poston R, Panayi GS. Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann Rheum Dis. 1993;52(12):870–875. | |
Melchiorri C, Meliconi R, Frizziero L, et al. Enhanced and coordinated in vivo expression of inflammatory cytokines and nitric oxide synthase by chondrocytes from patients with osteoarthritis. Arthritis Rheum. 1998;41(12):2165–2174. | |
Massicotte F, Lajeunesse D, Benderdour M, et al. Can altered production of interleukin-1beta, interleukin-6, transforming growth factor-beta and prostaglandin E(2) by isolated human subchondral osteoblasts identify two subgroups of osteoarthritic patients. Osteoarthritis Cartilage. 2002;10(6):491–500. | |
Sohn DH, Sokolove J, Sharpe O, et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Res Ther. 2012;14(1):R7. | |
Martel-Pelletier J, McCollum R, DiBattista J, et al. The interleukin-1 receptor in normal and osteoarthritic human articular chondrocytes. Identification as the type I receptor and analysis of binding kinetics and biologic function. Arthritis Rheum. 1992;35(5):530–540. | |
Sadouk MB, Pelletier JP, Tardif G, Kiansa K, Cloutier JM, Martel-Pelletier J. Human synovial fibroblasts coexpress IL-1 receptor type I and type II mRNA. The increased level of the IL-1 receptor in osteoarthritic cells is related to an increased level of the type I receptor. Lab Invest. 1995;73(3):347–355. | |
Séguin CA, Bernier SM. TNFalpha suppresses link protein and type II collagen expression in chondrocytes: role of MEK1/2 and NF-kappaB signaling pathways. J Cell Physiol. 2003;197(3):356–369. | |
Guerne PA, Carson DA, Lotz M. IL-6 production by human articular chondrocytes. Modulation of its synthesis by cytokines, growth factors, and hormones in vitro. J Immunol. 1990;144(2):499–505. | |
Ushiyama T, Chano T, Inoue K, Matsusue Y. Cytokine production in the infrapatellar fat pad: another source of cytokines in knee synovial fluids. Ann Rheum Dis. 2003;62(2):108–112. | |
Porée B, Kypriotou M, Chadjichristos C, et al. Interleukin-6 (IL-6) and/or soluble IL-6 receptor down-regulation of human type II collagen gene expression in articular chondrocytes requires a decrease of Sp1.Sp3 ratio and of the binding activity of both factors to the COL2A1 promoter. J Biol Chem. 2008;283(8):4850–4865. | |
Kwan Tat S, Padrines M, Théoleyre S, Heymann D, Fortun Y. IL-6, RANKL, TNF-alpha/IL-1: interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. 2004;15(1):49–60. | |
Scanzello CR, Umoh E, Pessler F, et al. Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthritis Cartilage. 2009;17(8):1040–1048. | |
Sun JM, Sun LZ, Liu J, Su BH, Shi L. Serum interleukin-15 levels are associated with severity of pain in patients with knee osteoarthritis. Dis Markers. 2013;35(3):203–206. | |
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. | |
Lubberts E, Joosten LA, van de Loo FA, van den Gersselaar LA, van den Berg WB. Reduction of interleukin-17-induced inhibition of chondrocyte proteoglycan synthesis in intact murine articular cartilage by interleukin-4. Arthritis Rheum. 2000;43(6):1300–1306. | |
Attur MG, Patel RN, Abramson SB, Amin AR. Interleukin-17 up-regulation of nitric oxide production in human osteoarthritis cartilage. Arthritis Rheum. 1997;40(6):1050–1053. | |
LeGrand A, Fermor B, Fink C, et al. Interleukin-1, tumor necrosis factor alpha, and interleukin-17 synergistically up-regulate nitric oxide and prostaglandin E2 production in explants of human osteoarthritic knee menisci. Arthritis Rheum. 2001;44(9):2078–2083. | |
Smelter E, Hochberg MC. New treatments for osteoarthritis. Curr Opin Rheumatol. 2013;25(3):310–316. | |
Olee T, Hashimoto S, Quach J, Lotz M. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J Immunol. 1999;162(2):1096–1100. | |
Möller B, Paulukat J, Nold M, et al. Interferon-gamma induces expression of interleukin-18 binding protein in fibroblast-like synoviocytes. Rheumatology (Oxford). 2003;42(3):442–445. | |
Joosten LA, Smeets RL, Koenders MI, et al. Interleukin-18 promotes joint inflammation and induces interleukin-1-driven cartilage destruction. Am J Pathol. 2004;165(3):959–967. | |
John T, Kohl B, Mobasheri A, Ertel W, Shakibaei M. Interleukin-18 induces apoptosis in human articular chondrocytes. Histol Histopathol. 2007;22(5):469–482. | |
Wojdasiewicz P, Poniatowski LA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:561459. | |
Ishii H, Tanaka H, Katoh K, Nakamura H, Nagashima M, Yoshino S. Characterization of infiltrating T cells and Th1/Th2-type cytokines in the synovium of patients with osteoarthritis. Osteoarthritis Cartilage. 2002;10(4):277–281. | |
van Meegeren ME, Roosendaal G, Jansen NW, et al. IL-4 alone and in combination with IL-10 protects against blood-induced cartilage damage. Osteoarthritis Cartilage. 2012;20(7):764–772. | |
Alaaeddine N, Di Battista JA, Pelletier JP, Kiansa K, Cloutier JM, Martel-Pelletier J. Inhibition of tumor necrosis factor alpha-induced prostaglandin E2 production by the antiinflammatory cytokines interleukin-4, interleukin-10, and interleukin-13 in osteoarthritic synovial fibroblasts: distinct targeting in the signaling pathways. Arthritis Rheum. 1999;42(4):710–718. | |
Iannone F, De Bari C, Dell’Accio F, et al. Interleukin-10 and interleukin-10 receptor in human osteoarthritic and healthy chondrocytes. Clin Exp Rheumatol. 2001;19(2):139–145. | |
Lacraz S, Nicod LP, Chicheportiche R, Welgus HG, Dayer JM. IL-10 inhibits metalloproteinase and stimulates TIMP-1 production in human mononuclear phagocytes. J Clin Invest. 1995;96(5):2304–2310. | |
Chadban SJ, Tesch GH, Foti R, Atkins RC, Nikolic-Paterson DJ. Interleukin-10 is a mesangial cell growth factor in vitro and in vivo. Lab Invest. 1997;76(5):619–627. | |
Mizuno S, Tateishi T, Ushida T, Glowacki J. Hydrostatic fluid pressure enhances matrix synthesis and accumulation by bovine chondrocytes in three-dimensional culture. J Cell Physiol. 2002;193(3):319–327. | |
Angele P, Yoo JU, Smith C, et al. Cyclic hydrostatic pressure enhances the chondrogenic phenotype of human mesenchymal progenitor cells differentiated in vitro. J Orthop Res. 2003;21(3):451–457. | |
Jovanovic D, Pelletier JP, Alaaeddine N, et al. Effect of IL-13 on cytokines, cytokine receptors and inhibitors on human osteoarthritis synovium and synovial fibroblasts. Osteoarthritis Cartilage. 1998;6(1):40–49. | |
Ding L, Heying E, Nicholson N, et al. Mechanical impact induces cartilage degradation via mitogen activated protein kinases. Osteoarthritis Cartilage. 2010;18(11):1509–1517. | |
Husa M, Liu-Bryan R, Terkeltaub R. Shifting HIFs in osteoarthritis. Nat Med. 2010;16(6):641–644. | |
Rogerson FM, Chung YM, Deutscher ME, Last K, Fosang AJ. Cytokine-induced increases in ADAMTS-4 messenger RNA expression do not lead to increased aggrecanase activity in ADAMTS-5-deficient mice. Arthritis Rheum. 2010;62(11):3365–3373. | |
Verma P, Dalal K. ADAMTS-4 and ADAMTS-5: key enzymes in osteoarthritis. J Cell Biochem. 2011;112(12):3507–3514. | |
Koshy PJ, Lundy CJ, Rowan AD, et al. The modulation of matrix metalloproteinase and ADAM gene expression in human chondrocytes by interleukin-1 and oncostatin M: a time-course study using real-time quantitative reverse transcription-polymerase chain reaction. Arthritis Rheum. 2002;46(4):961–967. | |
El Mansouri FE, Chabane N, Zayed N, et al. Contribution of H3K4 methylation by SET-1A to interleukin-1-induced cyclooxygenase 2 and inducible nitric oxide synthase expression in human osteoarthritis chondrocytes. Arthritis Rheum. 2011;63(1):168–179. | |
Hardy MM, Seibert K, Manning PT, et al. Cyclooxygenase 2-dependent prostaglandin E2 modulates cartilage proteoglycan degradation in human osteoarthritis explants. Arthritis Rheum. 2002;46(7):1789–1803. | |
Reginato AM, Sanz-Rodriguez C, Diaz A, Dharmavaram RM, Jimenez SA. Transcriptional modulation of cartilage-specific collagen gene expression by interferon gamma and tumour necrosis factor alpha in cultured human chondrocytes. Biochemical J. 1993;294(Pt 3):761–769. | |
Goldring MB, Fukuo K, Birkhead JR, Dudek E, Sandell LJ. Transcriptional suppression by interleukin-1 and interferon-gamma of type II collagen gene expression in human chondrocytes. J Cell Biochem. 1994;54(1):85–99. | |
Okazaki K, Li J, Yu H, Fukui N, Sandell LJ. CCAAT/enhancer-binding proteins beta and delta mediate the repression of gene transcription of cartilage-derived retinoic acid-sensitive protein induced by interleukin-1 beta. J Biol Chem. 2002;277(35):31526–31533. | |
Miyamoto M, Ito H, Mukai S, et al. Simultaneous stimulation of EP2 and EP4 is essential to the effect of prostaglandin E2 in chondrocyte differentiation. Osteoarthritis Cartilage. 2003;11(9):644–652. | |
Tan L, Peng H, Osaki M, et al. Egr-1 mediates transcriptional repression of COL2A1 promoter activity by interleukin-1beta. J Biol Chem. 2003;278(20):17688–17700. | |
Robbins JR, Thomas B, Tan L, et al. Immortalized human adult articular chondrocytes maintain cartilage-specific phenotype and responses to interleukin-1beta. Arthritis Rheum. 2000;43(10):2189–2201. | |
Deschner J, Hofman CR, Piesco NP, Agarwal S. Signal transduction by mechanical strain in chondrocytes. Curr Opin Clin Nutr Metab Care. 2003;6(3):289–293. | |
Fanning PJ, Emkey G, Smith RJ, Grodzinsky AJ, Szasz N, Trippel SB. Mechanical regulation of mitogen-activated protein kinase signaling in articular cartilage. J Biol Chem. 2003;278(51):50940–50948. | |
Knobloch TJ, Madhavan S, Nam J, Agarwal S Jr, Agarwal S. Regulation of chondrocytic gene expression by biomechanical signals. Crit Rev Eukaryot Gene Expr. 2008;18(2):139–150. | |
Sillat T, Barreto G, Clarijs P, et al. Toll-like receptors in human chondrocytes and osteoarthritic cartilage. Acta Orthop. 2013;84(6):585–592. | |
Campo GM, Avenoso A, D’Ascola A, et al. Hyaluronan differently modulates TLR-4 and the inflammatory response in mouse chondrocytes. Biofactors. 2012;38(1):69–76. | |
Liu-Bryan R, Terkeltaub R. Chondrocyte innate immune myeloid differentiation factor 88-dependent signaling drives procatabolic effects of the endogenous Toll-like receptor 2/Toll-like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein 1 in mice. Arthritis Rheum. 2010;62(7):2004–2012. | |
Marcu KB, Otero M, Olivotto E, Borzi RM, Goldring MB. NF-kappaB signaling: multiple angles to target OA. Curr Drug Targets. 2010;11(5):599–613. | |
Ijiri K, Zerbini LF, Peng H, et al. A novel role for GADD45beta as a mediator of MMP-13 gene expression during chondrocyte terminal differentiation. J Biol Chem. 2005;280(46):38544–38555. | |
Saito T, Fukai A, Mabuchi A, et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med. 2010;16(6):678–686. | |
Yang S, Kim J, Ryu JH, et al. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010;16(6):687–693. | |
Mobasheri A, Carter SD, Martin-Vasallo P, Shakibaei M. Integrins and stretch activated ion channels; putative components of functional cell surface mechanoreceptors in articular chondrocytes. Cell Biol Int. 2002;26(1):1–18. | |
Salter DM, Millward-Sadler SJ, Nuki G, Wright MO. Differential responses of chondrocytes from normal and osteoarthritic human articular cartilage to mechanical stimulation. Biorheology. 2002; 39(1–2):97–108. | |
Loeser RF, Forsyth CB, Samarel AM, Im HJ. Fibronectin fragment activation of proline-rich tyrosine kinase PYK2 mediates integrin signals regulating collagenase-3 expression by human chondrocytes through a protein kinase C-dependent pathway. J Biol Chem. 2003;278(27):24577–24585. | |
Dossumbekova A, Anghelina M, Madhavan S, et al. Biomechanical signals inhibit IKK activity to attenuate NF-kappaB transcription activity in inflamed chondrocytes. Arthritis Rheum. 2007;56(10):3284–3296. | |
Burrage PS, Huntington JT, Sporn MB, Brinckerhoff CE. Regulation of matrix metalloproteinase gene expression by a retinoid X receptor-specific ligand. Arthritis Rheum. 2007;56(3):892–904. | |
Ronzière MC, Aubert-Foucher E, Gouttenoire J, Bernaud J, Herbage D, Mallein-Gerin F. Integrin alpha1beta1 mediates collagen induction of MMP-13 expression in MC615 chondrocytes. Biochim Biophys Acta. 2005;1746(1):55–64. | |
Grall F, Gu X, Tan L, et al. Responses to the proinflammatory cytokines interleukin-1 and tumor necrosis factor alpha in cells derived from rheumatoid synovium and other joint tissues involve nuclear factor kappaB-mediated induction of the Ets transcription factor ESE-1. Arthritis Rheum. 2003;48(5):1249–1260. | |
Peng H, Tan L, Osaki M, et al. ESE-1 is a potent repressor of type II collagen gene (COL2A1) transcription in human chondrocytes. J Cell Physiol. 2008;215(2):562–573. | |
Miyaki S, Nakasa T, Otsuki S, et al. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum. 2009;60(9):2723–2730. | |
Martin MU, Wesche H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim Biophys Acta. 2002;1592(3):265–280. | |
Roman-Blas JA, Jimenez SA. NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage. 2006;14(9):839–848. | |
Anitua E, Carda C, Andia I. A novel drilling procedure and subsequent bone autograft preparation: a technical note. Int J Oral Maxillofac Implants. 2007;22(1):138–145. | |
Mifune Y, Matsumoto T, Takayama K, et al. The effect of platelet-rich plasma on the regenerative therapy of muscle derived stem cells for articular cartilage repair. Osteoarthritis Cartilage. 2013;21(1):175–185. | |
Mautner K, Colberg RE, Malanga G, et al. Outcomes after ultrasound-guided platelet-rich plasma injections for chronic tendinopathy: a multicenter, retrospective review. PM R. 2013;5(3):169–175. | |
Mei-Dan O, Carmont MR. The role of platelet-rich plasma in rotator cuff repair. Sports Med Arthrosc. 2011;19(3):244–250. | |
Coombes BK, Bisset L, Vicenzino B. Efficacy and safety of corticosteroid injections and other injections for management of tendinopathy: a systematic review of randomized controlled trials. Lancet. 2010; 376(9754):1751–1767. | |
van Ark M, Zwerver J, van den Akker-Scheek I. Injection treatments for patellar tendinopathy. Br J Sports Med. 2011;45(13):1068–1076. | |
Baksh N, Hannon CP, Murawski CD, Smyth NA, Kennedy JG. Platelet-rich plasma in tendon models: a systematic review of basic science literature. Arthroscopy. 2013;29(3):596–607. | |
Gosens T, Den Oudsten BL, Fievez E, van ‘t Spijker P, Fievez A. Pain and activity levels before and after platelet-rich plasma injection treatment of patellar tendinopathy: a prospective cohort study and the influence of previous treatments. Int Orthop. 2012;36(9):1941–1946. | |
Castelijns G, Crawford A, Schaffer J, Ortolano GA, Beauregard T, Smith RK. Evaluation of a filter-prepared platelet concentrate for the treatment of suspensory branch injuries in horses. Vet Comp Orthop Traumatol. 2011;24(5):363–369. | |
Sánchez M, Anitua E, Orive G, Mujika I, Andia I. Platelet-rich therapies in the treatment of orthopaedic sport injuries. Sports Med. 2009;39(5):345–354. | |
Dohan Ehrenfest DM, Rasmusson L, Albrektsson T. Classification of platelet concentrates: from pure platelet-rich plasma (P-PRP) to leucocyte- and platelet-rich fibrin (L-PRF). Trends Biotechnol. 2008;27:158–167. | |
Anitua E, Sánchez M, Orive G, Andía I. The potential impact of the preparation rich in growth factors (PRGF) in different medical fields. Biomaterials. 2007;28:4551–4560. | |
O’Shaughnessey K, Matuska A, Hoeppner J, et al. Autologous protein solution prepared from the blood of osteoarthritic patients contains an enhanced profile of anti-inflammatory cytokines and anabolic growth factors. J Orthop Res. 2014;32(10):1349–1355. | |
Trowbridge CC, Stammers AH, Woods E, Yen BR, Klayman M, Gilbert C. Use of platelet gel and its effects on infection in cardiac surgery. J Extra Corpor Technol. 2005;37(4):381–386. | |
Bielecki TM, Gazdzik TS, Arendt J, Szczepanski T, Król W, Wielkoszynski T. Antibacterial effect of autologous platelet gel enriched with growth factors and other active substances: an in vitro study. J Bone Joint Surg Br. 2007;89(3):417–420. | |
Bielecki T, Dohan Ehrenfest DM, Everts PA, Wiczkowski A. The role of leukocytes from L-PRP/L-PRF in wound healing and immune defense: new perspectives. Curr Pharm Biotechnol. 2012;13(7):1153–1162. | |
Cieslik-Bielecka A, Bielecki T, Gazdzik TS, Arendt J, Król W, Szczepanski T. Autologous platelets and leukocytes can improve healing of infected high-energy soft tissue injury. Transfus Apher Sci. 2009;41(1):9–12. | |
Moojen DJ, Everts PA, Schure RM, et al. Antimicrobial activity of platelet-leukocyte gel against Staphylococcus aureus. J Orthop Res. 2008;26(3):404–410. | |
Schneeberger AG, Yian E, Steens W. Injection-induced low-grade infection of the shoulder joint: preliminary results. Arch Orthop Trauma Surg. 2012;132(10):1387–1392. | |
Civinini R, Nistri L, Martini C, Redl B, Ristori G, Innocenti M. Growth factors in the treatment of early osteoarthritis. Clin Cases Miner Bone Metab. 2013;10(1):26–29. | |
Tang YQ, Yeaman MR, Selsted ME. Antimicrobial peptides from human platelets. Infect Immun. 2002;70(12):6524–6533. | |
Kubota S, Kawata K, Yanagita T, Doi H, Kitoh T, Takigawa M. Abundant retention and release of connective tissue growth factor (CTGF/CCN2) by platelets. J Biochem. 2004;136(3):279–282. | |
Bendinelli P, Matteucci E, Dogliotti G, et al. Molecular basis of anti-inflammatory action of platelet-rich plasma on human chondrocytes: mechanisms of NF-κB inhibition via HGF. J Cell Physiol. 2010; 225(3):757–766. | |
Assirelli E, Filardo G, Mariani E, et al. Effect of two different preparations of platelet-rich plasma on synoviocytes. Knee Surg Sports Traumatol Arthrosc. Epub June 19, 2014. | |
Fortier LA, Cole BJ. The anti-inflammatory and matrix restorative mechanisms of platelet-rich plasma in osteoarthritis: response to Patel and Dhillon. Am J Sports Med. 2014;42(6):NP31. | |
Stevens AL, Wishnok JS, White FM, Grodzinsky AJ, Tannenbaum SR. Mechanical injury and cytokines cause loss of cartilage integrity and upregulate proteins associated with catabolism, immunity, inflammation, and repair. Mol Cell Proteomics. 2009;8(7):1475–1489. | |
Cerza F, Carnì S, Carcangiu A, et al. Comparison between hyaluronic acid and platelet-rich plasma, intra-articular infiltration in the treatment of gonarthrosis. Am J Sports Med. 2012;40(12):2822–2827. | |
Bellamy N, Campbell J, Robinson V, Gee T, Bourne R, Wells G. Viscosupplementation for the treatment of the osteoarthritic knee. Cochrane Database Syst Rev. 2006;(2):CD005321. | |
Wobig M, Bach G, Beks P, et al. The role of elastoviscosity in the efficacy of viscosupplementation for osteoarthritis of the knee: a comparison of hylan G-F 20 and a lower-molecular-weight hyaluronan. Clin Ther. 1999;21(9):1549–1562. | |
Kon E, Mandelbaum B, Buda R, et al. Platelet-rich plasma intra-articular injection versus hyaluronic acid viscosupplementation as treatments for cartilage pathology: from early degeneration to osteoarthritis. Arthroscopy. 2011;27(11):1490–1501. | |
Sánchez M, Fiz N, Azofra J, et al. A randomized clinical trial evaluating plasma rich in growth factors (PGRF-Endoret) versus hyaluronic acid in the short-term treatment of symptomatic knee osteoarthritis. Arthroscopy. 2012;28(8):1070–1078. | |
Filardo G, Kon E, Di Martino A, et al. Platelet-rich plasma vs hyaluronic acid to treat knee degenerative pathology: study design and preliminary results of a randomized controlled trial. BMC Musculoskelet Disord. 2012;13:229. | |
Fernandes JC, Martel-Pelletier J, Pelletier JP. The role of cytokines in osteoarthritis pathophysiology. Biorheology. 2002;39:237–246. | |
Zelenka M, Schäfers M, Sommer C. Intraneural injection of interleukin-1beta and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain. 2005;116(3):257–263. | |
Woodell-May J, Matuska A, Oyster M, Welch Z, O’Shaughnessey K, Hoeppner J. Autologous protein solution inhibits MMP-13 production by IL-1β and TNFα-stimulated human articular chondrocytes. J Orthop Res. 2011;29:1320–1326. | |
Raeissadat SA, Sedighipour L, Rayegani SM, Bahrami MH, Bayat M, Rahimi R. Effect of Platelet-Rich Plasma (PRP) versus Autologous Whole Blood on Pain and Function Improvement in Tennis Elbow: A Randomized Clinical Trial. Pain Res Treat. 2014;2014:191525. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.