Back to Journals » Journal of Inflammation Research » Volume 18
Neutrophils Extracellular Traps Impair Lung Endothelial Proliferation in Sepsis via PLK1 Inhibition and Cell Cycle Arrest
Authors Zhu C, Qu M, Wu D, Shi Y, Zeng F, Miao C, Zhou D ![]()
Received 2 August 2025
Accepted for publication 16 December 2025
Published 27 December 2025 Volume 2025:18 Pages 18165—18180
DOI https://doi.org/10.2147/JIR.S556891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Anh Ngo
Chenyu Zhu,1– 3,* Mengdi Qu,1– 3,* Dan Wu,1– 3 Yuxin Shi,1– 3 Fu Zeng,1– 3 Changhong Miao,1– 3 Di Zhou1– 3
1Department of Anesthesiology, Zhongshan Hospital, Fudan University, Shanghai, People’s Republic of China; 2Shanghai Key Laboratory of Perioperative Stress and Protection, Zhongshan Hospital, Fudan University, Shanghai, People’s Republic of China; 3Department of Anesthesiology, Shanghai Medical College, Fudan University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Di Zhou, Department of Anesthesiology, Zhongshan Hospital, Fudan University, 180# Feng-Lin Road, Shanghai, 200032, People’s Republic of China, Email [email protected] Changhong Miao, Department of Anesthesiology, Zhongshan Hospital, Fudan University, 180# Feng-Lin Road, Shanghai, 200032, People’s Republic of China, Email [email protected]
Purpose: Sepsis continues to pose a significant threat to global health, characterized by elevated mortality rates. Pulmonary complications frequently develop in septic patients, with endothelial dysfunction correlating with adverse clinical outcomes. While overproduction of neutrophil extracellular traps (NETs) is implicated in vascular damage, their specific influence on the regenerative potential of pulmonary endothelial cells requires further elucidation. Our investigation aims to address this critical knowledge gap.
Patients and Methods: Clinical samples from sepsis patients and healthy controls were analyzed to establish the correlation between NETs and pulmonary endothelial injury. An in vivo sepsis model was generated through cecal ligation and puncture (CLP) in mice, with sham surgery animals serving as reference group. Human umbilical vein endothelial cells (HUVECs) were employed for in vitro assessment of NETs-mediated cell cycle modulation.
Results: Elevated NETs formation was observed in septic patients, showing positive association with inflammatory damage. CLP-induced mice demonstrated substantially increased NETs levels, pronounced pulmonary vascular permeability, and notable endothelial cell depletion. DNase I-mediated NETs degradation alleviated pulmonary inflammation and promoted endothelial recovery. Both experimental models revealed that excessive NETs release during sepsis compromises endothelial proliferation via polo-like kinase 1 (PLK1) pathway inhibition and subsequent G2/M phase arrest.
Conclusion: This study establishes that NETs accumulation in septic pulmonary injury hinders endothelial regeneration and vascular repair through PLK1 signaling suppression and G2/M cell cycle blockade.
Keywords: sepsis, NETs, endothelial dysfunction, polo like kinase 1, cell cycle
Introduction
Sepsis is a critical disorder presented as organ failure resulting from an excessive host immune reaction to pathogen.1 This syndrome continues to dominate as a primary reason for intensive care unit admissions worldwide. Epidemiological data from 2017 indicated approximately 48.9 million sepsis cases globally, with associated mortality reaching 11 million deaths - constituting nearly one-fifth (19.7%) of total global fatalities.2 Research has consistently shown Chinese demographic groups exhibit markedly elevated vulnerability compared to Western populations.2,3 Contemporary medical advances notwithstanding, treatment options remain predominantly supportive rather than targeted.4
In the pathophysiological cascade of sepsis, neutrophil granulocytes serve as frontline cellular defenders of innate immunity. Beyond traditional antimicrobial functions, activated neutrophils deploy neutrophil extracellular traps(NETs) containing dsDNA, MPO and NE proteins. While these webs initially contain microbial spread in mild infections, their overproduction during severe sepsis aggravates tissue destruction and disease severity.5,6 Sepsis-related acute pulmonary (SI-ALI) demonstrates mortality rates exceeding conventional lung injuries by 30%-40%.7,8 Emerging research implicates NETs in potentiating sepsis-induced lung pathology through diverse mechanisms including iron-dependent cell death, autophagic dysregulation, and immunocoagulation.9–14 The thromboinflammatory potential of NETs was particularly evident in critical COVID-19 cases through complement system interactions.15
Pulmonary endothelial cells form a semipermeable barrier in blood vessels, regulating fluid/protein exchange, maintaining antithrombotic and anti-inflammatory microvascular homeostasis, and ensuring normal pulmonary function.16 Sepsis-induced endothelial injury triggers interstitial and alveolar edema, leading to dyspnea, hypoxemia, and life-threatening respiratory failure requiring prolonged mechanical ventilation.17
Healthy endothelial cells exhibit robust regenerative capacity to restore function post-injury.18 Endothelial damage in sepsis involves both direct injury and impaired regenerative potential. Persistent endothelial regeneration dysfunction in sepsis-induced lung injury perpetuates vascular leakage, which results in poor clinical outcomes. We posit that enhancing endothelial proliferation potential rather than merely mitigating initial damage represents a critical therapeutic target for improving long-term prognosis. However, current research predominantly focuses on NETs-induced direct endothelial injury, while mechanisms underlying NETs-induced impaired endothelial regenerative potential remain underexplored.
In summary, this study investigates the impact of NETs on pulmonary endothelial cell proliferation during sepsis and elucidates the pathogenesis. We demonstrate that NETs inhibit endothelial proliferation through PLK1 downregulation and G2/M phase cell cycle arrest, as validated by in vivo and in vitro models. These findings advance the theoretical framework of sepsis pathogenesis and provide novel insights for therapeutic targets to enhance endothelial proliferation, thereby improving clinical outcomes in sepsis-induced lung injury.
Materials and Methods
Clinical Participants
The research protocol received ethical clearance from Zhongshan Hospital’s Institutional Review Board (number: B2023-409), with written consent acquired from all subjects. Seventeen ICU-admitted sepsis patients meeting Sepsis-3.0 criteria (documented infection plus SOFA score ≥2) were recruited between October and December 2023. An equal number of healthy individuals served as the reference cohort. Exclusion parameters comprised: age below 18 years, pregnancy status, and immunocompromised conditions. Venous blood samples were procured within 120 minutes post-diagnosis, with serum preserved at −80°C until analysis. Clinical parameters were retrospectively collected from medical records systems.
CLP Model
8–10 weeks male C57BL/6J mice (weight 20±2g, specific pathogen-free) were sourced from Shanghai Jihui Laboratory Animal Center and maintained in the Fudan University Animal Facility. The experimental design was authorized by the Institutional Animal Care and Use Committee at Fudan University (number: Y2022-340). All studies comply with the National Standards of the People’s Republic of China, Laboratory animal—Guideline for ethical review of animal welfare (GB/T 35892–2018). All rodents were acclimatized for seven days prior to experimentation under controlled environmental conditions (temperature 22±1°C, humidity 55±5%, 12-hour light/dark cycle) with ad libitum access to standard chow and sterile water.
CLP Mice Model and Treatments
The study employed three experimental cohorts: Sham-operated controls, CLP-induced sepsis models, and CLP subjects pretreated with DNaseI. Anesthetic administration involved injecting 1.25% tribromoethanol intraperitoneally (20 μL/g body weight). In the CLP cohort, surgical intervention comprised cecal ligation followed by perforation with a needle, allowing fecal leakage onto the intestinal surface prior to organ repositioning and abdominal closure. Postoperative resuscitation included 500 μL saline delivered via intraperitoneal injection. Sham controls received identical cecal exteriorization without ligation or perforation. DNaseI-pretreated animals were administered 5 mg/kg DNaseI (10104159001, Roche, Basel, Switzerland) intraperitoneally 3 hours pre-surgery to facilitate NET degradation. Terminal sample collection encompassed blood and pulmonary tissue procurement for downstream assays.
Neutrophil Separation and NETs Production
Human neutrophils were purified using a specialized isolation kit (LZS11131, TBD Sciences). In this procedure, peripheral blood was carefully overlaid onto a density gradient medium, followed by centrifugation at 3500 rpm for 30 min. The pellet containing neutrophils was harvested, subjected to erythrocyte lysis, and finally suspended in DMEM (BasalMedia, Shanghai, China) supplemented with 1% FBS (Sigma, St. Louis, MO, USA). Purified cells (1×106 cells/mL) were plated in 6-well dishes and activated with 125 nM PMA (MZ2401, MKbio) for 4 h. After removal of supernatants, NETs attached to the plates were gently rinsed, pelleted via centrifugation, and resuspended in 10% DMEM for further analysis.
Serum Components Quantification by ELISA
Concentrations of human IL-6 (EK106, MULTI SCIENCES, Hangzhou, China), IL-8 (CSB-E04641h, Cusabio, Wuhan, China), and MPO-DNA (ml085122-J, Mlbio Biology, Shanghai, China), as well as murine IL-6 (EK206, MULTI SCIENCES), TNF-α (EK282, MULTI SCIENCES), and MPO-DNA (ml332562, Mlbio Biology, Shanghai, China), were assessed using standardized ELISA kits. All assays were conducted in strict adherence to the providers’ guidelines.
dsDNA Quantification via PicoGreen Assay
Double-stranded DNA levels in murine blood samples were determined with the Quant-iTTM PicoGreen® dsDNA Reagent and Kits (Invitrogen), following the prescribed experimental protocol.
Tissue Processing and Histological Evaluation
Lungs were preserved in 4% PFA solution, subsequently embedded in paraffin blocks, and sliced into thin sections. Following xylene-based deparaffinization and gradual ethanol rehydration, tissue sections were subjected to hematoxylin and eosin (H&E) staining. Pulmonary damage was evaluated using a standardized grading system accounting for four pathological features: alveolar edema, hemorrhagic areas, leukocyte accumulation, and alveolar septal thickening (0 = normal; 1 = mild; 2 = moderate; 3 = severe). Cumulative scores from these parameters indicated overall lung injury severity.
Immunohistochemical Staining
Lung sections were pretreated with citrate buffer for antigen exposure and were blocked with 1% bovine serum albumin (WB6504, NCM Biotech, Shanghai, China) for 60 minutes at ambient temperature. Anti-CD31 antibodies (1:2000, ab182981, Abcam, MA, USA) were applied and maintained at 4°C overnight. After thorough washing, appropriate secondary antibodies were introduced and incubated at 37°C for 60 minutes prior to DAB development and hematoxylin nuclear staining. Mounted slides were examined microscopically, with CD31 expression quantified using ImageJ software.
Assessment of Vascular Permeability Using Evans Blue
Following CLP procedures, experimental mice were administered 20 mg/kg of 1% Evans blue dye (Sangon Biotech, Shanghai, China) through tail vein injection. After 120 minutes of systemic circulation, intravascular dye was eliminated by right ventricular perfusion with 50 mL ice-cold saline. Excised lung tissues were desiccated and weighed, then immersed in formamide (Sangon Biotech) at 60°C for 48 hours to extract Evans blue. Following centrifugation at 5000× g for 30 minutes, supernatant optical density was measured at 620 nm.
Endothelial Cell Culture and Experimental Protocols
HUVECs obtained from ATCC were maintained at 37°C with 5% CO2 in complete growth medium consisting of DMEM with 10% FBS, 1% penicillin/streptomycin, and 0.1% mycoplasma removal reagent. For experimental interventions, cells were exposed to medium containing neutrophil extracellular traps (NETs). Prior to cellular stimulation, NETs-containing medium was enzymatically digested using DNase I (EN0521, Thermo, Waltham, MA, USA) for 4 hours. PLK1 overexpression constructs and corresponding control lentiviruses were acquired from Shanghai Genechem Co., Ltd. (China), with transfection procedures executed per the supplier’s guidelines. Successful transfection was confirmed through quantitative PCR and immunoblotting analysis before proceeding with subsequent assays.
Assessment of Cellular Viability
The viability of HUVECs were assessed using a CCK-8 kit (C0037, Beyotime, Shanghai, China), with all experimental steps performed in strict compliance with the provided technical manual.
Evaluation of Proliferative Capacity
Cell proliferation rates were quantified via 5-ethynyl-2’-deoxyuridine (EdU) incorporation assay (C0071, Beyotime, Shanghai, China). Following a 2-hour incubation with 20 μM EdU labeling solution at 37°C, cells were fixed with 4% PFA for 15 minutes and permeabilized using 0.3% Triton X-100. The click reaction mixture was prepared according to the manufacturer’s specifications and allowed to react with samples under light-protected conditions for 30 minutes. After nuclear counterstaining with Hoechst 33342, fluorescence microscopy was employed for image acquisition. EdU-positive cells were enumerated under microscopic examination to determine proliferative capacity.
Cell Cycle Phase Determination
Cellular DNA content analysis was conducted with PI/RNase Staining buffer (550825, BD Biosciences). Following PBS washing, cells were preserved in 75% ethanol at 4°C for 12 hours. After removal of fixative, samples were treated with 500 μL staining solution and incubated at 37°C for 45 minutes. Cell cycle phase distribution (G1, S, G2) was quantified using a FACSCanto II flow cytometer (BD Biosciences).
Immunofluorescence
For NETs visualization, human neutrophils were seeded in 24-well plates. Following fixation with 4% paraformaldehyde, cells were permeabilized with 0.1% Triton X-100 for 20 minutes at room temperature and subsequently blocked for 1 hour. Primary antibodies against myeloperoxidase (1:100, ab90810, Abcam) and histone H3 (1:100, ab5103, Abcam) were applied for staining. Goat anti-mouse Cy3-conjugated secondary antibody (1:100, AS008, Abclonal, Wuhan, China) and goat anti-rabbit ABflo® 488 secondary antibody (1:250, AS053, ABclonal) were introduced and maintained at 37°C for 60 minutes for detection. Nuclear counterstaining was performed with DAPI.
For PLK1 detection, cells were cultured on sterile slides prior to processing. Fixation was achieved with 4% paraformaldehyde (20 min), followed by membrane permeabilization using 0.5% Triton X-100 (20 min) and blocking with 1% BSA (60 min). Primary anti-PLK1 antibody (1:250, 37–7100, Invitrogen, Carlsbad, CA, USA) was applied overnight at 4°C. After washing, goat anti-mouse Cy3-conjugated secondary antibody (1:100, AS008, Abcolonol) was introduced and maintained at 37°C for 60 minutes in darkness. Nuclear counterstaining was performed with DAPI prior to microscopic examination. Fluorescence intensity was quantified using ImageJ software.
Gene Expression Evaluation
RNA isolation was carried out with Trizol reagent (R0016, Beyotime, Shanghai, China). cDNA synthesis was performed using HiScript ® III All-in-one RT SuperMix (R333, Vazyme). Quantitative PCR amplification was executed with ChamQ Universal SYBR qPCR Master Mix (Q711, Vazyme) on a qTOWER3 thermal cycler (Analytikjena, Jena, Germany). Custom-designed primers (Tsingke Biotech, Beijing, China) were employed, with detailed sequences provided in Supplementary Table 1.
Western Blot
Cellular proteins were isolated using RIPA buffer (P0013B, Beyotime) supplemented with protease and phosphatase inhibitor cocktails. Following SDS-PAGE separation, proteins were electrotransferred onto PVDF membranes. After blocking with 5% BSA in TBST, membranes were probed with specific primary antibodies: anti-PLK1 (1:500, 4513, Cell Signaling Technology), anti-CyclinB1 (1:1000, 12231, Cell Signaling Technology), anti-p-CDC25C (1:250, 9529, Cell Signaling Technology), anti-CDC25C (1:1000, 4688, Cell Signaling Technology), anti-CDK1 (1:1000, A0220, Abcolonol), and anti-β-actin (1:1000, 4970, Cell Signaling Technology) at 4°C for 12 hours. Subsequent incubation with HRP-conjugated anti-rabbit IgG secondary antibody (1:2000, 7076, Cell Signaling Technology) was performed prior to ECL detection. Signal acquisition was conducted using a chemiluminescence imaging system (5200, Tanon), with band intensity quantified via ImageJ software.
Transcriptome Profiling by High-Throughput Sequencing
RNA was purified from both control and NETs-treated HUVECs using Trizol reagent (R0016, Beyotime, Shanghai, China). RNA integrity was verified by NanoDrop spectrophotometry (Thermo Fisher, USA), followed by polyadenylated RNA enrichment. Sequencing libraries were prepared and analyzed on the Illumina HiSeq X Ten platform. Transcripts exhibiting >2-fold expression alteration with statistical significance (P<0.05) were classified as differentially expressed genes. KEGG pathway and Gene Ontology analyses were performed, with data visualization including heatmap and volcano plot generation.
Computational Data Processing
Parametric data are represented as mean ± standard deviation (mean±SD), with between-group differences evaluated by unpaired t-test for two-group comparisons or one-way ANOVA for multiple groups. Non-parametric data are expressed as median (interquartile range) [median (P25, P75)] and analyzed using Mann–Whitney U-test. Statistical significance threshold was set at P<0.05. All experimental procedures were independently replicated three times. Data analysis and graphical representation were accomplished using SPSS 23.0 and GraphPad Prism 10.1.2 software.
Results
Increased NETs Formation and Endothelial Dysfunction in Sepsis-Induced Lung Injury Patients and Murine Models
This study enrolled 17 healthy controls and 17 sepsis patients. Sepsis patients exhibited significantly elevated neutrophils, IL-6, and IL-8, indicating aggravated inflammatory injury (Supplementary Table 2 and Figure 1A and B). Notably, myeloperoxidase-DNA (MPO-DNA) levels were markedly increased in sepsis patients (Figure 1C), suggesting enhanced NETs release. Furthermore, a positive correlation was observed between IL-6 and MPO-DNA levels in sepsis patients (Figure 1D), implying that NETs exacerbate inflammatory damage. Immunofluorescence co-staining of citrullinated histone H3 (citH3) and MPO in neutrophils directly visualized NETs release in sepsis patients (Figure 1E).
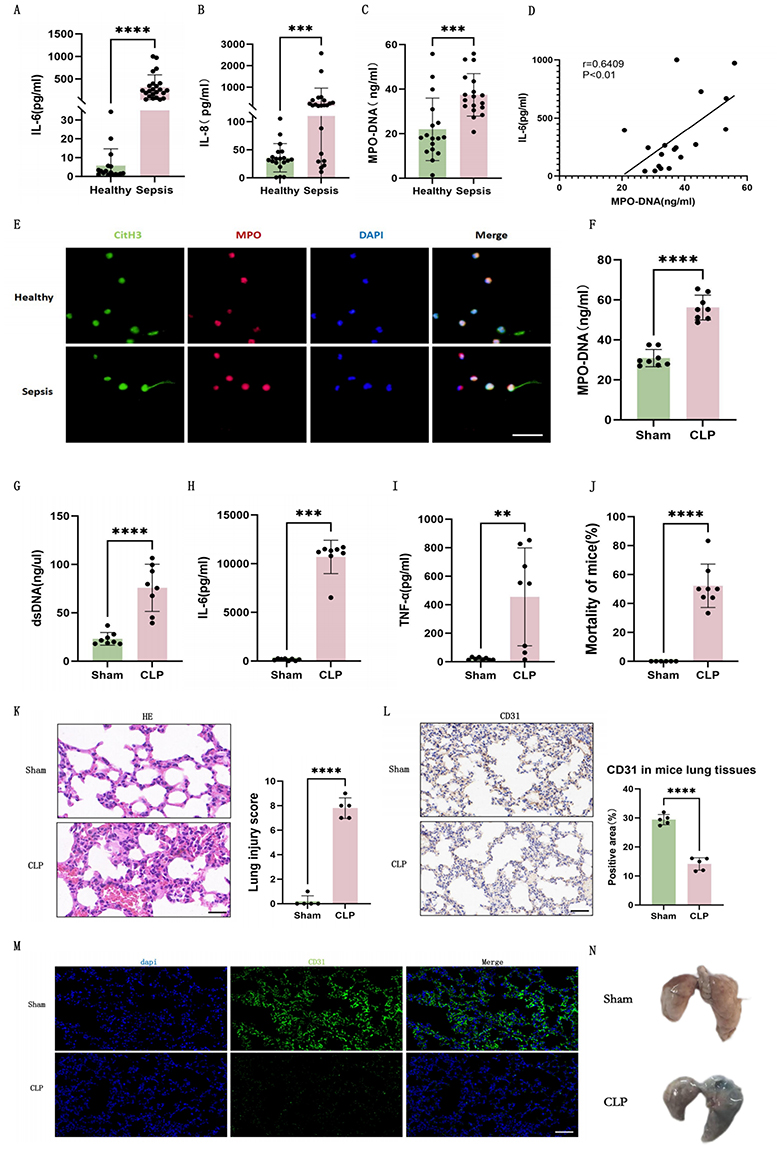 |
Figure 1 Increased NETs formation and endothelial dysfunction in sepsis-induced lung injury patients and murine models. (A) Plasma IL-6 levels in healthy controls (n=17) and sepsis patients (n=17). (B) Plasma IL-8 levels in healthy controls (n=17) and sepsis patients (n=17). (C) Plasma MPO-DNA levels in healthy controls (n=17) and sepsis patients (n=17). (D) Correlation analysis between plasma IL-6 and MPO-DNA levels in sepsis patients (n=17). (E) Immunofluorescence of NETs release in peripheral blood neutrophils from healthy controls and sepsis patients (green: CitH3, red: MPO, blue: DAPI, Scale bar: 30um). (F) Plasma MPO-DNA levels in mice (n=8). (G) Plasma dsDNA levels in mice (n=8). (H) Plasma IL-6 levels in mice (n=8). (I) Plasma TNF-α levels in mice (n=8). (J) Survival rates of mice (n=8). (K) H&E staining and lung injury scores in mice lung tissues (n=5, Scale bar: 25um). (L) Immunohistochemistry of CD31 and percentage of positive areas in lung tissues of mice (n=5, Scale bar: 50um). (M) Immunofluorescence of CD31 in lung tissues of mice (green: CD31, blue: DAPI, Scale bar: 50um). (N) EBA tracer measurement of mice lungs. Each bar represents the mean ± SD. Comparisons between groups were performed using unpaired t-tests. **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: cit-H3, citrullinated histone 3; CLP, cecal ligation and puncture; dsDNA, Double stranded DNA; HE, Hematoxylin Eosin; IL-6, interleukin-6; IL-8, interleukin-8; MPO, myeloperoxidase; TNF-α, tumor necrosis factor - α. |
We established a murine sepsis-induced lung injury model via CLP. CLP mice demonstrated increased plasma MPO-DNA and double-stranded DNA (dsDNA) levels (Figure 1F and G), consistent with enhanced NETs release observed in human patients. IL-6 and TNF-α were also elevated post-CLP (Figure 1H and I). The significantly higher mortality rate in CLP mice (Figure 1J) validated both model efficacy and severe organ dysfunction. Lung histopathology revealed severe pulmonary vascular hemorrhage (Figure 1K) while EBA tracer measurement revealed increased vascular permeability (Figure 1N) in CLP mice. These results confirmed endothelial dysfunction. Immunohistochemistry and immunofluorescence for CD31, a canonical endothelial marker, demonstrated reduced pulmonary endothelial cell density in CLP mice (Figure 1L and M).
Collectively, these findings indicated that both sepsis patients and CLP mice exhibit increased NETs formation and endothelial dysfunction.
NETs Degradation Alleviates Pulmonary Inflammation and Endothelial Injury
To further investigate the role of NETs in septic pulmonary endothelial injury, we administered DNaseI prior to CLP modeling to degrade NETs. Compared to CLP group, mice pretreated with DNaseI exhibited significantly reduced plasma MPO-DNA levels (Figure 2A). Decreases in IL-6 and TNF-α levels (Figure 2B and C) indicated attenuated systemic inflammation. H&E staining of lung tissues revealed diminished inflammatory infiltration, edema, and hemorrhage in the CLP+DNaseI group versus CLP group (Figure 2D), demonstrating that NETs degradation alleviates sepsis-induced lung injury. Immunohistochemistry of CD31 showed increased endothelial cell density in CLP+DNaseI group (Figure 2E), suggesting partial endothelial recovery. Furthermore, evans blue assays demonstrated reduced pulmonary vascular leakage in DNaseI-treated mice (Figure 2F). In summary, these findings indicate that NETs degradation mitigates endothelial damage in septic lung injury.
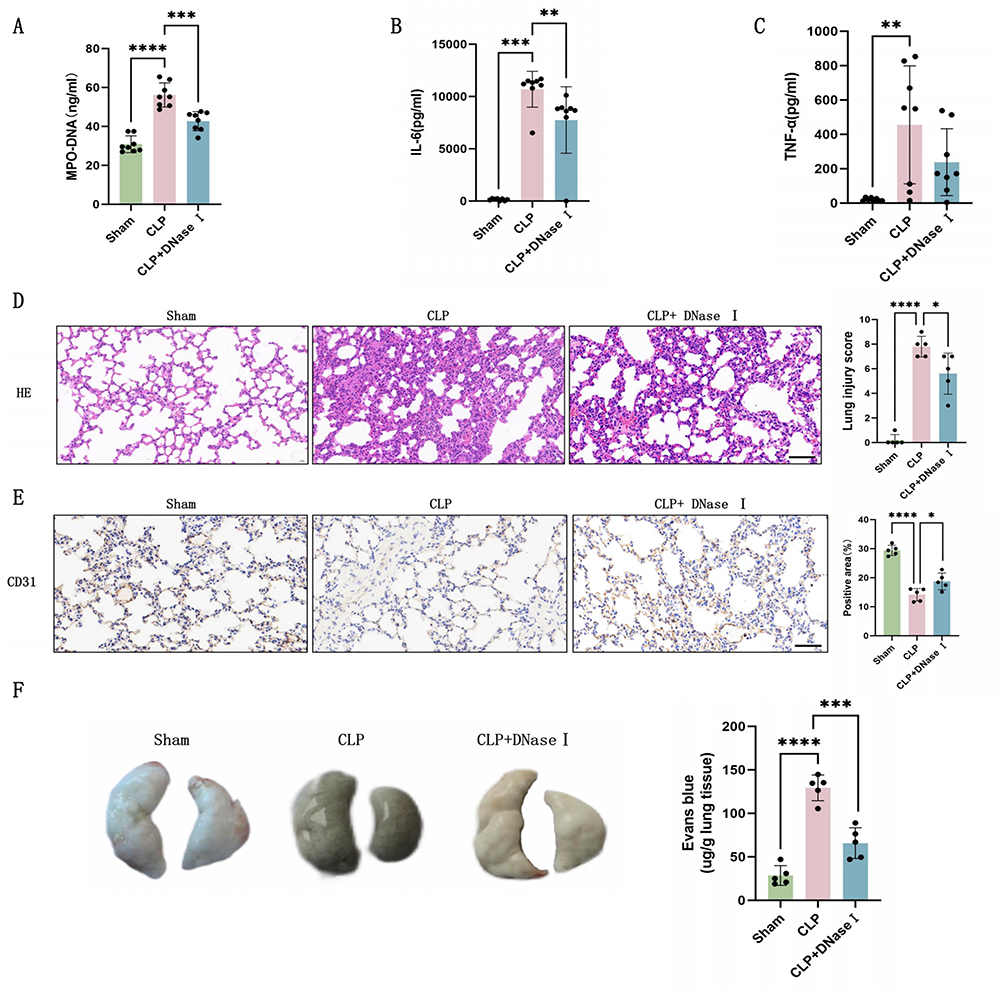 |
Figure 2 Degradation of NETs alleviates inflammation and endothelial injury. (A) Plasma MPO-DNA levels in mice (n=8). (B) Plasma IL-6 levels in mice (n=8). (C) Plasma TNF-α levels in mice (n=8). (D) H&E staining and lung injury scores in mice lung tissues (n=5, Scale bar: 50um). (E) Immunohistochemistry of CD31 and percentage of positive areas in lung tissues of mice (n=5, Scale bar: 50um). (F) Representative images and quantitative analysis of Evans blue vascular permeability analysis in mouse lung tissues (n=5). Each bar represents the mean ± SD. Comparisons between groups were performed using unpaired t-tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: CLP, cecal ligation and puncture; DNaseI, DeoxyribonucleaseI; dsDNA, Double stranded DNA; HE, Hematoxylin Eosin; IL-6, interleukin-6; MPO, myeloperoxidase; TNF-α, tumor necrosis factor - α. |
NETs Release Causes Endothelial Cell Cycle Arrest Leading to Delayed Division
To investigate the effects of NETs release on endothelial cells, we co-cultured HUVECs with NETs and conducted CCK-8 cell viability assay. Results showed that NETs caused significant poor cell viability (Figure 3A). Furthermore, Edu fluorescence staining revealed markedly reduced mitotic activity in HUVECs after NETs stimulation (Figure 3B). Further transcriptome sequencing demonstrated that cell cycle-related signaling pathways ranked second in both GO and KEGG analyses (Figure 3C and D).
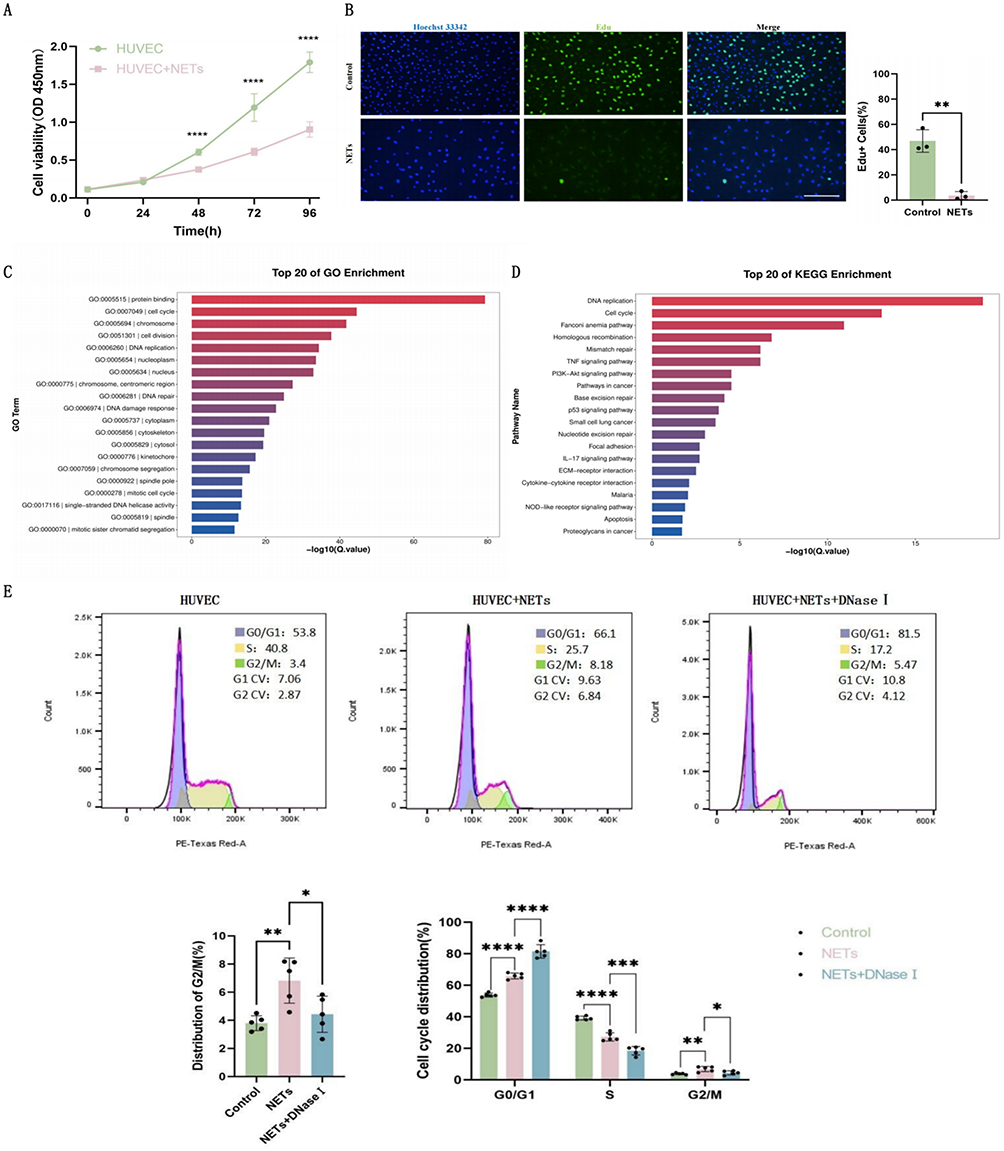 |
Figure 3 NETs release induces endothelial cell cycle arrest and proliferation inhibition. (A) CCK-8 cell viability assay of HUVECs (n=6). (B) Edu proliferation fluorescence images and positive cell ratio quantification of HUVECs (n=3, Scale bar: 100um). (C) GO enrichment analysis of HUVECs (24-hour NETs treatment) (n=3). (D) KEGG enrichment analysis of HUVECs (24-hour NETs treatment) (n=3). (E) Flow cytometric analysis of the cell cycle of HUVECs (n = 3). Each bar represents the mean ± SD. Comparisons between two groups were made using unpaired t-tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: DNaseI, DeoxyribonucleaseI; GO, Gene Ontology; HUVEC, human umbilical vein endothelial cells; KEGG, Kyoto Encyclopedia of Genes and Genomes; NETs, Neutrophil Extracellular Traps. |
We employed flow cytometric analysis to quantitatively assess cell cycle distribution patterns. Results demonstrated increased G2/M phase cell proportion in the NETs group, while the NETs+DNaseI group showed decreased G2/M phase proportion compared to the former (Figure 3E), indicating that NETs cause cell cycle G2/M phase blockade without initiating cell division and proliferation. This G2/M phase arrest was alleviated when NETs were degraded. Additionally, both G1 and S phase cell proportions increased in the NETs group, but showed no decrease in the NETs+DNaseI group compared to the former (Figure 3E), suggesting that the impact of NETs on G2/M phase is more important than that on G1 and S phase. Collectively, our findings indicate that NETs may impair endothelial proliferation by affecting the G2/M phase of the cell cycle.
NETs Inhibit PLK1 Pathway in CLP Mice Lung Endothelium and HUVECs
Further analysis of the transcriptome sequencing was conducted. Heatmap and volcano plot analysis of all cell cycle-related genes with statistically significant differences between groups revealed that PLK1, CDC25C, CCNB1, and CDK1 were significantly suppressed by NETs (Figure 4A and B). PLK1 signaling pathway has been extensively confirmed closely associated with the G2/M phase of the cell cycle. Its activation accelerates G2/M phase progression and promotes cell division, whereas its inhibition leads to G2/M phase arrest, delayed cell cycle progression, then suppressed proliferation. RT-qPCR results demonstrated time-dependent inhibition of PLK1, CDC25C, CCNB1, and CDK1 gene expression following NETs stimulation (Figure 4C). These findings collectively indicate that NETs release suppresses PLK1 pathway gene expression and this pathway likely mediates NETs-induced G2/M phase arrest in endothelial cells.
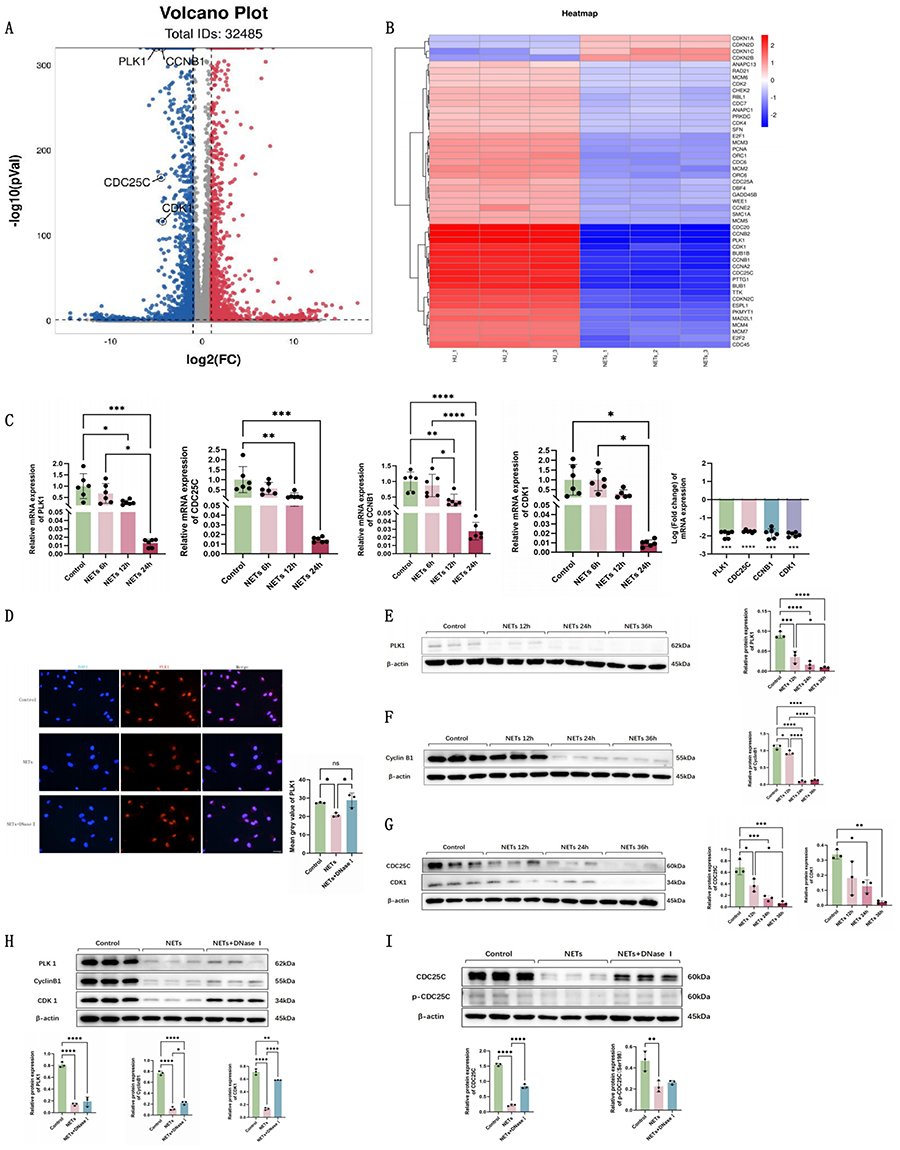 |
Figure 4 NETs release inhibits PLK1 pathway signaling in HUVECs. (A) Heatmap of DEGs implicated in cell cycle modulation after NETs stimulation (n=3). (B) Volcano plot analysis of DEGs implicated in cell cycle modulation after NETs stimulation (n=3). (C) RT-qPCR analysis of PLK1, CDC25C, CCNB1, and CDK1 gene expression changes in NETs-stimulated HUVECs (n=6). (D) PLK1 immunofluorescence images and fluorescence intensity quantification in HUVECs (n=3, Scale bar: 40um). (E–G) Representative Western blots and semi-quantification of PLK1, CDC25C, Cyclin B1, and CDK1 protein expression in HUVECs in different groups (n=3). (H and I) Representative Western blots and semi-quantification of PLK1, CDC25C, p-CDC25C, Cyclin B1, and CDK1 protein expression in HUVECs in different groups (n=3). Each bar represents the mean ± SD. Comparisons between two groups were made using unpaired t-tests.*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: CDK1, cyclin-dependent kinase 1; DNaseI, DeoxyribonucleaseI; HUVEC, human umbilical vein endothelial cells; NETs, Neutrophil Extracellular Traps; PLK1, polo like kinase 1. |
Protein expression of the PLK1 pathway in NETs-stimulated HUVECs was investigated. Immunofluorescence showed diminished PLK1 protein fluorescence intensity after NETs stimulation (Figure 4D). Western blot analysis confirmed time-dependent suppression of PLK1, CDC25C, Cyclin B1, and CDK1 protein expression (Figure 4E–G). The NETs+DNaseI group showed restored expression of PLK1, CDC25C, p-CDC25C, Cyclin B1, and CDK1 compared to the NETs group (Figure 4H and I).
The results were further confirmed in vivo. Lung endothelial cells (CD31+ CD45− Ep-CAM−) (Figure 5A) isolated by flow cytometry showed increased PLK1, p-CDC25C, and Cyclin B1 positive cells in the CLP+DNaseI group compared to CLP mice (Figure 5B–D). These results confirm that excessive NETs release during sepsis inhibits the PLK1 pathway in endothelial cells, and that NETs degradation can restore pathway protein expression.
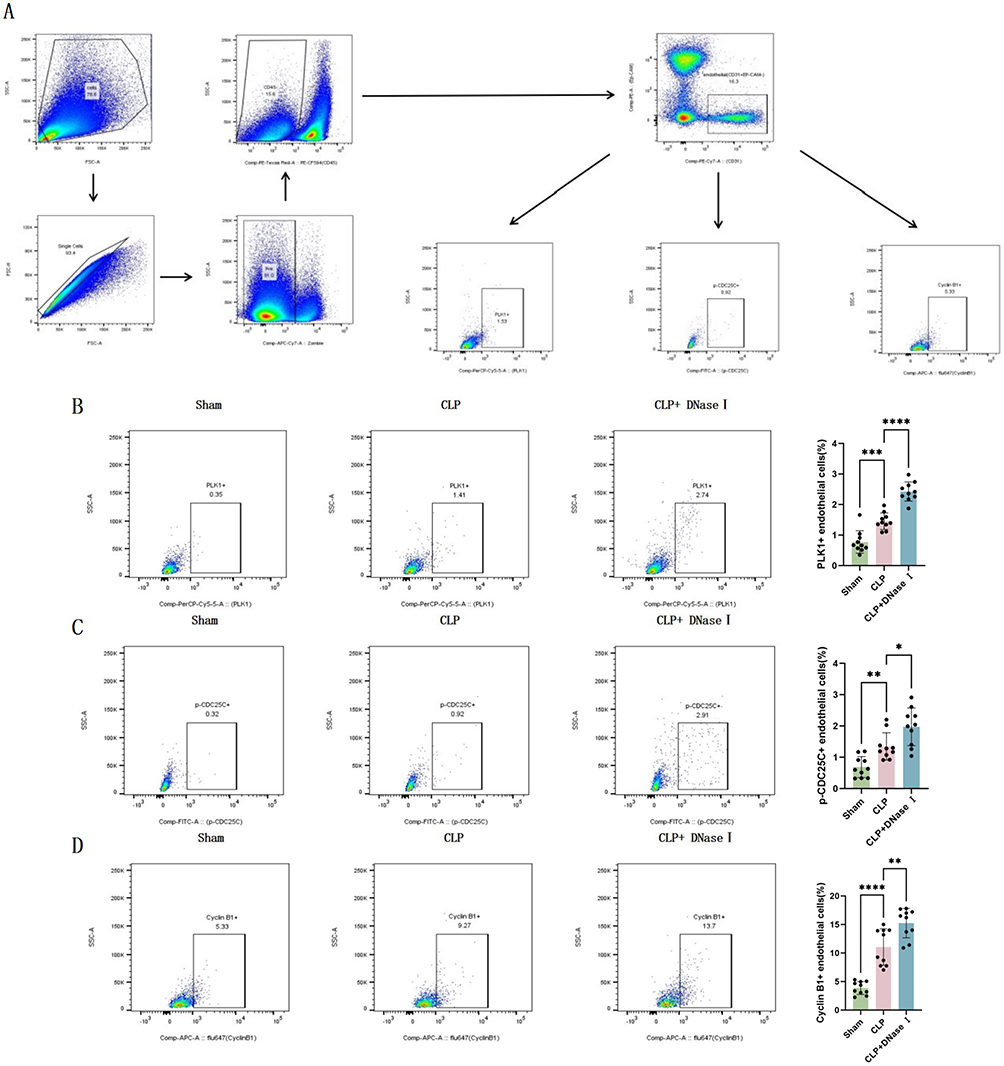 |
Figure 5 NETs release inhibits PLK1 pathway signaling in mouse lung endothelial cells. (A) Gating strategy for quantitative flow cytometry analysis of protein expression in mouse lung endothelial cells. (B) Proportion of PLK1-positive cells in mouse lung endothelial cells (n=10). (C) Proportion of p-CDC25C-positive cells in mouse lung endothelial cells (n=10). (D) Proportion of Cyclin B1-positive cells in mouse lung endothelial cells (n=10). Each bar represents the mean ± SD. Comparisons between two groups were made using unpaired t-tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: CLP, cecal ligation and puncture; DNaseI, DeoxyribonucleaseI; PLK1, polo like kinase 1. |
PLK1 Overexpression Endothelial Cells Exhibit Enhanced Proliferation Capacity and Alleviated G2/M Phase Arrest
To elucidate the mechanistic involvement of PLK1 signaling in NETs-mediated endothelial proliferative dysfunction, we established PLK1-overexpressing HUVECs through lentiviral transduction. The transfection efficiency was rigorously validated at both transcriptional and translational levels using PCR and WB, respectively (Figure 6A and B).
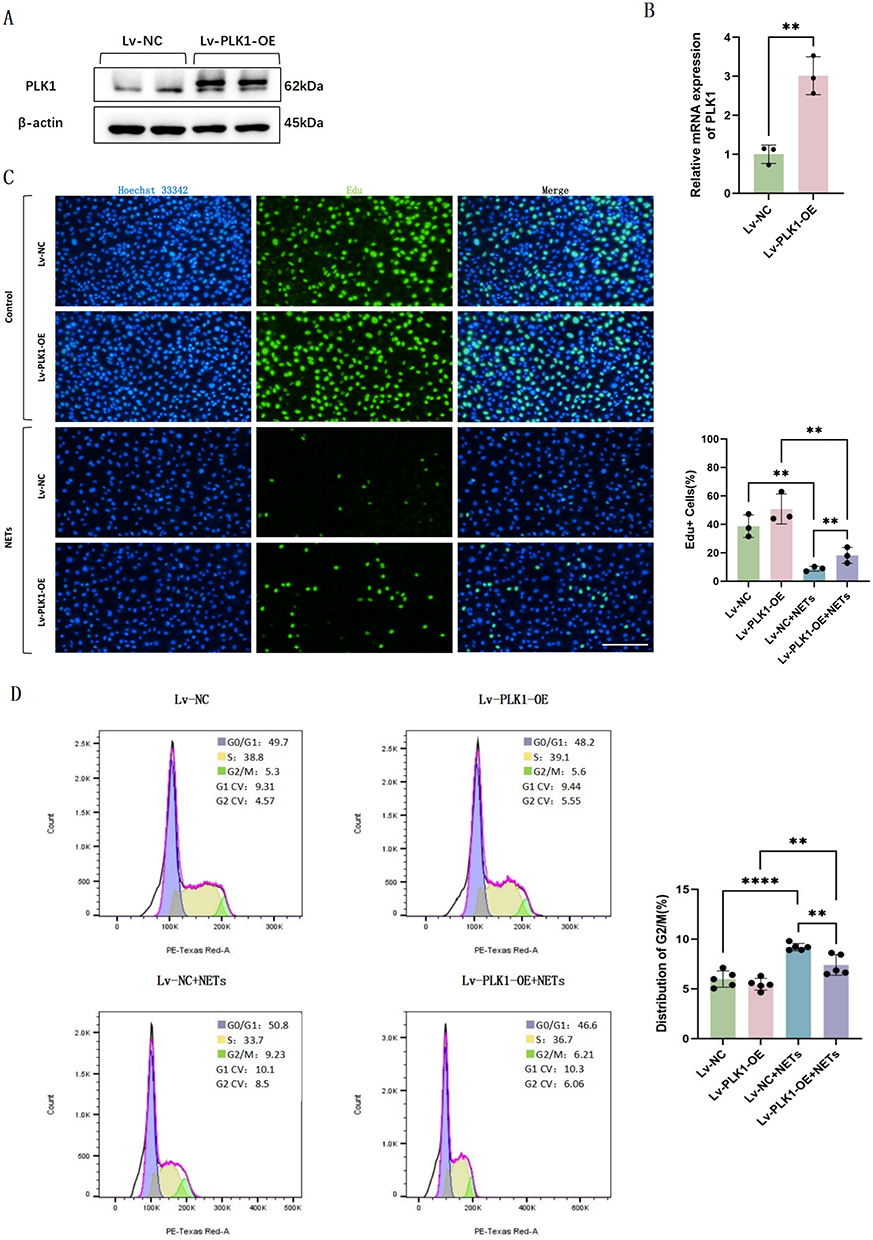 |
Figure 6 PLK1 overexpression endothelial cells exhibit enhanced proliferation capacity and alleviated G2/M phase arrest. (A) Western blot images of PLK1 expression in HUVECs transfected with Lv-NC or Lv-PLK1-OE. (B) Relative mRNA levels of PLK1 in HUVECs transfected with Lv-NC or Lv-PLK1-OE (n=3). (C) EdU fluorescence images and quantification of HUVECs (n=3). (D) Flow cytometric analysis of the cell cycle of HUVECs (n=5). Each bar represents the mean ± SD. Comparisons between two groups were made using unpaired t-tests. **p < 0.01, ****p < 0.0001. Abbreviations: HUVEC, human umbilical vein endothelial cells; NC, negative control; NETs, Neutrophil Extracellular Traps; OE, overexpression; PLK1, polo like kinase 1. |
Edu proliferation assay revealed restored proliferative potential in PLK1 overexpression endothelial cells compared to negative controls after NETs stimulation (Figure 6C). Cell cycle analysis further showed reduced G2/M phase proportions in PLK1 overexpression HUVECs versus negative controls after NETs stimulation, indicating more cells successfully transitioned from G2 to M phase to initiate subsequent division and proliferation (Figure 6D). These findings collectively suggest that PLK1 overexpression can activate endothelial cell proliferation after NETs release.
Discussion
During septic conditions, NETs exacerbate impairment of the pulmonary endothelial barrier, presenting clinically as sustained microvascular permeability and intra-alveolar bleeding —— pathological features strongly predictive of mortality outcomes. The reparative capacity of endothelial cells serves as a crucial prognostic indicator for clinical recovery. PLK1 is essential for the normal progression of the endothelial cell cycle, and intervention with its inhibitor TAK-960 in HUVEC cells leads to decreased cell viability and significant G2/M phase arrest (Supplementary Figure 1). Our research presents groundbreaking evidence that NETs impair endothelial proliferative potential in sepsis through modulation of the PLK1 cascade. Mechanistic studies reveal NETs-mediated suppression of the PLK1/CDC25C/CyclinB1-CDK1 axis induces G2/M cell cycle arrest, effectively blocking mitotic entry and cellular division. These findings illuminate potential therapeutic avenues for managing sepsis-induced pulmonary complications.
The vascular endothelium constitutes a selective biological interface between circulation and tissues, governing molecular transport through precisely regulated paracellular pathways. This barrier function assumes particular physiological significance in pulmonary circulation. Contrary to traditional views of endothelial quiescence, contemporary research reconceptualizes the endothelium as a dynamic, systemically integrated organ system orchestrating vascular homeostasis, remodeling, and senescence.16
Emerging evidence delineates multiple mechanisms of NETs-induced endothelial injury. Folco et al established that NETs components (IL-1α and cathepsin G) synergistically promote endothelial activation and prothrombotic states through surface erosion mechanisms.19 Aldabbous et al characterized NETs-mediated activation of the MPO/H2O2-TLR4/NF-κB axis in pulmonary arterial endothelial cells, driving inflammatory and angiogenic responses.20 Mussbacher et al further demonstrated NF-κB-dependent upregulation of VCAM-1, PECAM-1 and pro-angiogenic cytokines.21 Schmidt et al identified NETs-derived inflammatory mediators as direct disruptors of endothelial glycocalyx integrity,22 with Alphonsus et al revealing mast cell-mediated amplification through heparanase release.23 The procoagulant effects of NETs involve both anticoagulant pathway suppression and TF induction, with Kim et al demonstrating TLR2/4-dependent regulation of thrombomodulin/TF expression24 and Sugiyama et al implicating MPO-derived hypochlorous acid in TF upregulation.25
Endothelial cells possess robust regenerative capacity through three primary processes: 1) proliferation of surviving local ECs; 2) paracrine signaling and differentiation of endothelial progenitor cells; 3) reconnection of endothelial junctions.18 Numerous studies have elucidated mechanisms of endothelial proliferation post-injury. Following sepsis-induced vascular injury, the transcription factor FoxM1 expressed in pulmonary endothelial cells of young adult mice serves as a key mediator of endothelial proliferation. In contrast, aged mice exhibit deficient pulmonary endothelial regeneration due to FoxM1 loss,26,27 with HIF-1α acting as an upstream regulator of FoxM1-driven proliferation.28 Recent studies have elucidated molecular mechanisms governing endothelial regeneration. Miyagawa et al identified a BMPR2-Notch1-Myc signaling axis that modulates endothelial metabolism and epigenetic modifications to enhance proliferative capacity.29 Schober’s team revealed miR-126-5p maintains endothelial proliferation by negatively regulating Dlk1-mediated Notch1 inhibition.30 Using bone marrow transplantation models, Singhal et al demonstrated liver vascular repair occurs exclusively through resident endothelial proliferation rather than circulating progenitors.31 Zhao’s research team established FoxM1 as a master regulator of cell cycle progression in pulmonary endothelium through cyclin/CDK modulation.32 Itoh et al characterized the precise temporal dynamics of pial artery endothelial regeneration post-injury.33 McDonald’s work highlighted age-related decline in endothelial repair capacity linked to attenuated stress-response gene activation.34 Liu et al discovered endotoxin-induced Sox17 mediates cyclin E1-dependent endothelial regeneration during systemic inflammation.35
Despite comprehensive understanding of endothelial regeneration mechanisms and NETs-mediated vascular injury across various pathologies, only one prior investigation has addressed NETs effects on endothelial proliferation in sepsis, reporting p21-dependent cell cycle arrest.36 Our work reveals a novel PLK1-mediated pathway underlying NETs-induced proliferative impairment in septic endothelium. The G2/M transition requires accumulation of maturation-promoting factor (cyclinB1-CDK1 complex) beyond a critical threshold. Gheghiani’s studies in epithelial cells established PLK1 activation precedes and enables CDC25C phosphorylation for mitotic entry.37 Chiu et al identified p17 protein as a PLK1/CDK1 inhibitor inducing G2/M arrest.38 Gomes-da-Silva’s targeted PLK1 knockdown strategy enhanced chemosensitivity and G2/M accumulation in both cancer and endothelial cells.39 Li et al demonstrated arenobufagin’s antitumor mechanism through PLK1-CDC25C-CyclinB1/CDK1 axis inhibition.40 Our findings represent the first evidence of PLK1 pathway involvement in NETs-induced endothelial cell cycle arrest during sepsis.
This study has limitations. First, we did not distinguish between regenerating ECs derived from local residual cells versus circulating EPCs, leaving their relative contributions undefined. Second, clinical human samples validation was lacking, which leaves the feasibility of PLK1 pathway as a therapeutic target in sepsis patients unexplored. Finally, the proliferation of endothelial cells exhibits significant intra-organ heterogeneity. In heart, the proliferative activity of endothelial cells is significantly enhanced in the upper interventricular septum, the inner upper wall of the left ventricle, and the apex. In liver, e-cadherin-positive hepatic sinusoidal endothelial cells show a distinct proliferative advantage. In lungs, plasma membrane vesicle–associated protein-positive endothelial cells renew markedly faster than carbonic anhydrase 4-positive endothelial cells.41 Additionally, single-cell sequencing studies have confirmed that transcriptomic features of endothelial cells vary by tissue origin, and these differences likely contribute to distinct endothelial proliferative capacities.42 However, our study did not delve into the tissue specificity of endothelial proliferation.
Conclusion
In conclusion, our integrated cellular, murine, and clinical evidence demonstrates that NETs in septic lung injury suppress pulmonary endothelial proliferation via PLK1/CDC25C/CyclinB1-CDK1 pathway inhibition and G2/M phase arrest, resulting in persistent vascular leakage, poor prognosis, and high mortality. These findings provide novel therapeutic targets for improving endothelial regeneration in sepsis-associated pulmonary injury.
Abbreviations
ATCC, American Type Culture Collection; CDK1, cyclin-dependent kinase 1; cit-H3, citrullinated histone 3; CLP, cecal ligation and puncture; DNaseI, DeoxyribonucleaseI; dsDNA, Double stranded DNA; EC, endothelial cells; ELISA, enzyme linked immunosorbent assay; FBS, fetal bovine serum; GO, Gene Ontology; HE, Hematoxylin Eosin; HMEC-1, Human Microvascular Endothelial Cell line-1; HUVEC, human umbilical vein endothelial cells; IL-6, interleukin-6; IL-8, interleukin-8; IQR, interquartile range; KEGG, Kyoto Encyclopedia of Genes and Genomes; Lv, lentivirus; MPF, maturation promoting factor; MPO, myeloperoxidase; NE, neutrophil elastase; NETs, Neutrophil Extracellular Traps; OE, overexpression; PBS, phosphate buffer; PFA, paraformaldehyde; PLK1, polo like kinase 1; PMA, phorbol ester; PVDF, polyvinylidene fluoride; SI-ALI, sepsis induced acute lung injury; TNF-α, tumor necrosis factor - α.
Acknowledgments
We thank the patients who participated in the study.
Funding
This research was supported by the Natural Science Foundation of Shanghai Municipal Commission of Science and Technology (No. 23ZR1411400) and the National Natural Science Foundation of China (No. 82402503).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801. doi:10.1001/jama.2016.0287
2. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the global burden of disease study. Lancet. 2020;395(10219):200–211. doi:10.1016/S0140-6736(19)32989-7
3. Liu YC, Yao Y, Yu MM, et al. Frequency and mortality of sepsis and septic shock in China: a systematic review and meta-analysis. BMC Infect Dis. 2022;22(1):564. doi:10.1186/s12879-022-07543-8
4. Shen XF, Cao K, Jiang J, et al. Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med. 2017;21(9):1687–1697. doi:10.1111/jcmm.13112
5. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi:10.1126/science.1092385
6. Zhang H, Wang Y, Qu M, et al. Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis. Clin Transl Med. 2023;13(1):e1170. doi:10.1002/ctm2.1170
7. Li W, Li D, Chen Y, et al. Classic signaling pathways in alveolar injury and repair involved in sepsis-induced ALI/ARDS: new research progress and prospect. Dis Markers. 2022;2022:1–9. doi:10.1155/2022/6362344
8. Park I, Kim M, Choe K, et al. Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur Respir J. 2019;53(3):1800786. doi:10.1183/13993003.00786-2018
9. Qu M, Chen Z, Qiu Z. Neutrophil extracellular traps-triggered impaired autophagic flux via METTL3 underlies sepsis-associated acute lung injury. Cell Death Discovery. 2022;8(1). doi:10.1038/s41420-022-01166-3
10. Zhang H, Liu J, Zhou Y, et al. Neutrophil extracellular traps mediate m6A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int J Bio Sci. 2022;18(8):3337–3357. doi:10.7150/ijbs.69141
11. Zhang H, Wu D, Wang Y, et al. METTL3‐mediated N6‐methyladenosine exacerbates ferroptosis via m6A‐IGF2BP2‐dependent mitochondrial metabolic reprogramming in sepsis‐induced acute lung injury. Clin Transl Med. 2023;13(9):e1389. doi:10.1002/ctm2.1389
12. Wu D, Spencer CB, Ortoga L, et al. Histone lactylation-regulated METTL3 promotes ferroptosis via m6A-modification on ACSL4 in sepsis-associated lung injury. Redox Biol. 2024;74:103194. doi:10.1016/j.redox.2024.103194
13. Zhu S, Yu Y, Qu M, et al. Neutrophil extracellular traps contribute to immunothrombosis formation via the STING pathway in sepsis-associated lung injury. Cell Death Discovery. 2023;9(1):315. doi:10.1038/s41420-023-01614-8
14. Zhang H, Zhou Y, Qu M, et al. Tissue factor-enriched neutrophil extracellular traps promote immunothrombosis and disease progression in sepsis-induced lung injury. Front Cell Infect Microbiol. 2021;11:677902. doi:10.3389/fcimb.2021.677902
15. Zuo Y, Kanthi Y, Knight JS, et al. The interplay between neutrophils, complement, and microthrombi in COVID-19. Best Pract Res. 2021;35(1):101661. doi:10.1016/j.berh.2021.101661
16. Augustin HG, Koh GY. A systems view of the vascular endothelium in health and disease. Cell. 2024;187(18):4833–4858. doi:10.1016/j.cell.2024.07.012
17. Zou S, Jie H, Han X, et al. The role of neutrophil extracellular traps in sepsis and sepsis-related acute lung injury. Int Immunopharmacol. 2023;124:110436. doi:10.1016/j.intimp.2023.110436
18. Evans CE, Iruela-Arispe ML, Zhao YY. Mechanisms of endothelial regeneration and vascular repair and their application to regenerative medicine. Am J Pathol. 2021;191(1):52–65. doi:10.1016/j.ajpath.2020.10.001
19. Folco EJ, Mawson TL, Vromman A, et al. Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin-1α and cathepsin G. Arteriosclerosis Thrombosis Vasc Biol. 2018;38(8):1901–1912. doi:10.1161/ATVBAHA.118.311150
20. Aldabbous L, Abdul-Salam V, Mckinnon T, et al. Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arteriosclerosis Thrombosis Vasc Biol. 2016;36(10):2078–2087. doi:10.1161/ATVBAHA.116.307634
21. Mussbacher M, Salzmann M, Brostjan C, et al. Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front Immunol. 2019;10:85. doi:10.3389/fimmu.2019.00085
22. Schmidt EP, Yang Y, Janssen WJ, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nature Med. 2012;18(8):1217–1223. doi:10.1038/nm.2843
23. Alphonsus CS, Rodseth RN. The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia. 2014;69(7):777–784. doi:10.1111/anae.12661
24. Kim JE, Yoo HJ, Gu JY, et al. Histones induce the procoagulant phenotype of endothelial cells through tissue factor up-regulation and thrombomodulin down-regulation. PLoS One. 2016;11(6):e0156763.
25. Sugiyama S, Kugiyama K, Aikawa M, et al. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arteriosclerosis Thrombosis Vasc Biol. 2004;24(7):1309–1314. doi:10.1161/01.ATV.0000131784.50633.4f
26. Huang X, Zhao YY. Transgenic expression of foxm1 promotes endothelial repair following lung injury induced by polymicrobial sepsis in mice. PLoS One. 2012;7(11):e50094. doi:10.1371/journal.pone.0050094
27. Huang X, Zhang X, Machireddy N, et al. Endothelial FoxM1 reactivates aging-impaired endothelial regeneration for vascular repair and resolution of inflammatory lung injury. Sci Trans Med. 2023;15(709):eabm5755. doi:10.1126/scitranslmed.abm5755
28. Huang X, Zhang X, Zhao DX. Endothelial hypoxia-inducible factor-1α is required for vascular repair and resolution of inflammatory lung injury through forkhead box protein M1. Am J Pathol. 2019;189(8):1664–1679. doi:10.1016/j.ajpath.2019.04.014
29. Miyagawa K, Shi M, Chen PI, et al. Smooth muscle contact drives endothelial regeneration by BMPR2-Notch1–mediated metabolic and epigenetic changes. Circulation Res. 2019;124(2):211–224. doi:10.1161/CIRCRESAHA.118.313374
30. Schober A, Nazari-Jahantigh M, Wei Y. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nature Med. 2014;20(4):368–376. doi:10.1038/nm.3487
31. Singhal M, Liu X, Inverso D, et al. Endothelial cell fitness dictates the source of regenerating liver vasculature. J Exp Med. 2018;215(10):2497–2508. doi:10.1084/jem.20180008
32. Zhao YY, Gao XP, Zhao YD, et al. Endothelial cell–restricted disruption of FoxM1 impairs endothelial repair following LPS-induced vascular injury. J Clin Investig. 2006;116(9):2333–2343. doi:10.1172/JCI27154
33. Itoh Y, Toriumi H, Yamada S, et al. Resident endothelial cells surrounding damaged arterial endothelium reendothelialize the lesion. Arteriosclerosis Thrombosis Vasc Biol. 2010;30(9):1725–1732. doi:10.1161/ATVBAHA.110.207365
34. Mcdonald AI, Shirali AS, Aragón R, et al. Endothelial regeneration of large vessels is a biphasic process driven by local cells with distinct proliferative capacities. Cell Stem Cell. 2018;23(2):210–225.e6. doi:10.1016/j.stem.2018.07.011
35. Liu M, Zhang L, Marsboom G, et al. Sox17 is required for endothelial regeneration following inflammation-induced vascular injury. Nat Commun. 2019;10(1):2126. doi:10.1038/s41467-019-10134-y
36. Zhu S, Yu Y, Hong Q, et al. Neutrophil extracellular traps upregulate p21 and suppress cell cycle progression to impair endothelial regeneration after inflammatory lung injury. J Clin Med. 2024;13(5):1204. doi:10.3390/jcm13051204
37. Gheghiani L, Loew D, Lombard B, et al. PLK1 activation in late G2 sets up commitment to mitosis. Cell Rep. 2017;19(10):2060–2073. doi:10.1016/j.celrep.2017.05.031
38. Chiu HC, Huang WR, Liao TL, et al. Suppression of vimentin phosphorylation by the avian reovirus p17 through inhibition of CDK1 and Plk1 impacting the G2/M phase of the cell cycle. PLoS One. 2016;11(9):e0162356. doi:10.1371/journal.pone.0162356
39. Gomes-Da-Silva LC, Ramalho JS, Pedroso De Lima MC, et al. Impact of anti-PLK1 siRNA-containing F3-targeted liposomes on the viability of both cancer and endothelial cells. Eur J Pharm Biopharm. 2013;85(3):356–364. doi:10.1016/j.ejpb.2013.04.007
40. Li J, Ma R, Lv JL, et al. Telocinobufagin, a PLK1 suppressor that inhibits tumor growth and metastasis by modulating CDC25c and CTCF in HNSCC cells. Phytomedicine. 2024;127:155440. doi:10.1016/j.phymed.2024.155440
41. Han M, Liu Z, Liu L, et al. Spatiotemporal proliferative heterogeneity of intraorgan endothelial cells. Circulation Res. 2025;137(7):934–949. doi:10.1161/CIRCRESAHA.125.326748
42. Paik DT, Tian L, Williams IM, et al. Single-cell RNA sequencing unveils unique transcriptomic signatures of organ-specific endothelial cells. Circulation. 2020;142(19):1848–1862. doi:10.1161/CIRCULATIONAHA.119.041433
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.