Back to Journals » Nature and Science of Sleep » Volume 8
Neurophysiological basis of rapid eye movement sleep behavior disorder: informing future drug development
Authors Jennum P, Christensen JA, Zoetmulder M
Received 27 October 2015
Accepted for publication 2 February 2016
Published 15 April 2016 Volume 2016:8 Pages 107—120
DOI https://doi.org/10.2147/NSS.S99240
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Steven Shea
Poul Jennum, Julie AE Christensen, Marielle Zoetmulder
Department of Clinical Neurophysiology, Faculty of Health Sciences, Danish Center for Sleep Medicine, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark
Abstract: Rapid eye movement (REM) sleep behavior disorder (RBD) is a parasomnia characterized by a history of recurrent nocturnal dream enactment behavior and loss of skeletal muscle atonia and increased phasic muscle activity during REM sleep: REM sleep without atonia. RBD and associated comorbidities have recently been identified as one of the most specific and potentially sensitive risk factors for later development of any of the alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and other atypical parkinsonian syndromes. Several other sleep-related abnormalities have recently been identified in patients with RBD/Parkinson’s disease who experience abnormalities in sleep electroencephalographic frequencies, sleep–wake transitions, wake and sleep stability, occurrence and morphology of sleep spindles, and electrooculography measures. These findings suggest a gradual involvement of the brainstem and other structures, which is in line with the gradual involvement known in these disorders. We propose that these findings may help identify biomarkers of individuals at high risk of subsequent conversion to parkinsonism.
Keywords: motor control, brain stem, hypothalamus, hypocretin
Introduction
Rapid eye movement (REM) sleep behavior disorder (RBD) is a parasomnia characterized by a history of recurrent nocturnal dream enactment behavior and loss of skeletal muscle atonia and increased phasic muscle activity during REM sleep: REM sleep without atonia (RSWA). RBD was first described in animals by Jouvet in 19651 and in humans by Schenck et al in 1986 and Sforza et al in 1988.2–4 RBD is a complex, multidimensional parasomnia that is frequently linked with other sleep disorders (eg, untreated sleep apnea, narcolepsy with cataplexy of hypocretin-deficient type), a wide range of neurodegenerative disorders, and the pharmacotherapy of psychiatric and medical disorders (eg, antidepressants, beta-blockers). Imaging studies, clinico-electrophysiological and experimental models of RBD in cats and rats, and transgenic RBD mouse models5 have increased our knowledge of the underlying brainstem mechanisms of REM-atonia and REM sleep phasic motor activity. RSWA has been closely associated with hypocretin-deficient narcolepsy,6,7 and very strong associations have been identified between RBD and the alpha-synucleinopathies, primarily Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA).8–10 Schenck et al were the first to perform a follow-up study and reported that 38% of their original cohort of 29 patients had developed parkinsonism.11 Twenty years after diagnosis, 81% of that cohort had parkinsonism.8 Subsequent studies have reported that approximately half of the patients with idiopathic RBD (iRBD) develop a synucleinopathy within ~12 years of diagnosis of RBD.9,10 These studies include highly selected patients undergoing full video-polysomnography (PSG) recording from neurological sleep centers.
The finding that iRBD often heralds future parkinsonism has stimulated research on predictors of imminent parkinsonism in RBD. The most elusive goal is to enroll high-risk patients in therapeutic studies with promising neuroprotective (ie, disease-modifying) agents that could prolong, or ideally halt, the progression of iRBD to clinical parkinsonism/dementia.
Comorbid findings in RBD
Patients with iRBD typically present with a varying degree of nonmotor symptoms, including impaired olfactory function, cognitive function,12 and autonomic function determined by heart rate changes;13 cardiac 123I-metaiodobenzylguanidine (MIBG) uptake reduction;14,15 gastrointestinal abnormalities16; and electroencephalographic slowing.17 Although all of these nonmotor symptoms have been associated with parkinsonism, each of these symptoms individually has a too low specificity and sensitivity of predicting later development of parkinsonism. Hyposomnia and constipation, for instance, are often observed in the preclinical disease stage, but have a too low specificity for predicting PD.18 However, a combination of nonmotor symptoms, especially hyposomnia and RBD, has been found to increase risk of developing PD.18–20
Nevertheless, these findings generally support the assumption that RBD is part of a general progressive neurodegenerative process primarily involving the brainstem area, with later involvement of other brain structures.
Neurophysiology of REM sleep
REM sleep is characterized by: 1) tonic components: electroencephalogram (EEG), muscle atonia, and loss of thermoregulation; and 2) phasic components: REMs, muscle twitches occurring against a background of atonia, ponto-geniculo-occipital waves, as well as irregularities in breathing, heart rate, and blood pressure.
The EEG during REM sleep resembles that during wakefulness, which is characterized by low voltage and mixed frequency in the cerebral cortex, with 5–9 Hz waves in the hippocampus. Therefore, REM sleep is also called as “active” or “paradoxical” sleep.
There is sustained low muscle tone during REM sleep in most of the somatic muscles except those of the inner ear, eye, and diaphragm.21 Cranial muscles of the eyes, ears, and jaw, as well as muscles of the limb extremities may show phasic twitches/movements against a background of atonia during REM sleep. However, phasic activity in postural muscles is rarely seen.22
The orchestration of these tonic and phasic REM sleep suggests the existence of an “executive mechanism” that generates and maintains REM sleep.21,23,24
In the brainstem, there are at least two systems involved in wakefulness and nonrapid eye movement (NREM) and REM sleep: REM-off and REM-on systems.21,23,24
Neurons belonging to the REM-off system include the ventrolateral periaqueductal gray (vlPAG) and the dorsal deep mesencephalic reticular nucleus (dDpMe). These nuclei are active during wakefulness and NREM sleep, and are activated by projections from the wake-active noradrenergic locus coeruleus, serotonergic raphe nucleus, and the hypocretinergic neurons from the lateral thalamus. Furthermore, the REM-off vlPAG and dDpMe are the only pontomedullary structures containing a large number of GABAergic neurons projecting to the sublaterodorsal (SLD) nucleus, the structure critical for generating and maintaining REM sleep.25,26 When the vlPAG/dDpMe is inhibited by GABAergic projections from the ventrolateral preoptic nucleus and the lateral hypothalamus,23,27 a marked increase in REM sleep is seen in rats25 and cats.28,29
The REM-off system has reciprocal interactions with the REM-on system, and these two systems mutually inhibit each other by means of the neurotransmitter GABA.27 During wakefulness and NREM, the REM-off neurons send inhibitory GABAergic projections to structures belonging to the REM-on system to prevent the occurrence of REM sleep. During REM sleep, the REM-on neurons send inhibitory GABAergic/glycinergic projections to structures belonging to the REM-off system.30
Neurons in the caudal laterodorsal tegmental nucleus (cLDT) and SLD (cLDT-SLD) are regarded as critical sites for the generation and maintenance of REM sleep, the “executive mechanism”.5,31,32 These neurons activate the precoeruleus and parabrachial nucleus, which are involved in REM-EEG. In addition, the ventral part of the SLD induces sensory inhibition and motor atonia during REM sleep. The SLD contains glutamatergic neurons that directly project to inhibitory interneurons in lamina VIII of the spinal cord and to nuclei, inducing atonia in the ventromedial medulla.27,33 In turn, the spinal interneurons and the ventromedial medulla project GABAA/B/glycine to the spinal and cranial motoneurons, thereby hyperpolarizing the motor-facilitatory neurons and inducing atonia.5,27,31,34,35 Nuclei in the ventromedial medulla, belonging to the REM-on system, include the magnocellular reticular nucleus, gigantocellular nucleus (GiA, GiV), paragigantocellular nucleus (DPGi, LPGi), parvicellular nucleus, and the raphe magnus.26,31,36
Lesions of the SLD, such as a deletion of the Vglut2 in the cLDT-SLD, result in REM fragmentation, which is characterized by reductions in REM sleep epoch duration and an increase in the number of REM episodes. The SLD has direct projections to lamina VIII in the spinal cord, where they excite GABA/glycinergic interneurons. Elimination of GABA/glycinergic transmission from these interneurons in the ventral horn at the C3–C4 level in mice results in brief twitching and jerking movements, predominantly in the upper body and sporadically in the lower body.37 In addition, lesions of the SLD result in complex behaviors during REM sleep, symptoms consistent with the human syndrome of RBD.27 Therefore, it has been hypothesized that the SLD specifically functions to antagonize the phasic activity of postural muscles of complex behaviors driven by the motor cortex.38 In contrast, cell-body lesions of the ventromedial medulla seem to cause simple behaviors, such as rapid phasic jerking and twitching movements, during REM sleep in rats and cats.39,40 However, more complex behaviors, as seen with lesions of the SLD, are not observed. Therefore, the ventromedial medulla only partially mediates the SLD control of REM atonia.
Recent findings have revealed that glutamatergic neurons in the rostral parvocellular reticular formation are critical for phasic masseter activity during REM sleep.41
As SLD lesions apparently do not affect phasic masseter activity during REM sleep,41 it was hypothesized that the SLD is not the generator of phasic activity in this muscle and that it specifically antagonizes the complex activity of postural muscles driven by the motor cortex and simple behaviors controlled by the ventromedial medulla–spinal projections.38
Brainstem areas other than the SLD and the ventromedial medulla may be involved in the control of REM atonia. A study performed by Lai et al reported that lesions in the mesopontine junction cause periodic limb movements during REM sleep in cats.42 In addition to glutamate and GABA/glycine, other neurotransmitter systems may mediate REM atonia, by direct projections to the spinal cord or by acting on the vlPAG matter, lateral pontine tegmentum, SLD, or ventromedial medulla neurons, including monoaminergic, orexinergic, and melanin-concentrating hormones.33,43–45 In summary, animal studies indicate that the SLD is the critical region for the control of REM atonia, and lesions of the SLD give rise to the full phenotype of RBD, whereas lesions to the ventromedial medulla produce muscle jerks and twitches.
The occurrence of RBD is consistent with the findings of a study in 2003 by Braak et al,46 who described a staging system for the neuropathological development of PD. The aggregation of Lewy bodies is hypothesized to arise from the dorsal motor nucleus of the vagal nerve in the medulla and in the olfactory bulb, and to emerge through the coeruleus/subcoeruleus complex and the magnocellularis reticular nucleus until it involves the substantia nigra, which has been associated with the daytime motor symptoms of PD. Neuropathological studies of iRBD reveal the presence of Lewy bodies in the brain.47
Generally, the staging suggests that the accumulation of aggregated intracytoplasmic proteins has a general distribution, primarily including the upper area of the medulla oblongata and lower brainstem region with progressive involvement of other parts of the brainstem and midbrain. The joint involvement of the olfactory area suggests that specific parts of the brain are more prone to this, but the specific mechanism is not known in detail. The spreading pattern partly explains the progressive distribution of the involvement of nonmotor symptoms and other characteristics, eg, initial gastrointestinal symptoms (vagal nuclei/autonomic function) and loss of REM atonia (SLD nuclei).48 The picture of neuropathological spreading of Lewy bodies is probably more complex, as not all diagnosed PD patients have a history of recurrent dream enactment behavior and RBD and as other nonmotor symptoms occur with varying intensities prior to and in PD and other alpha-synucleinopathies.
RBD diagnosis
According to the third edition of the International Classification of Sleep Disorders (2012), the following criteria are required to make a diagnosis of RBD:
- Repeated episodes of sleep-related vocalization and/or complex motor behaviors;
- These behaviors are documented by PSG to occur during REM sleep, or based on clinical history of dream enactment, and are presumed to occur during REM sleep;
- Polysomnographic recordings demonstrating RSWA; and
- The disturbance not better explained as another sleep disorder, mental disorder, medication, or substance use.
According to the American Academy of Sleep Medicine, RSWA is defined by sustained muscle activity in REM sleep (Stage R) with 50% of the epoch having increased chin electromyography (EMG) amplitude, and/or excessive transient muscle activity, defined by the presence of five or more mini-epochs (a 30-second epoch is divided into ten 3-second mini-epochs) in an epoch featuring transient muscle activity lasting at least 0.5 seconds.22
A diagnosis of RBD cannot be made unless dream enactment and RSWA are both present, so a PSG is required to make the diagnosis. RBD has a wide spectrum of overt symptoms, such as twitching, jerking, shouting, and screaming, as well as more complex behaviors, including punching and escape behaviors, that may cause harm to some patients or bedpartners. The simultaneous video-PSG recording is essential for evaluating these movements and vocalizations appearing during REM sleep and to discount the presence of epileptiform activity in the EEG derivations in order to make a diagnosis of RBD.49
While violent and complex dream enactment behavior is rarely observed during a single night’s PSG recordings, an abnormal state of increased EMG tone during REM sleep and sparse limb jerks are common events.
Polysomnographic findings in RBD
PSG consists of a comprehensive and simultaneous recording of various body functions, including brain activity (electroencephalogram [EEG]), eye movements (electrooculography [EOG]), muscle activity (electromyogram [EMG]), and heart rhythm (electrocardiogram [ECG]) during sleep. Several studies have evaluated the involvement of different PSG findings. These abnormalities are presented later. A schematic representation of these PSG abnormalities and their potential neurophysiological and neuroanatomical findings is presented in Figure 1.
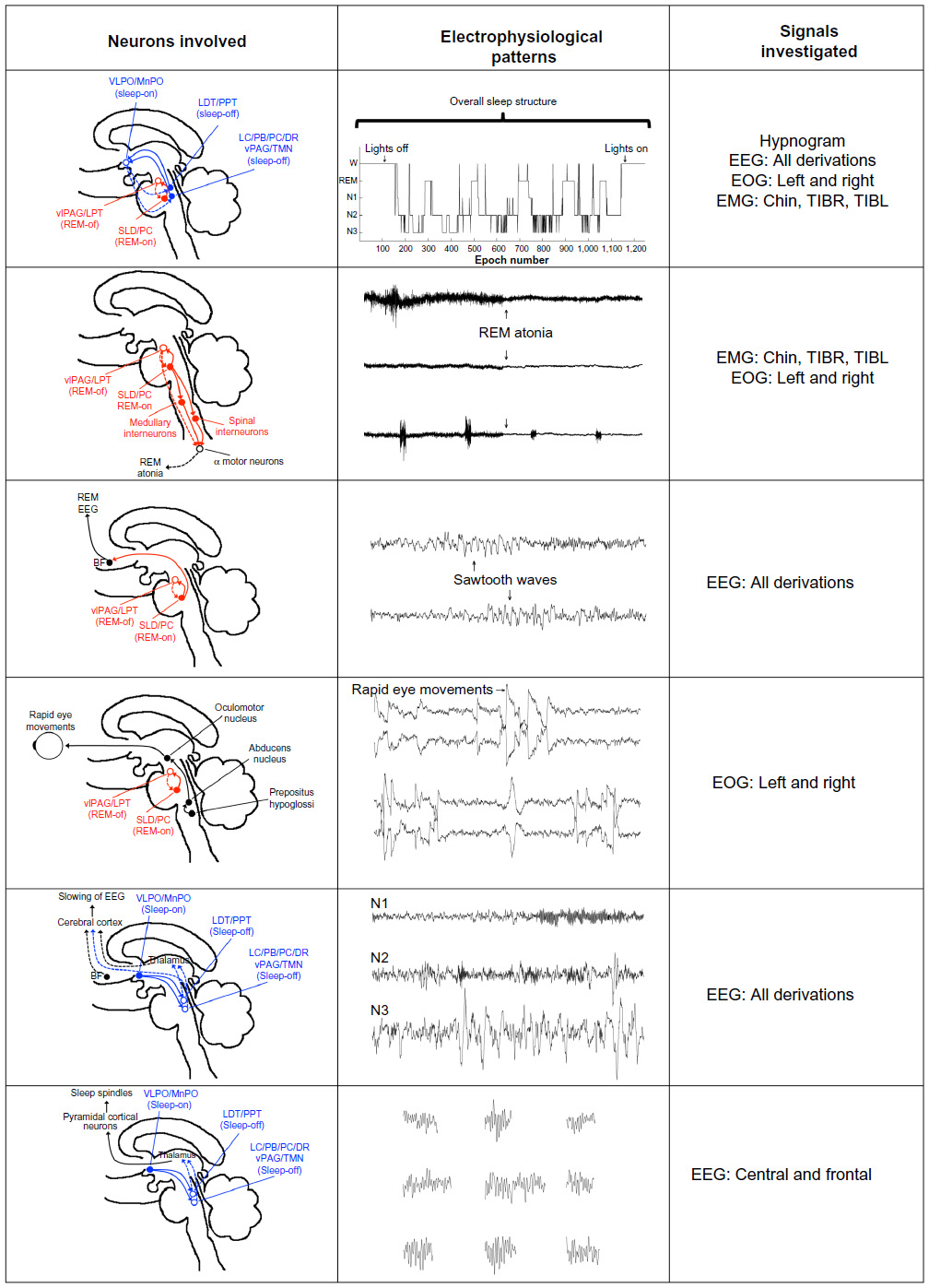 | Figure 1 Examples of potential PD biomarkers expressed in polysomnographic signals and their neuroanatomical correlates. |
Scoring RBD
Lapierre and Montplaisir were the first to present an objective scoring method, based on visual analysis of PSG studies.50 Since this initial description of RSWA, several methods for scoring of RBD have been proposed, analyzing RSWA with semiautomated or fully automated algorithms. Frauscher et al suggested a PSG montage quantifying “any” EMG activity (tonic, phasic, or both) in the mentalis muscle and phasic EMG activity in the right and left flexor digitorum superficialis muscles in the upper limbs, with a cutoff of 32% (when using 3-second mini-epochs) for the diagnosis of RBD.51 Ferri et al measured the amplitude of the submentalis muscle during sleep in 21 patients with iRBD, ten patients with MSA, and ten healthy controls, and provided practical indices for the objective evaluation of EMG atonia and EMG activations by means of a sleep atonia index.52 A subsequent study by Montplaisir et al53 attempted to identify cutoff values for tonic and phasic EMG activity in submentalis muscle and to assess the sensitivity and specificity of these values, taken separately or in combination, to diagnose patients with iRBD. They studied 80 patients with iRBD and 80 age- and sex-matched controls. Tonic and phasic EMG activities were visually identified in the chin, but not in the limbs. Completely correct classification of 81.9% was found for tonic chin EMG density ≥30%; 83.8% for phasic chin EMG density ≥15%, and 75.6% for ≥24 leg movements/hour of REM sleep. Another study by Frandsen et al defined a method for establishing a baseline in automatic quantifying submental motor activity during REM sleep in iRBD, PD, and controls.54 They found that no control had >30% of REM sleep with increased motor activity. However, patients with known RBD had activity as low as 4.5%. There is currently no comparison between the two methods, especially with respect to emphasizing the accuracy and the association with later neurodegenerative development.
The studies differ in their methods and definition of RBD EMG activity in the separation between patients with (i)RBD and healthy controls. Some studies differentiate between tonic and phasic EMG activity and use 20-second epochs for tonic and 2-second epochs for phasic,53 whereas others have included any EMG activity but use different methods to analyze EMG activity, either by separating sleep into 30-second and 3-second epochs51 or by distinguishing it from the background EMG activity.54 Although there is consensus that brief bursts of EMG activity and muscle twitches are phasic events, the challenge arises as to how to classify more complex behavioral events, such as a patient with RBD defending himself. It is possible that the difference between tonic and phasic may be more quantitative than qualitative, as suggested by Ramaligam et al, who proposed that the phasic movements are disinhibited in humans with RBD and that RBD results more from failure of the suppression of phasic activity of postural muscles than from dysfunction of tonic control.38 Other studies identified RSWA using the EEG and EOG channels for REM sleep detection, while the abnormally high muscle activity was detected from the EMG channels, and the submental was combined with the left and right anterior tibialis. RSWA was identified by considering it as an outlier problem, in which the number of outliers during REM sleep was used as a quantitative measure of muscle activity.55 There is no consensus in the medical literature about the scoring of EMG activity in iRBD, which suggests that a task-force is needed, especially as studies are beginning to focus on the role of RSWA without subjective complaints of dream-enactment behavior as a subclinical symptom of iRBD.
Is RSWA a marker for RBD?
In 2015, Stefani et al reported results of a follow-up study of subjects with isolated RSWA without dream enactment.56 After a mean of 8.6 years (SD =0.9), one of 14 participating subjects (7.3%) had progressed to RBD. Ten of 14 RSWA subjects (71.4%) were positive for at least one neurodegenerative biomarker. Substantia nigra hyperechogenicity and the presence of mild cognitive impairment (MCI) were both present in four of 14 subjects with isolated RSWA. Electromyographic activity measures increased significantly from baseline to follow-up PSG (any mentalis and both anterior tibialis muscles). The authors concluded that isolated RSWA may be an early biomarker of synuclein-mediated neurodegeneration.
A recent study reported the prevalence of iRBD to be 2.01% and subclinical RBD (ie, patients with isolated RSWA without dream enactment behavior) to be 4.95% in elderly Koreans.57 Whether RSWA without dream enactment is in fact a marker and comparable and as such a potential marker for later neurodegenerative conversion is not resolved and needs additional electrophysiological, clinical, and follow-up studies.
Periodic limb movements in RBD
Few studies have investigated the clinical significance of periodic leg movements of sleep (PLMS) in patients with RBD and the pathological association between these disorders. A high prevalence of PLMS is observed in patients with PD as well as with atypical parkinsonism.58 The occurrence of PLMS has been associated with the dopaminergic system, and the first-line treatment of idiopathic PLMS consists of dopaminergic medications, the same as in PD. In addition, studies with positron emission tomography (PET) and SPECT imaging suggest that sleep abnormalities in PD, including an increased prevalence of PLMS, are indirect manifestations of the primary striatal dopamine deficiency.59 Happe et al reported a negative correlation of PLMS and striatal [123I]β-CIT SPECT binding in patients with PD, which supports the hypothesis that the occurrence of excessive nocturnal movements in PD is dependent on the severity of the presynaptic dopaminergic dysfunction.60 Sasai et al compared iRBD patients with and without PLMS. RSWA/REM was found to be a factor significantly associated with the existence of PLMS during NREM and REM stages, but the duration of RBD morbidity was associated with PLMS only during the REM stage.61 Whether PLMS during REM sleep in iRBD involves the same structures mediating atonia needs to be explored further.
REMs during REM sleep
Electrolytic lesions of the pedunculopontine tegmental nucleus (PPT) reduce the phasic activity of eye movements and other phasic REM features in cats (Figure 1).62 Although this study did not determine which type of neurotransmitter (cholinergic or glutamatergic) is involved in REMs, it demonstrated that the PPT and surrounding regions contain the generators of REMs.62 A clinical study applying video-oculography in patients with clinically probable RBD showed that ~24% of these patients have abnormal eye movements compared with only 7% of patients without clinically the condition,63 suggesting brainstem or cerebellar dysfunction. This confirms the results of other studies that analyzed eye movements during REM sleep in iRBD and PD using an automatic algorithm.64
EEG-slowing
Animal studies have implicated the reciprocal projections between the glutamatergic projections of the SLD and the cholinergic projections of the PPN/LDT in initiating REM sleep. It is known that these structures initiate ponto-geniculo-occipital waves, which involve distinct cortical areas (Figure 1). Several studies have reported that patients with iRBD present with a slowing of the EEG during wakefulness and REM sleep. Fantini et al were the first to analyze the EEG in iRBD by doing a quantitative EEG analysis during wakefulness and REM sleep. In their study, patients with iRBD showed considerably higher theta power in frontal, temporal, and occipital regions, with lower beta power in the occipital region. During REM sleep, the patients with iRBD presented with lower beta power in the occipital regions than in controls, although the sleep architecture was similar in the two groups. They suggested that their results reflect an early sign of impaired cortical activity.65 In a subsequent study, Massicotte-Marquez et al obtained similar results from analyzing EEG recordings during wakefulness in patients with iRBD and controls.66 They reported higher delta and theta power in iRBD during wakefulness in all brain areas compared with controls. These results are in line with imaging studies showing decreased blood flow in the frontal, temporal, and parietal lobes in patients with iRBD.67 Moreover, Iranzo et al evaluated spectral power EEG activity during both wakefulness and REM sleep in iRBD subjects who later developed MCI, a transitional stage between normal cognitive function and dementia.68 They analyzed the right and left hemispheres and found that increased delta and theta activity was more marked in the central than in the occipital region and in the right than in the left hemisphere. Moreover, they observed that patients who went on to develop MCI had more severe EEG-slowing than those patients with iRBD who remained idiopathic. A subsequent study have confirmed the relation between EEG-slowing and the occurrence of MCI.69
The EEG spectral pattern of patients with iRBD, who developed MCI, as well as the cortical hypoperfusion, corresponds to the observations in early stage PD and DLB.70,71 Cortical EEG-slowing and the cognitive impairment occurring in patients with iRBD may be caused by damage of the brainstem structures that regulate REM sleep and activity in the neocortex.
On the other hand, NREM sleep does not seem to be affected. Latreille et al investigated slow wave characteristics in iRBD and controls based on automatic slow wave detection. They measured the slow wave density, amplitude, frequency, slope, and duration of negative and positive phases and found similar results in patients with iRBD and controls. They concluded that the level of synchronization of thalamocortical neurons during NREM sleep was similar in both groups.72
Sleep spindles
Changes have been reported in relation to the microstructure of sleep, especially in relation to sleep spindles. Christensen et al assessed sleep spindles in patients with iRBD, PD patients with RBD, PD patients without RBD, and controls. They measured the density of sleep spindles with an automatic algorithm in REM and NREM sleep, and found that patients with iRBD and PD patients with and without RBD had a markedly lower density of sleep spindles in NREM than did controls.73 They suggested that the lower density in sleep spindles in these patients might involve dysfunction in prethalamic fibers in alpha-synucleinopathies. However, in another study based on manually identified spindles, they found no association in PD patients between spindle density or morphology and disease duration or severity.74
Several studies suggest that sleep spindles and slow waves play a role in brain plasticity and are associated with cognitive function. Latreille et al investigated whether alterations in sleep spindles and slow waves at the baseline visit could predict development of dementia at follow-up in PD.75 They investigated 68 nondemented PD patients and 47 healthy controls with baseline-PSG and comprehensive neuropsychological assessment. Sleep spindles and slow waves were automatically detected during NREM sleep throughout the entire night. At follow-up, an average of 4.5 years later, 18 PD patients had developed dementia and 50 remained dementia free. Sleep spindle density and amplitude were lower in PD patients who converted to dementia compared with the patients who remained dementia free and controls, mostly in posterior cortical regions. Dementia-free PD patients were intermediate between dementia patients and controls, with lower baseline sleep spindle density in all cortical areas compared with controls. Moreover, the authors found that in demented PD patients, lower sleep spindle amplitude in parietal and occipital areas was associated with poorer visuospatial abilities. Although slow wave amplitude was lower in PD patients than in controls, no difference was observed between those who developed or did not develop dementia.
The aforementioned results demonstrate that there are NREM sleep EEG abnormalities in PD patients. Sleep spindle activity was particularly impaired in PD patients who developed dementia, with a more posterior topographic pattern. Sleep spindle alterations are associated with later development of dementia in PD, and thus, may serve as an additional marker of cognitive decline in these patients.
Sleep instability
The SLD is critical for inducing cortical activation and atonia during REM sleep. Although lesions of the SLD induce the RBD-like phenotype in rats, they do not seem to affect REM sleep time and sleep transitions.27 Instead, lesions involving the caudal part of the LDT, which lies dorsal to the SLD, lead to severe sleep fragmentation involving both REM and NREM sleep, and reduction in the amount of REM sleep.27 Sleep-wake fragmentation and reduction in REM sleep have also been reported in mice with lesions of the glutamatergic neurotransmission in this area and after large lesions of the PPT-LDT and SLD in cats.37,76 As the brainstem is affected in the early stages of the PD disease process,77 this may explain the sleep fragmentation seen in this disorder. Christensen et al evaluated sleep characteristics such as sleep stability and sleep transitions in patients with iRBD and PD.78 They determined the transitions and the stability measures based on the manually scored hypnogram as well as by a data-driven method. They found that the patients had less stable REM and NREM sleep, and more REM/NREM transitions than controls, and suggested that these are affected in iRBD and PD.78
Pathophysiologically, sleep fragmentation and abnormalities in sleep architecture in PD might originate from nocturnal motor phenomena, such as increased muscle tone, akinesia or tremor, or drug side-effects. Since iRBD is considered as a premotor stage to alpha-synucleinopathies, these motor phenomena do not occur, and hence, this instability may be explained by the neurodegenerative process itself, which involves the neurochemical systems responsible for sleep organization.
Heart rate variability
Lanfranchi et al studied heart rate variability (HRV) during REM and NREM sleep in ten patients with iRBD and found that differences normally observed between these sleep stages were not present in patients with iRBD, indicating a loss of cardiac autonomic innervation.79 Sorensen et al investigated heart rate response in relation to arousals and leg movements during sleep in eleven patients with iRBD, 14 PD patients with RBD, 16 PD patients without RBD, and 17 controls, and found a significantly lower heart rate response to leg movements, whereby the heart rate response in iRBD was intermediate with respect to the control and the parkinsonian groups.80
Cardiac function in iRBD has also been measured during waking ECG. A study by Valappil et al measured ECG during wakefulness in eleven patients with iRBD and eleven control subjects with idiopathic insomnia without RBD. HRV was determined from 5-minute segments of a single-channel ECG during PSG evaluations, using automatically detected R–R intervals during wakefulness. The authors were able to correctly classify 77.3% of subjects using discriminant analysis featuring the leave-one-out cross-validation method and suggested that HRV measured by routine ECGs could be used to screen for Lewy body disorders such as PD.81 These findings are in agreement with studies showing decreased cardiac autonomic dysfunction by means of MIBG-scanning.
Miyamoto et al used 123I-MIBG cardiac scintigraphy to investigate cardiac sympathetic denervation in patients with iRBD and observed reduced uptake in all 13 patients.82 These changes were also found in patients with PD and DLB,15 adding to the evidence supporting the hypothesis that iRBD is part of a continuum including PD and DLB.
Although cardiac denervation is shown in iRBD and in PD, the course of cardiac denervation over time seems to be heterogeneous and independent of the development of motor symptoms.83,84 Postuma et al analyzed PSG trace measures of beat-to-beat R–R variability in 21 patients with iRBD who developed neurodegenerative disease, including PD (eleven patients), MSA (one patient), and dementia (nine patients). They found that despite clear differences between patients with iRBD and controls, there were none for any measure between those who did or did not develop disease.85
The results from these studies suggest that HRV during both wakefulness and sleep is lower in patients with iRBD compared with control subjects, suggesting abnormalities of sympathetic and parasympathetic functions. Cardiac autonomic dysfunction is also impaired in PD, suggesting that impaired HRV may be an early sign of PD.
Brainstem reflexes
As the pathophysiology of RBD involves dysfunction of brainstem structures, neurophysiological investigation of brainstem reflexes may reveal abnormalities in patients with alpha-synucleinopathies presenting with RBD. The electric blink reflex is a neurophysiological technique measuring brainstem function through a reflex arc involving the fifth to seventh cranial nerves.86 Besides the trigeminofacial reflex arc, this reflex involves the brainstem reticular formation. Several investigations of the blink reflex have been performed in patients with neurodegenerative disorders. In patients with DLB, the electric blink reflex latency is delayed relative to controls, independent of the presence of RBD. The amplitude of the auditory blink reflex has been found to be normal in patients with MSA.87 However, these patients present with an exaggerated startle response based on the other muscles Kofler et al investigated.88 Abnormal startle responses have also been found in other parkinsonian disorders, including DLB.88 A case report of a patient with RBD had concurrent excessive startle response to visual stimuli,89 probably caused by a pontine lesion and subsequent affection of the bulbopontine reticular formation. Other methods have documented the involvement of brainstem. Zoetmulder et al evaluated the use of prepulse inhibition90,91 and found that the sensorimotor gating, as measured with prepulse inhibition, is markedly reduced in patients with MSA but not in patients with iRBD or PD, suggesting that striatal and brainstem dysfunction is more severely affected in these patients. There are, however, limited data relating to this topic.
Imaging
Functional imaging techniques, such as SPECT and PET, have been used in an attempt to detect abnormal functional alterations in patients with RBD predicting evolution toward the onset of alpha-synucleinopathies. A reduced striatal binding of radioligands, such as 123I-FP-CIT and 11C-DTBZ, has revealed subclinical nigrostriatal dopaminergic degeneration in patients with RBD before the appearance of PD.92–95 Iranzo et al96 reported that in patients with iRBD, serial (123) FP-CIT SPECT shows a decline in striatal tracer uptake that reflects progressive nigrostriatal dopaminergic dysfunction. The mean reduction in striatal (123) FP-CIT uptake from baseline to 3 years was 19.36% in the right putamen, 15.57% in the left putamen, 10.81% in the left caudate, and 7.14% in the right caudate. They suggested that serial (123) FP-CIT SPECT can be used to monitor the progression of nigrostriatal degeneration in iRBD and could be useful in studying the potential disease-modifying compounds in these patients. A 6-[18F]fluoro-l-dopa PET study assessing the rate of disease progression in PD scanning patients twice with a 5-year interval suggested that the disease process first affects the posterior putamen, followed by the anterior putamen and caudate nucleus.97 Brain imaging of iRBD patients with PET showed lower levels of dopamine transporters in the nigrostriatal pathways that further decreased over time until reaching the level when parkinsonism appeared.98 It is not clear to what extent the nigrostriatal dopamine system is directly associated with increased motor activity during REM sleep. A (123) FP-CIT SPECT by Eisensehr et al95 reported that muscle activity during REM sleep lasting persistently longer than 0.5 seconds was independently associated with reduction of striatal dopamine transporters. However, these results could not be reproduced in a subsequent (123) FP-CIT SPECT study by Kim et al,99 which indicates that this topic needs more investigation.
In addition to SPECT and PET scanning, imaging applying combined neuromelanin-sensitive, structural, and diffusion magnetic resonance imaging at 3T in PD patients with and without RBD showed a relationship between damage to the locus subcoeruleus and abnormal muscle tone during REM sleep.100
Imaging with [18F]-fluorodeoxyglucose PET in iRBD revealed a higher metabolic rate in the hippocampus/parahippocampus, cingulate, supplementary motor area, and pons, but a lower rate in the occipital cortex/lingual gyrus.101 This suggests that region-specific metabolic abnormalities exist in patients with RBD and regional metabolic activities are associated with clinical measures such as RBD duration and chin EMG activity. Few studies have investigated the brain regions that are active during RBD.
An investigation applying ictal SPECT with simultaneous PSG recordings showed increased perfusion in the supplementary motor area during a REM sleep behavior episode.102 These findings were confirmed in a subsequent study using ictal SPECT and simultaneous PSG reporting bilateral activation of the premotor (supplementary motor) areas, the interhemispheric cleft, the periaqueductal area, the dorsal and ventral pons, and the anterior lobe of the cerebellum in the four patients examined.103 These studies suggest a common motor pathway in RBD and localize the motor generators responsible for dream enactment behavior to include the supplementary motor pathway bypassing the basal ganglia. These findings confirm a previous study reporting the “normalization” of movements during REM sleep in patients with RBD, which suggests that the movements during RBD could be generated by the motor cortex and would involve the pyramidal tract bypassing the extrapyramidal system.104
Biomarkers
According to Stern, preclinical PD is suspected when genetic, molecular, or imaging biomarkers support the presence of PD-specific pathology, but when no clinical signs and symptoms are yet evident.105 However, identifying PD patients in the preclinical stage is very challenging.3 Premotor PD is defined by the presence of early nonmotor signs and symptoms due to extranigral PD pathology.3,49 On account of this, the challenge is to identify reliable biomarkers to confirm the diagnosis of premotor PD. Several biomarkers have already been identified in the preclinical stage of PD, including RBD, hyposmia, MCI, autonomic dysfunction, and imaging. However, it is still a matter of debate which combination of screening tests is most sensitive to predict conversion from iRBD to PD.
Moreover, there is limited data concerning the identification of molecular markers in RBD. In one study, elevated alpha-synuclein levels in cerebrospinal fluid and serum were suggested to correlate with probable RBD, but the presence of RSWA was not documented by video-PSG. As iRBD is a strong predictor of the later development of an alpha-synucleinopathy, the development of alpha-synuclein ligands used in imaging and the development of drugs antagonizing this protein should receive more focus. There are limited data regarding other neurodegenerative biomarkers, eg, Tau protein (t-Tau), phosphorylated Tau (p-Tau181) protein, α-synuclein, neurofilament light, and chitinase-3-like protein-1, also known as YKL-40. The inflammatory marker YKL-40 has been found to be lower in PD and atypical PD,106,107 whereas other studies have only found elevated levels in AD but not PD.108 RSWA/RBD is strongly associated with hypocretin-deficient narcolepsy type 1,7 but in nonnarcoleptic RBD and in LBD, hypocretin levels were found to be normal.109,110 However, this does not imply that the hypocretin system is not involved in the neurodegenerative process as a lower frequency of neurons has been identified in PD patients;111,112 the cerebrospinal fluid (CSF)-hcrt-1 level reflects the general cerebrospinal level, and it is likely that the CSF concentration may be normal or subnormal, despite the significant loss of hypothalamic hypocretinergic neurons.
Treatment of RBD episodes
The medical literature on the treatment of RBD reflects a lack of randomized, double-blind controlled trials. Clonazepam and melatonin have been the most commonly used drugs for the nocturnal treatment of RBD. The use of these drugs is justified on the basis of small case series,113 and a small controlled trial has been conducted only for melatonin,114 in which only a limited effect was proved. Studies have most frequently focused on clonazepam. However, side effects may limit the use of clonazepam, especially in elderly and demented patients.113 A single case report has been presented regarding the positive effect of hypnotics and sodium oxybate,115,116 but its findings are very limited. In addition, there is little information about the efficacy of medication for RBD in narcolepsy type 1. We also know little about other drugs.
The effect of antidepressants (SSRI, TCA) and sodium oxybate on RBD varies from positive to negative,117 although the evidence is limited. As depression is associated with RBD, it is a strong confounder. Future studies need to address the effect of medications on outcome measures such as the improvement of the PSG features of RBD including RSWA, improvement of the quality of sleep, and prevention of injury.
In PD, a number of pathways and mechanisms have been the target for pharmacological intervention, including mitochondrial dysfunction, oxidative stress, inflammation, protein handling, and prion-like processes.118 However, none of these interventions appear to be effective in delaying the disease process in PD.119 Therefore, increased understanding about the pathophysiological mechanism in both iRBD and PD is necessary to be able to develop effective treatment regimens. As follow-up studies have convincingly shown that iRBD is a strong marker for later development of an alpha-synucleinopathy, patients with iRBD may be enrolled in future neuroprotective pharmacological studies.
Physical activity, physiotherapy, and social activities have been shown to modify the symptoms of PD.120–122 However, there are currently no controlled studies investigating the potential effect of these activities in reducing the conversion rate from iRBD to PD. Therefore, these treatment options are not considered in the management of RBD at present.
Discussion
The current identification of sleep-related abnormalities and the possible relation to neurodegenerative disorders raises a significant number of research questions. One of the most important issues is that the current studies identifying RBD as a risk factor for later parkinsonian conversion are based on identifying and defining the population at risk. The current studies have identified patients in high-level neurological sleep clinics. It has still not been settled whether this may influence the potential effect on the conversion, eg, if patients identified in broader clinical settings with simpler methods influence the diagnostic pattern and prognosis. The presence of other factors, eg, medication, alcohol, and comorbidities, may also influence the risk association.
Although there are currently no drugs available to prevent or delay the disease process for parkinsonian conversion, any sensitive and specific biological markers would be important in the future for identifying high-risk individuals with the aim of managing the risk. Owing to the complex mechanism of wake–sleep regulation, knowledge of these mechanisms and abnormalities is central to our understanding of the progressive nature of alpha-synucleinopathies. Owing to the early involvement and specific physiological pattern, identification of RBD and probably RSWA is crucial for the development and identification of biomarkers and potential development of disease-modifying medication and interventions.
A number of other symptoms and methods have been identified in RBD/PD, including imaging techniques, testing for olfactory and autonomic abnormalities, molecular techniques, and other sleep measures. It is likely that combinations of such approaches may enable patients to be stratified into specific risk associations that are of use in risk management.
One of the major challenges in defining those at risk is the potential relation between RBD (as defined by video-confirmed evaluation of REM sleep-associated behavior and the presence of simultaneous RSWA) and minor episodes of muscle activity and RSWA. Recently, it was suggested that these minor episodes be identified as REM behavior events.123 Questionnaires’ information may show up intra- and interindividual variations.124 Current prospective studies and most of the comorbidity studies rely on the classic definition of video-confirmed RBD. The extent to which dream content influences the diagnostic value of RBD is not known, but it may reflect the extent of pathology in the brainstem. Recent studies of day-to-day variation suggest that not only sleep patterns but also motor activity varies between the recorded days. NREM motor phenomena in patients with RBD are not taken into consideration,125 and in many patients with advanced neurodegenerative diseases, the EEG shows major abnormalities, with slowing, pathological microevents (eg, K-complexes, sleep spindles). As a consequence, sleep scoring is often difficult and subject to significant interscorer variability.126
PD, DLB, and MSA are complex neurodegenerative disorders, and their progress, severity, and symptom profile vary greatly between patients. It is not fully established whether iRBD is a precursor of only one PD subgroup. In analyzing iRBD patients with the aim of identifying those at the highest risk of developing PD, it might be unrealistic to expect that a single biomarker will be sufficient. By combining multiple biomarkers, the sensitivity of identifying a person with preclinical PD will increase, due to the fact that more aspects of this complex disease will be revealed. Optimally, a combination of biomarkers expressing alterations in different areas or mechanisms of the brain would indicate the stage of the disease and the pace and direction in which it is progressing. This would not only help identify the patients at the highest risk of developing PD, and thereby possibly facilitate PD treatment before PD diagnosis, but also provide insight into treatment efficiency and thereby help personalized treatment.
We believe that several additional research questions need to be addressed in order to understand brainstem involvement and the associated physiological, imaging, and molecular findings. This is fundamental to our understanding of how to identify high-risk individuals and to the development of risk-reducing agents, including protective medication, for these devastating diseases.
Acknowledgment
There was no funding for this study.
Author contributions
PJ conceived the study, and all authors (PJ, JAEC, and MZ) contributed substantially to the article. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work and have no further financial disclosures.
References
Jouvet M. Paradoxical sleep – a study of its nature and mechanisms. Prog Brain Res. 1965;18:20–62. | |
Schenck CH, Bundlie SR, Ettinger MG, Mahowald MW. Chronic behavioral disorders of human REM sleep: a new category of parasomnia. Sleep. 1986;9:293–308. | |
Schenck CH, Montplaisir JY, Frauscher B, et al. Rapid eye movement sleep behavior disorder: devising controlled active treatment studies for symptomatic and neuroprotective therapy – a consensus statement from the International Rapid Eye Movement Sleep Behavior Disorder Study Group. Sleep Med. 2013;14:795–806. | |
Sforza E, Zucconi M, Petronelli R, Lugaresi E, Cirignotta F. REM sleep behavioral disorders. Eur Neurol. 1988;28:295–300. | |
Brooks PL, Peever JH. Impaired GABA and glycine transmission triggers cardinal features of rapid eye movement sleep behavior disorder in mice. J Neurosci. 2011;31:7111–7121. | |
Mattarozzi K, Bellucci C, Campi C, et al. Clinical, behavioural and polysomnographic correlates of cataplexy in patients with narcolepsy/cataplexy. Sleep Med. 2008;9:425–433. | |
Knudsen S, Gammeltoft S, Jennum PJ. Rapid eye movement sleep behaviour disorder in patients with narcolepsy is associated with hypocretin-1 deficiency. Brain. 2010;133:568–579. | |
Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a parkinsonian disorder or dementia in 81% of older males initially diagnosed with idiopathic REM sleep behavior disorder (RBD): 16 year update on a previously reported series. Sleep Med. 2013;14(8):744–748. | |
Iranzo A, Fernández-Arcos A, Tolosa E, et al. Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: study in 174 patients. PLoS One. 2014;9:e89741. | |
Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-Marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology. 2009;72:1296–1300. | |
Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology. 1996;46:388–393. | |
Terzaghi M, Zucchella C, Rustioni V, Sinforiani E, Manni R. Cognitive performances and mild cognitive impairment in idiopathic rapid eye movement sleep behavior disorder: results of a longitudinal follow-up study. Sleep. 2013;36:1527–1532. | |
Sorensen GL, Mehlsen J, Jennum P. Reduced sympathetic activity in idiopathic rapid-eye-movement sleep behavior disorder and Parkinson’s disease. Auton Neurosci. 2013;179:138–141. | |
Kashihara K, Imamura T, Shinya T. Cardiac 123I-MIBG uptake is reduced more markedly in patients with REM sleep behavior disorder than in those with early stage Parkinson’s disease. Parkinsonism Relat Disord. 2010;16:252–255. | |
Miyamoto T, Miyamoto M, Suzuki K, Nishibayashi M, Iwanami M, Hirata K. 123I-MIBG cardiac scintigraphy provides clues to the underlying neurodegenerative disorder in idiopathic REM sleep behavior disorder. Sleep. 2008;31:717–723. | |
Unger MM, Möller JC, Mankel K, et al. Patients with idiopathic rapid-eye-movement sleep behavior disorder show normal gastric motility assessed by the 13C-octanoate breath test. Mov Disord. 2011;26:2559–2563. | |
Petit D, Gagnon JF, Fantini ML, Ferini-Strambi L, Montplaisir J. Sleep and quantitative EEG in neurodegenerative disorders. J Psychosom Res. 2004;56:487–496. | |
Postuma RB, Gagnon JF, Montplaisir J. Clinical prediction of Parkinson’s disease: planning for the age of neuroprotection. J Neurol Neurosurg Psychiatry. 2010;81:1008–1013. | |
Ross GW, Abbott RD, Petrovitch H, Tanner CM, White LR. Pre-motor features of Parkinson’s disease: the Honolulu-Asia Aging Study experience. Parkinsonism Relat Disord. 2012;18(suppl 1): S199–S202. | |
Noyce AJ, Bestwick JP, Silveira-Moriyama L, et al. PREDICT-PD: identifying risk of Parkinson’s disease in the community: methods and baseline results. J Neurol Neurosurg Psychiatry. 2014;85:31–37. | |
Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–1042. | |
Iber C. The American Academy Sleep Medicine Manual for the Scoring of Sleep and Associated Events. Westchester, IL: American Academy of Sleep Medicine; 2007. | |
Luppi PH, Clement O, Fort P. Paradoxical (REM) sleep genesis by the brainstem is under hypothalamic control. Curr Opin Neurobiol. 2013;23(5):786–792. | |
Saper CB. The neurobiology of sleep. Continuum (Minneap Minn). 2013;19:19–31. | |
Sapin E, Lapray D, Bérod A, et al. Localization of the brainstem GABAergic neurons controlling paradoxical (REM) sleep. PLoS One. 2009;4:e4272. | |
Verret L, Leger L, Fort P, Luppi PH. Cholinergic and noncholinergic brainstem neurons expressing Fos after paradoxical (REM) sleep deprivation and recovery. Eur J Neurosci. 2005;21:2488–2504. | |
Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–594. | |
Sastre JP, Buda C, Kitahama K, Jouvet M. Importance of the ventrolateral region of the periaqueductal gray and adjacent tegmentum in the control of paradoxical sleep as studied by muscimol microinjections in the cat. Neuroscience. 1996;74:415–426. | |
Crochet S, Onoe H, Sakai K. A potent non-monoaminergic paradoxical sleep inhibitory system: a reverse microdialysis and single-unit recording study. Eur J Neurosci. 2006;24:1404–1412. | |
Verret L, Fort P, Gervasoni D, Léger L, Luppi PH. Localization of the neurons active during paradoxical (REM) sleep and projecting to the locus coeruleus noradrenergic neurons in the rat. J Comp Neurol. 2006;495:573–586. | |
Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P, Luppi PH. The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci. 2002;16:1959–1973. | |
Clement O, Sapin E, Berod A, Fort P, Luppi PH. Evidence that neurons of the sublaterodorsal tegmental nucleus triggering paradoxical (REM) sleep are glutamatergic. Sleep. 2011;34:419–423. | |
Siegel JM. The neurobiology of sleep. Semin Neurol. 2009;29:277–296. | |
Lai YY, Siegel JM. Pontomedullary glutamate receptors mediating locomotion and muscle tone suppression. J Neurosci. 1991;11:2931–2937. | |
Mileykovskiy BY, Kiyashchenko LI, Kodama T, Lai YY, Siegel JM. Activation of pontine and medullary motor inhibitory regions reduces discharge in neurons located in the locus coeruleus and the anatomical equivalent of the midbrain locomotor region. J Neurosci. 2000;20:8551–8558. | |
Luppi PH, Gervasoni D, Boissard R, et al. Brainstem structures responsible for paradoxical sleep onset and maintenance. Arch Ital Biol. 2004;142:397–411. | |
Krenzer M, Anaclet C, Vetrivelan R, et al. Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS One. 2011;6:e24998. | |
Ramaligam V, Chen MC, Saper CB, Lu J. Perspectives on the rapid eye movement sleep switch in rapid eye movement sleep behavior disorder. Sleep Med. 2013;14:707–713. | |
Vetrivelan R, Fuller PM, Tong Q, Lu J. Medullary circuitry regulating rapid eye movement sleep and motor atonia. J Neurosci. 2009;29:9361–9369. | |
Webster HH, Friedman L, Jones BE. Modification of paradoxical sleep following transections of the reticular formation at the pontomedullary junction. Sleep. 1986;9:1–23. | |
Anaclet C, Pedersen NP, Fuller PM, Lu J. Brainstem circuitry regulating phasic activation of trigeminal motoneurons during REM sleep. PLoS One. 2010;5:e8788. | |
Lai YY, Hsieh KC, Nguyen D, Peever J, Siegel JM. Neurotoxic lesions at the ventral mesopontine junction change sleep time and muscle activity during sleep: an animal model of motor disorders in sleep. Neuroscience. 2008;154:431–443. | |
Vetrivelan R, Chang C, Lu J. Muscle tone regulation during REM sleep: neural circuitry and clinical significance. Arch Ital Biol. 2011;149:348–366. | |
Hassani OK, Henny P, Lee MG, Jones BE. GABAergic neurons intermingled with orexin and MCH neurons in the lateral hypothalamus discharge maximally during sleep. Eur J Neurosci. 2010;32:448–457. | |
Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Muscle tone facilitation and inhibition after orexin-a (hypocretin-1) microinjections into the medial medulla. J Neurophysiol. 2002;87:2480–2489. | |
Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003 Mar–Apr;24(2):197–211. | |
Boeve BF, Silber MH, Ferman TJ, et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med. 2013;14:754–762. | |
Boeve BF. REM sleep behavior disorder: updated review of the core features, the REM sleep behavior disorder-neurodegenerative disease association, evolving concepts, controversies, and future directions. Ann N Y Acad Sci. 2010;1184:15–54. | |
Boeve BF. Predicting the future in idiopathic rapid-eye movement sleep behaviour disorder. Lancet Neurol. 2010;9:1040–1042. | |
Lapierre O, Montplaisir J. Polysomnographic features of REM sleep behavior disorder: development of a scoring method. Neurology. 1992;42:1371–1374. | |
Frauscher B, Iranzo A, Gaig C, et al; SINBAR (Sleep Innsbruck Barcelona) Group. Normative EMG values during REM sleep for the diagnosis of REM sleep behavior disorder. Sleep. 2012;35:835–847. | |
Ferri R, Manconi M, Plazzi G, et al. A quantitative statistical analysis of the submentalis muscle EMG amplitude during sleep in normal controls and patients with REM sleep behavior disorder. J Sleep Res. 2008;17:89–100. | |
Montplaisir J, Gagnon JF, Fantini ML, et al. Polysomnographic diagnosis of idiopathic REM sleep behavior disorder. Mov Disord. 2010;25:2044–2051. | |
Frandsen R, Nikolic M, Zoetmulder M, Kempfner L, Jennum P. Analysis of automated quantification of motor activity in REM sleep behaviour disorder. J Sleep Res. 2015;24(5):583–590. | |
Kempfner J, Sorensen GL, Nikolic M, Frandsen R, Sorensen HB, Jennum P. Rapid eye movement sleep behavior disorder as an outlier detection problem. J Clin Neurophysiol. 2014;31:86–93. | |
Stefani A, Gabelia D, Högl B, et al. Long-term follow-up investigation of isolated rapid eye movement sleep without atonia without rapid eye movement sleep behavior disorder: a pilot study. J Clin Sleep Med. 2015;11(11):1273–1279. | |
Kang SH, Yoon IY, Lee SD, Han JW, Kim TH, Kim KW. REM sleep behavior disorder in the Korean elderly population: prevalence and clinical characteristics. Sleep. 2013;36:1147–1152. | |
Terzaghi M, Arnaldi D, Rizzetti MC, et al. Analysis of video- polysomnographic sleep findings in dementia with Lewy bodies. Mov Disord. 2013;28:1416–1423. | |
Hilker R, Burghaus L, Razai N, Jacobs AH, Szelies B, Heiss WD. Functional brain imaging in combined motor and sleep disorders. J Neurol Sci. 2006;248:223–226. | |
Happe S, Pirker W, Klosch G, Sauter C, Zeitlhofer J. Periodic leg movements in patients with Parkinson’s disease are associated with reduced striatal dopamine transporter binding. J Neurol. 2003;250:83–86. | |
Sasai T, Inoue Y, Matsuura M. Clinical significance of periodic leg movements during sleep in rapid eye movement sleep behavior disorder. J Neurol. 2011;258:1971–1978. | |
Shouse MN, Siegel JM. Pontine regulation of REM sleep components in cats: integrity of the pedunculopontine tegmentum (PPT) is important for phasic events but unnecessary for atonia during REM sleep. Brain Res. 1992;571:50–63. | |
Kim YE, Yang HJ, Yun JY, Kim HJ, Lee JY, Jeon BS. REM sleep behavior disorder in Parkinson disease: association with abnormal ocular motor findings. Parkinsonism Relat Disord. 2014;20:444–446. | |
Christensen JA, Koch H, Frandsen R, et al. Classification of iRBD and Parkinson’s disease patients based on eye movements during sleep. Conf Proc IEEE Eng Med Biol Soc. 2013;2013:441–444. | |
Fantini ML, Gagnon JF, Petit D, et al. Slowing of electroencephalogram in rapid eye movement sleep behavior disorder. Ann Neurol. 2003;53:774–780. | |
Massicotte-Marquez J, Carrier J, Décary A, et al. Slow-wave sleep and delta power in rapid eye movement sleep behavior disorder. Ann Neurol. 2005;57:277–282. | |
Hanyu H, Inoue Y, Sakurai H, et al. Regional cerebral blood flow changes in patients with idiopathic REM sleep behavior disorder. Eur J Neurol. 2011;18:784–788. | |
Iranzo A, Isetta V, Molinuevo JL, et al. Electroencephalographic slowing heralds mild cognitive impairment in idiopathic REM sleep behavior disorder. Sleep Med. 2010;11:534–539. | |
Sasai T, Matsuura M, Inoue Y. Electroencephalographic findings related with mild cognitive impairment in idiopathic rapid eye movement sleep behavior disorder. Sleep. 2013;36:1893–1899. | |
Bonanni L, Thomas A, Tiraboschi P, Perfetti B, Varanese S, Onofrj M. EEG comparisons in early Alzheimer’s disease, dementia with Lewy bodies and Parkinson’s disease with dementia patients with a 2-year follow-up. Brain. 2008;131:690–705. | |
Morita A, Kamei S, Mizutani T. Relationship between slowing of the EEG and cognitive impairment in Parkinson disease. J Clin Neurophysiol. 2011;28:384–387. | |
Latreille V, Carrier J, Montplaisir J, Lafortune M, Gagnon JF. Non-rapid eye movement sleep characteristics in idiopathic REM sleep behavior disorder. J Neurol Sci. 2011;310:159–162. | |
Christensen JA, Kempfner J, Zoetmulder M, et al. Decreased sleep spindle density in patients with idiopathic REM sleep behavior disorder and patients with Parkinson’s disease. Clin Neurophysiol. 2014;125:512–519. | |
Christensen JA, Nikolic M, Warby SC, et al. Sleep spindle alterations in patients with Parkinson’s disease. Front Hum Neurosci. 2015;9:233. | |
Latreille V, Carrier J, Lafortune M, et al. Sleep spindles in Parkinson’s disease may predict the development of dementia. Neurobiol Aging. 2015;36:1083–1090. | |
Webster HH, Jones BE. Neurotoxic lesions of the dorsolateral pontomesencephalic tegmentum-cholinergic cell area in the cat. II Effects upon sleep-waking states. Brain Res. 1988;458:285–302. | |
Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. | |
Christensen JA, Jennum P, Koch H, et al. Sleep stability and transitions in patients with idiopathic REM sleep behavior disorder and patients with Parkinson’s disease. Clin Neurophysiol. 2015;127(1):537–543. | |
Lanfranchi PA, Fradette L, Gagnon JF, Colombo R, Montplaisir J. Cardiac autonomic regulation during sleep in idiopathic REM sleep behavior disorder. Sleep. 2007;30:1019–1025. | |
Sorensen GL, Kempfner J, Zoetmulder M, Sorensen HB, Jennum P. Attenuated heart rate response in REM sleep behavior disorder and Parkinson’s disease. Mov Disord. 2012;27:888–894. | |
Valappil RA, Black JE, Broderick MJ, et al. Exploring the electrocardiogram as a potential tool to screen for premotor Parkinson’s disease. Mov Disord. 2010;25:2296–2303. | |
Miyamoto T, Miyamoto M, Inoue Y, Usui Y, Suzuki K, Hirata K. Reduced cardiac 123I-MIBG scintigraphy in idiopathic REM sleep behavior disorder. Neurology. 2006;67:2236–2238. | |
Miyamoto T, Miyamoto M, Iwanami M, Hirata K. Follow-up study of cardiac 123I-MIBG scintigraphy in idiopathic REM sleep behavior disorder. Eur J Neurol. 2011;18:1275–1278. | |
Oguri T, Tachibana N, Mitake S, Kawanishi T, Fukuyama H. Decrease in myocardial 123I-MIBG radioactivity in REM sleep behavior disorder: two patients with different clinical progression. Sleep Med. 2008;9:583–585. | |
Postuma RB, Lanfranchi PA, Blais H, Gagnon JF, Montplaisir JY. Cardiac autonomic dysfunction in idiopathic REM sleep behavior disorder. Mov Disord. 2010;25:2304–2310. | |
Basso MA, Evinger C. An explanation for reflex blink hyperexcitability in Parkinson’s disease. II Nucleus raphe magnus. J Neurosci. 1996;16:7318–7330. | |
Valldeoriola F, Valls-Solé J, Tolosa E, Nobbe FA, Muñoz JE, Martí J. The acoustic startle response is normal in patients with multiple system atrophy. Mov Disord. 1997;12:697–700. | |
Kofler M, Müller J, Wenning GK, et al. The auditory startle reaction in parkinsonian disorders. Mov Disord. 2001;16:62–71. | |
Peter A, Hansen ML, Merkl A, Voigtländer S, Bajbouj M, Danker-Hopfe H. REM sleep behavior disorder and excessive startle reaction to visual stimuli in a patient with pontine lesions. Sleep Med. 2008;9:697–700. | |
Zoetmulder M, Biernat HB, Nikolic M, Korbo L, Friberg L, Jennum PJ. Prepulse inhibition is associated with attention, processing speed, and 123I-FP-CIT SPECT in Parkinson’s disease. J Parkinsons Dis. 2014;4:77–87. | |
Zoetmulder M, Biernat HB, Nikolic M, Korbo L, Jennum PJ. Sensorimotor gating deficits in multiple system atrophy: comparison with Parkinson’s disease and idiopathic REM sleep behavior disorder. Parkinsonism Relat Disord. 2014;20:297–302. | |
Albin RL, Koeppe RA, Chervin RD, et al. Decreased striatal dopaminergic innervation in REM sleep behavior disorder. Neurology. 2000;55:1410–1412. | |
Iranzo A, Valldeoriola F, Lomeña F, et al. Serial dopamine transporter imaging of nigrostriatal function in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study. Lancet Neurol. 2011;10:797–805. | |
Miyamoto M, Miyamoto T. Neuroimaging of rapid eye movement sleep behavior disorder: transcranial ultrasound, single-photon emission computed tomography, and positron emission tomography scan data. Sleep Med. 2013;14:739–743. | |
Eisensehr I, Linke R, Noachtar S, Schwarz J, Gildehaus FJ, Tatsch K. Reduced striatal dopamine transporters in idiopathic rapid eye movement sleep behaviour disorder. Comparison with Parkinson’s disease and controls. Brain. 2000;123(pt 6):1155–1160. | |
Iranzo A, Valldeoriola F, Lomeña F, et al. Serial dopamine transporter imaging of nigrostriatal function in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study. Lancet Neurol. 2011 Sep;10(9):797–805. | |
Nurmi E, Ruottinen HM, Bergman J, et al. Rate of progression in Parkinson’s disease: a 6-[18F]fluoro-L-dopa PET study. Mov Disord. 2001;16:608–615. | |
Miyamoto T, Orimo S, Miyamoto M, et al. Follow-up PET studies in case of idiopathic REM sleep behavior disorder. Sleep Med. 2010;11:100–101. | |
Kim YK, Yoon IY, Kim JM, et al. The implication of nigrostriatal dopaminergic degeneration in the pathogenesis of REM sleep behavior disorder. Eur J Neurol. 2010;17:487–492. | |
García-Lorenzo D, Longo-Dos Santos C, Ewenczyk C, et al. The coeruleus/subcoeruleus complex in rapid eye movement sleep behaviour disorders in Parkinson’s disease. Brain. 2013;136:2120–2129. | |
Ge J, Wu P, Peng S, et al. Assessing cerebral glucose metabolism in patients with idiopathic rapid eye movement sleep behavior disorder. J Cereb Blood Flow Metab. 2015;35(12):2062–2069. | |
Dauvilliers Y, Boudousq V, Lopez R, et al. Increased perfusion in supplementary motor area during a REM sleep behaviour episode. Sleep Med. 2011;12:531–532. | |
Mayer G, Bitterlich M, Kuwert T, Ritt P, Stefan H. Ictal SPECT in patients with rapid eye movement sleep behaviour disorder. Brain. 2015;138:1263–1270. | |
De Cock VC, Vidailhet M, Leu S, et al. Restoration of normal motor control in Parkinson’s disease during REM sleep. Brain. 2007;130:450–456. | |
Stern MB. The preclinical detection of Parkinson’s disease: ready for prime time? Ann Neurol. 2004;56:169–171. | |
Olsson B, Constantinescu R, Holmberg B, Andreasen N, Blennow K, Zetterberg H. The glial marker YKL-40 is decreased in synucleinopathies. Mov Disord. 2013;28:1882–1885. | |
Magdalinou NK, Paterson RW, Schott JM, et al. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 2015;86:1240–1247. | |
Wennström M, Surova Y, Hall S, et al. The inflammatory marker YKL-40 is elevated in cerebrospinal fluid from patients with Alzheimer’s but not Parkinson’s disease or dementia with Lewy bodies. PLoS One. 2015;10:e0135458. | |
Anderson KN, Vincent A, Smith IE, Shneerson JM. Cerebrospinal fluid hypocretin levels are normal in idiopathic REM sleep behaviour disorder. Eur J Neurol. 2010;17:1105–1107. | |
Baumann CR, Dauvilliers Y, Mignot E, Bassetti CL. Normal CSF hypocretin-1 (orexin A) levels in dementia with Lewy bodies associated with excessive daytime sleepiness. Eur Neurol. 2004;52:73–76. | |
Thannickal TC, Lai YY, Siegel JM. Hypocretin (orexin) cell loss in Parkinson’s disease. Brain. 2007;130:1586–1595. | |
Fronczek R, Overeem S, Lee SY, et al. Hypocretin (orexin) loss in Parkinson’s disease. Brain. 2007;130:1577–1585. | |
Aurora RN, Zak RS, Maganti RK, et al; Standards of Practice Committee; American Academy of Sleep Medicine. Best practice guide for the treatment of REM sleep behavior disorder (RBD). J Clin Sleep Med. 2010;6:85–95. | |
Kunz D, Mahlberg R. A two-part, double-blind, placebo-controlled trial of exogenous melatonin in REM sleep behaviour disorder. J Sleep Res. 2010;19:591–596. | |
Anderson KN, Shneerson JM. Drug treatment of REM sleep behavior disorder: the use of drug therapies other than clonazepam. J Clin Sleep Med. 2009;5:235–239. | |
Shneerson JM. Successful treatment of REM sleep behavior disorder with sodium oxybate. Clin Neuropharmacol. 2009;32:158–159. | |
Onofrj M, Luciano AL, Thomas A, Iacono D, D’Andreamatteo G. Mirtazapine induces REM sleep behavior disorder (RBD) in parkinsonism. Neurology. 2003;60:113–115. | |
Schapira AH, Olanow CW, Greenamyre JT, Bezard E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: future therapeutic perspectives. Lancet. 2014;384:545–555. | |
Henchcliffe C, Severt WL. Disease modification in Parkinson’s disease. Drugs Aging. 2011;28:605–615. | |
Cruickshank TM, Reyes AR, Ziman MR. A systematic review and meta-analysis of strength training in individuals with multiple sclerosis or Parkinson disease. Medicine (Baltimore). 2015;94:e411. | |
Tomlinson CL, Herd CP, Clarke CE, et al. Physiotherapy for Parkinson’s disease: a comparison of techniques. Cochrane Database Syst Rev. 2014;6:CD002815. | |
Shanahan J, Morris ME, Bhriain ON, Saunders J, Clifford AM. Dance for people with Parkinson disease: what is the evidence telling us? Arch Phys Med Rehabil. 2015;96:141–153. | |
Sixel-Doring F, Trautmann E, Mollenhauer B, Trenkwalder C. Rapid eye movement sleep behavioral events: a new marker for neurodegeneration in early Parkinson disease? Sleep. 2014;37:431–438. | |
Sixel-Doring F, Schweitzer M, Mollenhauer B, Trenkwalder C. Intraindividual variability of REM sleep behavior disorder in Parkinson’s disease: a comparative assessment using a new REM sleep behavior disorder severity scale (RBDSS) for clinical routine. J Clin Sleep Med. 2011;7:75–80. | |
Mayer G, Kesper K, Ploch T, et al. Quantification of tonic and phasic muscle activity in REM sleep behavior disorder. J Clin Neurophysiol. 2008;25:48–55. | |
Jensen PS, Sorensen HB, Leonthin HL, Jennum P. Automatic sleep scoring in normals and in individuals with neurodegenerative disorders according to new international sleep scoring criteria. J Clin Neurophysiol. 2010;27:296–302. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.