Back to Journals » Drug Design, Development and Therapy » Volume 13
Neuroprotective effects of melatonin and celecoxib against ethanol-induced neurodegeneration: a computational and pharmacological approach
Authors Al Kury LT ![]() , Zeb A
, Zeb A ![]() , Abidin ZU, Irshad N
, Abidin ZU, Irshad N ![]() , Malik I, Alvi AM
, Malik I, Alvi AM ![]() , Khalil AAK, Ahmad S, Faheem M
, Khalil AAK, Ahmad S, Faheem M ![]() , Khan AU, Shah FA
, Khan AU, Shah FA ![]() , Li S
, Li S
Received 1 March 2019
Accepted for publication 27 June 2019
Published 2 August 2019 Volume 2019:13 Pages 2715—2727
DOI https://doi.org/10.2147/DDDT.S207310
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yan Zhu
Lina T Al Kury,1 Alam Zeb,2 Zain Ul Abidin,2 Nadeem Irshad,3 Imran Malik,2 Arooj Mohsin Alvi,2 Atif Ali Khan Khalil,4 Sareer Ahmad,4 Muhammad Faheem,2 Arif-Ullah Khan,2 Fawad Ali Shah,2 Shupeng Li5
1College of Natural and Health Sciences, Zayed University, Abu Dhabi, United Arab Emirates; 2Riphah Institute of Pharmaceutical Sciences, Riphah International University, Islamabad, Pakistan; 3Department of Pharmacy, Quaid-I-Azam University, Islamabad, Pakistan; 4Rehman Medical Institute, Peshawar, Pakistan; 5State Key Laboratory of Oncogenomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, People’s Republic of China
Purpose: Melatonin and celecoxib are antioxidants and anti-inflammatory agents that exert protective effects in different experimental models. In this study, the neuroprotective effects of melatonin and celecoxib were demonstrated against ethanol-induced neuronal injury by in silico, morphological, and biochemical approaches.
Methods: For the in silico study, 3-D structures were constructed and docking analysis performed. For in vivo studies, rats were treated with ethanol, melatonin, and celecoxib. Brain samples were collected for biochemical and morphological analysis.
Results: Homology modeling was performed to build 3-D structures for IL1β), TNFα, TLR4, and inducible nitric oxide synthase. Structural refinement was achieved via molecular dynamic simulation and processed for docking and postdocking analysis. Further in vivo experiments showed that ethanol induced marked neuronal injury characterized by downregulated glutathione, glutathione S-transferase, and upregulated inducible nitric oxide synthase. Additionally, ethanol increased the expression of TNFα and IL1β. Finally, neuronal apoptosis was demonstrated in ethanol-intoxicated animals using caspase 3 and activated JNK staining. On the other hand, melatonin and celecoxib treatment ameliorated the biochemical and immunohistochemical alterations induced by ethanol.
Conclusion: These results demonstrated that ethanol induced neurodegeneration by activating inflammatory and apoptotic proteins in rat brain, while melatonin and celecoxib may protect rat brain by downregulating inflammatory and apoptotic markers.
Keywords: simulation, docking, cortex, hippocampus, ethanol, neurodegeneration
Corrigendum for this paper has been published.
Introduction
Alcohol abuse and alcoholism are associated with serious health issues across the world. Alcoholics are prone to numerous neurological problems, including learning deterioration, memory deficits, and depression, indicating that ethanol exposure is detrimental to brain development and neuronal proliferation and migration. Alcoholism induces neuronal susceptibility by triggering cascades of pathological events that overwhelm the physiological defense capacity of the brain and subsequently make neuronal cells prone to apoptosis and neurodegeneration.1,2 Studies have shown that neuroinflammation occupies a prominent position in ethanol-induced neurodegeneration, as it links to many apoptotic and transduction pathways. Ethanol elevates inflammatory protein levels and alters the antioxidant defense system in the brain.3 Toll-like receptors (TLRs) have been studied extensively in ethanol-induced neurodegeneration, as they are linked to many downstream cytokine targets, such as TNFα, IL1β, IL6, and MCP1. Rapid activation of astrocytes and microglial cells also release inflammatory cytokines, followed by infiltration of inflammatory cells.5 Furthermore, these inflammatory factors induce oxidative stress, amass calcium concentration, and inhibit the electron-transport chain.4 Release of these inflammatory factors aggravates the preexisting situation and renders neuronal cells prone to death,6 whereas attenuating these inflammatory markers will assist in neuronal survival.7
Melatonin is a potent antioxidant secreted by the pineal gland. As a powerful free-radical scavenger, melatonin diminishes neuronal death provoked by various neurotoxic substances, and its neuroprotective properties have been demonstrated in different neurodegenerative disorders and ischemic models.7–10 The amphiphilic nature of melatonin enables expedient blood–brain barrier permeation, which makes it an ideal candidate drug for brain disorders.11,12 Furthermore, melatonin interacts with dozens of receptors and signaling and transducing pathways, giving it multiple promising biological activities.13 Finally, its low toxicity profile and clinical safety records make it an ideal candidate as a neuroprotective agent. Celecoxib is a US Food and Drug Administration–approved COX2 inhibitor prescribed in the management of various painful and inflammatory conditions. Several studies have demonstrated the anti-inflammatory, antiapoptotic, and antioxidant potential of celecoxib.14,15 The neuroprotective effects of celecoxib have also been observed in various neurodegenerative models, such as lipopolysaccharide-induced cognitive deficit and 6-hydroxydopamine-induced degeneration of nigrostriatal neurons.16,17 In this study, we examined the neuroprotective effects of celecoxib and melatonin against ethanol-induced inflammatory and oxidative damage in adult rat brain.
Methods
Bioinformatic resources
Sequence information for IL1β, HO1, COX2, Nrf2, TLR4, inducible nitric oxide synthase (iNOS), and TNFα in rats was downloaded in FASTA format from the online UniProt database (http://www.uniprot.org). Sequences of these proteins were aligned in basic local alignment searching tool (BLAST) to probe the closest identical template. The BLAST sequenced the proteins against the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB), which resulted in several protein templates. The template showing high sequence identity was selected for homology modeling.18 Homology modeling was performed using the online Swiss-Model server. FASTA sequences of selected templates were utilized for modeling.19 Models generated were further refined by molecular dynamic simulation using Gromacs version 5.0.6 with CHARMM27 force-field parameterization.15,20 In short, a dodecahedron box filled with a TIP3P water model was prepared for each protein. CHARMM27 force-field parameters were applied for atomic representation. Systems were neutralized by Na+ and/or Cl– counterions.21 Neutralized systems were further exposed to energy minimization in Gromacs. Energy-minimization parameters were optimized to 50,000 steps at 10 kJ/mol. Well-minimized systems were verified for energy minimization by calculating the potential energy of the system, and protein coordinates were saved as per the PDB for the corresponding protein model.
Models constructed were examined for structural stability and quality by validation procedures, such as Procheck22 (http://deposit.pdb.org/validate) and ProSA https://prosa.services.came.sbg.ac.at/prosa.php).23 Procheck deals with Ramachandran plots and divides protein residues into φ- and ψ-angles. ProSA validates the quality of the generated model by measuring a quality-score plot. Models then proceeded to docking studies. For docking, model proteins and ligands were prepared as PDB and Mol2, files respectively. Melatonin and celecoxib were first converted to PDBQT format using AutoDock Tools (1.5.6rc2). Both proteins and ligands were then passed through AutoDock Vina docking software, which interprets docking results in the form of binding energy (E-value). Well-docked poses of ligand in each target protein were further analyzed in Discovery Studio Visualizer in terms of ligand-pose orientation and molecular interactions.
Chemicals
Mouse anti-pJNK, mouse anti-TNFα, mouse anti-COX2, mouse anti–caspase 3, an ABC Elite kit, and 3,3′-diaminobenzidine peroxidase were purchased from Santa Cruz Biotechnology. The secondary antibody goat antimouse was obtained from Abcam. PBS tablets, formaldehyde, proteinase K, H2O2, and mounting media were obtained from BDH (Germany). Melatonin was purchased from Alfa Asar. Glutathione (GSH), trichloroacetic acid, 1-chloro-2,4-dinitrobenzene, N-(1-naphthyl)ethylenediamine dihydrochloride and 5,5ʹ-dithiobis(2-nitrobenzoic acid) were purchased from Sigma-Aldrich. All other chemicals, including celecoxib, were obtained from local sources after ensuring high analytical grade.
Animal grouping and drug treatment
Adult male Sprague Dawley rats weighing 260–300 g aged 9–12 weeks were obtained from the local breeding facility of Riphah International University. Experiments were carried out in accordance with approved guidelines, and all experimental protocols were approved by the research and ethics committee of Riphah Institute of Pharmaceutical Sciences, Riphah International University, Pakistan. We did not use any blind allocation of rats. Instead, we randomly divided the rats. We adhered to criteria to assign similar-weight animals to the same group under the same experimental condition. Rats were divided into five groups (ten per group): control group (normal saline 1 mL/kg intraperitoneally for 11 consecutive day, ethanol group (2 g/kg intraperitoneally for 11 consecutive days), ethanol + melatonin group (50 mg/kg for 11 consecutive days), ethanol + celecoxib group (50 mg/kg for 11 consecutive days), and (5) ethanol + celecoxib + melatonin group. At the end of treatment, rats were killed using diethyl ether inhalation. Brain tissue collected was dissolved in PBS (approximately 5% w:v, pH 7.4) and homogenized. The homogenate was centrifuged (3,500 g, 15 minutes) and supernatant collected for further biochemical analysis. For histological preparation, tissue samples were fixed in 4% paraformaldehyde, imbedded in paraffin, and 4 μm coronary sections taken with a rotary microtome in five rats per group.
H&E staining
Tissue sections on coated slides were deparaffinized with absolute xylene (100%) and rehydrated with ethyl alcohol (from 100% [absolute] to 70%). Slides were rinsed with distilled water and immersed in hematoxylin for 10 minutes. Slides were then kept under running water in a glass jar for 10 minutes and treated with 1% HCl and 1% ammonia water. The slides were added to eosin solution for 5–10 minutes. After an appropriate time, the slides were rinsed in water and air-dried. Dried slides were dehydrated in graded ethyl alcohol (70%, 95%, and 100%), cleared with xylene, and mounted with glass coverslips. The slides were imaged with light microscopy (Olympus) and analyzed with ImageJ. The number of images per slide was five, focusing specifically on neuronal cell size and shape, inflammatory infiltrated cells, and vacuolation. TIF images were optimized to the same threshold intensity for all groups.
Oxidative enzyme analysis
GSH was quantified according to previously reported protocols. Briefly, diluted tissue homogenate was mixed with PBS and freshly prepared 5,5ʹ-dithiobis(2-nitrobenzoic acid) solution and absorbance measured at 412 nm. GSH S-transferase (GST) levels were assayed according to previous protocols with minor modifications. Briefly, equal proportions of GST and 1-chloro-2,4-dinitrobenzene were diluted with 0.1 M PBS (pH 6.5). Optical density was determined at 340 nm after serial dilution form tissue homogenate.24
Immunohistochemical analysis
Immunohistochemical staining was performed as described previously, with few modifications.25 After deparaffinization, slides were processed for the antigen-retrieval step (enzymatic method), then washed with PBS. The endogenous peroxidase was quenched by applying 3% H2O2 in methanol for 10 minutes. Slides were incubated with 5% normal goat serum containing 0.1% Triton X-100. After blocking, slides were incubated overnight with mouse anti-JNK (pJNK), mouse anti–caspase 3, mouse anti-TNFα, and mouse anti-COX2 antibodies (dilution 1:100, Santa Cruz Biotechnology). The following morning, after being washed with 0.1 M PBS, slides were incubated in biotinylated secondary antibody (dilution 1:50) according to the origin of the primary antibody and serum used. Following secondary antibody treatment, slides were incubated with the ABC Elite kit for 1 hour in a humidified chamber, then washed with 0.1 M PBS, stained in 3,3′-diaminobenzidine peroxidase solution, washed with distilled water, dehydrated in a graded ethanol series, fixed in xylene, and coverslipped in mounting medium. Immunohistochemical TIF images were captured with light microscopy.
Statistical analysis
Morphological data were analyzed using ImageJ. Data are presented as means ± SEM. Data were analyzed by one-way ANOVA followed by post hoc Bonferroni multiple comparison using GraphPad Prism 5.
Results
Homology-modeling and validation process
The BLAST of rat IL1β, TNFα, TLR4, and iNOS identified murine IL1β, mouse TNFα, crystal structure of mouse TLR4, and iNOS complex with 7-nitroindazole as the best templates with reference to RCSB and were further processed for Swiss modeling. Generated models for each target were ranked based on global-model quality-estimation score, and the topmost models were chosen for further analysis.19 Selected models were further subjected to energy minimization via molecular dynamic simulation in Gromacs, and resultant 3-D structures are shown in Figure 1A.
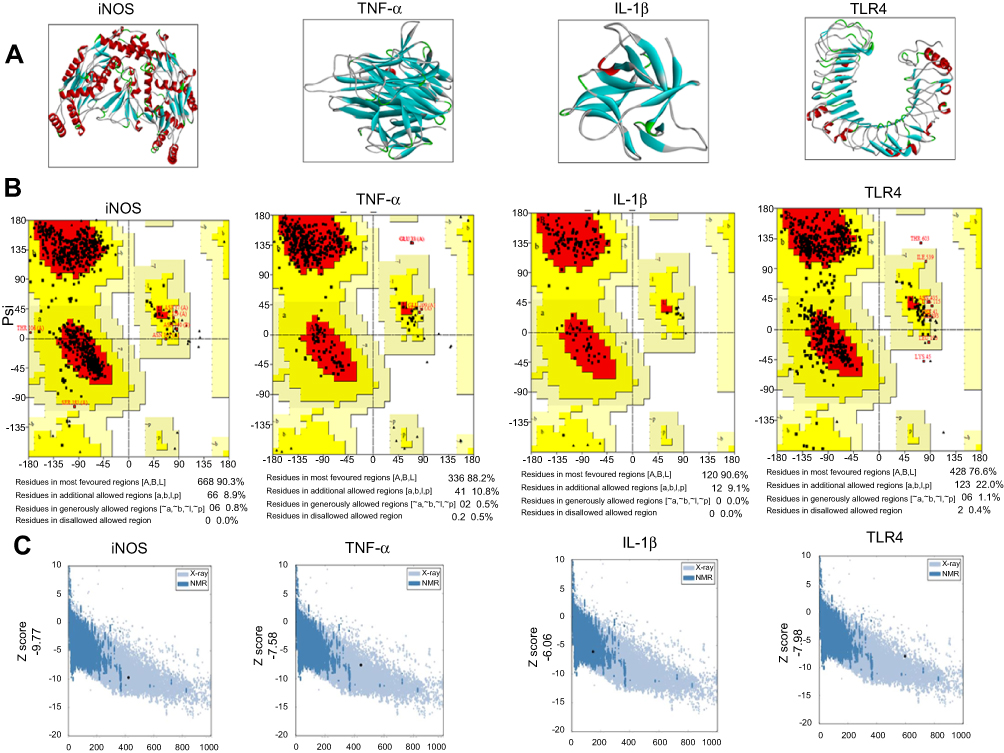 |
Figure 1 Computational analysis of IL1 β, TNFα, TLR4, iNOS residues. Notes: (A) Tertiary structures of iNOS, TNFα, IL1β, and TLR4. (B) Homology-modeling and tertiary-structure validation. Ramachandran plots for iNOS, TNFα, IL1β, and TLR4. (C) ProSA findings of iNOS, TNFα, IL1β, and TLR4. (D) Tertiary structures of COX2, Nrf2, HO1. (E) Sequence alignment of COX2 (rat accession number P35355 [PDB 1PXX]) and HO1 (rat accession number P06762 [PDB 1DVE]) by Clustal Omega. (F) Melatonin- and celecoxib-ligand structures drawn in ChemSketch and converted to PDB format by pymol.hgcghdc.Abbreviation: iNOS, inducible nitric oxide synthase. |
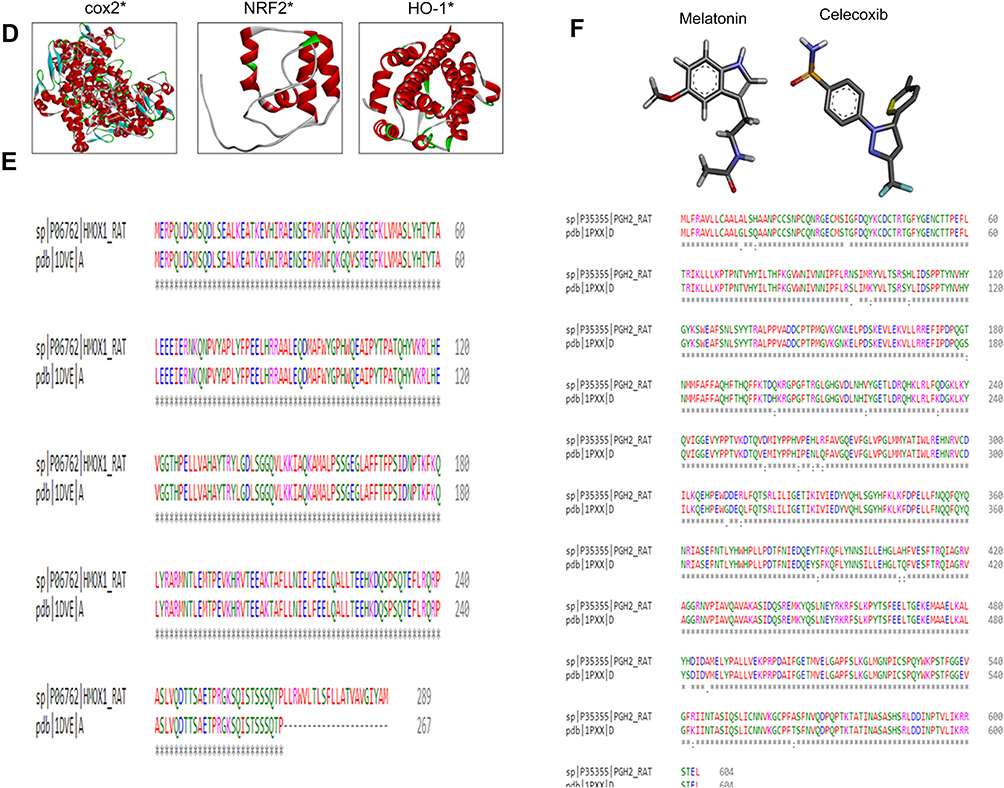 |
Figure 1 (Continued) |
The stereochemical integrity of these models was evaluated using Procheck.22 Ramachandran plots showed amino -acid residues of IL1β, TNFα, TLR4, and iNOS distributed within respective regions (Figure 1B). Briefly, amino -acid residues were 90.3%, 88.2%, 90.6%, and 76.6% in the favored regions for iNOS, TNFα, IL1β, and TLR4, respectively (Figure 1B). These models were further validated by the ProSA server for potential errors in terms of Z-scores and residual energy.23 Z-scores generally showed model quality, and negative residual energy values confirmed model uniformity. Z-scores for IL1β (−6.06), TNFα (−7.58), iNOS (−9.77), and TLR4 (−7.98) are shown in Figure 1C. Fortunately, BLAST analysis of COX2 (UniProt accession number P35355), Nrf2 (UniProt accession number O54968), and HO1 (UniProt accession number P06762) identified the chain A crystal structure of diclofenac bound to the COX active site (PDB 1PXX), human chain A, Nrf2 (PDB 2LZ1), and chain A, crystal structure of rat HO1 in complex with heme (PDB 1DVE) as the best identical-sequence templates (100% sequence identity) in the RCSB PDB, and are represented in the computational analysis in Figure 1D. To further validate the computational results, protein sequences were aligned by Clustal Omega.24 The results revealed that the entire sequences of these proteins were identical and conserved (Figure 1E). The structure of melatonin and celecoxib were retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/search). The 2-D structures were drawn in ChemSketch, subsequently converted to 3-D, and saved as Mol2 files in Discovery Studio Visualizer (Figure 1F).
Docking studies
Melatonin and celecoxib ligands were docked in catalytic active pockets of iNOS, IL1β, TNFα, COX2, Nrf2, HO1, and TLR4, as shown in Figures 2 and 3. Table 1 shows the binding energies and amino-acid residues involved in polar contacts. As demonstrated, three hydrogen bonds were formed between melatonin and iNOS (Figure 2, A and B). Pyrrole rings in melatonin formed two hydrogen bonds with Val395 and Pro396 of iNOS, with nitrogen in pyrrole rings as a hydrogen-bond donor (HBD), and oxygen in the methoxy group as a hydrogen-bond acceptor (HBA; Figure 2B). Similarly, one hydrogen bond was formed between the amide group (consisting of both an HBD and HBA) and the amino acid Ile392 (Figure 2B). Apart from this, van der Waals and electrostatic interactions were also observed, which further provided stability to the melatonin–iNOS complex.26 Docking results of melatonin and IL1β are represented in Figure 2, D–F. Pyrrole rings in melatonin formed one hydrogen bond with Leu67 of IL1β, and three hydrogen bonds were formed between the amide group (HBD and HBA) and amino acids Leu62 and Lys65 (Figure 2E). Figure 2, G–I shows the docking results of melatonin with TNFα, which revealed that pyrrole rings in melatonin formed one hydrogen bond with Ser98. Moreover, two hydrogen bonds were formed between the carbonyl group of the amide group (HBA) and Ser98 and Tyr114 (Figure 2H). Figure 2, J–L shows docking results of melatonin with COX2 and the formation of one hydrogen bonding between the amino group of amide and amino acid Gly522 (Figure 2K). Figure 2, M–O shows the docking results of melatonin with Nrf2, resulting in two hydrogen-bond formations: one between the amide group of melatonin and Ile33 of Nrf2, and the other between the methoxy group (HBA) and His32 (Figure 2N). Docking results for TLR4 and HO1 are shown in Figure 2, P–U.
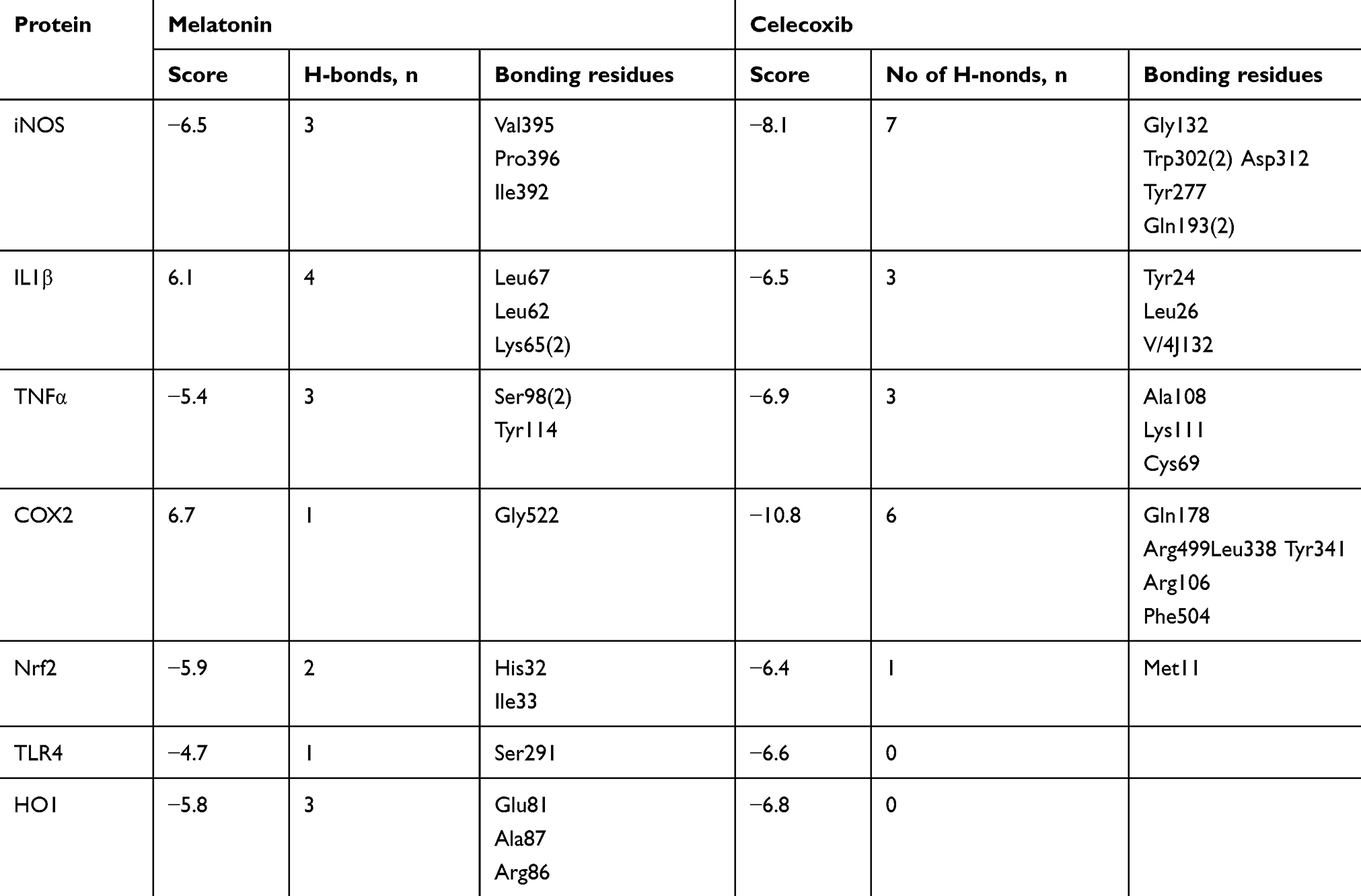 |
Table 1 Binding energy values after docking |
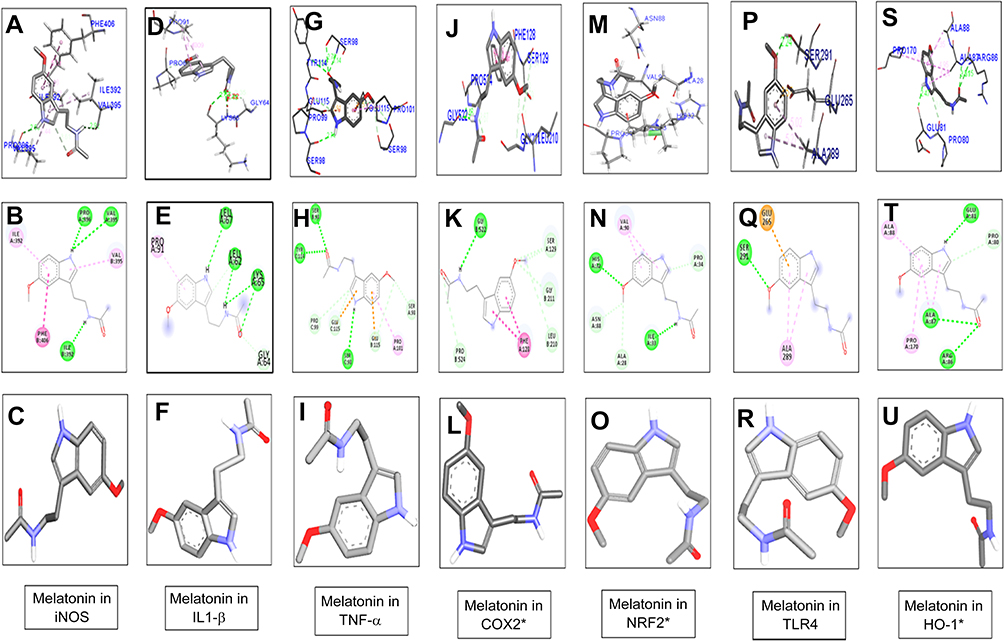 |
Figure 2 Docking results show the best pose of melatonin that fitted to iNOS, IL1β, TNFα, COX2, Nrf2, TLR4, and HO1.P Notes: Postdocking analyses visualized by Discovery Studio Visualizer in both 2-D and 3-D poses. Interaction between melatonin and iNOS (A, B), IL1β (D, E), TNFα (G, H), COX2 (J, K), Nrf2 (M, N), TLR4 (P, Q), and HO1 (S, T). 3-D poses (A, D, G, J, M, P, S) and 2-D (B, E, H, K, N, Q, T). (C, F, I, L, O, R, U) represents the best pose of melatonin that fitted to iNOS, IL1β, TNFα, COX2, Nrf2, TLR4, and HO1, respectively.Abbreviation: iNOS, inducible nitric oxide synthase. |
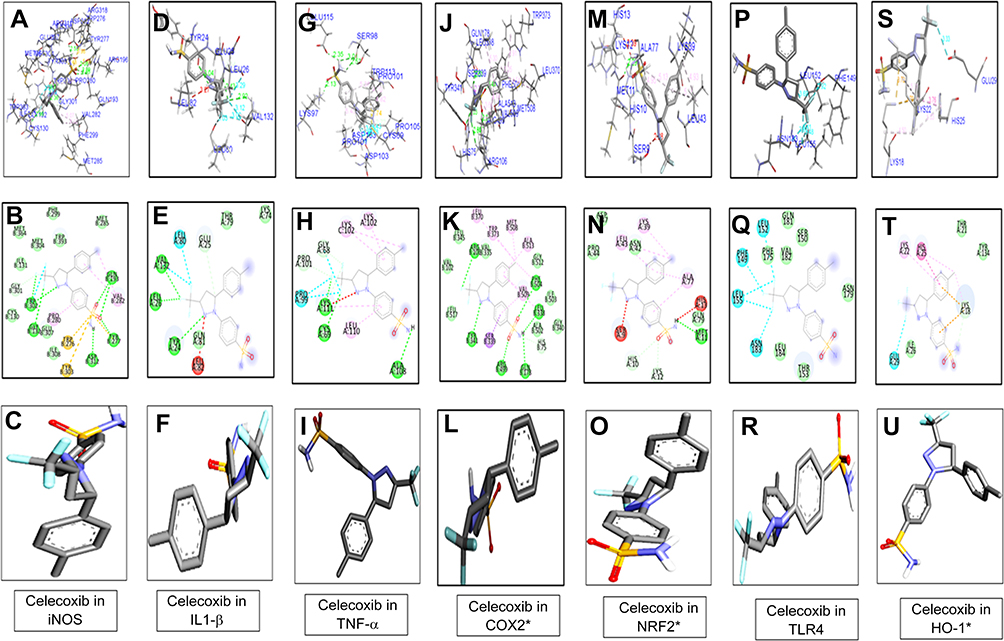 |
Figure 3 Docking results showed the best pose of celecoxib that fitted to iNOS, IL1-β, TNFα, COX2, Nrf2, TLR4, HO1. Post-docking analysis were visualized by Discovery Studio Visualizer in both 2-D and 3-D poses. Interaction between celecoxib and iNOS were shown by panel (A, ), IL1β by panel (D, E), TNFα by panel (G, H), COX2 by panel (J, K), Nrf2 by panel (M, N), TLR4 by panel (P, Q), and HO1 by panel (S, T). 3-D poses were shown by panel (A, D, G, J, M, P, S) and 2-D by panel (B, E, H, K, N, Q, T). Panel (C, F, I, L, O, R, U) represents the best pose of celecoxib that fits in iNOS, IL1β, TNFα, COX2, Nrf2, TLR4, HO1 respectively. Abbreviation: iNOS, inducible nitric oxide synthase. |
Effect of melatonin and celecoxib on ethanol-induced oxidative enzyme changes
The effect of melatonin and celecoxib on oxidative enzymes were then explored. As shown in Table 2, ethanol administration increased iNOS expression (P<0.001), but reduced the activity of GSH and GST in brain cortical tissue (P<0.001). Melatonin and celecoxib treatment attenuated the toxic effects of ethanol on oxidative enzymes (Table 2).
 |
Table 2 Effect of melatonin and celecoxib on oxidative enzymes |
Effect of melatonin and celecoxib on ethanol-induced neurodegeneration
To further examine the neuroprotective effects of melatonin and celecoxib, H&E staining was performed to examine neuronal loss after ethanol administration. Ethanol produced robust cellular changes in highly prone areas of brain, ie, the cortex and hippocampus (Figure 4A), while melatonin and celecoxib treatment attenuated this damage. As shown, ethanol induced aberrant morphological features, including alterations in neuronal size and shape, color staining (cytoplasmic eosinophilia/pyknosis and basophilic nature of nuclei), and vacuolation, whereas significantly more intact cells were observed in melatonin- and celecoxib-treated groups (P<0.05, Figure 4B).
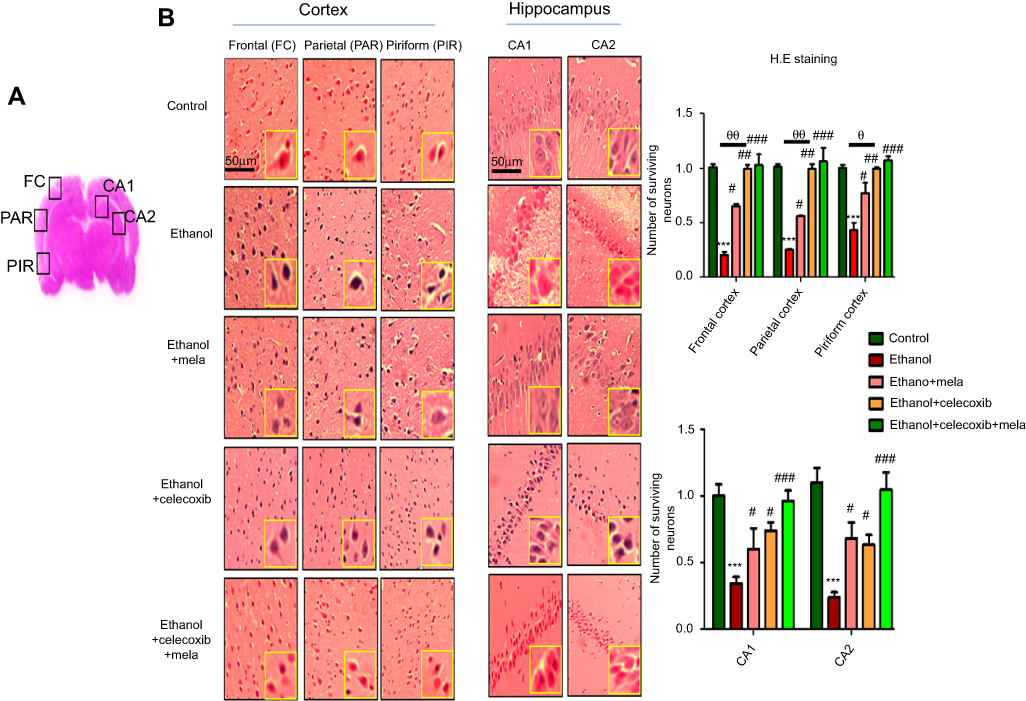 |
Figure 4 Effect of melatonin and celecoxib on neuronal cell death. Notes: (A) Regions of interest analyzed; (B) H&E staining showing the extent of surviving neurons in the cortex and hippocampus. Bar 50 μm, magnification 20×, n=5/group. Surviving neurons were marked by cytoplasmic swelling, scalloped neurons with intense cytoplasmic eosinophilia, and nuclear basophilia. These changes resulted from neuronal necrosis. Some cells had a shrunken appearance, along with pyknotic nuclei. Intensive neuropil vacuolation can be seen in the ethanol-only group. *Significant difference relative to control; #significant difference relative to ethanol group; θsignificant difference relative to ethanol + melatonin. Data presented as means ± SEM. Data analyzed by one-way ANOVA followed by post hoc Bonferroni multiple comparison using GraphPad Prism 5 software. ***P<0.001; #/θP<0.05; ##/θθP<0.01. |
Melatonin and celecoxib attenuated ethanol-induced neuronal apoptosis
p38 MAPK and JNKs are important members of the MAPK family involved in apoptotic pathways.27 JNK (as pJNK) activates apoptotic cell death by transcriptional and posttranscriptional modification.28,29 JNK is critical in death receptor–initiated extrinsic and mitochondrion-elicited intrinsic apoptotic pathways, and is responsible for a significant degree of apoptosis in various degenerative models.30,31 To reveal the possible involvement of JNK in the protective effects of melatonin against ethanol-induced apoptosis, immunohistochemical analysis was performed. The results showed that ethanol markedly activated JNK and caspase 3 in various cortical segments compared to the control group (P<0.001, Figure 5). Treatment with melatonin and celecoxib significantly reversed these changes and reduced expression of pJNK and caspase 3 (P<0.05, Figure 5).
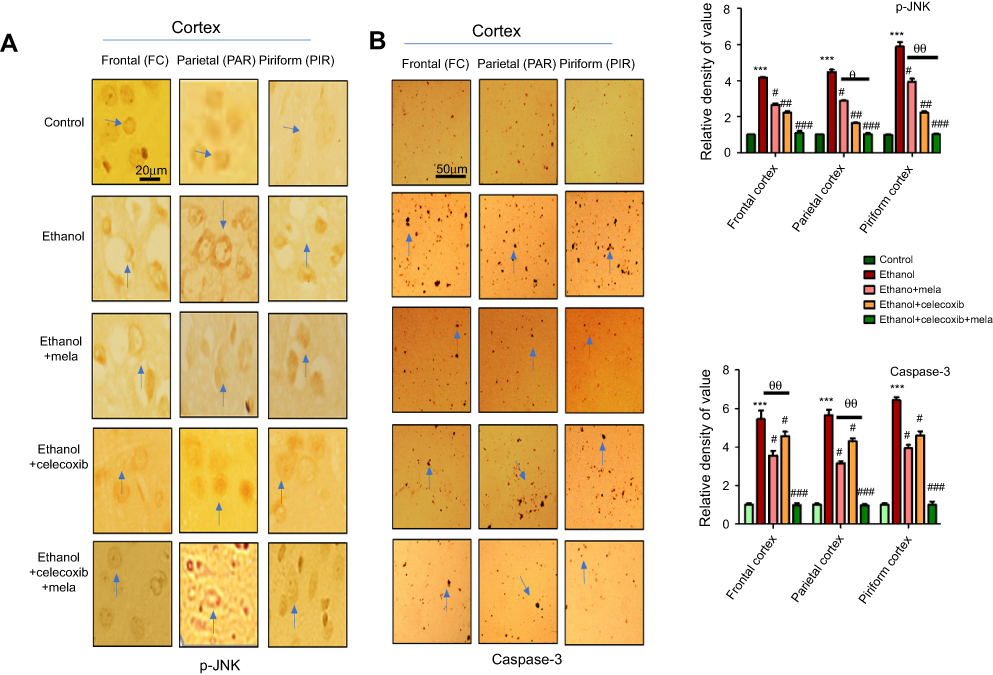 |
Figure 5 Effect of melatonin and celecoxib on apoptotic markers. Notes: (A) Immunohistochemistry results for pJNK. Bar 20 μm, magnification 40×. (B) Immunohistochemistry results for caspase 3 in frontal, partial, and pyriform cortices. Bar μm, magnification 20×, n=5/group. Both pJNK and caspase 3 exhibited cytoplasmic localization in the three performed. Histograms show comparatively higher expression of pJNK and caspase 3 in various segments of the ethanol-only group. *Significant difference relative to control; #significant difference relative to ethanol group; θsignificant difference relative to ethanol + melatonin. Data presented as means ± SEM. Data analyzed by one-way ANOVA followed by post hoc Bonferroni multiple comparison using GraphPad Prism 5 software. ***/###P<0.001; #/θP<0.05; ##/θθP<0.01. |
Effect of melatonin and celecoxib on inflammatory factors
JNK is involved in both adaptive and innate immunoresponses, as it can be activated by various proinflammatory cytokines, such as TNFα, IL1β, and TLR ligands. Moreover, a feedback mechanism exists between inflammatory cytokines and activated JNK, in which activation of one pathway will activate the other, suggesting critical roles of p38/JNK in inflammation.32,33 We thus investigated these inflammatory factors and found elevated expression of TNFα and COX2 after ethanol treatment in various cortical segments. Treatment with melatonin and celecoxib reversed this activation and significantly reduced the expression of TNFα and COX2 (Figure 6, P<0.05).
 |
Figure 6 Effect of melatonin and celecoxib on inflammatory cytokines. Notes: (A) Immunohistochemistry results for COX2; (B) immunohistochemistry results for TNFα in frontal, partial and pyriform cortices, Bar 20 μm, magnification 40×, n=5/group. Both COX2 and TNFα exhibited cytoplasmic localization in the three experiments performed. Histograms indicate comparative expression of COX2 and TNFα in various segments of the ethanol-only group. *Significant difference relative to control; #significant difference relative to the ethanol group; θsignificant difference relative to ethanol + melatonin group. Data presented as means ± SEM. Data analyzed by one-way ANOVA followed by post hoc Bonferroni multiple comparison using GraphPad Prism 5. **/##/θθP<0.01; #/*P<0.05; ***/###P<0.001. Arrows indicates neuronal cells, where COX2 and TNF expression were significantly modulated, further these arrowed cells are shown in high magnification in the same panel. |
Discussion
In this study, we compared the expression of inflammatory proteins in ethanol-, melatonin-, and celecoxib-treated groups. The focus of the study was to perform bioinformatic analysis and further determine the relative effects of melatonin and celecoxib. Since no 3-D structures for iNOS, TNFα, IL1β, or TLR4 are reported in the PDB for rats, homology modeling was performed to build 3-D structures. The stability of these structures was further assessed by molecular dynamic simulation, which is used to infer the real behavior of atoms under specified environmental conditions and is a well-practiced discipline in biological systems to explore the stability and physiological orientation and/or confirmation of biomolecules. Herein, Gromacs was used to minimize the energy of each modeled structure. We found rat COX2, Nrf2, and HO1 showed 100% sequence coverage with mouse COX2 (PDB 1PXX), human Nrf2 (PDB 2LZ1) and dimeric rat HO1 (PDB 1DVE), respectively, which was further aligned by Clustal Omega. The Omega analysis suggested that sequences of COX2, Nrf2, and HO1 in these species were conserved. Melatonin and celecoxib structures were drawn in ChemSketch and converted to PDB format with Pymol. Docking analysis was performed with Autodock Vina and binding energy evaluated. The interaction of docked poses of ligand (melatonin and celecoxib) in proteins was further visualized by Discovery Studio. Intermolecular interactions play an important role in stabilizing ligand(s) energetically in target protein(s). Therefore, molecular interactions were elaborated in terms of hydrogen bonding, van der Waals forces, and electrostatic interactions. Thorough analysis of computational docking showed that melatonin and celecoxib bound each target protein by forming H-bonds and other hydrophobic interactions. Hydrogen-bond interactions are important for internal stabilization, recognition, and molecular movement.34–36 Several studies have demonstrated the significance of hydrogen bonding in ligand–protein complex stability.37–39 The most stable interaction of hydrogen bond is observed at a bond distance of 2.6–3.2 A°. We speculate that the formation of H-bonds between proteins and ligands (melatonin and celecoxib) supports the correspondent complex stability.
The molecular mechanism underlying ethanol-induced neuronal damage is marked by complicated (complex) pathophysiological processes. Ethanol exposure leads to a cascade of events, such as glutamate excitotoxicity, energy failure, and formation of toxic radicals. The present study demonstrated that coadministration of melatonin and celecoxib attenuated neurodegeneration in the ethanol-intoxicated brain by downregulating inflammatory markers and reduced oxidative stress, similarly to previous reports.40 In line with docking studies (Table 1), a relatively higher antioxidant effect for celecoxib was observed in the ethanol-administered group (Table 2), which could partially be explained by the higher binding affinity of celecoxib with inflammatory proteins. Consistently with our results, several other studies have shown that melatonin and celecoxib attenuation activates the MAPK–p38–JNK pathway and inflammation by downregulating the pNFκB–pIKKβ pathway. Moreover, both agents are known to increase Nrf2 nuclear translocation and regulate the expression of genes involved in antioxidant response,7,41 which was further supported by the iNOS, GSH, and GST enzyme-activity changes in our study.
Inflammation worsens clinical prognosis and compromises therapeutic outcomes in many experimental models by triggering both resident and peripheral cells to migrate to the brain parenchyma, contributing to inflammatory pathogenesis. Release of inflammatory mediators induces ethanol-induced neuronal death by several mechanisms. First, activation of TLR4 on glial cells stimulates stress-related kinases, such as JNK and p38–MAPK,42 which triggers the mitochondrial apoptotic pathway. Second, following the activation of these kinases, inflammatory cytokines are produced, largely by activated microglia, astrocytes, and neurons, within an hour after ethanol insult.43 Third, activated nuclear transcriptional machinery like NFκB ultimately elicits the production of proinflammatory factors, such as TNFα, IL1β, and NO, eventually aggregating the damage from the initial pathological stimuli. It has been reported that NFκB activation by TLR4 triggers iNOS and COX2 production.42 Both COX2 and iNOS are toxic mediators of inflammatory cascades whose activity can be downregulated by inhibiting TLR4.44 We demonstrated attenuated expression of COX2, TNFα, and iNOS by melatonin and celecoxib, indicating their anti-inflammatory potential.
Our results are in accordance with previously reported data showing that melatonin and celecoxib prevented COX2 and iNOS overexpression in different models.45–47 Furthermore, the effects of celecoxib and melatonin on oxidative enzymes in this study are in line with previously reported data.48 The detailed molecular mechanisms underlying melatonin's and celecoxib's neuroprotective effects in ethanol-induced neurodegeneration are yet to be explored, but could be partially attributed to neuroinflammatory pathways and free radical–scavenging properties, as demonstrated previously.48 Consistent studies have demonstrated protective effects of melatonin and celecoxib in several related experimental models. Recent data have shown that melatonin and/or celecoxib may exert beneficial effects on synaptic receptors, inhibiting glutamate surge and calcium overload in brain.8,49 In heart tissue, melatonin and celecoxib attenuate inflammation-induced cardiac hypertrophy.50,51 Melatonin and celecoxib execute their effects by several mechanisms, such as inflammasomes and Wnt and BMP pathways.52–54 Moreover, further studies are required to investigate whether similar mechanisms are involved to mediate the effects of melatonin and celecoxib in ethanol-induced neurodegeneration.
Conclusion
Ethanol administration activates several proinflammatory mediators, including iNOS, TNFα, and COX2, and is further linked to ROS generation. Melatonin and celecoxib cotreatment synergistically attenuated ethanol-induced oxidative stress and the inflammatory cascade, eventually accounting for its neuroprotective effects against neuronal apoptosis.
Acknowledgment
This research was supported by the Start-Up Research Grant Program (SRGP), Ministry of Higher Education Commission (HEC), Pakistan (21-1615/SRGP/HEC).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Eckardt MJ, File SE, Gessa GL, et al. Effects of moderate alcohol consumption on the central nervous system. Alcohol Clin Exp Res. 1998;22:998–1040. doi:10.1111/j.1530-0277.1998.tb03695.x
2. Zeigler DW, Wang CC, Yoast RA, et al. The neurocognitive effects of alcohol on adolescents and college students. Prev Med. 2005;4023–4032.
3. Reddy VD, Padmavathi P, Kavitha G, et al. Alcohol induced oxidative/nitrosative stress alters brain mitochondrial membrane properties. Mol Cell Biochem. 2013;37:539–547.
4. Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122:2049–2063. doi:10.1053/gast.2002.33613
5. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17796–17808.
6. Qin L, Crews FT. NADPH oxidase and reactive oxygen species contribute to alcohol induced microglial activation and neurodegeneration. J Neuroinflammation. 2012;95.
7. Ali T, Rehman SU, Shah FA, et al. Acute dose of melatonin via Nrf2 dependently prevents acute ethanol-induced neurotoxicity in the developing rodent brain. J Neuroinflammation. 2018;15119.
8. Shah FA, Liu G, Al Kury LT, et al. Melatonin protects MCAO-induced neuronal loss via NR2A mediated prosurvival pathways. Front Pharmacol. 2019;10:297. doi:10.3389/fphar.2019.00297
9. Shah FA, Zeb A, Ali T, et al. Identification of proteins differentially expressed in striatum by melatonin in middle cerebral artery occlusion model-a proteomic and in silico approach. Front Neurosci. 2018;12:888. doi:10.3389/fnins.2018.00888
10. Espinar A, Garcia-Oliva A, Isorna EM, et al. Neuroprotection by melatonin from glutamate-induced excitotoxicity during development of the cerebellum in the chick embryo. J Pineal Res. 2000;28:81–88.
11. Chen HY, Chen TY, Lee MY, et al. Melatonin decreases neurovascular oxidative/nitrosative damage and protects against early increases in the blood–brain barrier permeability after transient focal cerebral ischemia in mice. J Pineal Res. 2006;41:175–182. doi:10.1111/j.1600-079X.2006.00352.x
12. Yeleswaram K, McLaughlin LG, Knipe JO, et al. Pharmacokinetics and oral bioavailability of exogenous melatonin in preclinical animal models and clinical implications. J Pineal Res. 1997;22:45–51. doi:10.1111/j.1600-079X.1997.tb00302.x
13. Cutando A, Lopez-Valverda A, Vicente DEJ, et al. Action of melatonin on squamous cell carcinoma and other tumors of the oral cavity (Review). Oncol Lett. 2014;7:923–926. doi:10.3892/ol.2014.1813
14. Takahashi T, Ogawa Y, Kitaoka K, et al. Selective COX-2 inhibitor regulates the MAP kinase signaling pathway in human osteoarthritic chondrocytes after induction of nitric oxide. Int J Mol Med. 2005;15:213–219.
15. Hamdulay SS, Wang B, Birdsey GM, et al. Celecoxib activates PI-3K/Akt and mitochondrial redox signaling to enhance heme oxygenase-1-mediated anti-inflammatory activity in vascular endothelium. Free Radic Biol Med. 2010;48:1013–1023. doi:10.1016/j.freeradbiomed.2010.01.017
16. Hunter RL, Dragicevic N, Seifert K, et al. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem. 2007;100:1375–1386. doi:10.1111/j.1471-4159.2006.04334.x
17. Sanchez-Pernaute R, Ferree A, Cooper O, et al. Selective COX-2 inhibition prevents progressive dopamine neuron degeneration in a rat model of Parkinson’s disease. J Neuroinflamm. 2004;16.
18. Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi:10.1016/S0022-2836(05)80360-2
19. Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:252–258. doi:10.1093/nar/gku340
20. Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. doi:10.1016/j.softx.2015.06.001
21. Zoete V, Cuendet MA, Grosdidier A, et al. SwissParam: A fast force field generation tool for small organic molecules. J Comp Chem. 2011;32:2359–2368. doi:10.1002/jcc.21816
22. Laskowski RA, MacArthur MW, Moss DS, et al. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. doi:10.1107/S0021889892009944
23. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:407–410. doi:10.1093/nar/gkm290
24. Chenna R, Sugawara H, Koike T, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi:10.1093/nar/gkg500
25. Shah FA, Park DJ, Koh PO. Identification of proteins differentially expressed by quercetin treatment in a middle cerebral artery occlusion model: a proteomics approach. Neurochem Res. 2018;1–16.
26. Bosshard HR, Marti DN, Jelesarov I. Protein stabilization by salt bridges: concepts, experimental approaches and clarification of some misunderstandings. J Mol Recog. 2004;17:1–16. doi:10.1002/jmr.657
27. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi:10.1152/physrev.00028.2011
28. Xia Z, Dickens M, Raingeaud J, et al. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi:10.1126/science.270.5244.1945
29. Tournier C, Hess P, Yang DD, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi:10.1126/science.288.5467.870
30. Okuno S, Saito A, Hayashi T, et al. The C-Jun N-terminal protein kinase signaling pathway mediates bax activation and subsequent neuronal apoptosis through interaction with bim after transient focal cerebral ischemia. J Neurosci. 2004;24:7879–7887. doi:10.1523/JNEUROSCI.0553-04.2004
31. Krajewski S, Mai JK, Krajewska M, et al. Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci. 1995;15:6364–6376. doi:10.1523/JNEUROSCI.15-10-06364.1995
32. Tu YF, Tsai YS, Wang LW, et al. Overweight worsens apoptosis, neuroinflammation and blood-brain barrier damage after hypoxic ischemia in neonatal brain through JNK hyperactivation. J Neuroinflamm. 2011;8:2094–2098. doi:10.1186/1742-2094-8-40
33. Nakajima K, Hirose H, Taniguchi M, et al. Involvement of BNIP1 in apoptosis and endoplasmic reticulum membrane fusion. Embo J. 2004;23:3216–3226. doi:10.1038/sj.emboj.7600333
34. Jeffrey GA, Saenger W. Hydrogen bonding in biological structures. Springer Sci Bus Media. 2012.
35. Baker E, Hubbard R. Hydrogen bonding in globular proteins. Prog Biophys Mol Biol. 1984;44:97–179. doi:10.1016/0079-6107(84)90007-5
36. Desiraju G, Steiner T. The weak hydrogen bond in chemistry and structural biology. IUCR Monographs Crystallogr. 1999;123:929–1021.
37. Sarkhel S, Desiraju GR. Hydrogen bonds in protein–ligand complexes: strong and weak interactions in molecular recognition. Proteins. 2004;54:247–259. doi:10.1002/prot.10567
38. Panigrahi SK, Desiraju GR. Strong and weak hydrogen bonds in the protein–ligand interface. Proteins. 2007;67:128–141. doi:10.1002/prot.21253
39. Glusker J. Intermolecular interactions around functional groups in crystals: data for modeling the binding of drugs to biological macromolecules. Acta Crystallogr D. 1995;51:418–427. doi:10.1107/S0907444995003313
40. Padillo FJ, Ruiz-Rabelo JF, Cruz A, et al. Melatonin and celecoxib improve the outcomes in hamsters with experimental pancreatic cancer. J Pineal Res. 2010;49:264–270. doi:10.1111/j.1600-079X.2010.00791.x
41. Al-Rashed F, Calay D, Lang M, et al. Celecoxib exerts protective effects in the vascular endothelium via COX-2-independent activation of AMPK-CREB-Nrf2 signalling. Sci Rep. 2018;8:6271. doi:10.1038/s41598-018-24548-z
42. Yao L, Kan EM, Lu J, et al. Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: role of TLR4 in hypoxic microglia. J Neuroinflamm. 2013;10:23. doi:10.1186/1742-2094-10-23
43. Rothwell NJ. Functions and mechanisms of interleukin 1 in the brain. Trends Pharmacol Sci. 1991;12:430–436. doi:10.1016/0165-6147(91)90623-Z
44. Yeligar SM, Machida K, Kalra VK. Ethanol-induced HO-1 and NQO1 are differentially regulated by HIF-1alpha and Nrf2 to attenuate inflammatory cytokine expression. J Biol Chem. 2010;285:35359–35373. doi:10.1074/jbc.M110.138636
45. El-Shenawy SM, Abdel-Salam OM, Baiuomy AR, et al. Studies on the anti-inflammatory and anti-nociceptive effects of melatonin in the rat. Pharmacol Res. 2002;46:235–243. doi:10.1016/S1043-6618(02)00094-4
46. Romana-Souza B, Santos JS, Bandeira LG, et al. Selective inhibition of COX-2 improves cutaneous wound healing of pressure ulcers in mice through reduction of iNOS expression. Life Sci. 2016;153:82–92. doi:10.1016/j.lfs.2016.04.017
47. Mayo JC, Sainz RM, Tan DX. Anti-inflammatory actions of melatonin and its metabolites, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) and N1-acetyl-5-methoxykynuramine (AMK), in macrophages. J Neuroimmunol. 2005;165:139–149. doi:10.1016/j.jneuroim.2005.05.002
48. Othman GQ. AL-ALWACHI SN. Influences of cyclooxygenase-2 inhibitor, melatonin and their combination on hepatocarcinogenesis in N-Itrosodiethylamine and partially hepatectomy treated rats. J Duhok Univ. 2009;12:118–124.
49. Lin TY, Lu CW, Wang CC, et al. Cyclooxygenase 2 inhibitor celecoxib inhibits glutamate release by attenuating the PGE2/EP2 pathway in rat cerebral cortex endings. J Pharmacol Exp Ther. 2014;351:134–145. doi:10.1124/jpet.114.217372
50. Tengattini S, Reiter RJ, Tan DX, et al. Cardiovascular diseases: protective effects of melatonin. J Pineal Res. 2008;44:16–25.
51. Zhang C, Wang F, Zhang Y, et al. Celecoxib prevents pressure overload‐induced cardiac hypertrophy and dysfunctin by inhibiting inflammation, apoptosis and oxidative stress. J Cell Mol Med. 2016;20:116–127. doi:10.1111/jcmm.12709
52. Huang C, Chen Y, Liu H, et al. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin E2 and down-regulating the Wnt pathway activity. Oncotarget. 2017;8:115254–115264. doi:10.18632/oncotarget.v8i70
53. Park KH, Kang JW, Lee EM, et al. Melatonin promotes osteoblastic differentiation through the BMP/ERK/Wnt signaling pathways. J Pineal Res. 2011;51:187–194. doi:10.1111/j.1600-079X.2011.00875.x
54. Hua KF, Chou JC, Ka SM, et al. Cyclooxygenase-2 regulates NLRP3 inflammasome-derived IL-1β production. J Cell Physiol. 2015;230:863–874. doi:10.1002/jcp.24815
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.