")
Back to Journals » Biologics: Targets and Therapy » Volume 11
New developments in the treatment of multiple myeloma –
clinical utility of daratumumab
Authors McEllistrim C, Krawczyk J, O'Dwyer ME
Received 25 January 2017
Accepted for publication 15 March 2017
Published 11 April 2017 Volume 2017:11 Pages 31—43
DOI https://doi.org/10.2147/BTT.S97633
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Doris Benbrook
Cian McEllistrim,1 Janusz Krawczyk,1 Michael E O’Dwyer1,2
1Department of Hematology, University Hospital Galway, 2Apoptosis Research Centre, Biomedical Sciences, National University of Ireland Galway, Galway, Ireland
Abstract: Multiple myeloma is a clonal disorder of plasma cells that is currently considered incurable. CD38 is a 46 kDa type II transmembrane glycoprotein that is highly expressed on myeloma cells. Daratumumab is a first in-class human IgG1 monoclonal antibody that targets CD38, and has antimyeloma effects through several mechanisms. Single-agent trials show surprising activity in heavily pretreated myeloma patients. Trials in the relapsed setting, where daratumumab is added to lenalidomide and dexamethasone or bortezomib and dexamethasone, have demonstrated significantly improved progression-free survival with acceptable toxicity. In this review, we discuss the mechanism of action, pharmacology and pharmacokinetics of daratumumab and review the available clinical data in detail. We examine how daratumumab interferes with transfusion testing due to the expression of CD38 on the red blood cells, leading to potential difficulties releasing blood products. Daratumumab also affects disease assessments in multiple myeloma, including serum protein electrophoresis, immunofixation and flow cytometry. Strategies to mitigate these effects are discussed. The optimal use of daratumumab has yet to be decided, and several trials are ongoing in the relapsed and upfront setting. We discuss the potential upfront role of this exciting therapy, which has significant potential for increased minimal residual disease negativity and improved progression-free survival even in high-risk groups.
Keywords: multiple myeloma, monoclonal antibodies, daratumumab, immunotherapy, CD38, minimal residual disease
Introduction
Multiple myeloma (MM) accounts for ~1% of malignancies in the USA.1 Although consistent improvements in progression-free survival (PFS) and overall survival (OS) have been made in the last decade, myeloma remains, generally, an incurable condition. Measured in months, the OS for patients who are refractory to proteasome inhibitors and immunomodulatory drugs (IMiDs) is poor. New therapies that act in distinct ways are needed for this patient group. Therapies that target tumor antigens are an attractive option following the success of anti-CD20 antibodies in non-Hodgkin lymphomas. Outside lymphoma, monoclonal antibodies against other targets in hematologic malignancy have enjoyed mixed success.
CD38 is a 46 kDa type II transmembrane glycoprotein that is highly expressed on the myeloma cells, but also heterogeneously expressed on other myeloid, lymphoid and non-hematopoietic tissues.2 It is a rational target for antimyeloma therapy. The first in-class human IgG1 monoclonal antibody daratumumab targets CD38 and has antimyeloma effects through several mechanisms (see below). Daratumumab has received breakthrough therapy designation on two occasions by the Food and Drug Administration for the treatment of relapsed myeloma,3 and has shown activity both as monotherapy and in combination with standard of care (SOC) therapies.
In this article, after initially examining the preclinical data and pharmacokinetics (PKs) of daratumumab, we review the available clinical data, safety profile, practicalities of drug administration and future directions for monoclonal antibodies in myeloma treatment.
Mechanism of action and preclinical studies of daratumumab
de Weers et al developed a panel of antibodies against CD38, from which daratumumab stood out, particularly in its ability to induce complement-dependent cytotoxicity (CDC).4 Daratumumab was shown to bind to a unique CD38 epitope and, furthermore, to induce tumor killing by other anti-Fc-mediated effector functions, for example, CDC, natural killer (NK) cell-mediated antibody-dependent cell-mediated cytotoxicity (ADCC) and apoptosis upon secondary cross-linking.5,6 In vitro induction of programmed cell death through FcγR-mediated cross-linking of myeloma cell lines, as well as in vivo cross-linking by the inhibitory FcγRIIb as well as the activating FcγRs inducing programmed cell death have been observed.7 ADCC killing by peripheral blood mononuclear cells (PBMCs) has been demonstrated in the presence of daratumumab, regardless of the source of PBMCs (healthy volunteer or patient). Subsequently, daratumumab entered clinical development for MM treatment.
Preclinical studies sought to investigate the enhancement of ADCC by combining daratumumab with IMiD treatment, based on the observation that thalidomide and lenalidomide enhanced the ADCC effect mediated by rituximab.5 ADCC against the myeloma cell line UM-9 was significantly increased by pretreating PBMCs with lenalidomide. Pretreatment of myeloma cells with lenalidomide did not improve ADCC when daratumumab was added. Similarly, enhanced killing was found when myeloma cells were studied in a physiologic environment. CDC was not increased by the combination. Effector cell activity, in summary, is enhanced synergistically to promote ADCC in the presence of daratumumab. NK-cell activation by lenalidomide has also been shown to add to the synergism of the daratumumab/lenalidomide combination in lenalidomide-resistant cell lines.8 Blockade of KIR inhibitory receptors may increase NK-cell lysis by daratumumab. The anti-killer-cell immunoglobulin-like receptor antibody, IPH2102, has also shown synergism with daratumumab with increased daratumumab-mediated ADCC of MM cells.9 The addition of lenalidomide by these investigators further improved MM-cell lysis, beyond what could be expected through additive effects. This may be explained by separate mechanisms of increasing NK-cell cytotoxicity – lenalidomide stimulates the proliferation and activation of NK cells, while IPH2102 blocks KIR inhibitory signaling of NK cells. However, despite NK cells’ ability to induce ADCC following daratumumab treatment in vitro, emerging evidence suggests that the contribution of NK cells as effectors in vivo is less certain. Patient NK-cell numbers decline rapidly following daratumumab treatment and full recovery can take up to 6 months following the discontinuation of daratumumab treatment.10 Thus, other effector cells may be more important. Cytometry by time-of-flight on samples from single-agent and combination studies again showed significant reduction in NK cells upon daratumumab treatment, with residual NK cells likely to be hypofunctional based on surface marker expression.11
There is increasing awareness of the importance of macrophages as effectors of antibody therapies for cancer.12 Using mouse macrophages as effector cells and CD38-positive Burkitt’s lymphoma Daudi cells, daratumumab has been shown to induce rapid phagocytosis.13 This observation was repeated using patient myeloma cells and macrophages derived from peripheral blood monocytes isolated from healthy donors. Phagocytosis was induced in 11/12 MM patients ex vivo. Using a subcutaneous (SC) and a leukemic intravenous (IV) xenograft tumor model, the authors also demonstrated that phagocytosis was an important in vivo mechanism of daratumumab activity.
An important recent paper provides a strong rationale for combining cyclophosphamide with monoclonal antibodies in hematologic malignancies based on enhanced antibody-dependent cellular phagocytosis.14 Indeed, many of the approved regimens involving monoclonal antibodies in hematologic malignancies contain cyclophosphamide, such as rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone in B-cell lymphoma and fludarabine, cyclophosphamide, rituximab in chronic lymphocytic leukemia. This may have important implications for the use of daratumumab in combination, given the potential importance of antibody-dependent cellular phagocytosis to the mechanism of action of daratumumab in vivo. Cyclophosphamide proved highly synergistic with alemtuzumab in a murine model of B-cell lymphoma/leukemia by activating phagocytic macrophages. Importantly, the doses of cyclophosphamide required were less than would be expected to induce significant DNA damage. Of note, there was a short temporal relationship between cyclophosphamide exposure and synergy with alemtuzumab of up to 2 days. The authors concluded that cyclophosphamide treatment alters the abundance and functionality of macrophages in a rapid, but transient time window during combination therapy. Recent work in our lab has sought to establish the existence of this phenomenon in MM.15 We found that exposure of macrophages to tissue culture–conditioned media from cyclophosphamide, lenalidomide and bortezomib treated cells significantly enhanced daratumumab-specific clearance of MM1S cells (P<0.01), with upregulation of Fc gamma receptors and downregulation of CD47 (the so-called “don’t eat me” antigen) which could enhance phagocytosis. This forms part of the rationale for an ongoing phase Ib study of weekly cyclophosphamide, bortezomib and dexamethasone (CyBorD) with daratumumab (NCT02955810).16
An unexpected benefit of treatment with daratumumab was the observation by Krejcik et al that daratumumab treatment could reduce local immune suppression within the bone marrow microenvironment, facilitating the expansion of positive immune effector cells, thereby contributing to the antimyeloma response.17 Specifically, they examined the effects of daratumumab on other immune cell populations that express CD38, namely, myeloid-derived suppressor cells and regulatory B cells. These are associated with decreased immune function and disease progression. Using peripheral blood and bone marrow from relapsed/refractory patients enrolled in two daratumumab monotherapy studies (Gen501 and SIRIUS), immune profiling and assessment of functional activity were performed. Samples from 148 patients across both studies were analyzed by flow cytometry, functional assays and T-cell receptor sequencing. They demonstrated a reduction of CD38-expressing immunosuppressive cells with induction of helper and cytotoxic T-cell expansion. Robust T-cell increases, increased CD8+:CD4+ ratios, increased antiviral responses and increased T-cell clonality were all observed after daratumumab treatment within this heavily pretreated, relapsed and refractory patient population which would not normally be expected to have strong immune responses. Reduction in immunosuppressive subsets of regulatory B cells and regulatory T cells, with increases in total T cells, as well as a shift toward higher CD8:CD4 and effector-naïve ratios have also been demonstrated using cytometry by time-of-flight.11 The clinical relevance of this immunomodulatory effect of daratumumab is still unknown, but suggests a role for PD-1/PDL-1 inhibitors, given that myeloma cells are known to express PDL-1, which could suppress T-cell activation.18 The ability of daratumumab to promote adaptive T-cell responses was explored further using peripheral blood samples from patients enrolled in the phase 3 POLLUX study (daratumumab, lenalidomide and dexamethasone [DRd] vs Rd).19,20 T-cell repertoire profiles between both groups, measured by T-cell receptor beta sequencing, were similar at baseline. T-cell receptor beta clonality changed significantly in the DRd group between cycles 1 and 3, compared to the Rd group. It was also observed that baseline T-cell receptor richness was associated with improved PFS in DRd, but not in Rd-treated patients. It seems that in patients who respond to daratumumab, an improved adaptive immune response is important. Reduction of immunosuppression through modulation of the enzymatic activity of CD38 and subsequent decreases in adenosine levels may also be important.21
CD38 expression levels have been shown to correlate with response to therapy, but are not the sole determinant of response.22 Daratumumab also rapidly depletes CD38 levels on myeloma cells in the bone marrow niche and in the circulation. This is the case for those who achieved ≥partial response (PR) or <PR in heavily pretreated cohorts from single-agent studies. Nijhof et al22 raise the possibility that the pressure to maintain myeloma cells in a CD38-negative state offers a clinical benefit. Increased CD38 expression leads to increased adherence to bone marrow stromal cells (through interaction with CD31). Therefore, loss of CD38 may reduce myeloma cell growth and survival. These authors also demonstrate the return of CD38 to baseline levels, 6 months after treatment cessation, which could provide a rationale for retreating selected patients.
Resistance of myeloma cells following progressive lines of therapy is therapeutically challenging. Nijhof et al investigated whether resistance to steroids, anthracyclines, alkylators and novel agents (lenalidomide and bortezomib) predicted resistance to daratumumab.23 Myeloma cells along with autologous effector cells from newly diagnosed and relapsed patients were treated with daratumumab. No difference in ADCC or CDC was observed. CD38 expression levels were found to be positively associated with response to daratumumab by ADCC/CDC. In addition, levels of complement inhibitory proteins (e.g., CD55 and CD59) were increased at the time of progression. They further postulated that upregulation of CD38 expression, mediated by all-trans retinoic acid (ATRA) would enhance daratumumab-mediated killing. Small doses of ATRA (as little as 10 nM) were observed to increase CD38 expression 1.9- to 4.4-fold in myeloma cell lines and in cells from 26 MM patients. Maximum effects were seen at 48 hours. In addition, ATRA treatment decreased CD55 and CD59 expression on myeloma cells from patients who had developed daratumumab resistance to pretreatment levels. CDC and, to a lesser extent, ADCC-mediated killing by daratumumab were enhanced in cell lines, patient samples and a humanized mouse model. This may also offer a potential mechanism of overcoming resistance.
The histone deacetylase inhibitor panobinostat has also been shown to upregulate CD38 expression in MM cell lines and primary myeloma cells.24 CD38 expression showed an up to 4-fold increase in primary myeloma cells treated with panobinostat. Increased daratumumab-mediated ADCC was observed in myeloma cell lines and primary myeloma cells treated with panobinostat, compared to untreated cells. This preclinical model displays the synergism of the two drugs, which may warrant further clinical evaluation as another possible means of overcoming resistance.
Efficacy and safety of daratumumab
Daratumumab has been studied in a number of single-agent and combination studies. A summary of the current clinical trials is presented in Table 1, with selected ongoing studies included in Table 2.
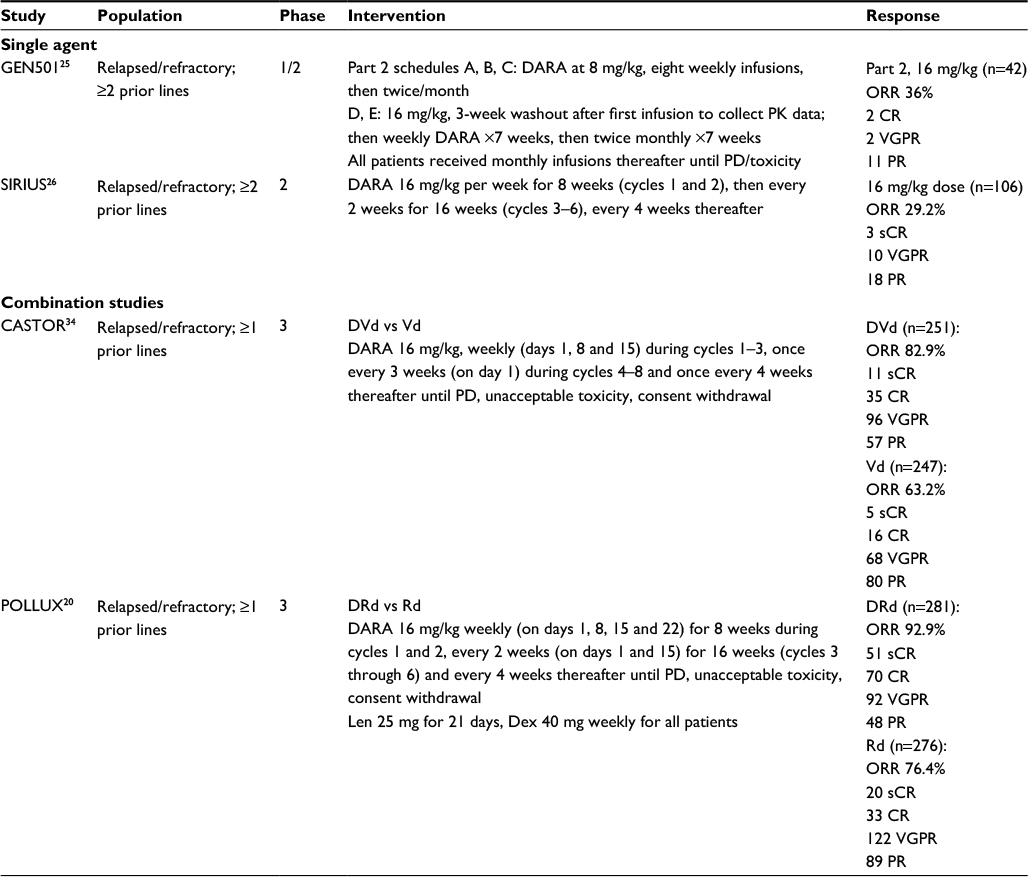 | Table 1 Clinical trials involving daratumumab Abbreviations: CR, complete response; DARA, daratumumab; Dex, dexamethasone; DRd, dara, lenalidomide and dexamethasone; DVd, daratumumab, bortezomib and dexamethasone; Len, lenalidomide; ORR, overall response rate; PD, progressive disease; PK, pharmacokinetics; PR, partial response; Rd, lenalidomide, dexamethasone; sCR, stringent complete response; Vd, bortezomib and dexamethasone; VGPR, very good partial response. |
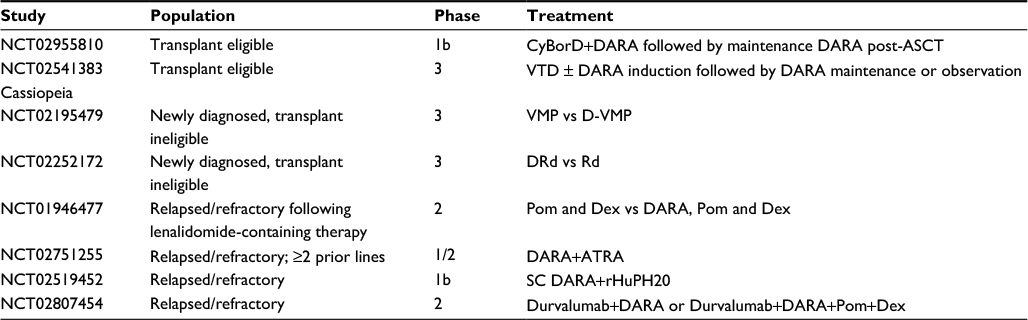 | Table 2 Selected trials evaluating daratumumab, currently recruiting Abbreviations: ASCT, autologous stem cell transplant; ATRA, all-trans retinoic acid; CyBorD, cyclophosphamide, bortezomib and dexamethasone; DARA, daratumumab; Dex, dexamethasone; DRd, daratumumab, lenalidomide and dexamethasone; D-VMP, DARA, bortezomib, melphalan and dexamethasone; Pom, pomalidomide; rHuPH20, recombinant human hyaluronidase; Rd, lenalidomide, dexamethasone; SC, subcutaneous; VMP, bortezomib, melphalan, prednisolone; VTD, bortezomib, thalidomide and dexamethasone. |
Single-agent trials
In the phases 1–2, open-label, GEN501 trial, Lokhorst et al used single-agent daratumumab in a relapsed/refractory cohort that had received a minimum of two prior lines of therapy.25 The part 1 dose-escalation phase enrolled 32 heavily pretreated patients (median number of prior lines was 6; 75% were refractory to both lenalidomide and bortezomib). Ten cohorts received doses of 0.005–24 mg of daratumumab per kilogram body weight. After the first full dose, a washout period of 3 weeks to collect safety and PK data was instituted. Thereafter, six full, weekly infusions were administered. The maximum tolerated dose was not reached. They reported a 33% overall response rate (ORR) (all PRs) in patients receiving doses of ≥4 mg/kg. No responses were seen at doses of ≤2 mg/kg. Seventy-two patients were enrolled in part 2 to dose cohorts of 8 and 16 mg/kg. Patients in schedules A, B and C received daratumumab at a dose of 8 mg/kg in eight, once-weekly infusions and then in twice-monthly infusions for 16 weeks. In schedules D and E, patients were treated with daratumumab at a dose of 16 mg/kg, and after the first infusion, they had a 3-week washout period to allow for the collection of PK data. They were then treated weekly for 7 weeks and then twice monthly for 14 weeks. Patients received monthly daratumumab thereafter, until progression or unacceptable toxicity. The median number of prior treatments in both cohorts was 4, with 63% and 64% of patients being double refractory in the respective dosing cohorts. The ORR in patients receiving 16 mg/kg was 36% (two complete responses [CR], two very good partial responses [VGPR] and 11 PR). This was 10% in the 8 mg/kg cohort ORR (three PR). The median duration of response in the 8 mg/kg cohort was estimated at 6.9 months and was not reached in the 16 mg/kg cohort. In patients who achieved a response with the 16 mg/kg dose, 65% were progression free at 12 months.
Among the 72 subjects treated in part 2 of study GEN501, 71% had infusion-related reactions (IRRs) which were all grade 1 or 2, with one grade 3 exception. Importantly, no one discontinued treatment because of IRRs. Reactions were typically during the first infusion. Patients who received a schedule of 8 mg/kg in 100 mL over 6 hours (Schedule C) had lower IRR rates than those who received higher concentrations. Serious adverse events (AEs) were reported in 40% of subjects in the 8 mg/kg group and 33% of subjects in the 16 mg/kg group). The most frequently reported serious AEs were infection-related events (17% in the 8 mg cohort, 10% in the 16 mg cohort). Neutropenia, which occurred in 12% of patients (five totally) in the 16 mg cohort, was the most common hematologic AE.
The SIRIUS phase 2 study of daratumumab monotherapy, in the multiple relapsed setting, was reported by Lonial et al.26 Thirty-four patients were randomly allocated to receive 8 or 16 mg/kg, with an additional 25 recruited to the 16 mg arm after the first interim analysis (the 8 mg dose did not meet the prespecified ORR for continuation). The schedules were as follows: daratumumab 16 mg/kg per week for 8 weeks (cycles 1 and 2), then every 2 weeks for 16 weeks (cycles 3–6), then every 4 weeks thereafter, or 8 mg/kg continuously. Part 2 enrolled 65 patients to receive 16 mg/kg and 106 patients were included in the final analysis at this dose. Median prior therapies were 5, 97% were refractory to their last line of therapy and 82% were double refractory. Overall response in this group was 29.2% (3 stringent complete response [sCR], 10 VGPR, 18 PR). Responses were noted regardless of risk stratification or prior lines of therapy. A median PFS of 3.7 months was reported with a median response duration of 7.4 months, with follow-up ongoing.
Again, daratumumab was well tolerated in this study with no discontinuations due to drug-related, treatment-emergent AEs. In the 16 mg/kg group, the most common hematologic treatment-emergent AEs of any grade (≥20%) were: anemia (35 [33%] patients), thrombocytopenia (27 [25%] patients) and neutropenia (24 [23%] patients). Grade 3 or higher anemia and thrombocytopenia occurred more frequently in nonresponders (24 [32%] and 18 [24%] of 75 patients, respectively) than in responders (1 [3%] and 2 [6%] of 31 patients, respectively). Grade 3 or higher neutropenia rates were similar in nonresponders (nine [12%]) and responders (four [13%]).
Further follow-up data have also been presented in a combined analysis of 148 GEN501/SIRIUS patients who received a 16 mg/kg dose of daratumumab at a median follow-up of 20.7 months.27 Responses deepened across the studies with continued daratumumab exposure. The combined ORR for the data set was 31.1% (95% confidence interval [CI]: 23.7–39.2). In patients who achieved a PR, or better, the median OS was not evaluable. Thirty-six of the 46 patients who responded to daratumumab were alive at a median follow-up of 20.7 months. A survival benefit was also appreciated in patients who had only achieved a minimal response or stable disease, with a median OS of 18.5 months.
Three studies presented at the 2016 American Society of Hematology (ASH) conference attempt to demonstrate the real-world applicability of daratumumab. The early access program for relapsed/refractory MM enrolled 348 patients in the USA.28 Inclusion criteria mirrored the SIRIUS study. Patients were treated for a median of 1.9 months. The most common AEs were IRRs (56%), mainly during the first dose. Only 8% were >grade 3. Anemia and thrombocytopenia were the most common grade ≥3 events observed (14% and 15%, respectively). Quality of life measures remained stable while patients were on daratumumab. A further aspect of this study sought to reduce IRR by administering montelukast 30 minutes prior to infusion. This was found to shorten the first infusion time in the 50 patients who received montelukast, compared to the 298 patients who did not.29
Using propensity score matching and multivariate Cox regression analyses, the effect of daratumumab vs SOC from an International Myeloma Foundation cohort suggests a significant gain in OS with daratumumab.30 A hazard ratio (HR) of 0.43 (95% CI: 0.32–0.59) is quoted. Historical registry data of heavily pretreated refractory patients from the Czech Republic have been used to perform an adjusted comparison with daratumumab monotherapy studies.31 A multivariate Cox proportional hazards regression was performed to adjust for relevant confounding errors. The PFS for daratumumab vs physician’s choice was 4.0 and 5.6 months with an OS of 20.1 and 11.9 months, respectively. It must be stated that comparisons across trials and using historical data have inherent biases and increased risk of Type 1 errors.
These studies demonstrate a surprising level of single-agent activity in an extremely challenging cohort of patients, surpassing that seen in other single-agent trials in refractory myeloma.32 Despite achieving a similar median PFS to pomalidomide/dexamethasone and carfilzomib as monotherapy in relapsed/refractory MM, treatment with daratumumab monotherapy has translated into a significantly longer OS. The reasons for this are at present unclear, but could include the subsequent development of an antitumor immune response or potentially increased sensitivity to treatment given at progression following daratumumab treatment. These findings prompted the Food and Drug Administration to grant accelerated approval of daratumumab monotherapy in patients who had received three or more prior lines of therapy.33
Combination trials
Several combination trials in upfront or relapsed settings are ongoing. Studies of combinations with lenalidomide and with bortezomib have recently been published and report encouraging results.20,34 Early-phase clinical trials demonstrated the safety and efficacy of daratumumab in combination with proteasome inhibitors and immunomodulatory agents, leading to phase 3 trials.
The phase 3, randomized, open-label, multicenter CASTOR trial enrolled 498 relapsed or refractory patients to receive either daratumumab, bortezomib (V) and dexamethasone (d) vs Vd alone, with PFS as the primary endpoint.34 All patients had at least one prior line of therapy, attained at least a PR to one prior line of therapy, had measurable and documented progression as per the International Myeloma Working Group criteria following their last cycle of treatment and were excluded if they had disease that was refractory to bortezomib or other proteasome inhibitors. Randomization was on a 1:1 basis. All patients received up to eight cycles (21 days per cycle) of bortezomib (1.3 mg/m2 IV; days 1, 4, 8, and 11) and dexamethasone, and in addition, patients in the DVd arm received weekly daratumumab 16 mg/kg (days 1, 8 and 15) during cycles 1–3, once every 3 weeks (on day 1) during cycles 4–8 and once every 4 weeks thereafter until withdrawal from the study or progression. A total of 498 patients were enrolled (treatment arm n=251; control n=247). Patients were refractory to their last line of therapy in 31.3% of cases; 65.5% had prior bortezomib and 61.2% had undergone prior autologous stem cell transplantation. The median number of prior therapies was 2. A maximum of eight cycles were received by 79.8% in the DVd group and 57.4% in the Vd group. The estimated 12-month PFS rate in the treatment group was 60.7%, compared to 26.9% in the control. Median PFS was not reached in the treatment arm and was 7.2 months in the control. ORR was 82.9% in the treatment group (11 sCR, 35 CR, 96 VGPR, 57 PR) and 63.2% in the control (5 sCR, 16 CR, 68 VGPR, 80 PR).
Data on a subgroup analysis of this trial, based on prior lines of therapy and outcomes based on cytogenetics, were recently presented at ASH.35 PFS was also significantly longer in patients with high-risk cytogenetics who received DVd vs Vd (HR, 0.46; 95% CI: 0.22–0.97; P=0.0367). The estimated 12-month PFS rates were 63.2% vs 26.7%. Patients who were bortezomib-naïve had significantly improved PFS with DVd (estimated 12-month PFS rates were 72% vs 28%).36 In patients who had previously received bortezomib, the estimated 12-month PFS rates were 55% vs 27%, with significantly higher rates of VGPR. Patients who were refractory to lenalidomide again had a significantly longer PFS (median: 10.3 vs 4.4 months) with higher rates of ≥VGPR (54% vs 12%; P<0.0001).
The combination DVd was well tolerated. IRRs were again common, reported in 45.3% of patients (98.2% during the first infusion). Most events were grade 1 or 2, with 8.4% grade 3 and no grade 4 reactions. Dyspnea, cough and bronchospasm were the most commonly documented reactions. Two patients discontinued treatment because of IRRs. Hematologic toxicities were more common in the treatment group (grade 3 or 4 thrombocytopenia 45.3% vs 32.9%, neutropenia 12.8% vs 4.2% and anemia 14.4% vs 16.0%). Peripheral sensory neuropathy of any grade was more common in the daratumumab arm (47.3% vs 37.6% [4.5% and 6.8% grade 3 or 4]). Grade 3 and 4 infection rates were similar in both groups (21.4% and 19.0%). AEs leading to death were similar in both groups and related to general deteriorations in patient’s overall health, with three reports of pneumonia (one DVd, two Vd), two ischemic strokes (two DVd, zero Vd) and respiratory failure (two DVd, zero Vd).
Daratumumab in combination with lenalidomide and dexamethasone has been evaluated in a phase 1/2 study in a relapsed/refractory population.37 Safety was the primary endpoint, with efficacy assessments also performed. A standard 3+3 dose escalation study was performed in part 1 (daratumumab 2, 4, 8 and 16 mg/kg). Daratumumab was administered weekly for two cycles, twice weekly for cycles 3–6 and every 4 weeks thereafter, until progression or unacceptable toxicity. Patients received dexamethasone (40 mg/week) and lenalidomide (25 mg/day, days 1–21). Twelve patients with a median of three prior lines of therapy (92.3% double refractory) were enrolled and no dose-limiting toxicities (DLTs) were observed. The recommended phase 2 dose of daratumumab was 16 mg/kg and 32 patients were enrolled. The ORR in part 2 was 81.3% (eight sCR, three CR, nine VGPR, six PR). At a median follow-up of 15.6 months, median PFS and OS were not reached. Eleven patients had received prior lenalidomide, but only one was considered refractory.
In part 2 IRRs occurred in 56% of patients, mainly during first infusions and more commonly with accelerated infusions (500 mL over 3 hours). Most were grade 1/2 (grade 3, 6.3%). One patient with laryngeal edema discontinued treatment. Grade 3 or 4 infections occurred in 15.6% of patients in part 2. Three deaths were reported, two due to progressive disease and one due to viral pneumonia.
The phase 3 POLLUX study randomly allocated 569 relapsed/refractory patients who had received at least one prior line of therapy to receive either daratumumab, lenalidomide and dexamethasone (treatment) or lenalidomide and dexamethasone (control). The daratumumab schedule was as follows: 16 mg/kg weekly (on days 1, 8, 15 and 22) for 8 weeks during cycles 1 and 2, every 2 weeks (on days 1 and 15) for 16 weeks (cycles 3 through 6) and every 4 weeks thereafter. The primary endpoint was PFS. Median prior line of therapy was 1 (PIs 85.6%, IMiDs 55.2%, both 43.9%). An autograft had been performed in 63.3% of patients. The rate of PFS at 12 months was 85.7% (95% CI: 80.9–89.4) in the daratumumab group (n=281), as compared with 63.2% (95% CI: 57.1–68.8) in the control group (n=276). The evaluable ORR was 92.9% (51 sCR, 70 CR, 92 VGPR, 48 PR) in the daratumumab group vs 76.4% in the control group (20 sCR, 33 CR, 122 VGPR, 89 PR).
Data on minimal residual disease (MRD) from both the POLLUX and CASTOR trials using next-generation sequencing of B-cell receptor (Ig) have recently become available.38 Assessment for both trials was performed at suspected CR and at 3 and 6 months (POLLUX) and at 6 and 12 months (CASTOR) if CR was maintained. Significantly higher MRD rates were observed in both trials with the addition of daratumumab at sensitivities of 10−4, 10−5 and 10−6. A >3-fold increase in MRD negativity at all thresholds was observed with daratumumab, regardless of the SOC regimen. This translated to low PFS event rates and suggests that deeper clinical responses induced by daratumumab may lead to improved survival, but we need more mature data to make this assertion. Very significantly, MRD negativity was seen in patients with high-risk fluorescence in situ hybridization (FISH) abnormalities, such as del17p, suggesting the potential of daratumumab-based combinations to overcome high-risk features, at least in some patients.
Chari et al have reported on 77 patients with relapsed/refractory myeloma recruited into the daratumumab, pomalidomide and dexamethasone arms of their phase 1b study (NCT01998971).39 Patients had received ≥2 prior lines of therapy, including lenalidomide and bortezomib. The median number of prior therapies was 3.5. Patients received daratumumab weekly for two cycles, fortnightly for four cycles and every 4 weeks until progression, along with pomalidomide 4 mg 21/28 days and 40 mg dexamethasone weekly. Median follow-up was only 72 days, and safety and tolerability were the primary endpoints. Little additional toxicity was seen with the addition of daratumumab, other than expected IRRs. Of 53 evaluable patients (>1 post-baseline assessment), the ORR was 58.5% (3 sCR, 1 CR, 12 VGPR, 15 PR). An ORR of 57.5% was noted in 40 double-refractory patients. Retrospective data from Emory University Hospital using this combination have also been presented.40 In 19 daratumumab- and pomalidomide-naïve patients with a median of three prior lines of treatment, they reported an ORR of 89%. The ORR of patients refractory to daratumumab or pomalidomide (n=22) was 40.9% and of those refractory to both (n=12) was 33.3%. This last group is interesting, as recovery of CD38 on discontinuation of daratumumab may support rechallenging some patients.
Daratumumab in combination with pomalidomide and dexamethasone has also been compared to single-agent daratumumab in the relapsed/refractory setting.41 The single-agent part of this study reports similar efficacy to previous single-agent trials in similar populations (n=25; ORR 28%; one CR, one VGPR, five PR). The combination study showed acceptable safety and increased responses (n=39; ORR 41%; 2 CR, 1 VGPR, 13 PR). High-risk patients, defined by gene expression profiling, also exhibit an ORR of 21%. The combination was well tolerated. IRRs were mild, with only 2% of patients experiencing a grade 3 reaction. Also, 8% of patients developed grade 3 or 4 pneumonia with this combination.41 While this is a small, nonrandomized study, it again demonstrates the safety and tolerability of daratumumab in combination regimens.
The Intergroupe Francophone du Myélome treated 64 heavily pretreated patients with daratumumab and dexamethasone.42 Daratumumab at a dose of 16 mg/kg was administered weekly for two cycles, fortnightly for four cycles and monthly thereafter. All patients were refractory to lenalidomide, pomalidomide and bortezomib, with a median of six prior lines of therapy. Similar toxicities to other trials were observed. Responses were reported for 40 patients with an ORR of 25% (eight PR, two VGPR) after a median of two cycles.
Overall, the safety and tolerability of daratumumab with doublet and triplet regimens is impressive, as is the depth of the responses achieved. The addition of daratumumab to SOC regimens adds significantly to PFS in both the CASTOR and POLLUX trials, with a 3-fold increase in conversion to MRD status. It should be borne in mind that bortezomib and dexamethasone were stopped after eight cycles of the CASTOR trial, leaving patients on either single-agent daratumumab or no therapy. This has obvious implications for the loss of synergism between bortezomib and daratumumab. Also, this design differs from the POLLUX trial and others where the backbone has been continued.43 Superior responses have been observed with earlier use of daratumumab, that is, after one prior line. The combination of daratumumab, lenalidomide and dexamethasone does appear to stand out when compared to other active combinations in the relapsed setting.44
Both the POLLUX and CASTOR studies demonstrate that the best responses to daratumumab-based therapy are seen in less-heavily pretreated patients, with the greatest benefit clearly seen in patients who have had only one prior therapy. Given this, it is likely that even more benefit would be observed in front-line treatment. Limited data are available on the upfront use of daratumumab with SOC regimens. However, several trials are ongoing (Table 2). Data presented at ASH in 2014 illustrated the feasibility of combining daratumumab with SOC regimens. Specifically, newly diagnosed patients (irrespective of transplant eligibility) received daratumumab (16 mg/kg) in combination with Vd or bortezomib, thalidomide and dexamethasone (VTD). No additional toxicities were observed in 17 newly diagnosed patients, though the median duration of treatment was short (44 days). To date, there is no evidence that daratumumab adversely affects stem cell mobilization, but this will need to be carefully monitored, given the presence of CD38 on hematopoietic progenitors.45
PK and pharmacology of daratumumab
A population PK analysis was performed on 223 patients enrolled in the GEN501 and MMY2002 studies.46 Concentration measurements were taken from 2,572 samples. The dose ranged from 0.1 to 24 mg/kg, with 150 of these patients receiving 16 mg/kg. Elimination of daratumumab was found to be nonlinear,25 demonstrating target-mediated drug disposition. Higher doses and multiple doses resulted in decreased clearance. The clearance and volume of distribution of clinical compartment (V1) increased with increasing bodyweight, indicating that there was relatively consistent daratumumab exposure across a range of bodyweights. The bodyweight-based dosing employed for daratumumab thus seems appropriate. No clinically significant effect on daratumumab exposure was observed when age, race, renal impairment, mild hepatic impairment and disease characteristics were considered. Formal studies on patients with renal impairment are lacking. The 8 mg/kg dose is probably less than the through threshold for target saturation, and while a dose of 24 mg/kg would likely be tolerated, it could not be expected to provide extra clinical benefit. The 16 mg/kg dose demonstrated PKs consistent with target saturation across the dosing intervals.
The mean half-life in part 2 of the GEN501 study at a dose of 16 mg/kg was estimated at 9.0±4.3 days after the first dose of 16 mg/kg and 10.6±9.0 days after varying numbers of repeat doses.
PK in the phase 1/2 DRd study was consistent with daratumumab monotherapy.37 Throughout the first two cycles of once-weekly dosing, daratumumab concentrations appeared to accumulate. Concentrations began to decrease slightly with less-frequent dosing. Anti-daratumumab antibodies were not detected in this study.
Population PK analysis, mainly using data from the CASTOR and POLLUX trials, was performed by Xu et al.47 PK was observed to be similar in single-agent and combination studies, and none of the intrinsic or extrinsic factors affected PK significantly.
SC administration of daratumumab is also under investigation.48 A cohort of 41 relapsed/refractory patients were given SC daratumumab along with recombinant human hyaluronidase enzyme (rHuPH20) to aid systemic absorption. In this small, phase 1b study, serum trough concentration of daratumumab was similar to, or greater than, IV daratumumab. The combination was well tolerated, with lower rates of IRRs observed.
Disease assessments and daratumumab
The ability to evaluate disease response is integral to the management of MM by assessment/quantification of M-protein by serum protein electrophoresis and immunofixation (IFE).49 The increasing use of monoclonal antibodies in myeloma presents a challenge for disease assessments. Daratumumab can be detected on serum protein electrophoresis and IFE assays, and presents a challenge when it migrates closely with endogenous monoclonal proteins. This is particularly important when the M-protein is IgG Kappa (as approximately 50% are)50 or Kappa light chains. Daratumumab is readily detectable at the end of the weekly dosing period.51 One strategy to address this is the daratumumab-specific immunofixation electrophoresis reflex assay (DIRA), relying on a highly specific antibody that binds daratumumab to alter its migration on IFE. This has been validated and can distinguish myeloma M-protein from daratumumab. A commercially available kit, based on DIRA, has also been evaluated: Hydrashift 2/4 daratumumab (Sebia).52 This test successfully automated the displacement of daratumumab, overcoming interference. IgG Kappa bands often appear during treatment of non-IgG Kappa myeloma with daratumumab; these can be effectively identified with DIRA. It should be noted that a recent clarification of the International Myeloma Working Group response criteria states that CR requires the disappearance of the original M-protein on immunofixation.53
In a small study, daratumumab has been shown to interfere with multiparameter flow cytometry evaluation of bone marrow aspirates.54 Virtually, no CD38 or CD138 events were detected in two patients treated with daratumumab. This was in contrast to the aspirate morphology which demonstrated abnormal plasma cells and the core biopsy which labeled CD38 or CD138 positively by immunohistochemistry. Using antibodies that bind to different CD38 epitopes may address this issue. The use of a multiepitope CD38 antibody proposed by EuroFlow-IMF may improve the identification by flow cytometry of CD38 on plasma cells together with cytoplasmic staining in patients undergoing treatment with daratumumab.55,56 Alternatively, other plasma cell markers may be used as a substitute, such as CD229, CD269 (BCMA) and CD319 (SLAMF7).56 If available, molecular assessments of MRD should not be affected by daratumumab.
Interference with blood compatibility testing
Given that CD38 is expressed on human red blood cells (RBCs),57 it is not surprising that daratumumab interferes with blood compatibility testing, leading to positive antibody screens.58 This has the potential to delay the provision of cross-match compatible RBC units owing to pan-reactivity on routine serologic testing. Direct binding of daratumumab to CD38 on endogenous RBCs has been demonstrated by several mechanisms.58 Adsorption methods do not eliminate this interference. Indirect antiglobulin tests may remain positive for up to 6 months after daratumumab exposure.59 Methods of negating this effect have been explored. The reducing agent dithiothreitol (DTT) has been shown to denature CD38.60 Chapuy et al validated a DTT-based method to resolve this issue. Twenty-five laboratories successfully identified an initially unknown antibody masked by daratumumab using a DTT method.61 A limitation of this method is DTT’s destruction of antibodies in the Kell (K) blood group system. Unless patients are known to be K+, K− units should be provided.
The above study was conducted in academic medical centers and reference transfusion laboratories. The DTT method may be impractical in smaller centers and other methods, such as providing phenotypically matched blood, are described.62 Prior to commencing therapy, all patients should be typed and screened and phenotyping or genotyping should be considered63 (as recommended by the American Association of Blood Banks). Patients should be issued with a patient alert card detailing the issues arising with transfusion.
Practical management of daratumumab administration and IRRs
As previously discussed, IRRs typically occur during the first infusions, are usually grade 1/2, with very rare discontinuations. The rate of IRRs has been shown to be influenced by the infusion rate. Our approach is as follows: first doses of daratumumab are administered in 1,000 mL at 50 mL/hr; in the absence of a reaction, the rate is increased by 50 mL/hr up to 200 mL/hr, second infusions are in 500 mL and start at rates of 50 mL/hr, while subsequent infusions start at 100 mL/hr up to 200 mL/hr.64 All patients receive premedications ~1 hr preinfusion: IV corticosteroid (methylprednisolone 100 mg or equivalent dose), diphenhydramine 25–50 mg (or equivalent) and paracetamol 650–1000 mg IV or orally. For subsequent infusions, 60 mg methylprednisolone 100 mg (or equivalent) is used. Furthermore, evidence shows that montelukast may be beneficial.29 For the prevention of delayed IRRs, oral corticosteroid (20 mg methylprednisolone or equivalent) should be administered on each of the 2 days following all infusions. For patients with a history of chronic obstructive pulmonary disease, we consider giving short- and long-acting bronchodilators and inhaled corticosteroids. For patients with suspected chronic obstructive pulmonary disease, we perform pulmonary function testing and exclude those with a forced expiratory volume in 1 second <50% of the predicted normal. With careful monitoring, we think that it is feasible to treat most patients with respiratory disease.
Should IRRs occur despite this, we immediately interrupt the infusion and institute symptomatic management. Once symptoms resolve, the infusion can be restarted at a lower rate (see Table 3). An IRR has not caused the discontinuation of daratumumab therapy to date. All patients receive antiviral prophylaxis for herpes zoster reactivation and seasonal influenza vaccination, where feasible.
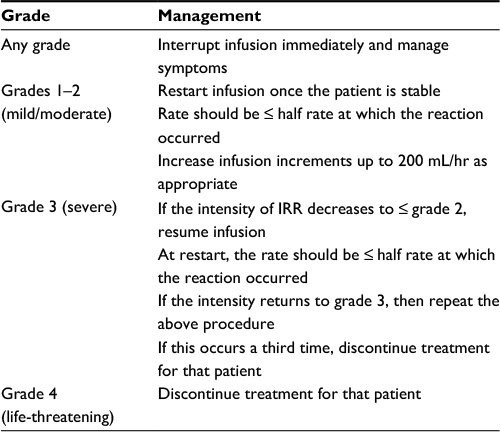 | Table 3 Management of IRR Abbreviation: IRR, infusion-related reaction. |
Conclusion
Further improvements to existing myeloma treatment strategies need to be based on the rational combination of synergistic agents, rather than ad hoc combinations of novel agents with proven regimens. Preclinical observations underpin several ongoing studies in this way. Given the number of new drugs, and indeed drug classes, carefully planned clinical trials are required to determine how best to integrate these into the treatment of MM. A risk-adapted approach is likely to become increasingly important, but the encouraging activity of daratumumab in high-risk patients bodes well for the future.
The addition of daratumumab to effective backbone regimens offers a significant advantage to relapsed/refractory patients and can be delivered safely. It represents another step forward in myeloma treatment following more than a decade of advancements. There are still many questions regarding the usage of daratumumab. Induction of deeper responses measured by MRD assessments is associated with increased PFS.65 This may justify the high cost of triplet and quadruplet combinations including daratumumab in the upfront setting. Patients with high risk, whose disease achieves less depth of response and relapse early, may achieve MRD negativity and improved PFS with the potential for breaks in their treatment. Perhaps, frontline daratumumab may obviate the need for autologous stem cell transplant in certain lower-risk groups or facilitate a fixed duration of maintenance therapy, likely guided by MRD status. Within a few years, results from ongoing frontline studies should help address some of these questions.
In conclusion, daratumumab is a “game changer” with the potential to change the natural history of myeloma in much the same way as monoclonal antibodies do in lymphoma, forming an important addition to the myeloma doctor’s arsenal.
Disclosure
MEO has received research support and honoraria from Janssen. The authors report no other conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Lin P, Owens R, Tricot G, Wilson CS. Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am J Clin Pathol. 2004;121(4):482–488. | ||
FDA Grants breakthrough therapy designation for daratumumab in combination with standard of care for multiple myeloma – The ASCO Post [Internet]. [cited October 7, 2016]. Available from: http://www.ascopost.com/News/43775. Accessed October 7, 1026. | ||
de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186(3):1840–1848. | ||
van der Veer MS, de Weers M, van Kessel B, et al. Towards effective immunotherapy of myeloma: enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica. 2011;96(2):284–290. | ||
Jansen JHM, Boross P, Overdijk MB, Bueren JJL van, Parren PWHI, Leusen JHW. Daratumumab, a human CD38 antibody induces apoptosis of myeloma tumor cells via Fc receptor-mediated crosslinking. Blood. 2012;120(21):2974–2974. | ||
Overdijk MB, Jansen JHM, Nederend M, et al. The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via Fcγ receptor-mediated cross-linking. J Immunol. 2016;197(3):807–813. | ||
Nijhof IS, Groen RWJ, Noort WA, et al. Preclinical evidence for the therapeutic potential of CD38-targeted immuno-chemotherapy in multiple myeloma patients refractory to lenalidomide and bortezomib. Clin Cancer Res. 2015;21(12):2802–2810. | ||
Nijhof IS, Lammerts van Bueren JJ, et al. Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica. 2015;100(2):263–268. | ||
Casneuf T, Xu Steven Xu, Adams III H, et al. Pharmacodynamic relationship between natural killer cells and daratumumab exposure in relapsed/reffractory multiple myeloma [Internet]. [cited January 3, 2017]. Available from: http://learningcenter.ehaweb.org/eha/2016/21st/133273/tineke.casneuf.pharmacodynamic.relationship.between.natural.killer.cells.and.html?f=m3. Accessed January 3, 2017. | ||
Adams H, Stevenaert F, Krejcik J, et al. High-parameter mass cytometry (CyTOF) evaluation of relapsed/refractory multiple myeloma (MM) Pts (Pts) treated with daratumumab supports immune modulation as a novel mechanism of action. Blood. 2016;128(22):4521–4521. | ||
Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs. 2015;7(2):303–310. | ||
Overdijk MB, Verploegen S, Bögels M, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs. 2015;7(2):311–321. | ||
Pallasch CP, Leskov I, Braun CJ, et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell. 2014;156(3):590–602. | ||
Rigalou A, Ryan A, Natoni A, Chiu C, Sasser K, O’Dwyer ME. Potentiation of anti-myeloma activity of daratumumab with combination of cyclophosphamide, lenalidomide or bortezomib via a tumor secretory response that greatly augments macrophage-induced ADCP. Blood. 2016;128(22):2101–2101. | ||
Cyclophosphamide-Bortezomib-Dexamethasone (CyBorD) With Daratumumab (DARA) – Full Text View – ClinicalTrials.gov [Internet]. [cited January 10, 2017]. Available from: https://clinicaltrials.gov/ct2/show/NCT02955810. Accessed January 11, 2017. | ||
Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128(3):384–394. | ||
Rosenblatt J, Avigan D. Targeting the PD-1/PD-L1 axis in multiple myeloma: a dream or a reality? Blood. 2017;129(3):275–279. | ||
Chiu C, Casneuf T, Axel A, et al. Daratumumab in combination with lenalidomide plus dexamethasone induces clonality increase and T-Cell expansion: results from a Phase 3 randomized study (POLLUX). Blood. 2016;128(22):4531–4531. | ||
Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319–1331. | ||
Horenstein AL, Chillemi A, Zaccarello G, et al. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology [September 1, 2013; Internet]. [cited February 28, 2017];2(9). Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3850273/ | ||
Nijhof IS, Casneuf T, van Velzen J, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood. 2016;128(7):959–970. | ||
Nijhof IS, Groen RWJ, Lokhorst HM, et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia. 2015;29(10):2039–2049. | ||
Garcia-Guerrero E, Gogishvili T, Danhof S, et al. Panobinostat induces upregulation of CD38 and augments the anti-myeloma efficacy of daratumumab in pre-clinical models. Blood. 2016;128(22):4481–4481. | ||
Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373(13):1207–1219. | ||
Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016;387(10027):1551–1560. | ||
Usmani SZ, Weiss BM, Plesner T, et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood. 2016;128(1):37–44. | ||
Chari A, Mark TM, Krishnan A, et al. Results of an early access treatment protocol (EAP) of daratumumab in United States patients with relapsed or refractory multiple myeloma. Blood. 2016;128(22):2133–2133. | ||
Chari A, Mark TM, Krishnan A, et al. Use of montelukast to reduce infusion reactions in an early access treatment protocol of daratumumab in United States patients with relapsed or refractory multiple myeloma. Blood. 2016;128(22):2142–2142. | ||
Kumar SK, Durie BGM, Su Z, et al. Adjusted comparisons suggest daratumumab is associated with prolonged survival compared with standard of care therapies in patients with heavily pre-treated and highly refractory multiple myeloma. Blood. 2016;128(22):4517–4517. | ||
Hájek R, Jelinek T, Maisnar V, et al. Comparative effectiveness of daratumumab monotherapy Vs a real-world historical control from the Czech republic in heavily pretreated and highly refractory multiple myeloma patients. Blood. 2016;128(22):3332–3332. | ||
Rajkumar SV. Daratumumab in multiple myeloma. Lancet. 2016;387(10027):1490–1492. | ||
Press Announcements – FDA approves Darzalex for patients with previously treated multiple myeloma [Internet]. [cited November 3, 2016]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm472875.htm. Accessed November 3, 2016. | ||
Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754–766. | ||
Mateos MV, Estell J, Barreto W, et al. Efficacy of daratumumab, bortezomib, and dexamethasone Vs bortezomib and dexamethasone in relapsed or refractory myeloma based on prior lines of therapy: updated analysis of castor. Blood. 2016;128(22):1150–1150. | ||
Chanan-Khan AA, Lentzsch S, Quach H, et al. Daratumumab, bortezomib and dexamethasone Vs bortezomib and dexamethasone alone for relapsed or refractory multiple myeloma based on prior treatment exposure: updated efficacy analysis of castor. Blood. 2016;128(22):3313–3313. | ||
Plesner T, Arkenau HT, Gimsing P, et al. Phase 1/2 study of daratumumab, lenalidomide, and dexamethasone for relapsed multiple myeloma. Blood. 2016;128(14):1821–1828. | ||
Avet-Loiseau H, Casneuf T, Chiu C, et al. Evaluation of minimal residual disease (MRD) in relapsed/refractory multiple myeloma (RRMM) patients treated with daratumumab in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Blood. 2016;128(22):246–246. | ||
Chari A, Lonial S, Suvannasankha A, et al. Open-label, multicenter, phase 1b study of daratumumab in combination with pomalidomide and dexamethasone in patients with at least 2 lines of prior therapy and relapsed or relapsed and refractory multiple myeloma. Blood. 2015;126(23):508–508. | ||
Nooka AK, Joseph N, Boise LH, Gleason C, Kaufman JL, Lonial S. Clinical efficacy of daratumumab, pomalidomide and dexamethasone in relapsed, refractory myeloma patients: utility of retreatment with daratumumab among refractory patients. Blood. 2016;128(22):492–492. | ||
Branca A, Buros A, Yoon D, et al. Daratumumab single agent and daratumumab plus pomalidomide and dexametasone in relapsed/refractory multiple myeloma: a real life retrospective evaluation. Blood. 2016;128(22):4516–4516. | ||
Boyle EM, Petillon MO, Herbaux C, et al. Daratumumab in combination with dexamethasone in resistant or refractory multiple myeloma: primary results of the IFM2014–04 trial. Blood. 2016;128(22):2138–2138. | ||
Rajkumar SV, Kyle RA. Progress in myeloma – A monoclonal breakthrough. N Engl J Med. 2016;375(14):1390–1392. | ||
Beurden-Tan CHYV, Franken M, Blommestein H, Groot CAU-D, Sonneveld P. Systematic literature review and network meta-analysis of treatments for relapsed/refractory multiple myeloma patients. Blood. 2016;128(22):2144–2144. | ||
Malavasi F, Deaglio S, Damle R, Cutrona G, Ferrarini M, Chiorazzi N. CD38 and chronic lymphocytic leukemia: a decade later. Blood. 2011;118(13):3470–3478. | ||
Yan X, Clemens PL, Puchalski T, et al. Target-mediated drug disposition of daratumumab following intravenous infusion in relapsed or refractory multiple myeloma after prior proteasome inhibitors and immunomodulatory drugs: a population pharmacokinetic analysis. Blood. 2015;126(23):4222. | ||
Xu XS, Liao S, Dimopoulos MA, et al. Population pharmacokinetic and exposure-response analyses for daratumumab in combination therapies for patients with multiple myeloma who have received 1 or more prior lines of therapy. Blood. 2016;128(22):3340–3340. | ||
Usmani SZ, Nahi H, Mateos MV, et al. Open-label, multicenter, dose escalation phase 1b study to assess the subcutaneous delivery of daratumumab in patients (pts) with relapsed or refractory multiple myeloma (PAVO). Blood. 2016;128(22):1149–1149. | ||
Rajkumar SV, Harousseau JL, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117(18):4691–4695. | ||
Moreau P, van de Donk NWCJ, San Miguel J, et al. Practical considerations for the use of daratumumab, a novel CD38 monoclonal antibody, in myeloma. Drugs. 2016;76(8):853–867. | ||
McCudden C, Axel AE, Slaets D, et al. Monitoring multiple myeloma patients treated with daratumumab: teasing out monoclonal antibody interference. Clin Chem Lab Med. 2016;54(6):1095–1104. | ||
Caillon H, Irimia A, Simon JS, et al. Overcoming the interference of daratumumab with immunofixation electrophoresis (IFE) using an industry-developed dira test : hydrashift 2/4 daratumumab. Blood. 2016;128(22):2063–2063. | ||
Durie BG, Miguel JF, Blade J, Rajkumar SV. Clarification of the definition of complete response in multiple myeloma. Leukemia. 2015;29(12):2416–2417. | ||
Perincheri S, Torres R, Tormey CA, Smith BR, Rinder HM, Siddon AJ. Daratumumab interferes with flow cytometric evaluation of multiple myeloma. Blood. 2016;128(22):5630–5630. | ||
Anti-human CD38 multi-epitope reagent [Internet]. [cited January 4, 2017]. Available from: http://www.cytognos.com/index.php/en/reagents/antibodies/1446-anti-human-cd38-multiepitope-reagent. Accessed January 4, 2017. | ||
van de Donk NW, Moreau P, Plesner T, et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood. 2016;127(6):681–695. | ||
Albeniz I, Demir Ö, Türker-Şener L, Yalçintepe L, Nurten R, Bermek E. Erythrocyte CD38 as a prognostic marker in cancer. Hematology. 2007;12(5):409–414. | ||
Chapuy CI, Nicholson RT, Aguad MD, et al. Resolving the daratumumab interference with blood compatibility testing. Transfusion. 2015;55(6 Pt 2):1545–1554. | ||
Oostendorp M, Lammerts van Bueren JJ, Doshi P, et al. When blood transfusion medicine becomes complicated due to interference by monoclonal antibody therapy. Transfusion. 2015;55(6 Pt 2):1555–1562. | ||
Berthelier V, Laboureau J, Boulla G, Schuber F, Deterre P. Probing ligand-induced conformational changes of human CD38. Eur J Biochem. 2000;267(10):3056–3064. | ||
Chapuy CI, Aguad MD, Nicholson RT, et al. International validation of a dithiothreitol (DTT)-based method to resolve the daratumumab interference with blood compatibility testing. Transfusion. 2016;56(12):2964–2972. | ||
Hannon JL, Clarke G. Transfusion management of patients receiving daratumumab therapy for advanced plasma cell myeloma. Transfusion. 2015;55(11):2770. | ||
AABB Association Bulletin. Mitigating the anti-CD38 interference with serologic testing [Internet]. [cited October 19, 2016]. Available from: https://www.aabb.org/programs/publications/bulletins/Documents/ab16–02.pdf. Accessed October 19, 2016. | ||
Voorhees PM, Weiss B, Usmani S, et al. Management of infusion-related reactions following daratumumab monotherapy in patients with at least 3 lines of prior therapy or double refractory multiple myeloma (MM): 54767414MMY2002 (Sirius). Blood. 2015;126(23):1829–1829. | ||
Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328–e346. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.