")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Neuroprotective effect of N-acetylcysteine against cisplatin-induced toxicity in rat brain by modulation of oxidative stress and inflammation
Authors Abdel-Wahab WM , Moussa FI
Received 17 October 2018
Accepted for publication 7 February 2019
Published 11 April 2019 Volume 2019:13 Pages 1155—1162
DOI https://doi.org/10.2147/DDDT.S191240
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Georgios Panos
Wessam M Abdel-Wahab,1,2 Farouzia I Moussa1
1Department of Zoology, Faculty of Science, University of Alexandria, Alexandria, Egypt; 2Department of Basic Sciences/Biology Unit, Deanship of Preparatory Year and Supporting Studies, Imam Abdulrahaman Bin Faisal University, Dammam, Saudi Arabia
Background: Neurotoxicity is a major obstacle to the effectiveness of cisplatin (CDDP) in cancer chemotherapy. Oxidative stress and inflammation are considered to be the major mechanisms involved in CDDP-induced neurotoxicity. The rationale of our study was to investigate the efficacy of N-acetylcysteine (NAC) at two different doses in the management of CDDP-induced toxicity in rat brain by monitoring its antioxidant and anti-inflammatory effects.
Methods: Thirty-five male rats were divided into five groups (n=7) as follows: control group (0.5 mL saline), NAC100 group (100 mg/kg), CDDP group (8 mg/kg), NAC50-CDDP group (50 mg/kg NAC and 8 mg/kg CDDP), and NAC100-CDDP group (100 mg/kg NAC and 8 mg/kg CDDP). NAC was administered for 20 consecutive days, while CDDP was injected once on day 15 of the treatment protocol.
Results: The neurotoxicity of CDDP was evidenced by a marked increase in acetylcholinesterase and monoamine oxidase activities. It also induced oxidative stress as indicated by increased levels of lipid peroxidation, nitric oxide, and protein carbonyl with a concomitant decline in reduced glutathione, glutathione peroxidase, glutathione S-transferase, superoxide dismutase, and catalase in the brain. Moreover, CDDP enhanced the synthesis of pro-inflammatory cytokines such as tumor necrosis factor-α, interleukin-1β, and interleukin-6. Treatment with NAC at the two selected doses significantly attenuated CDDP-induced changes in the brain cholinergic function, improved the brain oxidant/antioxidant status, and also reversed the overproduction of pro-inflammatory cytokines in brain and serum.
Conclusion: NAC could serve as an appropriate and safe complementary therapeutic agent to attenuate the toxicity of CDDP in the brain and therefore improve its outcomes in chemotherapy.
Keywords: cisplatin, neurotoxicity, brain, N-acetylcysteine, rats
Introduction
Platinum-derived chemotherapies are widely used for the treatment of several tumors.1 As one of the leading platinum chemotherapies, cisplatin (CDDP) has been extensively prescribed since it was approved by the US Food and Drug Administration in 1978.2 However, the adverse treatment reactions and the multiple iatrogenic toxicities constrain the therapeutic usefulness of CDDP.3 Neurotoxicity has been identified as one of the most serious problems associated with CDDP chemotherapy.4 About 40%–50% of patients receiving CDDP therapy develop moderate or severe neuropathy which may result in discontinuation or even terminating the course of the chemotherapy.5 Many studies reported on peripheral neuropathy of CDDP; however, few reports focused on its central neurotoxicity.6–8
Different mechanisms have been presumed for the neurotoxic effect of CDDP, including oxidative injury, activation of pro-inflammatory cytokines, and apoptosis.9 Interstrand and intrastrand cross-linking of DNA is also believed to be one of the important mechanisms involved in the toxicity of CDDP.10 Furthermore, free oxygen radicals are known to induce oxidative DNA damage.11 CDDP can also affect the mitochondria through the formation of adducts with mitochondrial DNA which evokes morphological alterations and induces failure in energy production.12 The therapeutic role played by CDDP cannot be abandoned. Therefore, finding out a new and safe adjunct therapy to protect against the neurotoxicity of CDDP is a prime goal of preclinical studies. Since oxidative stress and inflammation are key hallmarks in the development of CDDP neurotoxicity, using an agent possessing antioxidant and anti-inflammatory activities may be promising against the drug-related toxicity.
N-acetylcysteine (NAC) is thiolic amino acid that has been reported to scavenge free radicals, replenish reduced glutathione (GSH) and prevent its depletion, and inhibit lipid peroxidation (LPO).13 It can also restore the deterioration in the pro-oxidant/antioxidant balance via its metal-chelation activity.14 NAC possesses neurovascular-protective actions and shows a positive effect in the treatment of neurological disorders through its ability to downregulate inflammation and oxidative stress.15,16 Furthermore, NAC has been shown to be able to cross the blood–brain barrier and improve brain dysfunctions.17,18 It also increases the level of brain-derived neurotrophic factor which supports the survival of existing neurons and can stimulate neurogenesis.16 We previously demonstrated the protective effect of NAC, either alone or combined with taurine, against the nephrotoxicity of CDDP in rats.19 In the current study, we aimed to further validate the neurotoxicity of CDDP in rat brain. Hitherto, the protective effect of NAC against the toxicity of CDDP in the brain of rats was not reported. Therefore, the main goal of our study was to investigate, for the first time, the effectiveness of NAC at two different doses in the management of CDDP-induced brain injury in rats. Markers of neurotoxicity, oxidant/antioxidant indices, and pro-inflammatory mediators were monitored to have an insight into the possible mechanism(s) by which NAC possibly protects the brain against CDDP toxicity in rats.
Materials and methods
Chemicals and drugs
CDDP (Cis-diaminodicholoroplatinum II) and NAC (the N-acetyl derivative of cysteine) were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Both agents were dissolved in physiological saline. The assay kits for tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) were obtained from RayBiotech (Norcross, GA, USA). All other reagents and assay kits used in this study were purchased from Biodiagnostic Co. (Giza, Egypt).
Animals and experimental design
Thirty-five adult male rats (weighing 190–210 g) were used in the study. Rats were housed in cages at a temperature of 22°C±2°C and under a 12-hour light/dark cycle. Food and water were available to the rats throughout the study. All procedures and animal handling conformed to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The experimental protocol of the study was approved by the Institutional Animal Care and Use Committee of Alexandria University, Egypt (Approval number AU04190126301). Rats were acclimatized to the laboratory conditions for 7 days, and then they were randomly assigned to five experimental groups (n=7). Group 1 (untreated control) was injected with 0.5 mL saline once daily throughout the experimental study. Group 2 (NAC100 group) was administered with 100 mg/kg of NAC. Group 3 (CDDP group) received a single dose of CDDP (8 mg/kg). Group 4 (CDDP-NAC50) received 50 mg/kg of NAC and 8 mg/kg of CDDP. Group 5 (CDDP-NAC100) was administered with 100 mg/kg of NAC and 8 mg/kg of CDDP. The doses of NAC and CDDP were selected based on the literature.18,20 Both CDDP and NAC were injected intraperitoneally in 0.9% saline. CDDP was injected once on day 15 of the study, while the administration of NAC started 14 days before CDDP administration and continued thereafter for 5 more days.
Separation of serum and preparation of whole-brain homogenate
Twenty-four hours after completion of the treatment protocol, rats were sacrificed under anesthesia for collecting blood and brain samples. Serum was separated by centrifugation at 3,000 rpm for 15 minutes. The collected sera were used for measuring the levels of TNF-α, IL-1β, and IL-6. The whole brain was isolated, freed from any adherent fatty tissues, and washed with physiological saline. Brain extract was prepared by homogenization of 1 g of the brain tissue in 10 volumes of cold potassium phosphate buffer (pH 7.4) containing 0.1 mM ethylenediaminetetraacetic acid (EDTA) using Potter-Elvehjem type glass-Teflon homogenizer. The homogenate was centrifuged at 4,000 rpm for 20 minutes and the clear supernatant was separated and stored at −20°C for further assays.
Determination of acetylcholinesterase and monoamine oxidase activities in the brain
The activity of acetylcholinesterase (AChE) in the brain, expressed as μg/mg protein, was measured according to the method of Ellman et al.21 Monoamine oxidase (MAO) activity was assayed in the brain based on the enzymatic oxidation of benzylamine hydrochloride and was expressed as nmol/mg protein.22
Assessment of brain oxidant and antioxidant status
Brain LPO (as malondialdehyde [MDA]) was determined according to the method of Ohkawa et al.23 It was expressed as nmol/mg protein. Protein carbonyl (PC), an indicator of protein oxidation, was measured based on the reaction with 2,4-dinitrophenylhydrazine.24 The carbonyl content was measured at 430 nm and expressed as nmol/mg protein. Nitric oxide level was assayed based on the reaction of nitrite content with Griess reagent to form a colored compound that absorbs light at 540 nm.25 The results were expressed as nmol/mg protein. The total antioxidant capacity of the brain was determined based on inhibition of the blue-green color of ABTS (2,2′-azino-bis [3-ethylbenzothiazoline-6-sulfonic acid]) radical by the antioxidants in the sample.26 Inhibition of the color formation was proportional to the concentration of total antioxidants, and results were expressed as mmol Trolox equivalent/mg protein. GSH content was determined in the brain using Elman’s reagent (5, 5′-Dithiobis [2-nitrobenzoic acid]) and the results were expressed as mmol/mg protein.27 The activity of glutathione-S-transferase (GST) was determined by measuring the conjugation of 1-chloro-2,4-dinitrobenzene to GSH and the subsequent increase in absorbance at 340 nm.28 The GST activity was directly proportional to the rate of increase. The activity of glutathione peroxidase (GPx) was measured indirectly by coupled reaction with glutathione reductase.29 Catalase (CAT) activity was determined by monitoring the decline in absorbance due to the degradation of hydrogen peroxide (H2O2) at 240 nm.30 Superoxide dismutase (SOD) activity was determined using nitro blue tetrazolium dye.31 The activities of GST, GPx, CAT, and SOD were expressed as U/mg protein.
Measurement of pro-inflammatory cytokines in the brain homogenate and the serum
The levels of TNF-α, IL-1β, and IL-6 were determined in both the brain and the serum according to the method illustrated in the ELISA kit. Absorbance was measured at 450 nm, and results were expressed as pg/mg protein when measured in the brain and as pg/mL when measured in the serum.
Data analysis
Statistical analysis was performed with the Statistical Package for Social Sciences version 17.0 for Windows. The normality of distribution of samples was tested by quantile–quantile plot. Data showed normal distribution and were presented as mean ± standard error of the mean (SEM). Differences among the experimental groups were tested by one-way ANOVA followed by Fisher’s least significance difference post hoc analysis. P<0.05 was considered to be statistically significant.
Results
NAC reduced the activities of acetylcholinesterase and monoamine oxidase in the brain of rats treated with CDDP
Figure 1A and B illustrates the effect of NAC on the activity of AChE and MAO in the brain of rats administered CDDP. Intraperitoneal injection of 8 mg/kg of CDDP significantly enhanced the activities of AChE (127.4%, P=0.023) and MAO (139.5%, P=0.016) in the brain compared to the control. Both doses of NAC significantly protected against the increase in AChE and MAO activities as compared to the CDDP group. Furthermore, 100 mg/kg of NAC was found to be more effective in improving these enzymes as compared with 50 mg/kg. Administration of NAC alone at a higher dose (100 mg/kg) induced a nonsignificant change in AChE and MAO activities compared to the control group.
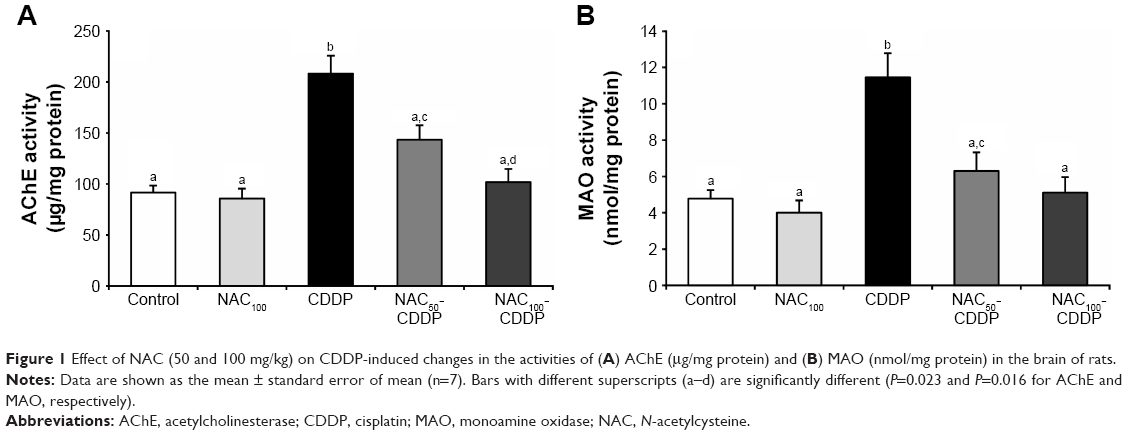 | Figure 1 Effect of NAC (50 and 100 mg/kg) on CDDP-induced changes in the activities of (A) AChE (μg/mg protein) and (B) MAO (nmol/mg protein) in the brain of rats. |
Effect of NAC on CDDP-induced alteration in the brain oxidative status
The oxidative status of the brain was evaluated by assessing the levels of MDA, NO, and PC in the tissue (Table 1). Administration of CDDP provoked a statistically significant increase in the brain level of MDA (161.4%, P=0.006), NO (115.9%, P=0.002), and PC (175.8%, P<0.001) as compared to the control group. These CDDP-induced changes in the oxidative status were significantly and dose-dependently reversed upon administration of NAC. Treatment with 100 mg/kg of NAC decreased the levels of MDA, NO, and PC by 55.5%, 45.2%, and 52% while 50 mg/kg of NAC reduced them by 38.3%, 35%, and 29.5%, respectively, compared with the CDDP group. Administration of the higher dose of NAC without CDDP was safe and showed nonsignificant change in the levels of MDA, NO, and PC when compared to the control.
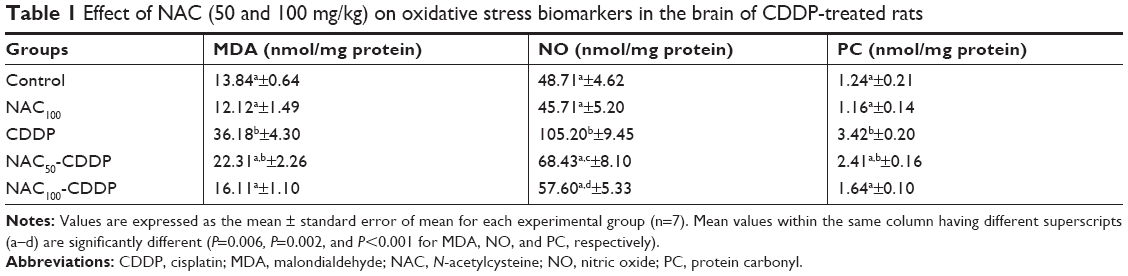 | Table 1 Effect of NAC (50 and 100 mg/kg) on oxidative stress biomarkers in the brain of CDDP-treated rats |
NAC modulated CDDP-induced change in the brain antioxidant status
The influence of NAC on CDDP-induced change in the brain antioxidant status is illustrated in Figure 2 and Table 2. Intraperitoneal injection of 8 mg/kg of CDDP significantly decreased the brain total antioxidant capacity (TAC) (51.3%, P=0.003) and the GSH level (58.8%, P=0.007) as shown in Figure 2. Furthermore, a significant reduction was reported in the activity of GPx (63.6%, P=0.002), GST (60.6%, P=0.046), SOD (67.9%, P=0.002), and CAT (66.1%, P=0.018) relative to the control group (Table 2). Treatment with NAC significantly improved the antioxidant status of the brain as shown by the increase in the levels of these antioxidants compared with CDDP group. Comparing the effect of the two doses used in the treatment, the higher dose of NAC was found to be more effective in improving the antioxidant status of the brain. It is also apparent that administration of 100 mg/kg of NAC without CDDP did not induce a significant change in the TAC, the level of GSH, and the activities of GPx, GST, SOD, and CAT when compared with the control.
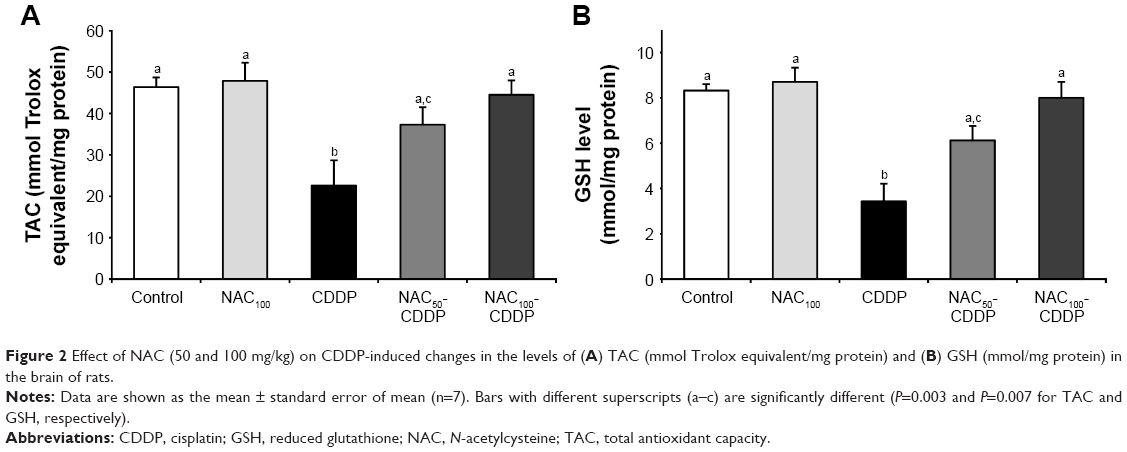 | Figure 2 Effect of NAC (50 and 100 mg/kg) on CDDP-induced changes in the levels of (A) TAC (mmol Trolox equivalent/mg protein) and (B) GSH (mmol/mg protein) in the brain of rats. |
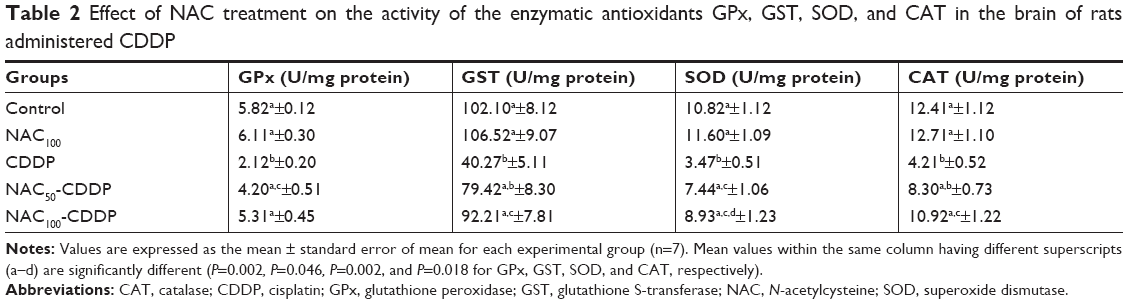 | Table 2 Effect of NAC treatment on the activity of the enzymatic antioxidants GPx, GST, SOD, and CAT in the brain of rats administered CDDP |
NAC attenuated CDDP-induced increase in the brain and serum pro-inflammatory cytokines
According to Table 3, intraperitoneal injection of 8 mg/kg of CDDP provoked a marked neuro-inflammation. It significantly enhanced the levels of TNF-α (162.3%, P=0.003), IL-1β (147.2%, P=0.012), and IL-6 (113.3%, P=0.033) in the brain tissue and also in the serum (159.1%, 125.1%, and 137.8%, P<0.001) relative to the control group. Treatment with NAC exerted an anti-inflammatory action. The dosage of 50 mg/kg of NAC significantly decreased the CDDP-induced elevation in the brain TNF-α, IL-1β, and IL-6 by 32.2%, 41.7%, and 35.1%, respectively, compared to the CDDP group. Furthermore, treatment with 100 mg/kg of NAC was found to be more effective in modulating the neuro-inflammation as shown by a decrease in the levels of TNF-α (47.2%), IL-1β (51.5%), and IL-6 (44.3%), respectively, compared with the CDDP group. Similar improvement was recorded in the serum levels of these pro-inflammatory cytokines. It is worth mentioning that the administration of 100 mg/kg of NAC was safe and induced nonsignificant change in the level of the measured pro-inflammatory cytokines as compared to the control group.
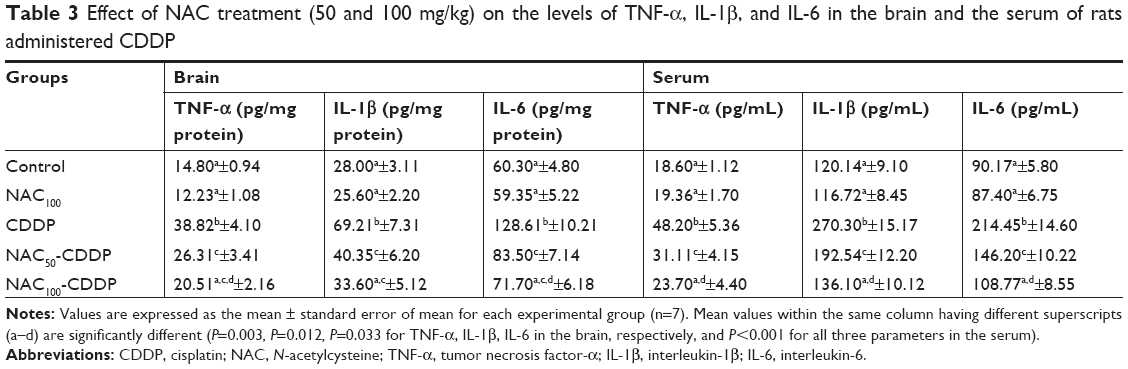 | Table 3 Effect of NAC treatment (50 and 100 mg/kg) on the levels of TNF-α, IL-1β, and IL-6 in the brain and the serum of rats administered CDDP |
Discussion
Neurotoxicity is a frequent adverse effect of chemotherapies including CDDP. Clarification of mechanisms implicated in CDDP neurotoxicity is important to find out an effective complementary therapy. Different mechanisms have been proposed; however, oxidative injury and inflammation seem to be the basic mechanisms in the neurotoxicity of CDDP.9 Results of the present study pointed out to a strong neurotoxic effect for CDDP in the brain of rats which was evidenced by the enhanced activities of AChE and MOA, altered oxidative/antioxidative status, and enhanced production of pro-inflammatory cytokines. Because of its antioxidative and anti-inflammatory properties, the neuroprotective effect of NAC against CDDP-induced brain injury was investigated in the present study for the first time. Two different doses of NAC were selected (50 and 100 mg/kg). Results showed that the two doses of NAC, especially the higher one, were effective in attenuating the neurotoxic effect of CDDP in the brain. It effectively improved the cholinergic dysfunction, restored the deterioration in the brain oxidant/antioxidant status, and suppressed the neuro-inflammation.
Cholinergic dysfunction is a primary sign of neurotoxicity. Herein, the toxicity of CDDP in the brain was characterized by a significant increase in AChE activity which agrees with previous studies.32,33 Increased production of ROS may be one of the reasons behind the increase in AChE activity. ROS have been found to enhance the peroxidation of the plasma membrane which affects the integrity and functionality of the cholinergic system.34 Moreover, CDDP in our study significantly enhanced the activity of MAO in the brain. MAO was reported to catalyze the oxidative deamination of monoamine neurotransmitters which generates H2O2 as a byproduct.35 Therefore, the enhanced activity of MAO may an important mechanism in increasing the production of H2O2, thereby augmenting oxidative stress. Treatment with NAC in the present study restored the normal activity of AChE and MAO. NAC has been reported to be an AChE inhibitor.36 The ameliorative effect of NAC may be largely attributed to its antioxidant action and its ability to quench free radicals.
The brain is at high risk of oxidative damage because of its high utilization of oxygen, high content of non-heme iron which contributes to the production of ROS, and high content of polyunsaturated fatty acids.37 It is well established that CDDP can disturb the redox homeostasis. It can enhance the generation of ROS which subsequently attack and damage multiple subcellular macromolecules such as lipids and proteins.38 Therefore, oxidative modification of lipids and proteins may be one of the mechanisms behind the observed CDDP-induced neurotoxicity. This hypothesis was further confirmed in the present study by the enhanced level of MDA (the end product of LPO) and PC (a reliable indicator for protein oxidation) in the brain. These results are in agreement with earlier reports.20,39 NO is a strong damaging free radical when produced in excess. It can combine with the available superoxide radical to form peroxynitrite, a more powerful oxidant. This radical can actively react with lipids, proteins, DNA, and can also inactivate GSH and GPx.39 The involvement of NO in the toxicity of CDDP has been reported and was attributed to the enhancement of inducible NO synthase.40 Results from our study showed a marked increase in NO level in the brain of CDDP-administered rats which agrees with Aydin et al.20 CDDP-induced changes in MDA, PC, and NO levels were significantly alleviated with NAC treatment. The ameliorative effect of NAC may be largely attributed to its ability to eradicate free radicals. Evidence supports the potent radical scavenging activity of NAC as being a sulfhydryl donor that can directly react with free radicals and/or its metal chelating activity.13,14 These beneficial effects of NAC may interpret its capability to restore the normal oxidant status of the brain in our study.
Cellular antioxidants, both enzymatic and nonenzymatic, are necessary to combat oxidative stress. In our study, the antioxidant status of the brain was significantly suppressed as evidenced by the decrease in the TAC, the level of GSH, as well as the activities of GPx, GST, SOD, and CAT in the brain upon administration of CDDP. Similar inhibition in the antioxidant defense system was reported in previous studies.33,41 These results support the hypothesis that the mechanism of CDDP-induced neurotoxicity is associated with depletion of the endogenous antioxidants. Suppression of the brain antioxidant status may be a result of the enhanced production of oxidants as mentioned earlier. The brain antioxidant status was significantly improved upon NAC treatment. These results support the notion that the protective effect of NAC is mostly mediated through its antioxidant activity. NAC has been reported to potentiate the cellular antioxidant defense system by replenishing the production of GSH.13
Pro-inflammatory cytokines are key mediators implicated in the pathophysiological processes associated with brain injury.42 Among the pro-inflammatory cytokines, TNF-α, IL-1β, and IL-6 are extremely potent and considered to be the major cytokines because they can also initiate a cascade of events and affect the release of other cytokines.43 Massive production of pro-inflammatory cytokines has been shown to be involved in several neuropathological conditions.44 Neuro-inflammation may be another possible mechanism in CDDP-induced toxicity in the brain of rats. This was clearly evidenced in our study by the increase in the levels of TNF-α, IL-1β, and IL-6 in both the brain and the serum which agrees with earlier report.7 Nuclear factor-kappa B (NF-κB) is an essential factor which induces the transcription of the pro-inflammatory genes and thus plays a key role in the inflammation.45 Systemic injection of CDDP has been found to be associated with upregulation of NF-κB which in turn initiates the overproduction of pro-inflammatory cytokines.33,46 Oxidative stress and inflammation may be interrelated mechanisms in the development of CDDP neurotoxicity. CDDP-induced overproduction of ROS has been found to accelerate the release of the pro-inflammatory cytokines.47 In our study, high levels of pro-inflammatory cytokines induced by CDDP showed a good response to treatment with NAC as evidenced by the decline in the levels of TNF-α, IL-1β, and IL-6. Similar anti-inflammatory effects of NAC were reported in previous studies.48,49 NAC has been reported to protect against the proliferation of cytokines, increase neuronal survival, and decrease apoptosis.14 NAC was also reported to have the ability to downregulate NF-κB.13 Therefore, the anti-inflammatory effect of NAC by deactivating NF-κB and its antioxidant activity by enhancing GSH may be important mechanisms behind the reported improvement in the pro-inflammatory cytokines.
A limitation of the present study is the lack of investigation of the gene expression of AchE, TNF-α, IL-1β, and IL-6 using PCR and the protein level of the AChE using Western blot. Therefore, future studies are needed to confirm the biochemical results obtained in our study using molecular tools and more immunohistochemical techniques.
Conclusion
In conclusion, CDDP induced neurotoxicity in the brain of rats which was evidenced by oxidative injury and neuro-inflammation. Treatment with NAC at the two selected doses (50 and 100 mg/kg) provided effective protection against the toxicity of CDDP in the brain. It modulated the CDDP-induced oxidative stress, improved the antioxidant defense system, and downregulated the overproduction of pro-inflammatory cytokines. The mechanisms by which NAC provides protection are largely dependent on the interplay between its antioxidant and anti-inflammatory activities. Based on these results, NAC may serve as a complementary therapeutic agent to enhance the outcomes of CDDP chemotherapy. Further preclinical studies are needed to identify the best strategy for its clinical application. It is also important to determine whether NAC compromises the anticancer activity of CDDP or not.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang J, Wang L, Xing Z, et al. Status of bi- and multi-nuclear platinum anticancer drug development. Anticancer Agents Med Chem. 2010;10(4):272–282. | ||
von Hoff DD, Rozencweig M. Cis-Diamminedichloroplatinum (II): a metal complex with significant anticancer activity. Adv Pharmacol Chemother. 1979;16:273–298. | ||
Avan A, Postma TJ, Ceresa C, et al. Platinum-induced neurotoxicity and preventive strategies: past, present, and future. Oncologist. 2015;20(4):411–432. | ||
Mcwhinney SR, Goldberg RM, Mcleod HL. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009;8(1):10–16. | ||
Wolf S, Barton D, Kottschade L, Grothey A, Loprinzi C. Chemotherapy-induced peripheral neuropathy: prevention and treatment strategies. Eur J Cancer. 2008;44(11):1507–1515. | ||
Almutairi MM, Alanazi WA, Alshammari MA, et al. Neuro-protective effect of rutin against cisplatin-induced neurotoxic rat model. BMC Complement Altern Med. 2017;17(1):472. | ||
Chen C, Zhang H, Xu H, Zheng Y, Wu T, Lian Y. Ginsenoside Rb1 ameliorates cisplatin-induced learning and memory impairments. J Ginseng Res. 2017;1–9. | ||
Owoeye O, Adedara IA, Farombi EO. Pretreatment with taurine prevented brain injury and exploratory behaviour associated with administration of anticancer drug cisplatin in rats. Biomed Pharmacother. 2018;102:375–384. | ||
Jaggi AS, Singh N. Mechanisms in cancer-chemotherapeutic drugs-induced peripheral neuropathy. Toxicology. 2012;291(1–3):1–9. | ||
Mcdonald ES, Randon KR, Knight A, Windebank AJ. Cisplatin preferentially binds to DNA in dorsal root ganglion neurons in vitro and in vivo: a potential mechanism for neurotoxicity. Neurobiol Dis. 2005;18(2):305–313. | ||
Kawai Y, Nakao T, Kunimura N, Kohda Y, Gemba M. Relationship of intracellular calcium and oxygen radicals to cisplatin-related renal cell injury. J Pharmacol Sci. 2006;100(1):65–72. | ||
Podratz JL, Knight AM, Ta LE, et al. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol Dis. 2011;41(3):661–668. | ||
Samuni Y, Goldstein S, Dean O, Berk M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta. 2013;1830(8):4117–4129. | ||
Giampreti A, Lonati D, Ragghianti B, et al. N-acetyl-cysteine as effective and safe chelating agent in metal-on-metal Hip-Implanted patients: two cases. Case Reports in Orthopedics. 2016;2016(1):1–7. | ||
Bhatti J, Nascimento B, Akhtar U, et al. Systematic review of human and animal studies examining the efficacy and safety of N-acetylcysteine (NAC) and N-acetylcysteine amide (NACA) in traumatic brain injury: impact on neurofunctional outcome and biomarkers of oxidative stress and inflammation. Front Neurol. 2017;8:744. | ||
Bavarsad Shahripour R, Harrigan MR, Alexandrov AV. N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav. 2014;4(2):108–122. | ||
Farr SA, Poon HF, Dogrukol-Ak D, et al. The antioxidants α-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem. 2003;84(5):1173–1183. | ||
Prakash A, Kalra JK, Kumar A. Neuroprotective effect of N-acetylcysteine against streptozotocin-induced memory dysfunction and oxidative damage in rats. J Basic Clin Physiol Pharmacol. 2015;2015(26):13–23. | ||
Abdel-Wahab WM, Moussa FI, Saad NA. Synergistic protective effect of N-acetylcysteine and taurine against cisplatin-induced nephrotoxicity in rats. Drug Des Devel Ther. 2017;11:901–908. | ||
Aydin D, Peker EG, Karakurt MD, et al. Effects of Ginkgo biloba extract on brain oxidative condition after cisplatin exposure. Clin Invest Med. 2016;39(6):27511. | ||
Ellman GL, Courtney KD, Andres V, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7(2):88–95. | ||
Holt A, Sharman DF, Baker GB, Palcic MM. A continuous spectrophotometric assay for monoamine oxidase and related enzymes in tissue homogenates. Anal Biochem. 1997;244(2):384–392. | ||
Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95(2):351–358. | ||
Levine RL, Garland D, Oliver CN, et al. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–478. | ||
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126(1):131–138. | ||
Miller NJ, Rice-Evans C, Davies MJ, Gopinathan V, Milner A. A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin Sci. 1993;84(4):407–412. | ||
Beutler E, Duron O, Kelly BM. Improved method for the determination of blood glutathione. J Lab Clin Med. 1963;61:882–888. | ||
Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249(22):7130–7139. | ||
Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70(1):158–169. | ||
Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–126. | ||
Kakkar P, Das B, Viswanathan PN. A modified spectrophotometric assay of superoxide dismutase. Indian J Biochem Biophys. 1984;21(2):130–132. | ||
Chtourou Y, Gargouri B, Kebieche M, Fetoui H. Naringin abrogates cisplatin-induced cognitive deficits and cholinergic dysfunction through the down-regulation of AChE expression and iNos signaling pathways in hippocampus of aged rats. J Mol Neurosci. 2015;56(2):349–362. | ||
Jangra A, Kwatra M, Singh T, et al. Edaravone alleviates cisplatin-induced neurobehavioral deficits via modulation of oxidative stress and inflammatory mediators in the rat hippocampus. Eur J Pharmacol. 2016;791:51–61. | ||
Melo JB, Agostinho P, Oliveira CR. Involvement of oxidative stress in the enhancement of acetylcholinesterase activity induced by amyloid beta-peptide. Neurosci Res. 2003;45(1):117–127. | ||
Ooi J, Hayden MR, Pouladi MA. Inhibition of excessive monoamine oxidase A/B activity protects against stress-induced neuronal death in Huntington disease. Mol Neurobiol. 2015;52(3):1850–1861. | ||
Costa M, Bernardi J, Costa L, et al. N-acetylcysteine decreases mice brain acetyl cholinesterase activity: An in vitro kinetic study. Enz Eng. 2015;5:135. | ||
Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97(6):1634–1658. | ||
Marullo R, Werner E, Degtyareva N, et al. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS One. 2013;8(11):e81162. | ||
Abdel Moneim AE. The neuroprotective effects of purslane (Portulaca oleracea) on rotenone-induced biochemical changes and apoptosis in brain of rat. CNS Neurol Disord Drug Targets. 2013;12(6):830–841. | ||
Güleç M, Iraz M, Yılmaz HR, Özyurt H, Temel I. The effects of Ginkgo biloba extract on tissue adenosine deaminase, xanthine oxidase, myeloperoxidase, malondialdehyde, and nitric oxide in cisplatin-induced nephrotoxicity. Toxicol Ind Health. 2006;22(3):125–130. | ||
Abdel Moneim AE. Azadirachta indica attenuates cisplatin-induced neurotoxicity in rats. Indian J Pharmacol. 2014;46(3):316–321. | ||
Kadhim H, Duchateau J, Sebire G. Cytokines and brain injury. J Intensive Care Med. 2008;23(4):236–249. | ||
Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signaling and inflammatory disease. Biochim Biophys Acta. 2014;1843(11):2563–2582. | ||
Kuhad A, Chopra K. Curcumin attenuates diabetic encephalopathy in rats: behavioral and biochemical evidences. Eur J Pharmacol. 2007;576(1–3):34–42. | ||
Baldwin AS. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu Rev Immunol. 1996;14(1):649–683. | ||
So H, Kim H, Lee JH, et al. Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF-kappaB. J Assoc Res Otolaryngol. 2007;8(3):338–355. | ||
Akman T, Akman L, Erbas O, Terek MC, Taskiran D, Ozsaran A. The preventive effect of oxytocin to cisplatin-induced neurotoxicity: an experimental rat model. Biomed Res Int. 2015;2015(4):1–5. | ||
Chen G, Shi J, Hu Z, Hang C. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm. 2008;2008(5):1–8. | ||
da Silva ND, Roseguini BT, Chehuen M, et al. Effects of oral N-acetylcysteine on walking capacity, leg reactive hyperemia, and inflammatory and angiogenic mediators in patients with intermittent claudication. Am J Physiol Heart Circ Physiol. 2015;309(5):H897–H905. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.