")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Neuropsychiatric disturbances in atypical parkinsonian disorders
Authors Belvisi D, Berardelli I, Suppa A , Fabbrini A, Pasquini M , Pompili M , Fabbrini G
Received 26 June 2018
Accepted for publication 31 July 2018
Published 9 October 2018 Volume 2018:14 Pages 2643—2656
DOI https://doi.org/10.2147/NDT.S178263
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Daniele Belvisi,1 Isabella Berardelli,2 Antonio Suppa,1,3 Andrea Fabbrini,3 Massimo Pasquini,3 Maurizio Pompili,2 Giovanni Fabbrini1,3
1IRCCS Neuromed, Pozzilli, Italy; 2Department of Neurosciences, Mental Health and Sensory Organs, Suicide Prevention Center, Sant’Andrea Hospital, Sapienza University of Rome, Rome, Italy; 3Department of Human Neurosciences, Sapienza University of Rome, Rome, Italy
Abstract: Multiple system atrophy (MSA), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD) are the most common atypical parkinsonisms. These disorders are characterized by varying combinations of autonomic, cerebellar and pyramidal system, and cognitive dysfunctions. In this paper, we reviewed the evidence available on the presence and type of neuropsychiatric disturbances in MSA, PSP, and CBD. A MedLine, Excerpta Medica, PsycLit, PsycInfo, and Index Medicus search was performed to identify all articles published on this topic between 1965 and 2018. Neuropsychiatric disturbances including depression, anxiety, agitation, and behavioral abnormalities have been frequently described in these disorders, with depression as the most frequent disturbance. MSA patients show a higher frequency of depressive disorders when compared to healthy controls. An increased frequency of anxiety disorders has also been reported in some patients, and no studies have investigated apathy. PSP patients may have depression, apathy, disinhibition, and to a lesser extent, anxiety and agitation. In CBD, neuropsychiatric disorders are similar to those present in PSP. Hallucinations and delusions are rarely reported in these disorders. Neuropsychiatric symptoms in MSA, PSP, and CBD do not appear to be related to the severity of motor dysfunction and are one of the main factors that determine a low quality of life. The results suggest that neuropsychiatric disturbances should always be assessed in patients with atypical parkinsonisms.
Keywords: atypical parkinsonisms, multiple system atrophy, progressive supranuclear palsy, corticobasal degeneration, neuropsychiatric disturbances, depression
Introduction
Atypical parkinsonisms (APs) are neurological conditions which differ from Parkinson’s disease (PD) mainly because of the wider involvement of the central nervous system.1 APs include multiple system atrophy (MSA), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD). These are relatively rare disorders characterized by progressive parkinsonism, a poor response to levodopa treatment, and clinical signs due to the involvement of the autonomic, cerebellar, and pyramidal systems.2 Cognitive dysfunctions are also present in these disorders. Patients affected by APs often complain of neuropsychiatric disturbances.3 It is however unclear whether each type of AP displays specific neuropsychiatric disorders or whether psychiatric disorders are part of the disease spectrum or are secondary to the severity of motor impairment. It is also unclear whether neuropsychiatric symptoms may be present in the early phase of the disease and how they impact the quality of life of these patients. To answer these questions, we reviewed the evidence available on neuropsychiatric disorders in patients with MSA, PSP, and CBD. For the purposes of the paper, we first summarized the most important clinical features of each clinical condition.
Methods
A systematic review of neuropsychiatric disturbances in MSA, PSP, and CBD was performed using MedLine, Excerpta Medica, PsycLit, PsycInfo, and Index Medicus search to identify all papers published on this topic between 1965 and 2018. The PRISMA statement for reporting systematic reviews was followed.4 The search terms used were: MSA OR PSP OR CBD OR CBS OR anxiety OR depression OR obsessive compulsive disorders OR hallucinations OR delusions OR psychosis OR delirium OR disinhibition OR abnormal behaviour OR disinhibition. We first reviewed the titles and abstracts and applied the selection criteria outlined previously with the exception of study design (Figure 1). Only articles published in English, peer-reviewed journals were considered and full articles were examined to consider their relevance based on the inclusion criteria. Possible discrepancies between the reviewers with regard to the inclusion criteria were resolved through consultation with the senior author (GF). In addition, reference lists of the papers were also examined. We excluded meta-analyses and systematic reviews, and studies which did not clearly report statistical analysis, diagnostic criteria, or the number of patients included. The principal reviewer (DB) checked all items. Then, three reviewers independently inspected all citations of the studies identified by the search and grouped them according to topic.
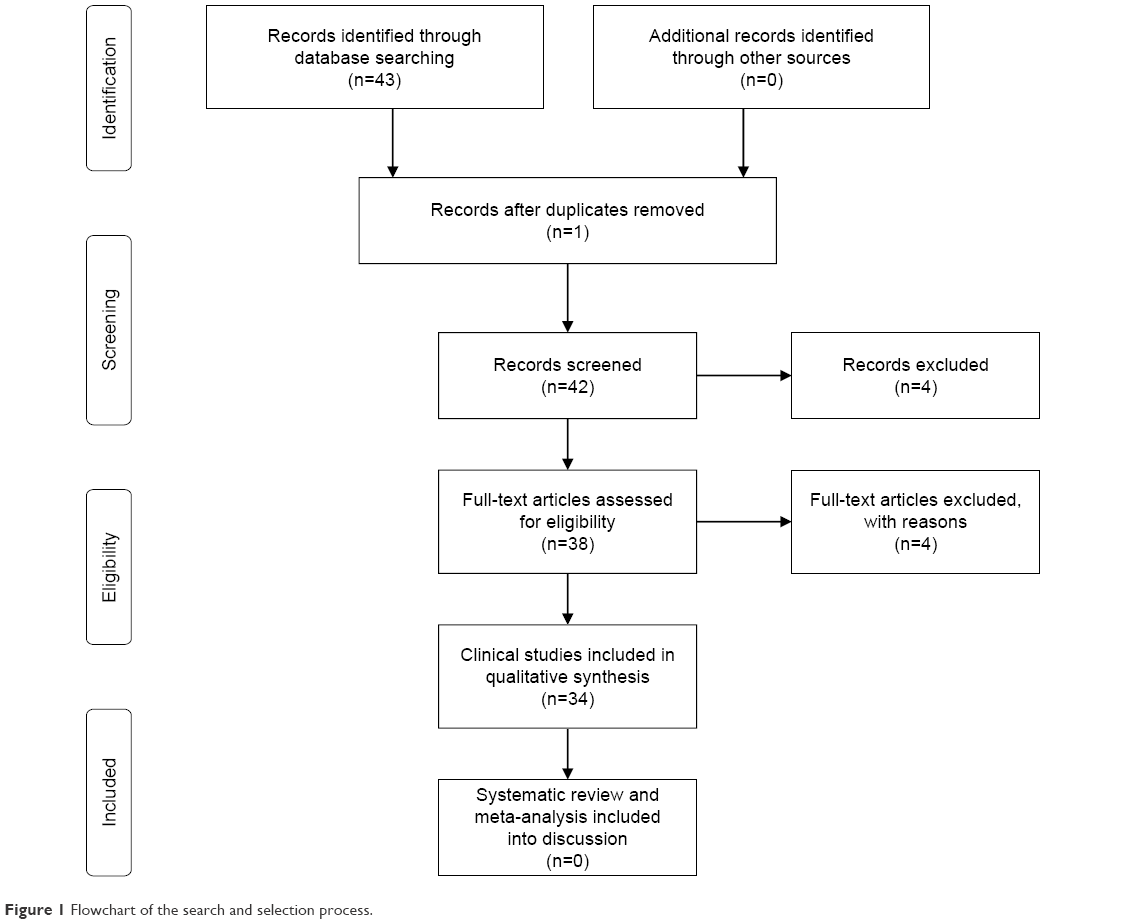 | Figure 1 Flowchart of the search and selection process. |
A quality assessment was performed as shown in Tables 1–3. Studies were rated for quality assessment using the following criteria: 1) pathological confirmation of the diagnosis: no confirmation: 0 points; confirmation: 2 points; 2) presence of a control group: no control group: 0 points; control group (either other disease or healthy controls [HC]): 1 point; both HC + other disease: 2 points; 3) number of subjects assessed: 0 points if N<20; 1 point if N between 20 and 100; 2 points if >100; 4) follow-up assessment: no follow-up: 0 points; follow-up: 1 point; 5) data presentation: unclear data presentation: 0 points; clear data presentation: 1 point; and 6) type of psychiatric assessment: no rating scales used: 0 points; use of rating scales for psychiatric symptoms: 1 point; diagnosis according to DSM criteria with structured interview and rating scales for psychiatric symptoms: 2 points.
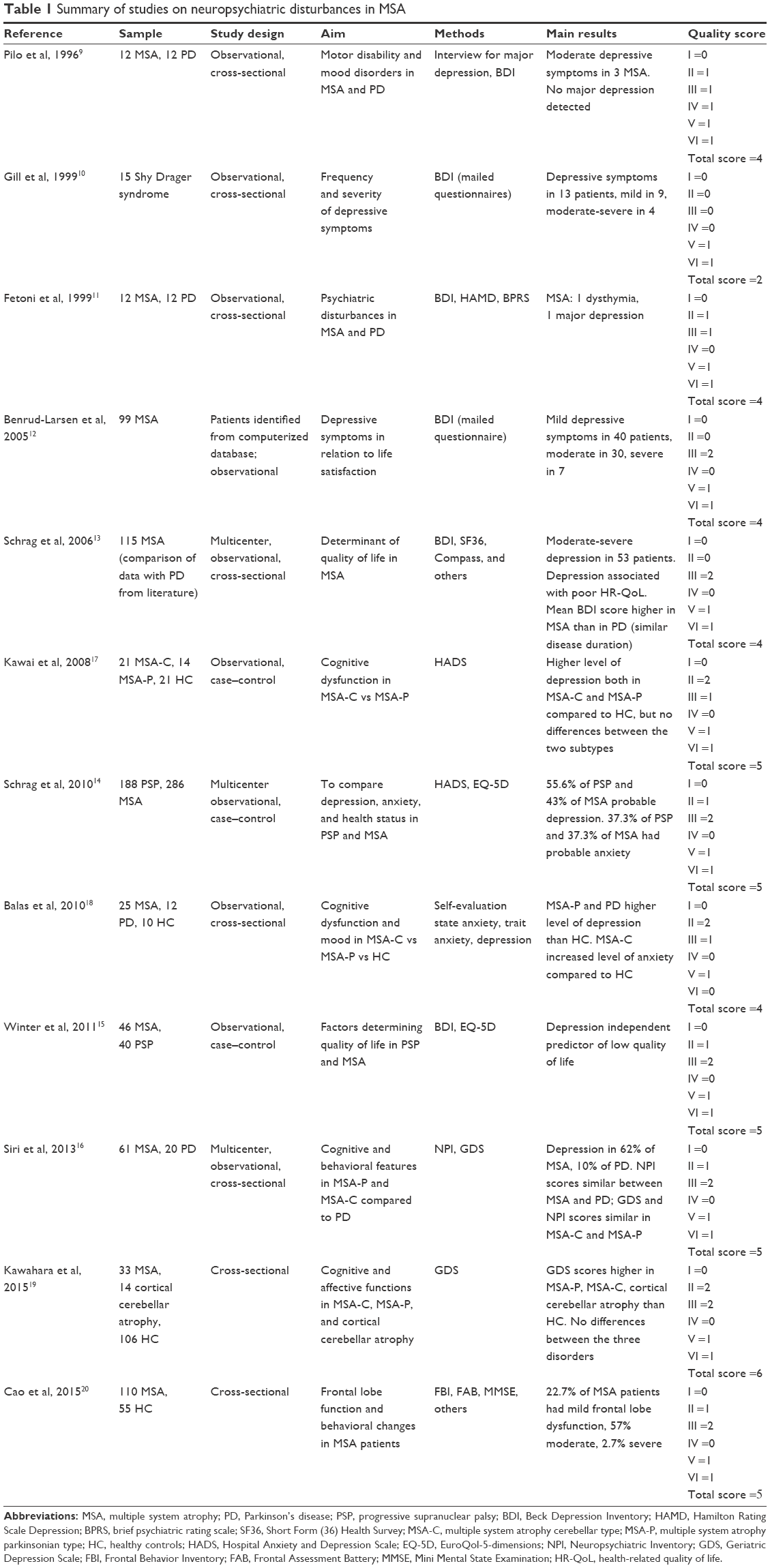 | Table 1 Summary of studies on neuropsychiatric disturbances in MSA |
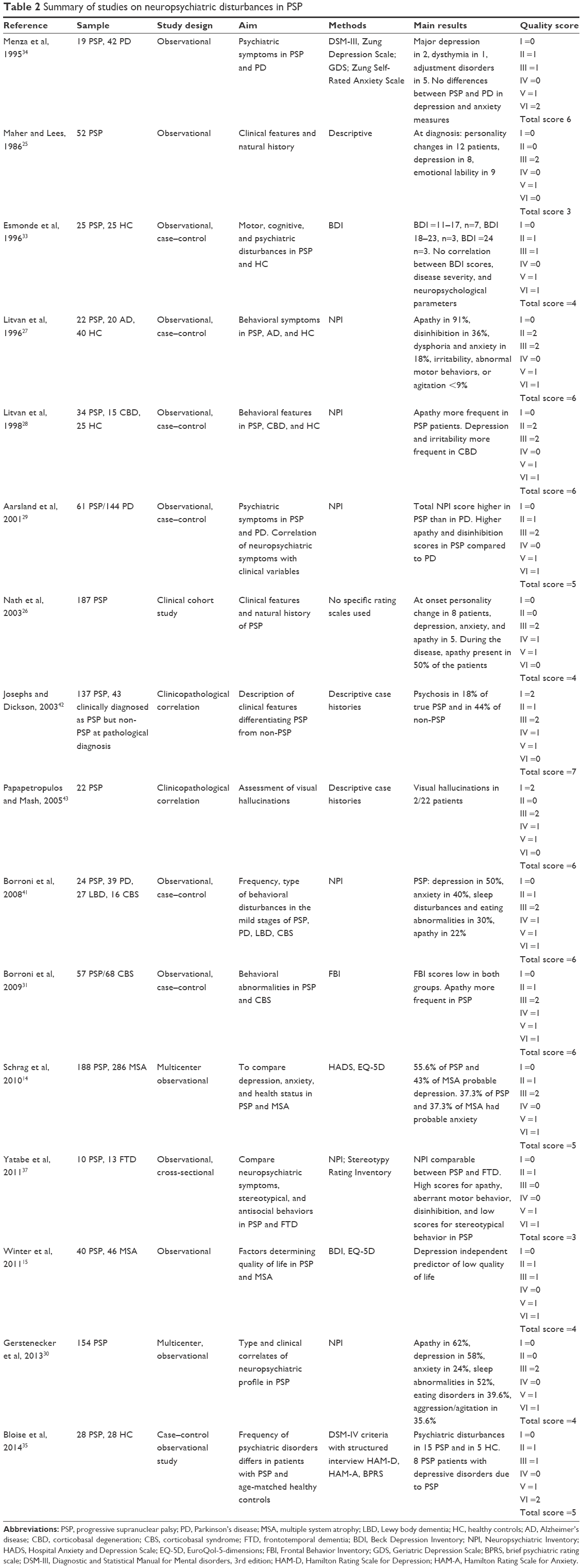 | Table 2 Summary of studies on neuropsychiatric disturbances in PSP |
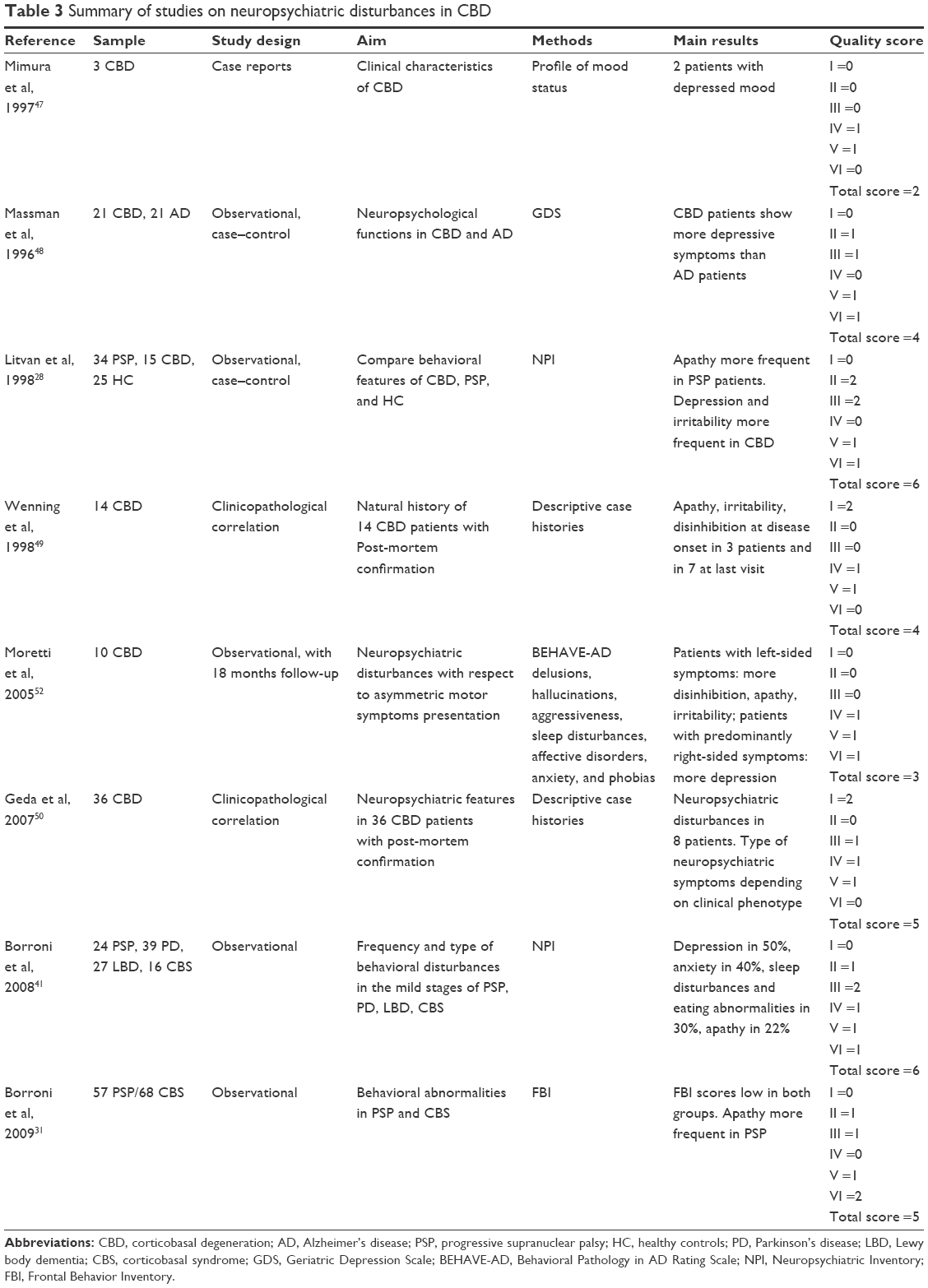 | Table 3 Summary of studies on neuropsychiatric disturbances in CBD |
MSA
MSA is a rare, sporadic progressive neurodegenerative disease. It is clinically characterized by a combination of parkinsonism, cerebellar dysfunction, pyramidal signs, and autonomic failure.5,6 MSA is classified in two variants on the basis of prominent clinical features: the parkinsonian type (MSA-P) and the cerebellar type (MSA-C).6,7 Pathologically, MSA is characterized by the accumulation of glial cytoplasmic inclusions of alpha-synuclein in the oligodendrocytes of several cortical and sub-cortical regions.8
Neuropsychiatric symptoms in MSA
Pilo et al (1996)9 reported findings of 12 patients with MSA and 12 patients with PD (but not HC) using the Beck Depression Inventory (BDI) and a non-structured interview to assess the features of major depressive disorders. None of the patients interviewed had a major depressive disorder, and only three MSA patients and three PD patients had BDI scores over 18. Using a BDI mailed questionnaire, Gill et al (1999)10 reported depressive symptoms in 13 of the 15 patients with Shy Drager syndrome (a form of MSA characterized by predominant autonomic dysfunction) in an uncontrolled study. Depressive symptoms were rated as mild in nine patients and moderate to severe in four and did not correlate with disability. Fetoni et al (1999)11 compared 12 patients with PD and 12 with possible MSA-P (with similar disability scores) using the Hamilton Rating Scale for Depression (HAM-D), supported by the DSM-IV criteria for depression, and the brief psychiatric rating scale (BPRS). No HC were assessed in this study. The authors found that only two of the MSA patients had a BDI score indicative of depression (one had a score of 10 and one a score of 18). The remaining ten patients had a score <7. When compared with PD patients, patients with MSA had higher scores on items indicating “blunted affect”, which suggests that MSA patients are emotionally indifferent to their condition.11
In comparison to the previously mentioned studies, which report findings from small numbers of patients, other authors assessed larger populations of MSA patients. Benrud-Larson et al (2005),12 using a computerized database of patients seen at the Mayo Clinic between 1998 and 2002, found that 40 of the 99 MSA patients they analyzed had BDI scores consistent with mild depression, 30 with moderate depression, and seven with severe depression. The authors did not compare the data with a control population. More than one third of the patients in their study were taking antidepressant medications. Similar findings emerged from a large, multicenter, multinational survey of the European MSA-Study Group conducted on 115 MSA patients, which detected moderate or severe depression, defined on the basis of a BDI score of 17 points or higher, in approximately half of the MSA patients studied.13 When the results obtained from MSA patients were compared with those of PD patients collected from the literature, Schrag et al concluded that there were no differences between the BDI scores of MSA and PD patients. Schrag et al (2010)14 used the Hospital Anxiety and Depression Scale (HADS) in a multicenter study to compare psychiatric disturbances in 286 MSA patients and 188 PSP patients. No HC were included in this study. The authors found that 43% of the MSA patients had probable depression and 37.3% had probable anxiety. Depressive symptoms measured with the BDI, appeared to be an independent predictor of quality of life in a study that compared 46 MSA patients with 40 PSP patients.15 In a recent multicenter study on 61 MSA patients and 20 PD patients used as controls, Siri et al (2013)16 found that the mean total Neuropsychiatric Inventory (NPI) value in MSA patients was not statistically different from that in PD patients (16.2±15.7 and 21.9±14.8, respectively), but that mild to severe depression, as assessed by the Geriatric Depression Scale (GDS), was present in 62% of MSA patients and in only 10% of PD patients.
Data on whether neuropsychiatric symptoms are more frequent in one of the two variants of MSA (MSA-P and MSA-C) are controversial. Kawai et al (2008),17 in a study in which the primary endpoint was to assess the degree of cognitive impairment, investigated 21 patients with MSA-C, 14 patients with MSA-P, and 21 age- and education-matched HC. Using the HADS, the authors found that patients with MSA-P and MSA-C had comparably higher levels of depression (8.7±3.8 and 9.1±3.8, respectively) than HC (4.6±2.7). Balas et al (2010)18 compared cognitive deficits and mood disorders in 15 patients with MSA-P, ten with MSA-C, 12 with PD, and ten HC. In this study, the presence of neuropsychiatric disturbances was evaluated by means of patient self-administered questionnaires (state anxiety, trait anxiety, depression). When compared with HC, MSA-P patients displayed abnormally increased levels of depression and anxiety, whereas MSA-C patients only displayed an increased level of anxiety. More recently, Kawahara et al (2015)19 examined cognition, affective ability, and vitality in 25 patients with MSA-C, eight with MSA-P, and 14 with cortical cerebellar atrophy. Both MSA-C and MSA-P patients had higher GDS scores than patients with cortical cerebellar atrophy. Cao et al (2015)20 investigated frontal cognitive and behavioral deficits using the Frontal Assessment Battery and Frontal Behavioral Inventory (FBI) in 110 Chinese patients with MSA (43 with MSA-P and 67 with MSA-C) and 55 HC. They found that 41.6% of the patients had a frontal lobe dysfunction, characterized mainly by deficits in inhibitory control. Logophenic aphasia was present in 60.9% of the sample and apathy in 43.6%. Higher FBI scores were associated with increased severity of disease, as assessed by the Unified Multiple System Rating Scale.
Patients with MSA only rarely manifest visual hallucinations, being reported in approximately 5% of cases compared to up of 75% of PD patients. Therefore, despite not excluding a diagnosis of MSA, visual hallucinations should at least raise suspicions of a different diagnosis.21
Table 1 provides a summary of studies assessing neuropsychiatric disturbances in MSA.
PSP
PSP is a rare neurological disorder characterized by usually symmetrical parkinsonism, supranuclear gaze palsy, axial rigidity, gait disturbances, postural instability with early falls, and fronto-limbic cognitive dysfunction.22 Clinicopathological studies have revealed that several clinical and pathological variants of PSP exist.23,24 Pathologically, PSP is characterized by abnormal hyperphosphorylated tau accumulation in several areas of the brain.22
Neuropsychiatric dysfunction in PSP
Neuropsychiatric alterations have been described in PSP patients even in the early phases of the disease. Neuro-behavioral abnormalities (personality changes, depressive symptoms, emotional lability) were already present at diagnosis in 31 out of 52 PSP patients in a study on the natural history of the disease.25 In another clinical cohort enrolled in a natural history study, Nath et al (2003)26 described personality changes, irritability, anhedonia, and depressed mood upon presentation in 15% of the 187 PSP patients studied. Neuropsychiatric disturbances at disease onset, as assessed by means of the NPI, were also found in two of the 22 PSP patients studied by Litvan et al (1996).27 More specific neuropsychiatric disturbances emerge in PSP patients during the disease course. The NPI detected apathy in 20 of the 22 patients studied by Litvan et al (1996).27 In their study, the apathy score in PSP patients was higher than that observed in 40 HC and 50 patients with Alzheimer’s disease (AD). Moreover, apathy was detected in 80% of the patients at some time during the course of the disease and was rated in most cases as moderate to severe. In a subsequent study on 34 PSP patients, Litvan et al (1998)28 confirmed, by means of the NPI, that apathy was the most frequently reported psychiatric symptom and that it was significantly more frequent in PSP patients than in 15 CBD patients and 25 HC. Similarly, 93 of the 187 PSP patients studied by Nath et al (2003)26 displayed apathy, although no formal rating scale was used in their study. When Aarsland et al (2001)29 compared the NPI scores in 61 PSP patients and 144 PD patients (though not in HC), they found that apathy and disinhibition scores were higher in PSP patients than in PD patients, whereas hallucination and delusion scores were higher in PD than in PSP patients.29 The NPI was also used in a more recent multicenter study on a sample of 154 PSP patients.30 The authors of that study confirmed that apathy, which was present in 62% of the sample, was the most frequent behavioral symptom in PSP; however, depressive symptoms, which were reported in 58% of the patients, also emerged as a significant feature. No relationship was detected either between apathy and depression scores or between neuropsychiatric disturbances and age, age at onset, education, disease duration, and severity of motor impairment (as measured by means of the Progressive Supranuclear Palsy Rating Scale). Using the FBI in 68 PSP and 57 CBD patients, Borroni et al (2009)31 found apathy in more than half of the PSP patients and in approximately one third of the CBD patients. Apathy in their study did not correlate with the severity of motor impairment as assessed by the UPDRS. In another study based on the Cambridge Behavioral Inventory, which is a caregiver-based questionnaire, the apathy scores assigned by caregivers were high in 35% of PSP patients despite low scores in the mood subtest.32
Other studies have demonstrated that depressive symptoms are the most frequent neuropsychiatric abnormality in PSP patients. Esmonde et al (1996)33 diagnosed depression, as measured by means of the BDI, in 13 of the 25 PSP patients they studied. Depressive symptoms were mild in seven patients, moderate in three, and severe in three (data on HC were not provided). Depression in their study did not correlate with other clinical or demographic parameters such as the severity of disease or cognitive impairment. When Schrag et al (2010)14 used the HADS to compare 188 PSP patients with 286 MSA patients, they found that 55.6% of the PSP patients had probable depression. Interestingly, the HADS depression subscore in their study was the factor more significantly associated with quality of life measures. Depression is a main finding in PSP patients even when a more conservative psychiatric assessment, such as the structured psychiatric interview, is used. When Menza et al (1995)34 used the Diagnostic and Statistical Manual for Mental disorders, 3rd edition criteria, they found that two of the 19 PSP patients studied had major depression, one had dysthymia, and five were diagnosed with adjustment disorders. However, when these 19 PSP patients were compared with a sample of 42 PD patients, no differences emerged between the two patient groups in either the frequency or the severity of psychiatric disturbances. Bloise et al (2014)35 conducted a case–control study using the structured clinical interview for DSM-IV (SCID-I) on 28 PSP patients and 28 age-matched HC. The authors found that psychiatric disturbances were significantly more frequent in PSP patients than in controls (53% vs 17.3%). It is noteworthy that eight patients were diagnosed as having mood disorders due to PSP.35 Supporting the hypothesis that depression may be part of the clinical spectrum of PSP, is the observation that no differences in the clinical demographic variables emerged between PSP patients with and those without depressive disorders or symptoms either in the study by Bloise et al (2014)35 or in the study by Gertstenecker et al (2013).30 Depression in PSP patients may be severe enough to lead, albeit very rarely, to a completed suicide.36
Anxiety, agitation, and irritability are also observed in PSP patients, though to a lesser extent than apathy and depression. Indeed, only three of the 19 PSP patients studied by Menza et al (1995)34 had anxiety disorders. This finding was confirmed by the fact that anxiety did not emerge as a major determinant of health status in the 188 PSP patients studied by Schrag et al (2010)14 by means of the HADS and that the presence of anxiety explained only 5% of the variance relative to health status. When Gerstenecker et al (2013)30 used the NPI, they found that significant anxiety was present in 24% of the PSP patients, agitation in 36%, irritability in 33%, and disinhibition in 32%. By contrast, no PSP patient was diagnosed as having an anxiety disorder in the case–control study by Bloise et al (2014),35 which suggests that the use of a structured clinical interview yields the most conservative results.
PSP patients also display neuropsychiatric symptoms that reflect frontal lobe dysfunction, such as emotional blunting, disinterest and disinhibition behaviors, stereotypies, rituals, and greedy eating habits. Disinhibition may be present in up to one third of PSP patients according to some studies.27,29 At least one antisocial behavior (physical assault, hypersexuality, neglect of the traffic rules, and public urination) was reported in five out of ten PSP patients in another study.37 By contrast, Borroni et al (2009)31 did not describe typical disinhibition behaviors in any of their PSP patients. In the only study that assessed the presence of obsessive compulsive personality disorders in PSP, Nicoletti et al (2016)38 compared 14 PSP patients with 15 MSA patients, 16 essential tremor patients, and 20 HC. Obsessive compulsive personality disorders were observed in five (35.7%) PSP patients, two (13.3%) MSA patients, two (12.5%) essential tremor patients, and two (10%) controls. PSP patients may also display disordered social interactions, such as problems in interacting and communicating with family members. Changes in emotion processing, which may be studied by using tests that assess facial expression recognition, may, at least in part, be responsible for these problems. Indeed, facial expression recognition for all negative emotions was found to be impaired in PSP patients,39 and recognition of sad and happy emotional faces was found to be impaired to a greater extent in PSP patients than in PD patients matched for age, disease duration, cognition, and depression.40
Hallucinations and delusions are rarely observed in PSP. None of the 24 PSP patients with a mean disease duration of approximately 3 years studied by Borroni et al (2008)41 displayed delusions or hallucinations. When pathologically confirmed cases of PSP were compared with clinically misdiagnosed PSP cases with other pathologies,42 psychosis was found to be the only behavioral abnormality that was less frequent in the pathologically confirmed PSP cases (18% vs 41%, P<0.01). Papapetropolous and Mash (2005)43 reported that only two of the 22 pathologically confirmed PSP patients displayed visual hallucinations during the disease course. Similar data emerged from a large, retrospective multicenter study based on a pathologically confirmed diagnosis, in which only ten of the 100 PSP patients had visual hallucinations during the disease course.44 When hallucinations are present, their characteristics are similar to those observed in PD.43
Table 2 provides a summary of studies assessing neuropsychiatric disturbances in PSP.
CBD
CBD is a rare, progressive neurodegenerative disorder with variable clinical presentation. Corticobasal syndrome (CBS), the most frequent clinical presentation of pathologically confirmed CBD, is characterized by an akinetic-rigid parkinsonian syndrome with a poor response to levodopa and a combination of asymmetric motor (rigidity, akinesia, dystonia, and myoclonus) and non-motor (apraxia, agnosia, cortical sensory loss, and alien limb phenomena) symptoms, which may be associated with cognitive-behavioral disorders.45,46 Other clinical presentations are possible. CBD is characterized by neuronal tau pathology that mainly affects the forebrain.45,46
Neuropsychiatric symptoms in CBD
When Mimura et al (1997)47 described the clinical features of three CBD patients, they found that two had a depressed mood (Table 3). In a study designed to compare the neuropsychological functions in 21 CBD patients and 21 AD patients, depressive symptoms were found to be more severe in CBD than in AD (GDS score 14.2 and 5.2, respectively).48 The GDS score in CBD patients correlated with disease duration.48 Litvan et al (1998)28 were the first to systematically investigate the frequency and type of neuropsychiatric disturbances in patients with CBD. By using the NPI, the authors found that depression was present in 73% of patients, apathy in 40%, irritability in 20%, and agitation in 20%. When they compared the NPI scores of CBD patients, PSP patients, and HC, the authors of that study found that depression was more frequent in CBD and that apathy was more frequent in PSP. Wenning et al (1998)49 clinically followed 14 CBD patients whose diagnosis was later confirmed at the post-mortem examination. The authors found that apathy, irritability, and disinhibition were present at disease onset in three patients and were observed in a further four patients at the last visit. A more recent study on a cohort of 36 neuropathologically confirmed CBD patients documented the presence of neuropsychiatric disturbances in 22% of the patients.50 The authors classified the patients in three subtypes according to their predominant clinical phenotype: 1) progressive aphasia/frontotemporal dementia (FTD); 2) FTD; and 3) CBS. These three groups displayed different neuropsychiatric traits: patients with an aphasia/FTD phenotype displayed compulsive behaviors, socially inappropriate and disinhibited behavior, and a voracious appetite, patients with an FTD phenotype displayed irritability, aggressivity, and impaired judgement, while those with a CBS phenotype mainly displayed depression. Borroni et al (2008)41 compared the presence and type of behavioral disturbances in CBD, PD, Lewy body dementia (LBD), and PSP patients. The NPI scores were indicative of depressive symptoms in 50% of the CBD patients. In a subsequent study, Borroni et al (2009)31 reported frontal lobe dysfunction symptoms in CBD patients whose score was similar to that observed in PSP patients. An analysis of the FBI subscores in their study revealed that spontaneity and language were impaired in CBD patients, but that they did not display disinhibition. Moretti et al (2005)52 investigated whether CBD patients with clearly asymmetric symptoms differ in terms of neuropsychiatric disturbances. When they used the Behavioral Pathology in AD rating scale to specifically measure seven items from the scale (delusions, hallucinations, activity disorders, aggressiveness, sleep disturbances, affective disorders, and anxiety and phobias), the authors observed that patients with left-sided symptoms displayed a greater degree of disinhibition, apathy and irritability, and a lower degree of depression than patients with predominantly right-sided symptoms.52 Lastly, visual hallucinations and delusions in CBD patients were not described either in the study by Litvan et al (1998)28 or in that by Geda et al (2007).50
Table 3 provides a summary of studies assessing neuropsychiatric disturbances in CBD.
Discussion
In the present paper, we reviewed the data from studies that assessed the frequency and type of neuropsychiatric disturbances in MSA, PSP, and CBD, which are the most frequent APs.51 The studies reviewed confirm that neuropsychiatric disturbances, a frequent finding in all three disorders: 1) are characterized by relatively heterogeneous clinical manifestations; 2) are not secondary to the severity of motor impairment; 3) may be present even at disease onset, before motor symptoms become manifest; and 4) may be detrimental to the quality of life of both patients and their caregivers.
Early studies on neuropsychiatric symptoms in MSA have provided conflicting results, probably because of limitations in the number of patients studied and in the methodology used to assess psychiatric symptoms.9,10 When using validated questionnaires such as the BDI and the NPI, MSA patients display moderate to severe depressive symptoms and affective detachment,11–16 which have a marked impact on quality of life.13–15,54 A position paper by the Neuropsychology Task Force of the MDS-MSA study group confirmed that approximately 37% of patients with MSA may manifest depression.53 Studies that have compared MSA with PD failed to show clear differences between these two conditions.9–11 Whether neuropsychiatric disturbances differ between the two variants of MSA (MSA-P and MSA-C) is unclear.16,17,19 The presence of depression in MSA in all the studies we reviewed appears to be independent of the severity of motor impairment, thereby suggesting that depression is part of the clinical spectrum of the disease rather than secondary to the severity of motor dysfunction.
Studies that adopted the structured psychiatric interview in PSP have consistently demonstrated that depressive disorders are more frequent in PSP patients than in controls, and are more frequent than anxiety disturbances.34,35 The observation that depression is due to PSP35 (according to the SCID) and that the severity of depressive symptoms does not correlate with the severity of motor impairment30,33,35 suggest that depression is part of the clinical spectrum of the disease and not merely a reactive process. Depression may also be a major determinant of subjective health status in PSP patients,14,15 albeit to a lesser degree than symptoms related to frontal lobe dysfunction.54 When less conservative diagnostic instruments, such as questionnaires, were used to measure neuropsychiatric symptoms, apathy was shown to be the most frequent symptom in PSP patients.27–31 As the presence and severity of apathy and depression are not correlated in PSP,14,30 these two symptoms should be considered separately.
PSP patients also display symptoms related to frontal lobe dysfunction. Disinhibition, changes in eating habits (voracious appetite), and socially inappropriate behaviors may be observed, together with increased levels of anxiety and agitation.14,27,30,41 The presence of these symptoms, which sometimes occur early in the disease course, may complicate the differential diagnosis not only with degenerative disorders such as FTD but also with primary psychiatric conditions. One noteworthy observation is that delusions and hallucinations are very rarely observed in PSP patients.42,43 This observation is in contrast to the high rate of hallucinations and delusions observed in synucleinopathies such as LBD, PD, and PD-dementia.21 The absence of visual hallucinations may thus serve as a useful diagnostic marker in uncertain cases that supports a diagnosis of PSP.
Owing to the variable clinical phenotypic presentation of CBD,55 studies on neuropsychiatric disturbances in this disorder are more difficult to interpret. If studies based on a pathologically confirmed diagnosis are considered,49,50 neuropsychiatric disturbances in CBD patients are characterized above all by depression and apathy, whereas disinhibition and behavioral abnormalities are less frequent. Studies designed to compare CBD and PSP patients have shown that depression is more common in CBD patients while apathy is more common in PSP patients.28,29 As in PSP, hallucinations and delusions are very rarely reported in CBD patients.
MSA patients manifest a decreased activity in all major neurotransmitter systems.56–59 Most of the aforementioned studies detected correlations between changes in neurotransmitter function and motor and vegetative symptoms, though it is unclear whether changes in neurotransmission also underlie neuropsychiatric symptoms in MSA. Other studies have shown that mood disturbances in MSA patients correlate with deficits in cognitive performances,18 thereby highlighting a possible pathophysiological link between frontal cognitive and behavioral/psychiatric disturbances in patients with MSA. These findings are in keeping with neuroimaging studies that have revealed frontal lobe dysfunction in patients with MSA.6
In PSP, apathy may result from auto-activation deficits and a lack of self-initiated behaviors, which may be due to the disconnection of the orbitofrontal cortex (OFC) and lateral prefrontal regions from the sub-cortical regions, including the pulvinar, dorsomedial and anterior nuclei of the thalamus, and superior and inferior culliculum.60 Depression in PSP may, at least in part, be due to the severe dopaminergic loss observed in this condition both in the striatum61 and in the extranigral A10 midbrain cell groups, which innervate the motor and limbic cortical and thalamic regions.62 Severe noradrenergic changes may be directly linked to cognitive and behavioral manifestations in PSP, such as rigidity and impulsivity.63 Cholinergic abnormalities may also be involved in neuropsychiatric abnormalities in PSP.64–66
In CBD, Huey et al (2015)67 found that reduced gray matter volume in the right medial ventral prefrontal cortex (PFC) was associated with increased anxiety and apathy, reduced volume in the right lateral ventral PFC with apathy and inappropriate repetitive behaviors, and reduced volume in the left lateral ventral PFC with poor performance in executive function tests. The same authors found that degeneration of the right medial OFC correlated with increased anxiety and apathy in patients with CBS, whereas degeneration of the lateral OFC was related to a greater extent with apathy and inappropriate behaviors.67 Clinicopathological studies have also shown that neuronal loss in the subthalamic nucleus and substantia nigra in patients with pathologically confirmed CBD whose initial symptoms fell within the neurobehavioral domain was less severe than that observed in patients who displayed motor symptoms from disease onset.68 Recent neuroimaging69–71 and neurophysiological findings72 suggest that asymmetric brain changes in patients with CBD may underlie the pathophysiology of asymmetric clinical symptoms, including neuropsychiatric and behavioral abnormalities. Finally, the loss of dopaminergic activity may explain the high frequency of depressive disorders observed in CBD patients.59,73
The review we conducted highlighted a number of limitations in the studies considered. First, few studies used a standardized psychiatric interview to diagnose psychiatric disorders. This is particularly true of the studies conducted on MSA, none of which assessed the presence of neuropsychiatric disorders through a structured interview. Another limitation that emerged is that not all the studies examined compared the results obtained in MSA, PSP, and CBD with those of HC or patients with other neurodegenerative disorders. Due to the relatively rare conditions investigated, several studies we reviewed enrolled only a limited number of patients. Lastly, the clinical assessment of the patients was followed by a pathological confirmation of the diagnosis in few studies. It should also be underlined that the majority of papers we reviewed only investigated the presence of neuropsychiatric symptoms in the different APs. Only a limited number of papers, mainly case series or case reports, have reported the efficacy of drug treatment in psychiatric disturbances. Future studies should therefore be designed to evaluate the efficacy and safety of different interventions in neuropsychiatric disturbances in APs.
Conclusion
We found that depression is the most frequent neuropsychiatric disturbance in all three disorders examined. Further studies are needed to understand whether other neuropsychiatric symptoms, such as apathy, anxiety, and behavioral disturbances, markedly affect MSA patients. Besides manifesting depression, PSP patients also display a high frequency of apathy, disinhibition, personality changes, and eating disorders; anxiety and irritability are two other symptoms that are relatively common in PSP. The neuropsychiatric profile of CBD patients is similar to that observed in PSP patients, although studies in which these two populations were compared suggest that depression is more common in CBD while apathy is more common in PSP. Finally, as hallucinations, delusions, and psychoses are rarely observed in all these disorders, the presence of such symptoms should raise the suspicion of a different diagnosis. We conclude that as the evidence regarding the burden exerted by neuropsychiatric symptoms on the quality of life of patients, as well as of their caregivers, is unequivocal, it should be assessed on a routine basis.
Disclosure
The authors report no conflicts of interest in this work.
References
Stamelou M, Hoeglinger GU. Atypical parkinsonism: an update. Curr Opin Neurol. 2013;26(4):401–405. | ||
Stamelou M, Bhatia KP. Atypical parkinsonism: diagnosis and treatment. Neurol Clin. 2015;33(1):39–56. | ||
Kertesz A, Mcmonagle P. Behavior and cognition in corticobasal degeneration and progressive supranuclear palsy. J Neurol Sci. 2010;289(1–2):138–143. | ||
Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700. | ||
Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci. 1999;163(1):94–98. | ||
Krismer F, Wenning GK. Multiple system atrophy: insights into a rare and debilitating movement disorder. Nat Rev Neurol. 2017;13(4):232–243. | ||
Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670–676. | ||
Papp MI, Lantos PL. Accumulation of tubular structures in oligodendroglial and neuronal cells as the basic alteration in multiple system atrophy. J Neurol Sci. 1992;107(2):172–182. | ||
Pilo L, Ring H, Quinn N, Trimble M. Depression in multiple system atrophy and in idiopathic Parkinson’s disease: A pilot comparative study. Biol Psychiatry. 1996;39(9):803–807. | ||
Gill CE, Khurana RK, Hibler RJ. Occurrence of depressive symptoms in Shy-Drager syndrome. Clin Auton Res. 1999;9(1):1–4. | ||
Fetoni V, Soliveri P, Monza D, Testa D, Girotti F. Affective symptoms in multiple system atrophy and Parkinson’s disease: response to levodopa therapy. J Neurol Neurosurg Psychiatry. 1999;66(4):541–544. | ||
Benrud-Larson LM, Sandroni P, Schrag A, Low PA. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord. 2005;20(8):951–957. | ||
Schrag A, Geser F, Stampfer-Kountchev M, et al. Health-related quality of life in multiple system atrophy. Mov Disord. 2006;21(6):809–815. | ||
Schrag A, Sheikh S, Quinn NP, et al. A comparison of depression, anxiety, and health status in patients with progressive supranuclear palsy and multiple system atrophy. Mov Disord. 2010;25(8):1077–1081. | ||
Winter Y, Spottke AE, Stamelou M, et al. Health-related quality of life in multiple system atrophy and progressive supranuclear palsy. Neurodegener Dis. 2011;8(6):438–446. | ||
Siri C, Duerr S, Canesi M, et al. A cross-sectional multicenter study of cognitive and behavioural features in multiple system atrophy patients of the parkinsonian and cerebellar type. J Neural Transm. 2013;120(4):613–618. | ||
Kawai Y, Suenaga M, Takeda A, et al. Cognitive impairments in multiple system atrophy: MSA-C vs MSA-P. Neurology. 2008;70(16 Pt 2):1390–1396. | ||
Balas M, Balash Y, Giladi N, Gurevich T. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369–375. | ||
Kawahara Y, Ikeda Y, Deguchi K, et al. Simultaneous assessment of cognitive and affective functions in multiple system atrophy and cortical cerebellar atrophy in relation to computerized touch-panel screening tests. J Neurol Sci. 2015;351(1–2):24–30. | ||
Cao B, Zhao B, Wei Q-Q, et al. The Global Cognition, Frontal Lobe Dysfunction and Behavior Changes in Chinese Patients with Multiple System Atrophy. PLoS One. 2015;10(10):e0139773. | ||
Bertram K, Williams DR. Visual hallucinations in the differential diagnosis of parkinsonism: Table 1. J Neurol Neurosurg Psychiatry. 2012;83(4):448–452. | ||
Colosimo C, Bak TH, Bologna M, Berardelli A. Fifty years of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2014;85(8):938–944. | ||
Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord. 2017;32(6):853–864. | ||
Williams DR, de Silva R, Paviour DC, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain. 2005;128(6):1247–1258. | ||
Maher ER, Lees AJ. The clinical features and natural history of the Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. 1986;36(7):1005–1008. | ||
Nath U, Ben-Shlomo Y, Thomson RG, Lees AJ, Burn DJ. Clinical features and natural history of progressive supranuclear palsy: a clinical cohort study. Neurology. 2003;60(6):910–916. | ||
Litvan I, Mega MS, Cummings JL, Fairbanks L. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology. 1996;47(5):1184–1189. | ||
Litvan I, Cummings JL, Mega M. Neuropsychiatric features of corticobasal degeneration. J Neurol Neurosurg Psychiatry. 1998;65(5):717–721. | ||
Aarsland D, Litvan I, Larsen JP. Neuropsychiatric symptoms of patients with progressive supranuclear palsy and Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 2001;13(1):42–49. | ||
Gerstenecker A, Duff K, Mast B, Litvan I; ENGENE-PSP Study Group. Behavioral abnormalities in progressive supranuclear palsy. Psychiatry Res. 2013;210(3):1205–1210. | ||
Borroni B, Alberici A, Agosti C, Cosseddu M, Padovani A. Pattern of behavioral disturbances in corticobasal degeneration syndrome and progressive supranuclear palsy. Int Psychogeriatr. 2009;21(3):463–468. | ||
Bak TH, Crawford LM, Berrios G, Hodges JR. Behavioural symptoms in progressive supranuclear palsy and frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2010;81(9):1057–1059. | ||
Esmonde T, Giles E, Gibson M, Hodges JR. Neuropsychological performance, disease severity, and depression in progressive supranuclear palsy. J Neurol. 1996;243(9):638–643. | ||
Menza MA, Cocchiola J, Golbe LI. Psychiatric Symptoms in Progressive Supranuclear Palsy. Psychosomatics. 1995;36(6):550–554. | ||
Bloise MC, Berardelli I, Roselli V, et al. Psychiatric disturbances in patients with progressive supranuclear palsy: A case-control study. Parkinsonism Relat Disord. 2014;20(9):965–968. | ||
Wiener J, Moran MT, Haut MW. Completed suicide in a case of clinically diagnosed progressive supranuclear palsy. Neurodegener Dis Manag. 2015;5(4):289–292. | ||
Yatabe Y, Hashimoto M, Kaneda K, et al. Neuropsychiatric symptoms of progressive supranuclear palsy in a dementia clinic. Psychogeriatrics. 2011;11(1):54–59. | ||
Nicoletti A, Luca A, Luca M, et al. Obsessive compulsive personality disorder in Progressive Supranuclear Palsy, Multiple System Atrophy and Essential Tremor. Parkinsonism Relat Disord. 2016;30:36–39. | ||
Ghosh BC, Rowe JB, Calder AJ, Hodges JR, Bak TH. Emotion recognition in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2009;80(10):1143–1145. | ||
Pontieri FE, Assogna F, Stefani A, et al. Sad and happy facial emotion recognition impairment in progressive supranuclear palsy in comparison with Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(7):871–875. | ||
Borroni B, Turla M, Bertasi V, et al. Cognitive and behavioral assessment in the early stages of neurodegenerative extrapyramidal syndromes. Arch Gerontol Geriatr. 2008;47(1):53–61. | ||
Josephs KA, Dickson DW. Diagnostic accuracy of progressive supranuclear palsy in the Society for Progressive Supranuclear Palsy Brain Bank. Mov Disord. 2003;18(9):1018–1026. | ||
Papapetropoulos S, Mash DC. Visual Hallucinations in Progressive Supranuclear Palsy. Eur Neurol. 2005;54(4):217–219. | ||
Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. 2014;14:1758–1766. | ||
Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496–503. | ||
Alexander SK, Rittman T, Xuereb JH, Bak TH, Hodges JR, Rowe JB. Validation of the new consensus criteria for the diagnosis of corticobasal degeneration. J Neurol Neurosurg Psychiatry. 2014;85(8):925–929. | ||
Mimura M, Whire RF, Albert ML. Corticobasal degeneration: neuropsychological and clinical correlates. J Neuropsychiatry Clin Neurosci. 1997;9(1):94–98. | ||
Massman PJ, Kreiter KT, Jankovic J, Doody RS. Neuropsychological functioning in cortical-basal ganglionic degeneration: Differentiation from Alzheimer’s disease. Neurology. 1996;46(3):720–726. | ||
Wenning GK, Litvan I, Jankovic J, et al. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry. 1998;64(2):184–189. | ||
Geda YE, Boeve BF, Negash S, et al. Neuropsychiatric features in 36 pathologically confirmed cases of corticobasal degeneration. J Neuropsychiatry Clin Neurosci. 2007;19(1):77–80. | ||
Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet. 1999;354(9192):1771–1775. | ||
Moretti R, Torre P, Antonello RM, Cattaruzza T, Cazzato G. Cognitive impairment in the lateralized phenotype of corticobasal degeneration. Dement Geriatr Cogn Disord. 2005;20(2–3):158–162. | ||
Stankovic I, Krismer F, Jesic A, et al. Cognitive impairment in multiple system atrophy: A position statement by the neuropsychology task force of the MDS multiple system atrophy (MODIMSA) study group. Mov Disord. 2014;29(7):857–867. | ||
Schrag A, Selai C, Davis J, et al. Health-related quality of life in patients with progressive supranuclear palsy. Mov Disord. 2003;18(12):1464–1469. | ||
Ling H, O’Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. 2010;133(7):2045–2057. | ||
Schmeichel AM, Buchhalter LC, Low PA, et al. Mesopontine cholinergic neuron involvement in Lewy body dementia and multiple system atrophy. Neurology. 2008;70(5):368–373. | ||
Benarroch EE. Brainstem in multiple system atrophy: clinicopathological correlations. Cell Mol Neurobiol. 2003;23(4/5):519–526. | ||
Benarroch EE, Schmeichel AM, Parisi JE. Depletion of mesopontine cholinergic and sparing of raphe neurons in multiple system atrophy. Neurology. 2002;59(6):944–946. | ||
Nikolaus S, Antke C, Müller HW. In vivo imaging of synaptic function in the central nervous system: I. Movement disorders and dementia. Behav Brain Res. 2009;204(1):1–31. | ||
Pagonabarraga J, Kulisevsky J, Strafella AP, Krack P. Apathy in Parkinson’s disease: clinical features, neural substrates, diagnosis, and treatment. Lancet Neurol. 2015;14(5):518–531. | ||
Hornykiewicz O, Shannak K. Brain monoamines in progressive supranuclear palsy – comparison with idiopathic Parkinson’s disease. J Neural Transm. 1994;42:219–229. | ||
Murphy KE, Karaconji T, Hardman CD, Halliday GM. Excessive dopamine neuron loss in progressive supranuclear palsy. Mov Disord. 2008;23(4):607–610. | ||
Murley AG, Rowe JB. Neurotransmitter deficits from frontotemporal lobar degeneration. Brain. 2018;141(5):1263–1285. | ||
Ruberg M, Javoy-Agid F, Hirsch E, et al. Dopaminergic and cholinergic lesions in progressive supranuclear palsy. Ann Neurol. 1985;18(5):523–529. | ||
Roy R, Niccolini F, Pagano G, Politis M. Cholinergic imaging in dementia spectrum disorders. Eur J Nucl Med Mol Imaging. 2016;43(7):1376–1386. | ||
Shinotoh H, Namba H, Yamaguchi M, et al. Positron emission tomographic measurement of acetylcholinesterase activity reveals differential loss of ascending cholinergic systems in Parkinson’s disease and progressive supranuclear palsy. Ann Neurol. 1999;46(1):62–69. | ||
Huey ED, Lee S, Brickman AM, et al. Neuropsychiatric effects of neurodegeneration of the medial versus lateral ventral prefrontal cortex in humans. Cortex. 2015;73:1–9. | ||
Ikeda C, Yokota O, Nagao S, et al. Corticobasal degeneration initially developing motor versus non-motor symptoms: a comparative clinicopathological study. Psychogeriatrics. 2014;14(3):152–164. | ||
Upadhyay N, Suppa A, Piattella MC, et al. MRI gray and white matter measures in progressive supranuclear palsy and corticobasal syndrome. J Neurol. 2016;263(10):2022–2031. | ||
Abe Y, Kimura N, Goto M, Aso Y, Matsubara E. Brain Perfusion in Corticobasal Syndrome with Progressive Aphasia. Dement Geriatr Cogn Dis Extra. 2016;6(1):133–141. | ||
Bharti K, Bologna M, Upadhyay N, et al. Abnormal Resting-State Functional Connectivity in Progressive Supranuclear Palsy and Corticobasal Syndrome. Front Neurol. 2017;8:1–9. | ||
Suppa A, di Stasio F, Marsili L, et al. Primary motor cortex LTP/LTD-like plasticity in probable corticobasal syndrome. J Neurophysiol. 2016;115(2):717–727. | ||
Klaffke S, Kuhn AA, Plotkin M, et al. Dopamine transporters, D2 receptors, and glucose metabolism in corticobasal degeneration. Mov Disord. 2006;21(10):1724–1727. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.