")
Back to Journals » Drug Design, Development and Therapy » Volume 8
New approaches to the management of schizophrenia: focus on aberrant hippocampal drive of dopamine pathways
Received 1 November 2013
Accepted for publication 6 March 2014
Published 2 July 2014 Volume 2014:8 Pages 887—896
DOI https://doi.org/10.2147/DDDT.S42708
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 8
Stephanie M Perez, Daniel J Lodge
Department of Pharmacology and Center for Biomedical Neuroscience, University of Texas Health Science Center, San Antonio, TX, USA
Abstract: Schizophrenia is a disease affecting up to 1% of the population. Current therapies are based on the efficacy of chlorpromazine, discovered over 50 years ago. These drugs block dopamine D2-like receptors and are effective at primarily treating positive symptoms in a subset of patients. Unfortunately, current therapies are far from adequate, and novel treatments require a better understanding of disease pathophysiology. Here we review the dopamine, gamma-aminobutyric acid (GABA), and glutamate hypotheses of schizophrenia and describe a pathway whereby a loss of inhibitory signaling in ventral regions of the hippocampus actually drives a dopamine hyperfunction. Moreover, we discuss novel therapeutic approaches aimed at attenuating ventral hippocampal activity in a preclinical model of schizophrenia, namely the MAM GD17 rat. Specifically, pharmacological (allosteric modulators of the α5 GABAA receptor), neurosurgical (deep brain stimulation), and cell-based (GABAergic precursor transplants) therapies are discussed. By better understanding the underlying circuit level dysfunctions in schizophrenia, novel treatments can be advanced that may provide better efficacy and a superior side effect profile to conventional antipsychotic medications.
Keywords: dopamine, GABA, glutamate, schizophrenia, hippocampus, MAM rat
Introduction
Schizophrenia is a severe psychiatric disorder for which there is currently no cure. Affecting approximately 1% of the population,1,2 schizophrenia is believed to be a developmental disorder with symptoms typically appearing during adolescence and early adulthood.3 There are three major symptom clusters associated with this disease: positive (psychosis, disorganized thinking and speech, hallucinations, delusions), negative (anhedonia, social withdrawal), and cognitive deficits (reversal learning, working memory deficits).4,5 Common pharmacological interventions for the treatment of schizophrenia include typical and atypical antipsychotic drugs which primarily act to decrease positive symptoms by the blockade of D2-type dopamine receptors.6,7 Unfortunately, antipsychotic drugs are not always effective at reducing positive symptoms,8,9 are largely ineffective at treating negative and cognitive deficits,10–13 and are associated with debilitating adverse side effects such as movement14,15 and metabolic disorders.4 For these reasons, the rates of discontinuance are relatively high4 and novel therapeutic approaches are required to better treat this devastating neuropsychiatric disease. Here we review some basic theories of schizophrenia and attempt to provide an integrated hypothesis that combines gamma-aminobutyric acid (GABA)ergic, glutamatergic, and dopaminergic theories to provide a framework by which therapeutic approaches targeting the hippocampus as a novel region can be established for the treatment of schizophrenia.
Antipsychotic drugs
While there are numerous adjunct therapies, antipsychotic drugs have been the first line of treatment for schizophrenia since the advent of chlorpromazine in the 1950s.16 Chlorpromazine largely replaced surgical and electroconvulsive approaches for the treatment of psychiatric symptoms and has been heralded as one of the most significant advances in the field of psychiatry.16,17 Since this time, there has been extensive research into the mechanisms underlying the effects of antipsychotic medications. It has been established that all antipsychotic drugs are dopamine D2-like receptor antagonists and produce effects by blocking dopamine receptors.6,18,19 The typical, or first generation, antipsychotic drugs are effective at reducing positive symptoms,20 likely attributable to their ability to block dopamine receptors in the mesolimbic dopamine system.21 Unfortunately, they also cause dopamine receptor blockade in the nigrostriatal, mesocortical, and tuberoinfundibular dopamine systems, which is associated with a number of debilitating extrapyramidal side effects including Parkinson’s-like symptoms (tremors, muscle rigidity, tardive dyskinesia) and hyperprolactinemia.22
Atypical or second-generation antipsychotics were approved by the FDA in the late 1980s, as they were believed to be more effective at treating symptoms of schizophrenia without the motor side effects.23 Much like typical antipsychotics, atypical antipsychotics bind to dopamine receptors; however, atypical antipsychotics also display an affinity for the serotonin-2A (5HT-2A) receptor.24,25 Indeed, the ratio of 5HT-2A to dopamine D2 receptor affinity distinguishes typical and atypical antipsychotic drugs better than any known pharmacological feature.24 Given that 5HT-2A receptors are located presynaptically on dopamine terminals in the nigrostriatal and mesocortical pathways, atypical antipsychotics augment dopamine release in these regions to counteract the effects of D2 antagonism.26–28 The net result is that atypical antipsychotics maintain efficacy against positive symptoms by transiently occupying D2 receptors and rapidly dissociating to allow for normal dopamine neurotransmission, while providing fewer extrapyramidal side effects.29 Additionally, 5HT-2A receptors are not only localized on dopamine terminals but are widely expressed throughout the cortex, where these receptors also contribute to the beneficial effects of atypical antipsychotics.30 Indeed, patients who take atypical antipsychotics also report mild improvements in cognition and reductions in suicidal thoughts and negative symptoms, which are not commonly observed with typical antipsychotics.30–35 It should be noted that while atypical antipsychotics produce fewer motor side effects associated with blockade of dopamine receptors, they often cause severe metabolic side effects, including weight gain and glucose intolerance.36
To evaluate the effectiveness of various antipsychotic drug treatments, a number of large trials have been undertaken, including the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, the Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS) and the European First-Episode Schizophrenia Trial (EUFEST).4,37 These studies, and others,38 have demonstrated that there are no dramatic differences in the efficacies of typical and atypical antipsychotic treatments.39 However, a striking observation was that the vast majority of patients (~74%), independent of the type of antipsychotic treatment, discontinued their treatment during Phase I due to ineffectiveness or intolerable side effects, clearly demonstrating that current therapeutic approaches are far from adequate.4 For these reasons, it is essential to identify novel therapeutic approaches for the treatment of schizophrenia. To do this, one must start with a more complete understanding of the pathophysiology of schizophrenia.
The dopamine hypothesis
The dopamine hypothesis of schizophrenia is one of the more predominant theories advanced to explain symptoms of the disease.40 It posits that hyperactivity of mesolimbic dopamine transmission underlies positive symptoms of schizophrenia.41–43 Observations that validate this hypothesis include the finding described above that all antipsychotic drugs bind to and block dopamine D2 receptors.20 In addition, indirect dopamine agonists, such as amphetamine, have been demonstrated to induce psychosis in the general population,44 whereas individuals with schizophrenia are significantly more sensitive to the dopamine-releasing effects of amphetamine.41,43 Additional evidence for the dopamine hypothesis stems from human imaging studies providing evidence for region-specific increases in dopamine transmission in individuals with schizophrenia, particularly in the associative areas of the striatum.45 Specifically, positron emission topography (PET) studies demonstrate a higher occupancy of striatal dopamine D2 receptors by dopamine in individuals with schizophrenia, predictive of antipsychotic effectiveness,46,47 whereas 18F-DOPA PET studies have demonstrated an elevated dopamine synthesis capacity in striatal regions.48,49 It should be noted that while studies examining D2 receptor availability are not entirely consistent, changes in presynaptic dopamine function are more reliable with a recent meta-analysis demonstrating an effect size of d=0.8.50
An association between aberrant dopamine neurotransmission is not only correlated with positive symptoms of the disease41,43 but has also been suggestive to be a contributing factor to cognitive deficits observed in patients.49,51 In contrast to that observed in the mesolimbic system, a decrease in dopamine transmission to the cortex may underlie deficits in working memory and executive function.52 Specifically, dopamine transmission in the dorsal lateral prefrontal cortex produces an inverted U-shaped dose response curve whereby both increases and decreases in dopamine signaling can impair working memory and executive function.53 Taken together, there is considerable evidence for aberrant dopamine signaling in individuals with schizophrenia that likely contributes to both positive and cognitive symptoms.40 However, it is important to note that there appears to be a lack of an overt pathology within the midbrain dopamine system.54 This has led to the suggestion that it is the regulation of dopamine neuron transmission that is dysfunctional in schizophrenia.55
The glutamate and GABA hypotheses
While considerable evidence indicates a dysregulation of dopamine signaling in schizophrenia, this is likely secondary to aberrant regulation by glutamatergic and GABAergic systems. The glutamate hypothesis of schizophrenia centers on hypofunctionality of glutamate signaling via N-methyl-D-aspartate (NMDA) receptors. Thus, NMDA receptor antagonists, such as phencyclidine (PCP) and ketamine, produce both psychotomimetic effects as well as cognitive impairments in humans.56,57 For this reason, considerable preclinical work has been performed following either acute or subchronic administration of NMDA receptor antagonists.58–61 Drugs like PCP inhibit signaling by the NMDA receptor and produce behavioral deficits in rodents that are analogous to positive (hyperlocomotion62) and negative (social interaction deficits63,64) symptoms as well as cognitive deficits (deficits in reversal learning and extradimensional set shifting65,66), which coincide with what is seen in the clinic. Additionally, perinatal PCP treatments have been used to model altered brain pathologies commonly observed in individuals with schizophrenia, such as altered expression of NMDA receptors and parvalbumin-containing interneurons.67 More recently, genetic studies have demonstrated the potential involvement of aberrant glutamatergic signaling in schizophrenia. Specifically, a transgenic mouse model with reduced NMDA receptor expression displays behaviors related to schizophrenia, such as increased locomotor activity and decreased social interaction, both of which were reversed by administration of the antipsychotic clozapine.68 Interestingly, it appears that the net effect of NMDA receptor antagonism is to augment glutamatergic transmission. While at first glance this may seem counterintuitive, it is thought that NMDA receptor antagonists preferentially alter GABAergic activity due to the more depolarized membrane potential and subsequent lack of Mg2+ block of the NMDA receptor.69,70 This would lead to increases in glutamatergic signaling. Indeed, magnetic resonance spectroscopy studies have demonstrated increased levels of glutamine in first-episode, untreated schizophrenia patients that provides in vivo evidence for enhanced glutamatergic activity.71
Deficits in GABAergic neurotransmission have been consistently demonstrated in post mortem studies of individuals with schizophrenia.72 These studies have shown alterations in GABAergic markers, including glutamic acid decarboxylase, parvalbumin (PV), somatostatin, and calretinin, to name a few.73–77 Various subtypes of interneurons exist that provide inhibitory control and modulation of cortical and hippocampal circuits.73 These interneuron subpopulations can be reliably distinguished by the neurochemicals they express. The cortex and hippocampus both contain interneurons that differentially express calcium binding proteins (PV, calretinin, calbindin) and neuropeptides (somatostatin, neuropeptide Y) with distinct cell types playing a vital role in inhibitory control over pyramidal cell activity in both regions.73,78–80 This is important, as it appears that the GABAergic dysfunction in schizophrenia is restricted to subpopulations of interneurons; ie, those containing PV or somatostatin.80–82 PV containing interneurons are fast firing, perisomatic targeting, and are thus situated to regulate pyramidal cell firing.73,77,83 In addition, these cells contribute to large scale neuronal oscillations within and between brain regions that are thought to be essential for cognitive function, and are known to be altered in individuals with schizophrenia.77,84,85
Information from genetics studies
Arguably, the most widely replicated biological factor in regard to schizophrenia is a family history of the disease.86 Over the years, researchers have studied the families of patients diagnosed with schizophrenia,87 along with fraternal and identical twin studies,88,89 in order to determine whether genetics plays a role in the disease. A quantitative meta-analysis of published twin studies of schizophrenia was conducted, and found that the heritability of the disease is very high (point estimate, 81%),90 consistent with prior summaries of literature on twins.91 It is important to note that no single gene has been identified as being solely responsible for the development of schizophrenia; rather, there are several candidate genes associated with an increased risk for the disease (ie, disrupted in schizophrenia 192–94 and neuregulin 195–97). Thus, genome-wide association studies (GWAS) have been instrumental in identifying candidate genes that may play a role in the heritability of schizophrenia. Specifically, GWAS examine the entire genome to detect common genetic variants in different individuals in order to implicate specific variants in disease risk. Because schizophrenia is a heterogeneous disease, rare variants with large effects have a very low frequency in the population as a whole, and are consequently not detectable by population-based GWAS.98 However, of relevance to the hypotheses detailed above, there have been reports implicating abnormalities in GABAergic and glutamatergic systems,99 as well as candidate genes involved in neurodevelopment (ie, EFHD1, RELN, ANK3, NRG1, etc,100–102). It should be noted, however, that individual results from GWAS have not identified overlapping polymorphisms nor do candidate genes consistently reach the level of genome-wide statistical significance,98,99,103 leaving many unanswered questions about the genetics of schizophrenia. This is not surprising, as it is commonly accepted that schizophrenia is associated with many genes that contribute modestly to the risk of developing the disease. However, when a combination of genetic alterations is placed in the context of environmental factors, they contribute to the pathophysiology of the disorder.
Towards an integrated hypothesis
Post mortem studies have identified structural alterations in human patients that are largely associated with cortical and hippocampal regions.104–106 These regions are also those that display consistent decreases in the expression of the interneuron marker PV,80,107 as well as deficits in activation during task performance, as measured by functional imaging studies.108 It is not currently understood how alterations within these specific neuronal systems contribute to the symptoms associated with schizophrenia. Thus, preclinical research is required that utilizes animal models that closely recapitulate several facets of the disease state. Animal models of schizophrenia can be generalized into three main categories: pharmacological, developmental, and genetic. The model which we typically utilize is a developmental disruption model that involves the administration of the mitotoxin methylazoxymethanol acetate (MAM) to pregnant dams on gestational Day 17.109 The offspring of these dams display anatomical, neurophysiological, and behavioral alterations consistent with those observed in individuals with schizophrenia.110 The exact mechanism as to how MAM produces a schizophrenia-like phenotype in rodents is not clear; however, this short-acting deoxyribonucleic acid (DNA)-alkylating agent is thought to alter cell division and gene expression (via DNA methylation).109,111
Given the significant evidence for mesolimbic dopamine hyperfunction in schizophrenia (see above), we have examined the regulation of dopamine neuron activity in the MAM model of schizophrenia.112–115 Using in vivo extracellular recordings from dopamine neurons in the ventral tegmental area (VTA), it has been previously demonstrated that distinct brain regions can differentially modulate discrete dopamine neuron activity states.116,117 Of relevance to schizophrenia are the ventral subfields of the hippocampus (vHipp) and the medial prefrontal cortex. Activation of the vHipp has been demonstrated to increase dopamine levels in the forebrain117 by increasing the number of spontaneously active dopamine neurons in the VTA (defined as population activity).118 In contrast, activation of the medial prefrontal cortex produces more selective alterations in dopamine neuron firing rate and pattern.116,119 Electrophysiological recordings in the MAM model of schizophrenia have demonstrated a pathological increase in dopamine neuron population activity without alterations in firing rate or pattern, thus implicating the vHipp.112–115 It is thought that alterations in dopamine neuron population activity provide a gain of function whereby phasic stimuli can be amplified or attenuated by hippocampal inputs in response to environmental context or novelty. A pathological increase in dopamine neuron population activity would therefore lead to an abnormally high salience being ascribed to previously innocuous stimuli and may contribute to the delusions and paranoia exhibited by individuals with schizophrenia. Indeed, recent evidence from human imaging studies supports the notion that aberrant hippocampal activity may underlie the dopamine dysregulation observed in individuals with schizophrenia.120 Specifically, elevated baseline activity is present in the anterior hippocampus of patients120,121 and in the analogous vHipp of rodent models.112 To demonstrate the involvement of the vHipp in this pathologically enhanced dopamine neuron activity in a rodent model, the vHipp was pharmacologically inactivated by the administration of the Na+-channel blocker, tetrodotoxin.112 While having no significant effect in control animals, tetrodotoxin inactivation of the vHipp completely reversed the elevated dopamine neuron population activity in MAM-treated rats.112 It is important to note that the effects of MAM on rodents are global and not simply restricted to the hippocampus. Therefore, it is likely that MAM produces alterations in afferents to the VTA and/or nucleus accumbens that also modulate dopamine activity and release in a freely behaving animal. Moreover, at face value, the hypothesis of hyperactivity within hippocampal subfields may appear inconsistent with one of the most well-known rodent models of schizophrenia, the neonatal ventral hippocampal lesion model.122 However, on the basis of work by Swerdlow et al, it has been suggested that behavioral abnormalities in the neonatal ventral hippocampal lesion model reflect, at least in part, aberrant function within spared elements of the hippocampal complex.123 Specifically, rats with large lesions encompassing the entire vHipp do not display the augmented responses to psychomotor stimulants consistently observed in rodent models and individuals with schizophrenia.123 These data suggest that the hypothesis of aberrant vHipp drive of the dopamine system may not be specific to the MAM model and may actually reflect a common pathophysiology in a number of diverse rodent models. Indeed, we have also demonstrated a similar observation in the subchronic phencyclidine rodent model (unpublished observations).
As mentioned above, evidence from postmortem studies suggests a decrease in PV interneuron function in individuals with schizophrenia.72,124 Given that these interneurons are well situated to regulate pyramidal cell activity,72 we posit that a decrease in PV interneuron function may actually underlie the hippocampal hyperactivity, and consequent dopamine neuron hyperfunction in schizophrenia. Indeed, a loss of PV function induced by degrading the extracellular matrix support structure of the PV interneurons was sufficient to increase hippocampal pyramidal neuron activity and produce enduring changes in dopamine system function.125 Similar results have been obtained by hippocampal selective knockdown of PV itself (unpublished observations). Taken together, we suggest that schizophrenia may be associated, at least in part, with a loss of interneuron control over hippocampal activity. This results in a pathologically enhanced hippocampal drive of the dopamine system that is likely associated with the positive symptoms of the disease (see Figure 1).
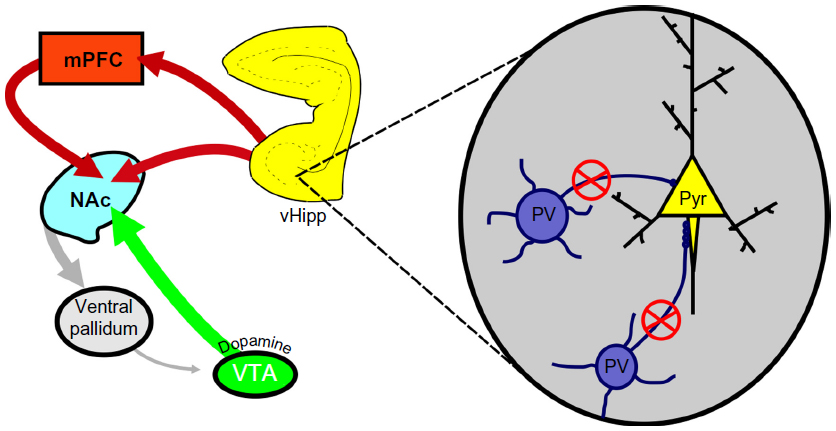 | Figure 1 PV-positive interneurons primarily synapse on the axon initial segment of pyramidal neurons of the vHipp (inset). |
The hippocampus as a potential therapeutic target
Given that a deficit in hippocampal GABAergic transmission may contribute to the pathophysiology of schizophrenia, it stands to reason that targeting this system may provide a novel approach for the treatment of schizophrenia. Indeed, we have evidence that pharmacological,126,127 neurosurgical,114 and cell-based therapies128 are effective in reversing aberrant neuron activity and behaviors in the MAM rodent model of schizophrenia.
There is considerable preclinical and clinical data demonstrating a robust augmentation of GABAergic transmission by allosteric modulators of the GABAA receptor. Indeed, the benzodiazepine and barbiturate classes of drug are widely utilized in this regard. Benzodiazepines have also been prescribed alone and in conjunction with antipsychotic medication for the treatment of schizophrenia, but the evidence as to its effectiveness as a treatment for schizophrenia is lacking and inconclusive to date,129 and may even be associated with an increased mortality.130 In addition, the extensive distribution of the GABAA receptor makes it difficult to target specific brain regions with conventional medications. The GABAA receptor is a pentameric structure consisting of a number of distinct subunits.131–133 Interestingly, the α5 subunit is preferentially expressed in the hippocampus134,135 leading to the suggestion that positive allosteric modulation of α5 subunit containing GABAA receptors may be effective in treating symptoms associated with schizophrenia. Indeed, the systemic administration of a selective allosteric modulator of the α5GABAA receptor, SH-053-2′F-R-CH3, attenuated hippocampal activity in the MAM-model of schizophrenia.126 Furthermore, this was associated with a restoration of aberrant dopamine neuron activity, as well as reversing the behavioral hyperactivity to amphetamine.126 These data provide preclinical evidence that allosteric modulators of the α5GABAA receptor may be an effective therapeutic approach to reverse dopamine hyperactivity by targeting aberrant hippocampal function.
In addition to the pharmacological targeting of hippocampal transmission, surgical approaches may also be effective at altering hippocampal activity. Deep brain stimulation (DBS), although still a very controversial treatment, has been gaining in popularity for the treatment of psychiatric disorders such as depression136 and obsessive compulsive disorder.137 It is thought that, in cortical regions, DBS acts to induce a functional lesion (likely by depolarization blockade of pyramidal neurons).138,139 For this reason, it was suggested that DBS of the vHipp may be a novel and effective therapeutic approach to treat the hippocampal hyperactivity thought to contribute to the symptoms of schizophrenia. Indeed, DBS of the anterior hippocampus has been demonstrated to be well tolerated in human patients (treated for epilepsy) with minimal side effects.140 We have recently demonstrated that that vHipp DBS in MAM-treated rats is able to reverse aberrant dopamine neuron activity and the behavioral hyper-responsivity to psychomotor stimulants, thought to be an index of positive symptomatology.114 Given that current approaches are often ineffective at reducing cognitive symptoms, we examined the effect of vHipp DBS in a working memory task analogous to the Cambridge Neuropsychological Test Automated Battery in patients.114 MAM-treated rats demonstrated deficits in reversal learning and extradimensional set shifting, consistent with deficits observed in patients.114,127 Interestingly, vHipp DBS was able to ameliorate these deficits in the MAM rodent model. These studies suggest that hippocampal DBS may represent a novel therapeutic approach, not only for the dopamine-dependent symptoms but also for the cognitive deficits.114 It should be noted that this is based on our recent preclinical study in rodents and that DBS treatments in humans are still experimental; while the hippocampus has been targeted in epilepsy, underlying differences in pathology suggest that effects in schizophrenia may be variable.
As mentioned earlier, a central hypothesis is that a decrease in PV expression underlies deficits in GABAergic signaling that contribute to the symptoms of the disease. Thus, restoring PV interneuron function would be expected to directly reverse the pathophysiology of the disorder. To test this hypothesis, we have used a cell-based therapy in an attempt to replace the deficient interneurons in the hippocampus.128 GABAergic precursor cells were obtained from the medial ganglionic eminence (MGE) and transplanted in the vHipp of MAM-treated rats. The transplanted cells were shown to display a GABAergic phenotype and migrate away from the site of injection to integrate within existing circuitry. In addition, MAM-treated rats transplanted with vHipp MGE neurons demonstrated a restoration of hippocampal function and consequent normalization of aberrant dopamine neuron activity.128 Finally, the increased sensitivity to amphetamine was also reversed following MGE transplants.128 Thus, interneuron transplants were effective in treating aberrant neurophysiology and behavior in the MAM model of schizophrenia and may provide the framework for a more permanent therapeutic approach for the treatment of schizophrenia.
Summary
For the last 50 years or so, the treatment of schizophrenia has relied on dopamine receptor antagonists that are not always effective and are often associated with debilitating side effects. For this reason, it is essential to develop novel therapeutic approaches. To do this, one must better understand the underlying pathophysiology of this disease. Here we report on an integrated hypothesis of schizophrenia that, while it may not be all encompassing, provides a potential novel site of intervention. Specifically, it is suggested that the aberrant dopamine system function, thought to underlie the positive symptoms of the disease, is secondary to hyperactivity within hippocampal subfields. Furthermore, this hyperactivity is likely attributable to a decrease in intrinsic GABAergic signaling by PV interneurons within the hippocampus. Therefore, approaches aimed at restoring hippocampal function may provide a novel approach for the treatment of schizophrenia. Indeed, here we provide preclinical data demonstrating the effectiveness of three distinct experimental approaches (pharmacological, neurosurgical, and cell-based) in a rodent model of schizophrenia. Future experiments in additional rodent and primate models are warranted before such novel approaches are translated to the clinic.
Disclosure
The authors report no conflicts of interest in this work.
References
McGrath J, Saha S, Welham J, El Saadi O, MacCauley C, Chant D. A systematic review of the incidence of schizophrenia: the distribution of rates and the influence of sex, urbanicity, migrant status and methodology. BMC Med. 2004;2:13. | |
Stilo SA, Murray RM. The epidemiology of schizophrenia: replacing dogma with knowledge. Dialogues Clin Neurosci. 2010;12:305–315. | |
DeLisi LE. The significance of age of onset for schizophrenia. Schizophren Bull. 1992;18:209–215. | |
Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–1223. | |
Carpenter WT Jr, Buchanan RW. Schizophrenia. N Engl J Med. 1994;330(10):681–690. | |
Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol. 1963;20:140–144. | |
Andén NE, Butcher SG, Corrodi H, Fuxe K, Ungerstedt U. Receptor activity and turnover of dopamine and noradrenaline after neuroleptics. Eur J Pharmacol. 1970;11(3):303–314. | |
Stone JM, Raffin M, Morrison P, McGuire PK. Review: The biological basis of antipsychotic response in schizophrenia. J Psychopharmacol. 2010;24(7):953–964. | |
Conley RR, Buchanan RW. Evaluation of treatment-resistant schizophrenia. Schizophr Bull. 1997;23(4):663–674. | |
Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, et al. World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13:318–378. | |
Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;149(1):2–44. | |
Javitt DC. Management of negative symptoms of schizophrenia. Curr Psychiatry Rep. 2001;3(5):413–417. | |
Tamminga CA, Buchanan RW, Gold JM. The role of negative symptoms and cognitive dysfunction in schizophrenia outcome. Int Clin Psychopharmacol. 1998;13 Suppl 3:S21–S26. | |
Davé M. Clozapine-related tardive dyskinesia. Biol Psychiatry. 1994;35(11):886–887. | |
Ertugrul A, Demir B. Clozapine-induced tardive dyskinesia: a case report. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(4):633–635. | |
López-Muñoz F, Alamo C, Cuenca E, Shen WW, Clervoy P, Rubio G. History of the discovery and clinical introduction of chlorpromazine. Ann Clin Psychiatry. 2005;17(3):113–135. | |
Altschule MD. Use of chlorpromazine and reserpine in mental disorders. N Engl J Med. 1956;254(11):515–519. | |
Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192(4238):481–483. | |
Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science. 1975;188(4194):1217–1219. | |
Miyamoto S, Duncan GE, Marx CE, Lieberman JA. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry. 2005:10(1):79–104. | |
Stevens JR. An anatomy of schizophrenia? Arch Gen Psychiatry. 1973;29(2):177–189. | |
Meltzer HY, Simonovic M, Gudelsky GA. Phencyclidine-induced inhibition of rat prolactin secretion: increased portal blood dopamine. Eur J Pharmacol. 1985;110(1):143–146. | |
Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789–796. | |
Meltzer HY. Clinical studies on the mechanism of action of clozapine: the dopamine-serotonin hypothesis of schizophrenia. Psychopharmacology (Berl). 1989;99 Suppl:S18–S27. | |
Abi-Dargham A, Laruelle M. Mechanisms of action of second generation antipsychotic drugs in schizophrenia: insights from brain imaging studies. Eur Psychiatry. 2005;20(1):15–27. | |
Frankle WG, Gil R, Hackett E, et al. Occupancy of dopamine D2 receptors by the atypical antipsychotic drugs risperidone and olanzapine: theoretical implications. Psychopharmacology (Berl). 2004;175(4):473–480. | |
Bressan RA, Erlandsson K, Jones HM, et al. Is regionally selective D2/D3 dopamine occupancy sufficient for atypical antipsychotic effect? an in vivo quantitative [123I]epidepride SPET study of amisulpride-treated patients. Am J Psychiatry. 2003;160(8):1413–1420. | |
Kapur S, Remington G. Dopamine D(2) receptors and their role in atypical antipsychotic action: still necessary and may even be sufficient. Biol Psychiatry. 2001;50(11):873–883. | |
Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47(1):27–38. | |
Meltzer HY, McGurk SR. The effects of clozapine, risperidone, and olanzapine on cognitive function in schizophrenia. Schizophren Bull. 1999;25(5):233–255. | |
Hasan A, Falkai P, Wobrock T, et al. World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13:318–378. | |
Hasan A, Falkai P, Wobrock T, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;149(1):2–44. | |
Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997;58(12):538–546. | |
Tollefson GD, Sanger TM. Negative symptoms: a path analytic approach to a double-blind, placebo- and haloperidol-controlled clinical trial with olanzapine. Am J Psychiatry. 1997;154(4):466–474. | |
Keefe RS, Silva SG, Perkins DO, Lieberman JA. The effects of atypical antipsychotic drugs on neurocognitive impairment in schizophrenia: a review and meta-analysis. Schizophr Bull. 1999;25(2):201–222. | |
Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs. 2005;19 Suppl 1:1–93. | |
Jones PB, Barnes TR, Davies L, et al. Randomized controlled trial of the effect on Quality of Life of second- vs first-generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1). Arch Gen Psychiatry. 2006;63(10):1079–1087. | |
Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951–962. | |
Leucht S, Kissling W, Davis JM. Second-generation antipsychotics for schizophrenia: can we resolve the conflict?Psychol Med. 2009;39(10):1591–1602. | |
Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III – the final common pathway. Schizophr Bull. 2009;35(3):549–562. | |
Laruelle M, Abi-Dargham A. Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. J Psychopharmacol. 1999;13(4):358–371. | |
Carlsson A, Waters N, Waters S, Carlsson ML. Network interactions in schizophrenia – therapeutic implications. Brain Res Brain Res Rev. 2000;31(2–3):342–349. | |
Abi-Dargham A. Do we still believe in the dopamine hypothesis? New data bring new evidence. Int J Neuropsychopharmacol. 2004;7 Suppl 1:S1–S5. | |
Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology (Berl). 1987;91(4):415–433. | |
Kegeles LS, Abi-Dargham A, Frankle WG, et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67(3):231–239. | |
Laruelle M, D’Souza CD, Baldwin RM, et al. Imaging D2 receptor occupancy by endogenous dopamine in humans. Neuropsychopharmacology. 1997;17(3):162–174. | |
Fujita M, Verhoeff NP, Varrone A, et al. Imaging extrastriatal dopamine D(2) receptor occupancy by endogenous dopamine in healthy humans. Eur J Pharmacol. 2000;387(2):179–188. | |
Egerton A, Chaddock CA, Winton-Brown TT, et al. Presynaptic striatal dopamine dysfunction in people at ultra-high risk for psychosis: findings in a second cohort. Biol Psychiatry. 2013;74(2):106–112. | |
McGowan S, Lawrence AD, Sales T, Quested D, Grasby P. Presynaptic dopaminergic dysfunction in schizophrenia: a positron emission tomographic [18F]fluorodopa study. Arch Gen Psychiatry. 2004;61(2):134–142. | |
Howes OD, Kambeitz J, Kim E, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry. 2012;69(8):776–786. | |
Meyer-Lindenberg A, Miletich RS, Kohn PD, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci. 2002;5(3):267–271. | |
Collins P, Roberts AC, Dias R, Everitt BJ, Robbins TW. Perseveration and strategy in a novel spatial self-ordered sequencing task for nonhuman primates: effects of excitotoxic lesions and dopamine depletions of the prefrontal cortex. J Cogn Neurosci. 1998;10(3):332–354. | |
Williams JM, Galli A. The dopamine transporter: a vigilant border control for psychostimulant action. Handb Exp Pharmacol. 2006215–06232. | |
Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122(Pt 4):593–624. | |
Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci. 2011;32(9):507–513. | |
Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199–214. | |
Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20(3):201–225. | |
Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301–1308. | |
Jenkins TA, Harte MK, Reynolds GP. Effect of subchronic phencyclidine administration on sucrose preference and hippocampal parvalbumin immunoreactivity in the rat. Neurosci Lett. 2010;471(3):144–147. | |
Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349–1352. | |
Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17(8):2921–2927. | |
Sturgeon RD, Fessler RG, Meltzer HY. Behavioral rating scales for assessing phencyclidine-induced locomotor activity, stereotyped behavior and ataxia in rats. Eur J Pharmacol. 1979;59(3–4):169–179. | |
Sams-Dodd F. Distinct effects of d-amphetamine and phencyclidine on the social behaviour of rats. Behav Pharmacol. 1995;6(1):55–65. | |
Sams-Dodd F. Phencyclidine-induced stereotyped behaviour and social isolation in rats: a possible animal model of schizophrenia. Behav Pharmacol. 1996;7(1):3–23. | |
Noda A, Noda Y, Kamei H, et al. Phencyclidine impairs latent learning in mice: interaction between glutamatergic systems and sigma(1) receptors. Neuropsychopharmacology. 2001;24(4):451–460. | |
Egerton A, Brett RR, Pratt JA. Acute delta9-tetrahydrocannabinol-induced deficits in reversal learning: neural correlates of affective inflexibility. Neuropsychopharmacology. 2005;30(10):1895–1905. | |
Wang CZ, Yang SF, Xia Y, Johnson KM. Postnatal phencyclidine administration selectively reduces adult cortical parvalbumin-containing interneurons. Neuropsychopharmacology. 2008;33(10):2442–2455. | |
Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98(4):427–436. | |
Csicsvari J, Hirase H, Czurko A, Buzsáki G. Reliability and state dependence of pyramidal cell-interneuron synapses in the hippocampus: an ensemble approach in the behaving rat. Neuron. 1998;21(1):179–189. | |
Maccaferri G, Dingledine R. Control of feedforward dendritic inhibition by NMDA receptor-dependent spike timing in hippocampal interneurons. J Neurosci. 2002;22(13):5462–5472. | |
Théberge J, Bartha R, Drost DJ, et al. Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry. 2002;159(11):1944–1946. | |
Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. | |
Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25(1):1–27. | |
Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35(1):57–67. | |
Curley AA, Lewis DA. Cortical basket cell dysfunction in schizophrenia. J Physiol. 2012;590(Pt 4):715–724. | |
Curley AA, Eggan SM, Lazarus MS, Huang ZJ, Volk DW, Lewis DA. Role of glutamic acid decarboxylase 67 in regulating cortical parvalbumin and GABA membrane transporter 1 expression: implications for schizophrenia. Neurobiol Dis. 2013;50:179–186. | |
Gonzalez-Burgos G, Lewis DA. NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull. 2012;38(5):950–957. | |
Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry. 2002;52(7):708–715. | |
Beneyto M, Morris HM, Rovensky KC, Lewis DA. Lamina- and cell-specific alterations in cortical somatostatin receptor 2 mRNA expression in schizophrenia. Neuropharmacology. 2012;62(3):1598–1605. | |
Hashimoto T, Volk DW, Eggan SM, et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23(15):6315–6326. | |
Woo TU, Whitehead RE, Melchitzky DS, Lewis DA. A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci U S A. 1998;95(9):5341–5346. | |
Pierri JN, Chaudry AS, Woo TU, Lewis DA. Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects. Am J Psychiatry. 1999;156(11):1709–1719. | |
Kawaguchi Y, Hama K. Fast-spiking non-pyramidal cells in the hippocampal CA3 region, dentate gyrus and subiculum of rats. Brain Res. 1987;425(2):351–355. | |
Gonzalez-Burgos G, Lewis DA. GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr Bull. 2008;34(5):944–961. | |
Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci. 2009;29(8):2344–2354. | |
Tsuang MT, Gilbertson MW, Faraone SV. The genetics of schizophrenia. Current knowledge and future directions. Schizophr Res. 1991;4(2):157–171. | |
Tsuang MT, Stone WS, Faraone SV. Schizophrenia: family studies and treatment of spectrum disorders. Dialogues in Clinical Neuroscience. 2000;2(4):381–391. | |
Cardno AG, Jones LA, Murphy KC, et al. Dimensions of psychosis in affected sibling pairs. Schizophr Bull. 1999;25(4):841–850. | |
Kendler KS. Twin studies of psychiatric illness. Current status and future directions. Arch Gen Psychiatry. 1993;50(11):905–915. | |
Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60(12):1187–1192. | |
McGuffin P, Owen MJ, Farmer AE. Genetic basis of schizophrenia. Lancet. 1995;346(8976):678–682. | |
Millar JK, Wilson-Annan JC, Anderson S, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9(9):1415–1423. | |
Debono R, Topless R, Markie D, Black MA, Merriman TR. Analysis of the DISC1 translocation partner (11q14.3) in genetic risk of schizophrenia. Genes Brain Behav. 2012;11(7):859–863. | |
Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10(1):40–68. | |
Craddock N, O’Donovan MC, Owen MJ. The genetics of schizophrenia and bipolar disorder: dissecting psychosis. J Med Genet. 2005;42(3):193–204. | |
Harrison PJ, Owen MJ. Genes for schizophrenia? Recent findings and their pathophysiological implications. Lancet. 2003;361(9355):417–419. | |
Stefansson H, Sigurdsson E, Steinthorsdottir V, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71(4):877–892. | |
Hosak L, Silhan P, Hosakova J. Genomic copy number variations: A breakthrough in our knowledge on schizophrenia etiology? Neuro Endocrinol Lett. 2012;33(2):183–190. | |
Jia P, Wang L, Meltzer HY, Zhao Z. Common variants conferring risk of schizophrenia: a pathway analysis of GWAS data. Schizophr Res. 2010;122(1–3):38–42. | |
Alkelai A, Lupoli S, Greenbaum L, et al. Identification of new schizophrenia susceptibility loci in an ethnically homogeneous, family-based, Arab-Israeli sample. FASEB J. 2011;25(11):4011–4023. | |
Alkelai A, Lupoli S, Greenbaum L, et al. DOCK4 and CEACAM21 as novel schizophrenia candidate genes in the Jewish population. Int J Neuropsychopharmacol. 2012;15(4):459–469. | |
Athanasiu L, Mattingsdal M, Kähler AK, et al. Gene variants associated with schizophrenia in a Norwegian genome-wide study are replicated in a large European cohort. J Psychiatr Res. 2010;44(12):748–753. | |
Gershon ES, Alliey-Rodriguez N, Liu C. After GWAS: searching for genetic risk for schizophrenia and bipolar disorder. Am J Psychiatry. 2011;168(3):253–256. | |
Brown R, Colter N, Corsellis JA, et al. Postmortem evidence of structural brain changes in schizophrenia. Differences in brain weight, temporal horn area, and parahippocampal gyrus compared with affective disorder. Arch Gen Psychiatry. 1986;43(1):36–42. | |
Bogerts B, Falkai P, Greve B, Schneider T, Pfeiffer U. The neuropathology of schizophrenia: past and present. J Hirnforsch. 1993;34(2):193–205. | |
Bogerts B, Lieberman JA, Ashtari M, et al. Hippocampus-amygdala volumes and psychopathology in chronic schizophrenia. Biol Psychiatry. 1993;33(4):236–246. | |
Gonzalez-Burgos G, Hashimoto T, Lewis DA. Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr Psychiatry Rep. 2010;12(4):335–344. | |
Hill K, Mann L, Laws KR, Stephenson CM, Nimmo-Smith I, McKenna PJ. Hypofrontality in schizophrenia: a meta-analysis of functional imaging studies. Acta Psychiatr Scand. 2004;110(4):243–256. | |
Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry. 2006;60(3):253–264. | |
Lodge DJ, Grace AA. Gestational methylazoxymethanol acetate administration: a developmental disruption model of schizophrenia. Behav Brain Res. 2009;204(2):306–312. | |
Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14(1):83–144. | |
Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27(42):11424–11430. | |
Perez SM, Lodge DJ. Aberrant dopamine D2-like receptor function in a rodent model of schizophrenia. J Pharmacol Exp Ther. 2012;343(2):288–295. | |
Perez SM, Shah A, Asher A, Lodge DJ. Hippocampal deep brain stimulation reverses physiological and behavioural deficits in a rodent model of schizophrenia. Int J Neuropsychopharmacol. 2013;16(6):1331–1339. | |
Perez SM, Lodge DJ. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol Psychiatry. 2013;18(11):1193–1198. | |
Floresco SB, Todd CL, Grace AA. Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J Neurosci. 2001;21(13):4915–4922. | |
Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6(9):968–973. | |
Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons – 1. Identification and characterization. Neuroscience. 1983;10(2):301–315. | |
Lodge DJ, Grace AA. The hippocampus modulates dopamine neuron responsivity by regulating the intensity of phasic neuron activation. Neuropsychopharmacology. 2006;31(7):1356–1361. | |
Schobel SA, Lewandowski NM, Corcoran CM, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66(9):938–946. | |
Heckers S, Rauch SL, Goff D, et al. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nat Neurosci. 1998;1(4):318–323. | |
Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology. 1993;9(1):67–75. | |
Swerdlow NR, Halim N, Hanlon FM, Platten A, Auerbach PP. Lesion size and amphetamine hyperlocomotion after neonatal ventral hippocampal lesions: more is less. Brain Res Bull. 2001;55(1):71–77. | |
Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res. 2002;55(1–2):1–10. | |
Shah A, Lodge DJ. A loss of hippocampal perineuronal nets produces deficits in dopamine system function: relevance to the positive symptoms of schizophrenia. Transl Psychiatry. 2013;3:e215. | |
Gill KM, Lodge DJ, Cook JM, Aras S, Grace AA. A novel α5GABA(A)R-positive allosteric modulator reverses hyperactivation of the dopamine system in the MAM model of schizophrenia. Neuropsychopharmacology. 2011;36(9):1903–1911. | |
Gastambide F, Cotel MC, Gilmour G, O’Neill MJ, Robbins TW, Tricklebank MD. Selective remediation of reversal learning deficits in the neurodevelopmental MAM model of schizophrenia by a novel mGlu5 positive allosteric modulator. Neuropsychopharmacology. 2012;37(4):1057–1066. | |
Perez SM, Lodge DJ. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol Psychiatry. 2013;18(11):1193–1198. | |
Volz A, Khorsand V, Gillies D, Leucht S. Benzodiazepines for schizophrenia. Cochrane Database Syst Rev. 2007:CD006391. | |
Tiihonen J, Suokas JT, Suvisaari JM, Haukka J, Korhonen P. Polypharmacy with antipsychotics, antidepressants, or benzodiazepines and mortality in schizophrenia. Arch Gen Psychiatry. 2012;69(5):476–483. | |
McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19(4):139–143. | |
Quirk K, Blurton P, Fletcher S, et al. [3H]L-655,708, a novel ligand selective for the benzodiazepine site of GABAA receptors which contain the alpha 5 subunit. Neuropharmacology. 1996;35(9–10):1331–1335. | |
Pritchett DB, Sontheimer H, Shivers BD, et al. Importance of a novel GABAA receptor subunit for benzodiazepine pharmacology. Nature. 1989;338(6216):582–585. | |
Heldt SA, Ressler KJ. Forebrain and midbrain distribution of major benzodiazepine-sensitive GABAA receptor subunits in the adult C57 mouse as assessed with in situ hybridization. Neuroscience. 2007;150(2):370–385. | |
Ramos B, Lopez-Tellez JF, Vela J, et al. Expression of alpha 5 GABAA receptor subunit in developing rat hippocampus. Brain Res Dev Brain Res. 2004;151(1–2):87–98. | |
Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651–660. | |
Greenberg BD, Pinto A, Mancebo M, Eisen J, Rasmussen SA. Obsessive-compulsive disorder: recognition across medical settings, and treatments from behavior therapy to neurosurgery. Med Health R I. 2006;89(5):162–165. | |
Benabid AL, Pollak P, Gervason C, et al. Long-term suppression of tremor by chronic stimulation of the ventral intermediate thalamic nucleus. Lancet. 1991;337(8738):403–406. | |
Wyckhuys T, De Smedt T, Claeys P, et al. High frequency deep brain stimulation in the hippocampus modifies seizure characteristics in kindled rats. Epilepsia. 2007;48(8):1543–1550. | |
Boon P, Vonck K, De Herdt V, et al. Deep brain stimulation in patients with refractory temporal lobe epilepsy. Epilepsia. 2007;48(8):1551–1560. | |
Grace AA, Floresco SB, Goto Y, Lodge DJ. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 2007;30(5):220–227. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.