Back to Journals » Vascular Health and Risk Management » Volume 11
New developments in atherosclerosis: clinical potential of PCSK9 inhibition
Authors Giunzioni I, Tavori H
Received 20 May 2015
Accepted for publication 22 July 2015
Published 24 August 2015 Volume 2015:11 Pages 493—501
DOI https://doi.org/10.2147/VHRM.S74692
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Daniel Duprez
Ilaria Giunzioni, Hagai Tavori
Knight Cardiovascular Institute, Center for Preventive Cardiology, Oregon Health and Science University, Portland, OR, USA
Abstract: Pro-protein convertase subtilisin/kexin type 9 (PCSK9) is a secreted 692-amino acid protein that binds surface low-density lipoprotein (LDL) receptor (LDLR) and targets it toward lysosomal degradation. As a consequence, the number of LDLRs at the cell surface is decreased, and LDL-cholesterol (LDL-C) clearance is reduced, a phenomenon that is magnified by gain-of-function mutations of PCSK9. In contrast, loss-of-function mutations of PCSK9 result in increased surface LDLR and improved LDL-C clearance. This provides the rationale for targeting PCSK9 in hypercholesterolemic subjects as a means to lower LDL-C levels. Monoclonal antibodies (mAbs) against PCSK9 that block its interaction with the LDLR have been developed in the past decade. Two companies have recently received the approval for their anti-PCSK9 mAbs by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) Regeneron/Sanofi, with alirocumab (commercial name – PRALUENT®) and, Amgen with evolocumab (commercial name – Repatha™). The introduction of anti-PCSK9 mAbs will provide an alternative therapeutic strategy to address many of the unmet needs of current lipid-lowering therapies, such as inability to achieve goal LDL-C level, or intolerance and aversion to statins. This review will focus on the kinetics of PCSK9, pharmacokinetics and pharmacodynamics of anti-PCSK9 mAbs, and recent data linking PCSK9 and anti-PCSK9 mAbs to cardiovascular events. Moreover, it will highlight the unanswered questions that still need to be addressed in order to understand the physiologic function, kinetics, and dynamics of PCSK9.
Keywords: PCSK9, LDLR, monoclonal antibodies, pharmacokinetics, cardiovascular risk
Introduction
Pro-protein convertase subtilisin/kexin type 9 (PCSK9) plays a fundamental role in low-density lipoprotein (LDL) metabolism through the post-transcriptional regulation of LDL receptor (LDLR).1–3 PCSK9 is mainly produced by the liver, intestine, and kidney and is synthesized as a precursor of 75 kDa, which undergoes autocatalytic cleavage in the endoplasmic reticulum to form the mature, secreted heterodimer. Once secreted, PCSK9 circulates in the plasma compartment in two different molecular forms, the 62 kDa form, which is the most active4–7 and predominantly present on LDL,8–10 and a 55 kDa form (produced by cleavage of the mature PCSK9 by furin), which is considered to be less active4–7 and is mainly present in the apolipoprotein B (apoB)-free plasma compartment.11 Mature PCSK9 directly binds the epidermal growth factor-like repeat A (EGF-A) domain of LDLR and acts as a chaperone, targeting LDLR toward intracellular degradation through an endosomal/lysosomal route.12 One study also suggested that PCSK9 might directly influence LDLR degradation intracellularly, preventing LDLR from reaching the cell surface.2
Gain-of-function mutations in PCSK9 account for 1%–3% of the individuals with familial hypercholesterolemia (FH) and are associated with early onset of cardiovascular diseases (CVDs).13 On the contrary, PCSK9 loss-of-function mutations reduce LDL-cholesterol (LDL-C) levels and significantly decrease CVD risk.14,15 A few individuals with no detectable levels of PCSK9 in plasma have been identified. Despite carrying extremely low LDL-C levels, these subjects are healthy, fertile, and have normal cognitive functions.16–18 Subjects with more common PCSK9 loss-of-function mutations14 have reduced LDL-C levels and CVD risk.15,19 These observations combined have provided the rationale for a safe and effective use of PCSK9 inhibitors to reduce LDL-C level and CVD risk.
Currently, statins are the most widely prescribed lipid-lowering drugs.20 Statins reduce LDL-C levels by inhibiting HMG-CoA reductase (also known as 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, or HMGCR), the rate-limiting step in cholesterol synthesis.21 The depletion of the intracellular cholesterol pool increases LDLR transcription, which in turn favors LDL clearance.22 LDLR upregulation under cellular cholesterol-depletion state is mediated by sterol regulatory element-binding protein 2 (SREBP2)-dependent mechanisms. Surprisingly, SREBP2 is also responsible for the regulation of PCSK9 expression.23 Thus, statin-mediated upregulation of PCSK9 should limit the LDL-C-lowering effect of these drugs.24
The current dogma (“cholesterol hypothesis”) is that the effect of lowering LDL-C on CVD risk is independent of the mechanism by which LDL-C is lowered.25 PCSK9 inhibition using monoclonal antibodies (mAbs) may help reach the goal of LDL-C reduction and may improve CVD risk in hypercholesterolemic individuals as either monotherapy or in addition to statins. The recently published results of the Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) confirmed that the administration of lipid-lowering agents such as ezetimibe on top of statins further reduced LDL-C levels and the CVD event rate compared to monotherapy.26 These data provide an encouraging platform for the likelihood that agents that act through LDL-lowering mechanisms other than HMGCR will also have cardiovascular (CV) benefits.
mAbs directed toward PCSK9 have shown their efficacy in reducing LDL-C levels, and a detailed summary of the phase III clinical trials with alirocumab (Odissey program), evolocumab (Proficio program) and bococizumab (Spire program) has been recently reviewed in another publication27 by the authors of the current review and others.28,29 However, despite the efficacy of PCSK9 antibodies on LDL-C reduction and their excellent safety profile,30 three central questions related to their effect and mechanism of action remain unanswered: 1) Is the effect of the blocking antibody evident within minutes from injection? This question is triggered by the knowledge that whereas the PCSK9-LDLR complex is formed in only a few minutes, degradation of LDLR instead takes several hours; 2) What are the pharmacokinetic and pharmacodynamic characteristics of the antibody–antigen (Ab–Ag) complex? This question is triggered by the knowledge that a portion of the Ab–Ag complex will reside on lipoproteins, which may direct clearance of the immune complex via unique pathways; 3) Do PCSK9 mAbs reduce atherosclerotic plaque burden and CVD events? This question is triggered by the knowledge that inhibiting PCSK9 not only drives down LDL-C levels but also prohibits PCSK9 function in the plaque. Recent comprehensive reviews have summarized the genetics, physiology, and cell biology of PCSK9,31–33 and the safety and tolerability of the anti-PCSK9 antibodies.28,30 This review will focus on the kinetics of PCSK9, the pharmacokinetics and pharmacodynamics of PCSK9 mAbs, and will provide an updated view of the possible link between PCSK9 inhibition and CVD events.
Kinetics of PCSK9
The mechanism of internalization of LDL by LDLR was first described in 1976 in human fibroblasts (Figure 1A).34 LDL binds to LDLR on coated pits, which then invaginate and get internalized as coated vesicles that expand to become endosomes, from where LDL is delivered to the lysosome while LDLR returns to the cell surface.35,36 The LDLR makes one round trip every 10–15 minutes for a total of over 100 trips in its 20-hour lifespan.37
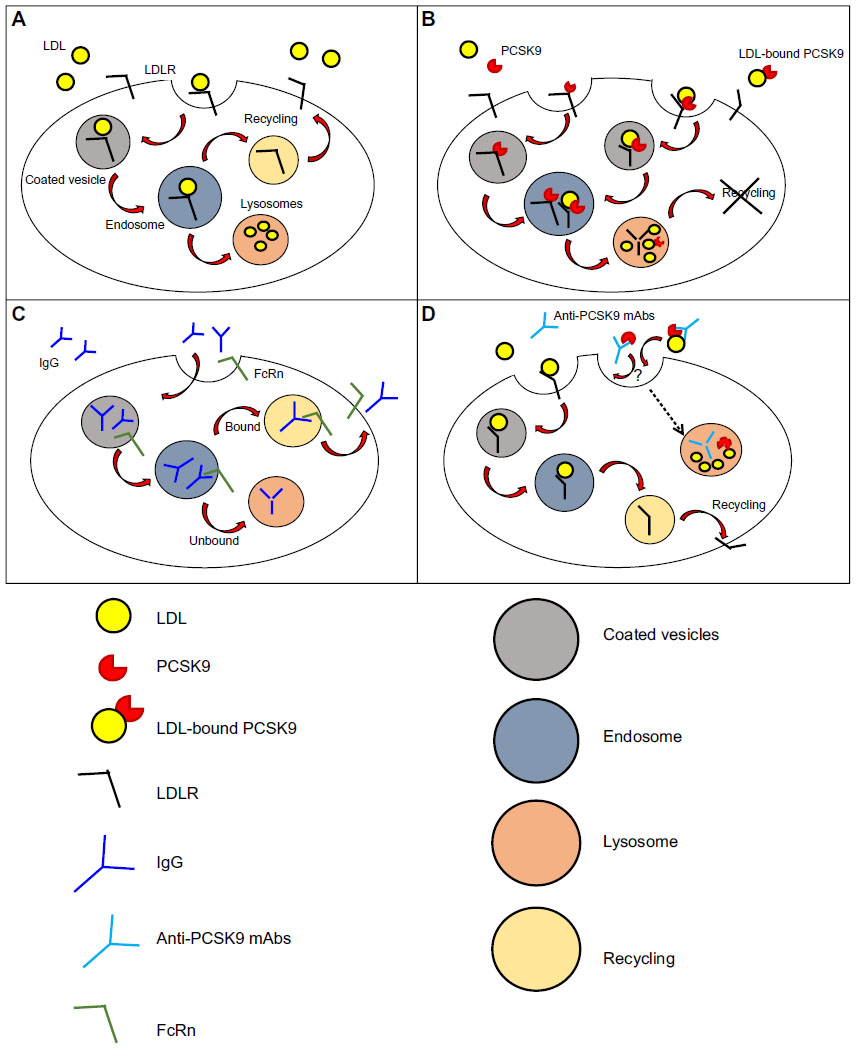 | Figure 1 Mechanism of action and clearance for PCSK9 and anti-PCSK9 antibodies. |
The discovery of PCSK9 has modified our view of lipoprotein metabolism, from a system under complete intracellular control to a system based on a secreted protein that competes with LDL to terminate the LDLR lifecycle (Figure 1B). The kinetics of wild-type (WT) PCSK9 binding to LDLR shows Kd values that range from 90 to 840 nM at neutral pH, and its affinity to LDLR becomes ~100-fold higher at lower pH with Kd values ranging from 1–8 nM.38–41 As a consequence of the increased affinity of PCSK9-LDLR complex at acidic pH, the LDLR in the late endosome fails to dissociate from the ligand, and is instead targeted to lysosomal degradation, apparently together with PCSK9.42 PCSK9 binding to LDLR has been described as biphasic, with a first rapid phase characterized by a half-time of 6.6 minutes, which accounts for 35% of the equilibrium binding and a second slow phase whose half-time is 94 minutes.43 Similarly, 25% of the PCSK9 bound to LDLR dissociates during the rapid phase with a half-time of 19 minutes, while the remaining PCSK9 dissociates slowly with a half-time of 297 minutes.43 Despite the rapid binding of PCSK9 and internalization of LDLR by hepatocytes, PCSK9-mediated degradation of LDLR in vitro has only been observed after several hours.44,45 It was further shown that, at least in mice, PCSK9 remains intact in the liver for up to 4 hours after its internalization,10 thus suggesting that other events might be required in order to allow PCSK9-mediated degradation of LDLR (or LDLR-mediated degradation of PCSK9).
In humans and mice, LDLR is a major regulator for PCSK9 levels10,46 and clearance.10,47 Under normal conditions (ie, the presence of two fully functional LDLR allele products), PCSK9 half-life in plasma is approximately 5 minutes. The presence of an additional copy of LDLR in the liver (induced by transgenic expression) reduces the half-life of PCSK9 by 50%, to 2.9 minutes, whereas in the absence of LDLR, the half-life of PCSK9 in serum is prolonged between 3–10 times above normal.10,47 It has recently been suggested that other proteins such as amyloid precursor protein and amyloid precursor protein-like protein-2 can mediate the internalization of PCSK9 in the absence of LDLR binding.48
We and others have shown that PCSK9 binds to LDL in mice and humans8,10,49 with Kd values ranging from 180–325 nM.49,50 PCSK9 binding to LDL is a common event for PCSK9 (25%–40% of PCSK9 is bound to LDL); however, it is a rare event for LDL, as on average only one in 500–1,000 LDL particles carries a PCSK9 molecule.33 The LDL particle carries predominantly the mature 62 kDa form of PCSK9,8–10 which is considered the most active form in plasma,4–7 whereas the rest of plasma PCSK9 is mainly present as the 55 kDa furin-cleaved fragment and is found in the apoB-free plasma compartment.11 It is unclear what drives this partial compartmentalization of the molecular forms of circulating PCSK9 and whether there is a significant functional correlation to this molecular distribution.
Pharmacokinetics and pharmacodynamics of anti- PCSK9 mAbs
The majority of commercially available antibodies are administered by intravenous, subcutaneous (SC), or intramuscular (IM) routes.51 Systemic absorption following SC and IM injections occurs via lymphatic circulation and via diffusion of antibodies to blood vessels in the proximity of the site of injection.51,52 Absorption following SC administration is relatively slow, and the time required to reach the peak of antibody maximal concentration in the blood varies between 2–8 days, with absolute bioavailability that ranges from 50%–100%, depending on presystemic catabolism and systemic absorption.53 Unlike small-molecule drugs, which are commonly eliminated with renal or hepatic routes, mAbs and immune complexes are cleared by different mechanisms, including fluid-phase pinocytosis and receptor-mediated endocytosis in phagocytes (Figure 1C).51,54 Phagocytes are key players in the elimination of endogenous IgGs.55 Internalization of IgGs in these cells is mediated by binding of the Fc fragment of the antibody to Fcγ-receptors expressed on the cell surface, and elimination occurs through a endosomal/lysosomal route.56 For mAbs that target cell surface antigens (eg, cetuximab and trastuzumab that target the EGF-receptor and HER-2 [human EGF-receptor 2], respectively57) “target-mediated disposition” is the most important elimination route and leads to internalization of the complex in the cell types harboring the antigen.58,59 Antibodies can also be cleared through nonspecific pinocytosis.60 Independently from the mechanism of internalization, a significant fraction of IgG is not targeted toward lysosomal degradation due to the protective action of the FcRn receptor, which redirects IgGs to the cell surface and releases them into the plasma.61
Several preclinical studies were conducted in mice and monkeys to evaluate the kinetics of the anti-PCSK9 antibodies and the therapeutic effect (ie, LDL-C reduction).62,63 A mAb from Pfizer (J16) was able to reduce the levels of LDL-C in normocholesterolemic and hypercholesterolemic cynomolgus macaques by 50%–80%.62 The reduction in LDL-C was achieved by day 3 after treatment (3 mg/kg), and serum LDL-C levels returned to baseline between 2.5–3 weeks after injection.62 A mAb from Merck (mAb1) was tested in mice and non-human primates to study pharmacokinetics and pharmacodynamics.63 The antibody administered in WT mice caused a reduction in total cholesterol that was evident 3 days after a single injection. Cholesterol levels progressively rose to background up to 12 days after injection, with a dose-dependent effect on duration. This antibody also reduced LDL-C in non-human primates 3 days after injection, with a maximum effect on LDL-C reduction at day 10. Serum antibody concentration was monitored during the study, and the half-life of the antibody was calculated to be 61 hours. Also, PCSK9 levels were monitored, and less than 3% of PCSK9 was detected as unbound protein by 15 minutes after injection. Low levels were maintained for 3 days and gradually returned to baseline over 14 days. The kinetics of free PCSK9 reflects the time for the LDLR to increase and internalize larger amount of LDL. However, the fate of the complex PCSK9-antibody was not investigated in this study.63
It has been reported that the average half-life of PCSK9 mAbs is 2.5–3 days63 and that the elimination of the complex PCSK9-mAbs is presumably mediated by PCSK9 through a mechanism similar to PCSK9-mediated degradation of LDLR (endosomal/lysosomal route) (Figure 1D).64 Antibodies can be engineered to escape lysosomal-mediated degradation and to prolong their half-life to 6 days, thus increasing the duration of the cholesterol-lowering effect.64–66 However, no additional information has been provided to fully understand the mechanisms of the antibody or Ab–Ag complexes’ internalization and clearance.
In humans, the three leading mAbs directed against PCSK9 are administered SC (alirocumab by Regeneron/Sanofi, evolocumab by Amgen, and bococizumab by Pfizer). A phase I study on 60 healthy individuals with LDL-C over 95 mg/dL and not receiving other lipid-lowering therapies was conducted to compare the pharmacokinetics and pharmacodynamics after single SC administration of alirocumab at three different injection sites (abdomen, upper arm, and thigh). The administration of alirocumab (75 mg) presented pharmacokinetics and pharmacodynamics profiles independently of the injection site, with a complete loss of free PCSK9 (unbound to the antibody) between day 3 and day 4, and maximal reduction in LDL-C achieved at day 15, thus offering different choices of SC injection site to increase patient compliance.67 Another phase I clinical trial further showed that alirocumab (150 mg SC) reduced free PCSK9 levels within a day and that the effect persisted for 10 days.68 After PCSK9 binding to the antibody, LDLR levels increased, and more LDL particles were internalized. As a consequence, LDL-C levels dropped, and a peak was reached after 14 days.68 However, from these studies,67,68 it is still not clear how the antibody is cleared from circulation. Moreover, since a large portion of PCSK9 is bound to LDL, it remains to be determined whether the affinity of the binding PCSK9 mAbs is affected by the presence of an LDL particle. Furthermore, it is unknown whether LDL-PCSK9-antibody complexes exist in plasma and to what extent these complexes affect antibody and PCSK9 clearance.
PCSK9, mAb, and cardiovascular events
Recently, several studies have correlated PCSK9 levels with parameters directly related to atherosclerosis progression. It has been shown that, in heterozygous (He)FH subjects, high PCSK9 levels are associated with carotid atherosclerosis as measured by carotid intima media thickness (CIMT).69 Moreover, a Pakistani cohort of 400 patients with chronic chest pain who underwent angiography but were not taking lipid-lowering drugs showed a correlation between PCSK9 levels and atheroma burden, independent of LDL-C levels or other CVD risk factors.70 Another study, presented at the 2014 AHA Scientific Sessions, reported an association between serum levels of PCSK9 and the amount of necrotic core tissue in coronary atherosclerotic plaques.71 In this study, PCSK9 levels did not correlate with plaque size or atherosclerotic burden, thus suggesting an involvement of inflammatory processes rather than LDL-C levels.71
The correlation between PCSK9 and atherosclerosis suggests that PCSK9 inhibition might reduce atherosclerosis development and CVD events. This effect was tested in a mouse model of hypercholesterolemia. Weekly administration of alirocumab (3 or 10 mg/kg) alone or in combination with atorvastatin (3.6 mg/kg/d) for 18 weeks decreased total cholesterol and triglycerides (TGs) in a dose-dependent manner. Combination therapy with atorvastatin further decreased cholesterol levels. More importantly, alirocumab dose-dependently decreased atherosclerotic lesion size (−71% at a dose of 3 mg/kg and −88% at a dose of 10 mg/kg). In addition, the PCSK9 inhibitor reduced monocyte recruitment and improved lesion composition by increasing smooth muscle cell and collagen content and by decreasing macrophage and necrotic core content.72 Moreover, we recently showed that PCSK9 of macrophage origin promotes lesion inflammation in mice, independently of systemic lipid changes,73 thus suggesting that therapies with PCSK9 mAbs might have direct local effects to block plaque development.
Except for those patients who are intolerant to statins, it is reasonable to expect that PCSK9 inhibition will be used as an additional therapy to statins in those patients who cannot achieve the goal of LDL-C reduction, including FH patients. Lifelong exposure to severely elevated LDL-C dramatically accelerates CVD, with clinical manifestations often occurring at a young age.74 The benefits of mAbs against PCSK9 in HeFH and receptor-defective homozygous (Ho)FH subjects have been documented.75,76 A recent study investigated the predictive value of PCSK9 in HeFH subjects with reduced LDLR function due to mutations in LDLR causing either defective transport of LDLR to the Golgi (D206E and D154N) or impairing the recycling of LDLR to the cell surface (V408M).77 PCSK9 reduced surface levels of LDLR in fibroblasts from HeFH patients carrying the different LDLR mutations, as it did in non-FH subjects. These data would suggest that HeFH subjects should benefit from PCSK9 inhibition therapy in terms of CV outcomes. In contrast, LDLR-negative HoFH subjects are not likely to respond to PCSK9 inhibition therapy.78
Two recent reports describe the results of studies with mAbs against PCSK9 and their potential effects on CVD events.79,80 The administration of alirocumab (150 mg biweekly) in 2,341 patients at high risk of CVD events (with LDL-C over 70 mg/dL) receiving the highest tolerated dose of statin together with other lipid-lowering agents reduced LDL-C levels by 61.9% after 24 weeks. The goal of LDL-C reduction below 70 mg/dL was achieved in 79.3% of patients, and the reduction in LDL-C was maintained for the duration of treatment (–56% after 78 weeks). Alirocumab also significantly reduced non-high density lipoprotein (non-HDL) cholesterol (–52.3%), apoB (–54%), total cholesterol (–37.5%), lipoprotein (a) (Lp[a]) (–25.6%), and fasting TGs (–17.3%). HDL cholesterol was increased by 4.6%, and apolipoprotein A-I (apoA1) was increased by 2.9%. In a post hoc safety analysis, the significant changes in plasma lipid profile induced by alirocumab were associated with a reduced rate of major CVD events (–48%).80
In two different randomized double-blind trials, the open-label study of long-term evaluation against LDL cholesterol 1 (OSLER-1) and the open-label study of long-term evaluation against LDL cholesterol 2 (OSLER-2), a total of 4,465 patients with hypercholesterolemia and various co-morbidities were included. The administration of 140 mg of evolocumab biweekly or 420 mg monthly in addition to lipid-lowering therapies reduced LDL-C (–61%), non-HDL cholesterol (–52%), apoB (–47.3%), total cholesterol (–36%), TGs (–12.6%), and Lp(a) (–25%), and also increased HDL cholesterol (+7%) and apoA1 (+4.2%). Evolocumab was associated with a 50% lower CVD event rate, further suggesting that anti-PCSK9 therapy will likely prove to reduce CVD risk. In both studies, the administration of PCSK9 inhibitors was safe overall. One of the major concerns was the increase in “neurocognitive” side effects.79
Despite the positive results seen with CVD events and the apparently good safety record, it is important to note that the above studies were not designed to evaluate CVD events or neurocognitive functions as primary or secondary endpoints. At the moment, four placebo-controlled trials (ClinicalTrials.gov number NCT01764633, NCT01663402, NCT01975376, and NCT01975389) are ongoing with the aim to provide proof of CV benefits. In addition, larger trials, including a dedicated neurocognitive substudy (ClinicalTrials.gov number, NCT02207634), will soon provide more detailed information on longer-term effects of these mAbs.
Conclusion
The discovery of PCSK9 has changed our understanding of body cholesterol metabolism from a process thought to be entirely regulated through intracellular processes to an autocrine/paracrine process that can be controlled by plasma components. Similar to LDL, PCSK9 serves as a ligand for the LDLR; thus, the latter is a major determinant for circulating PCSK9 levels. Humans with loss-of-function of PCSK9 have extremely low levels of plasma LDL-C, and even small LDL-C reductions due to common mutations in PCSK9 have been shown to reduce lifetime CVD events. It is anticipated that PCSK9 inhibition therapy will reduce atherosclerotic burden and CVD events, although trial results will not be available until 2018,79,80 probably after the FDA approves the commercial antibodies. Anti-PCSK9 mAbs are cleared from the circulation in a matter of few days, which gives enough time for them to block PCSK9-mediated degradation of LDLR, which in turn leads to reduction in plasma LDL-C levels. The exact pharmacokinetics/dynamics of the antibody, and more importantly of the Ab–Ag complexes, have not been fully studied with these specific antibodies. Furthermore, our current understating of PCSK9 and LDLR dynamics does provide full explanation as to the kinetics of the PCSK9-mediated LDLR degradation process. Even though PCSK9 is being widely investigated in clinical trials and shows promise as an effective lipid-lowering agent, it is important to remember that this mechanism of cholesterol regulation is relatively new, with several gaps in our basic understanding of its full physiologic function, kinetics, and dynamics.
In addition, another consideration that requires thought is the cost of the therapy. PCSK9 inhibitors are injected, generally once or twice a month. CVS Health Corporation indicated that estimates of annual pricing for PCSK9 inhibitors ranged from US$7,000 to US$12,000 per patient.81 Even if PCSK9 inhibitors are indicated for a very narrow patient population, the potential overall costs will be high. In addition, PCSK9 inhibitors are biologics; thus, unlike small molecule drugs, the introduction in the future of cheaper generics will not be simple. Thus, careful management of costs and careful selection of target patients will be necessary in order to contain future expenses.
Disclosure
The authors report no conflicts of interest in this work.
References
Lalanne F, Lambert G, Amar MJ, et al. Wild-type PCSK9 inhibits LDL clearance but does not affect apoB-containing lipoprotein production in mouse and cultured cells. J Lipid Res. 2005;46(6):1312–1319. | |
Poirier S, Mayer G, Poupon V, et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem. 2009; 284(42):28856–28864. | |
Zaid A, Roubtsova A, Essalmani R, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. 2008;48(2):646–654. | |
Lipari MT, Li W, Moran P, et al. Furin-cleaved proprotein convertase subtilisin/kexin type 9 (PCSK9) is active and modulates low density lipoprotein receptor and serum cholesterol levels. J Biol Chem. 2012; 287(52):43482–43491. | |
Essalmani R, Susan-Resiga D, Chamberland A, et al. In vivo evidence that furin from hepatocytes inactivates PCSK9. The J Biol Chem. 2011; 286(6):4257–4263. | |
Benjannet S, Rhainds D, Hamelin J, Nassoury N, Seidah NG. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J Biol Chem. 2006;281(41):30561–30572. | |
Han B, Eacho PI, Knierman MD, et al. Isolation and Characterization of the Circulating Truncated Form of PCSK9. J Lipid Res. 2014;55(7):1505–1514. | |
Fan D, Yancey PG, Qiu S, et al. Self-association of human PCSK9 correlates with its LDLR-degrading activity. Biochemistry. 2008; 47(6):1631–1639. | |
Sun H, Samarghandi A, Zhang N, Yao Z, Xiong M, Teng BB. Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler Thromb Vasc Biol. 2012; 32(7):1585–1595. | |
Tavori H, Fan D, Blakemore JL, et al. Serum proprotein convertase subtilisin/kexin type 9 and cell surface low-density lipoprotein receptor: evidence for a reciprocal regulation. Circulation. 2013;127(24):2403–2413. | |
Tavori H, Giunzioni I, Linton MF, Fazio S. Loss of plasma proprotein convertase subtilisin/kexin 9 (PCSK9) after lipoprotein apheresis. Circ Res. 2013;113(12):1290–1295. | |
Zhang DW, Lagace TA, Garuti R, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282(25):18602–18612. | |
Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–156. | |
Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37(2):161–165. | |
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–1272. | |
Cariou B, Ouguerram K, Zaïr Y, et al. PCSK9 dominant negative mutant results in increased LDL catabolic rate and familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 2009;29(12):2191–2197. | |
Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79(3):514–523. | |
Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193(2):445–448. | |
Kathiresan S; Myocardial Infarction Genetics Consortium. A PCSK9 missense variant associated with a reduced risk of early-onset myocardial infarction. N Engl J Med. 2008;358(21):2299–2300. | |
Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S1–S45. | |
Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation. 2000;101(2):207–213. | |
Brown MS, Dana SE, Goldstein JL. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts by lipoproteins. Proc Natl Acad Sci U S A. 1973;70(7):2162–2166. | |
Dubuc G, Chamberland A, Wassef H, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24(8):1454–1459. | |
Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res. 2008; 49(2):399–409. | |
Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 factorial mendelian randomization study. J Am Coll Cardiol. 2015;65(15):1552–1561. | |
DiNicolantonio JJ, Chatterjee S, Lavie CJ, Bangalore S, O’Keefe JH. Ezetimibe plus moderate-dose simvastatin after acute coronary syndrome: what are we IMPROVEing on? Am J Med. 2015;128(8):914.e1–914.e4. | |
Shapiro MD, Fazio S, Tavori H. Targeting PCSK9 for therapeutic gains. Curr Atheroscler Rep. 2015;17(4):499. | |
Cicero AF, Tartagni E, Ertek S. Efficacy and safety profile of evolocumab (AMG145), an injectable inhibitor of the proprotein convertase subtilisin/kexin type 9: the available clinical evidence. Expert Opin Biol Ther. 2014;14(6):863–868. | |
Dadu RT, Ballantyne CM. Lipid lowering with PCSK9 inhibitors. Nat Rev Cardiol. 2014;11(10):563–575. | |
Cicero AF, Tartagni E, Ertek S. Safety and tolerability of injectable lipid-lowering drugs: a review of available clinical data. Expert Opin Drug Saf. 2014;13(8):1023–1030. | |
Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res. 2014;114(6):1022–1036. | |
Stein EA, Raal F. Reduction of low-density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu Rev Med. 2014;65: 417–431. | |
Tavori H, Rashid S, Fazio S. On the function and homeostasis of PCSK9: reciprocal interaction with LDLR and additional lipid effects. Atherosclerosis. 2015;238(2):264–270. | |
Anderson RG, Goldstein JL, Brown MS. Localization of low density lipoprotein receptors on plasma membrane of normal human fibroblasts and their absence in cells from a familial hypercholesterolemia homozygote. Proc Natl Acad Sci U S A. 1976;73(7):2434–2438. | |
Anderson RG, Brown MS, Beisiegel U, Goldstein JL. Surface distribution and recycling of the low density lipoprotein receptor as visualized with antireceptor antibodies. J Cell Biol. 1982;93(3):523–531. | |
Anderson RG, Brown MS, Goldstein JL. Role of the coated endocytic vesicle in the uptake of receptor-bound low density lipoprotein in human fibroblasts. Cell. 1977;10(3):351–364. | |
Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431–438. | |
Fisher TS, Lo Surdo P, Pandit S, et al. Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation. J Biol Chem. 2007;282(28):20502–20512. | |
Cunningham D, Danley DE, Geoghegan KF, et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol. 2007;14(5):413–419. | |
Pearlstein RA, Hu QY, Zhou J, et al. New hypotheses about the structure-function of proprotein convertase subtilisin/kexin type 9: analysis of the epidermal growth factor-like repeat A docking site using WaterMap. Proteins. 2010;78(12):2571–2586. | |
Piper DE, Jackson S, Liu Q, et al. The crystal structure of PCSK9: a regulator of plasma LDL-cholesterol. Structure. 2007;15(5):545–552. | |
Zhang DW, Garuti R,Tang WJ, Cohen JC, Hobbs HH. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc Natl Acad Sci U S A. 2008;105(35):13045–13050. | |
Mousavi SA, Berge KE, Berg T, Leren TP. Affinity and kinetics of proprotein convertase subtilisin/kexin type 9 binding to low-density lipoprotein receptors on HepG2 cells. FEBS J. 2011;278(16):2938–2950. | |
Qian YW, Schmidt RJ, Zhang Y, et al. Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. J Lipid Res. 2007;48(7):1488–1498. | |
Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A. 2004;101(18):7100–7105. | |
Raal F, Panz V, Immelman A, Pilcher G. Elevated PCSK9 levels in untreated patients with heterozygous or homozygous familial hypercholesterolemia and the response to high-dose statin therapy. J Am Heart Assoc. 2013;2(2):e000028. | |
Grefhorst A, McNutt MC, Lagace TA, Horton JD. Plasma PCSK9 preferentially reduces liver LDL receptors in mice. J Lipid Res. 2008; 49(6):1303–1311. | |
DeVay RM, Shelton DL, Liang H. Characterization of proprotein convertase subtilisin/kexin type 9 (PCSK9) trafficking reveals a novel lysosomal targeting mechanism via amyloid precursor-like protein 2 (APLP2). J Biol Chem. 2013;288(15):10805–10818. | |
Kosenko T, Golder M, Leblond G, Weng W, Lagace TA. Low-density lipoprotein binds to proprotein convertase subtilisin/kexin type-9 (PCSK9) in human plasma and inhibits PCSK9-mediated LDL receptor degradation. J Biol Chem. 2013;288(12):8279–8288. | |
Romagnuolo R, Scipione CA, Boffa MB, Marcovina SM, Seidah NG, Koschinsky ML. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. J Biol Chem. 2015;290(18):11649–11662. | |
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–558. | |
Supersaxo A, Hein WR, Steffen H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm Res. 1990;7(2):167–169. | |
Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93(11):2645–2668. | |
Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M. Pharmacokinetics, pharmacodynamics and physiologically-based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet. 2013;52(2):83–124. | |
Waldmann TA, Strober W. Metabolism of immunoglobulins. Prog Allergy. 1969;13:1–110. | |
Comber PG, Gomez F, Rossman MD, Schreiber AD. Receptors for the Fc portion of immunoglobulin G (Fc gamma R) on human monocytes and macrophages. Prog Clin Biol Res. 1989;297:273–285. | |
Kawaguchi Y, Kono K, Mimura K, et al. Targeting EGFR and HER-2 with cetuximab- and trastuzumab-mediated immunotherapy in oesophageal squamous cell carcinoma. Br J Cancer. 2007;97(4):494–501. | |
Foltz IN, Karow M, Wasserman SM. Evolution and emergence of therapeutic monoclonal antibodies: what cardiologists need to know. Circulation. 2013;127(22):2222–2230. | |
Press OW, Hansen JA, Farr A, Martin PJ. Endocytosis and degradation of murine anti-human CD3 monoclonal antibodies by normal and malignant T-lymphocytes. Cancer Res. 1988;48(8):2249–2257. | |
Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507. | |
Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. 1996;93(11):5512–5516. | |
Liang H, Chaparro-Riggers J, Strop P, et al. Proprotein convertase substilisin/kexin type 9 antagonism reduces low-density lipoprotein cholesterol in statin-treated hypercholesterolemic nonhuman primates. J Pharmacol Exp Ther. 2012;340(2):228–236. | |
Chan JC, Piper DE, Cao Q, et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci U S A. 2009;106(24):9820–9825. | |
Chaparro-Riggers J, Liang H, DeVay RM, et al. Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J Biol Chem. 2012;287(14):11090–11097. | |
Igawa T, Ishii S, Tachibana T, et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol. 2010;28(11):1203–1207. | |
Henne KR, Ason B, Howard M, et al. Anti-PCSK9 antibody pharmacokinetics and low-density lipoprotein-cholesterol pharmacodynamics in nonhuman primates are antigen affinity-dependent and exhibit limited sensitivity to neonatal Fc receptor-binding enhancement. J Pharmacol Exp Ther. 2015;353(1):119–131. | |
Lunven C, Paehler T, Poitiers F, et al. A randomized study of the relative pharmacokinetics, pharmacodynamics, and safety of alirocumab, a fully human monoclonal antibody to PCSK9, after single subcutaneous administration at three different injection sites in healthy subjects. Cardiovasc Ther. 2014;32(6):297–301. | |
Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366(12):1108–1118. | |
Huijgen R, Fouchier SW, Denoun M, et al. Plasma levels of PCSK9 and phenotypic variability in familial hypercholesterolemia. J Lipid Res. 2012;53(5):979–983. | |
Walton TA, Nishtar S, Lumb PJ, et al. Pro-protein convertase subtilisin/kexin 9 concentrations correlate with coronary artery disease atheroma burden in a Pakistani cohort with chronic chest pain. Int J Clin Pract. 2015;69(7):738–742. | |
Cheng JM, Oemrawsingh R, Garcia-Gracia H, et al. Serum proprotein convertase subtilisin/kexin type 9 level is associated with coronary plaque inflammation and cardiovascular outcome independent from serum LDL level. Abstract presented at: American Heart Association Scientific Sessions 2014; November 7-11, 2014; Orlando, FL, USA. Abstract 130:A16101. | |
Kuhnast S, van der Hoorn JW, Pieterman EJ, et al. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J Lipid Res. 2014;55(10):2103–2112. | |
Giunzioni I, Tavori H, Covarrubias R, Major A, Linton MF, Fazio S. Macrophage expression of Pcsk9 influences atherosclerosis development. AHA Sci Session. 2013;128(2 Suppl):A19083. | |
Marks D, Thorogood M, Neil HA, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis. 2003;168(1):1–14. | |
Raal FJ, Honarpour N, Blom DJ, et al; TESLA Investigators. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385(9965):341–350. | |
Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the PCSK9 Monoclonal Antibody, AMG 145, in Homozygous Familial Hypercholesterolemia. Circulation. 2013;128(19):2113–2120. | |
Lambert G, Petrides F, Chatelais M, et al. Elevated plasma PCSK9 level is equally detrimental for patients with nonfamilial hypercholesterolemia and heterozygous familial hypercholesterolemia, irrespective of low-density lipoprotein receptor defects. J Am Coll Cardiol. 2014; 63(22):2365–2373. | |
Lambert G, Chatelais M, Petrides F, et al. Normalization of low-density lipoprotein receptor expression in receptor defective homozygous familial hypercholesterolemia by inhibition of PCSK9 with alirocumab. J Am Coll Cardiol. 2014;64(21):2299–2300. | |
Sabatine MS, Giugliano RP, Wiviott SD, et al; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500–1509. | |
Robinson JG, Farnier M, Krempf M, et al; ODYSSEY LONG TERM Investigators. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1489–1499. | |
CVS health raises concerns about the potential impact of new class of cholesterol drugs on the health care system [webpage on the Internet]. Woonsocket, RI: CVS Health. Available from: http://www.cvshealth.com/newsroom/press-releases/cvs-health-raises-concerns-about-potential-impact-new-class-cholesterol. Accessed July 2, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.