Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities
Authors Lozano D, Gonzales-Portillo G, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan C
Received 13 October 2014
Accepted for publication 3 December 2014
Published 8 January 2015 Volume 2015:11 Pages 97—106
DOI https://doi.org/10.2147/NDT.S65815
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Diego Lozano,* Gabriel S Gonzales-Portillo,* Sandra Acosta, Ike de la Pena, Naoki Tajiri, Yuji Kaneko, Cesar V Borlongan
Department of Neurosurgery and Brain Repair, University of South Florida Morsani College of Medicine, Tampa, FL, USA
*These authors contributed equally to this work
Abstract: Traumatic brain injury (TBI) is a serious public health problem accounting for 1.4 million emergency room visits by US citizens each year. Although TBI has been traditionally considered an acute injury, chronic symptoms reminiscent of neurodegenerative disorders have now been recognized. These progressive neurodegenerative-like symptoms manifest as impaired motor and cognitive skills, as well as stress, anxiety, and mood affective behavioral alterations. TBI, characterized by external bumps or blows to the head exceeding the brain’s protective capacity, causes physical damage to the central nervous system with accompanying neurological dysfunctions. The primary impact results in direct neural cell loss predominantly exhibiting necrotic death, which is then followed by a wave of secondary injury cascades including excitotoxicity, oxidative stress, mitochondrial dysfunction, blood–brain barrier disruption, and inflammation. All these processes exacerbate the damage, worsen the clinical outcomes, and persist as an evolving pathological hallmark of what we now describe as chronic TBI. Neuroinflammation in the acute stage of TBI mobilizes immune cells, astrocytes, cytokines, and chemokines toward the site of injury to mount an antiinflammatory response against brain damage; however, in the chronic stage, excess activation of these inflammatory elements contributes to an “inflamed” brain microenvironment that principally contributes to secondary cell death in TBI. Modulating these inflammatory cells by changing their phenotype from proinflammatory to antiinflammatory would likely promote therapeutic effects on TBI. Because neuroinflammation occurs at acute and chronic stages after the primary insult in TBI, a treatment targeting neuroinflammation may have a wider therapeutic window for TBI. To this end, a better understanding of TBI etiology and clinical manifestations, especially the pathological presentation of chronic TBI with neuroinflammation as a major component, will advance our knowledge on inflammation-based disease mechanisms and treatments.
Keywords: head trauma, chronic, inflammation, secondary cell death, regenerative medicine, stem cells
A Letter to the Editor has been received and published for this article.
Introduction
Traumatic brain injury (TBI) is an intracranial injury caused by an external force that impacts the head, exceeding the brain’s protective capacity. This force could be a violent blow, a bump, a projectile, or even a blast.1 Depending on the severity of the impact, it may lead to bruising, bleeding, brain dysfunction, or death.2–4 The extent of these effects may last temporarily, for a couple of days, or for the rest of a patient’s life.2–4
TBI can be classified as mild, moderate, or severe, depending on the damage to the brain,5,6 and because of this heterogeneity, accurately categorizing each TBI case has been difficult. To this end, the clinical test used most is the Glasgow coma scale, which assesses the level of consciousness of the patient, measuring motor responsiveness, verbal performance, and eye reflex, with a composite score from all three tests of 3–8 considered severe, a score of 9–13 considered moderate, and a score of 14–15 considered mild.1,7,8 The use of imaging modalities (eg, computed tomography or magnetic resonance imaging scans) and differential diagnosis of comorbidity factors (ie, an existing pathological condition such as endocrine diseases/problems such as hypopituitarism or growth hormone deficiency and diabetes insipidus)9–11 are equally important in determining TBI severity.12 A similar challenge exists in characterizing the pathological severity in animal models of TBI.5 However, both human and animal TBI brains present with the distinctive signature of a neuroinflammatory response.5,9–12
Mild TBI, sometimes referred to as concussion, is the most prevalent TBI.3,4,13,14 Although TBI has been traditionally considered an acute injury, accumulating clinical and laboratory evidence has recognized the chronic pathology of the disease. Indeed, TBI can manifest many symptoms of neurodegenerative disorders, such as Parkinson’s15–18 and Alzheimer’s disease.13,19
Accumulating laboratory and clinical evidence has implicated neuroinflammation in both acute and chronic stages of TBI,2–6 suggesting this secondary cell death pathway may be the key to the disease pathology and treatment, which is the major topic of this article.
TBI is a serious public health problem, contributing to 30% of deaths related to injury in the United States,3,4 with a mortality rate of around 50,000 individuals each year, 1.4 million receiving emergency treatment, and more than 235,000 requiring hospitalization.20,21 Among the survivors, around 85,000 suffer long-term complications, including chronic disability, leading to an estimated prevalence of 3.2–5.3 million Americans with TBI.20,22 The economic burden is approximately $4.5 billion from direct treatment at hospitals and long-term care, $20.6 billion lost as a result of work absence or disability, and $12.7 billion in lost income from premature death, all adding up to more than $37.8 billion.23 In addition, TBI affects not only the individual but also family members, friends, and even the community, which adds another cost to TBI. The significant extent of social and financial toll to the United States contributes to TBI’s designation as an urgent clinically unmet need.
TBI is prevalent in both military personnel and civilians.24–26 Hence, TBI will continue to be an issue even in peaceful times, necessitating the need for research to gain better insights into the disease. Here, we focus on neuroinflammation, which closely manifests immediately after TBI onset, and equally important, it persists in the chronic stages of the disease, making it an appealing target for understanding TBI pathology and its treatment (Figure 1).
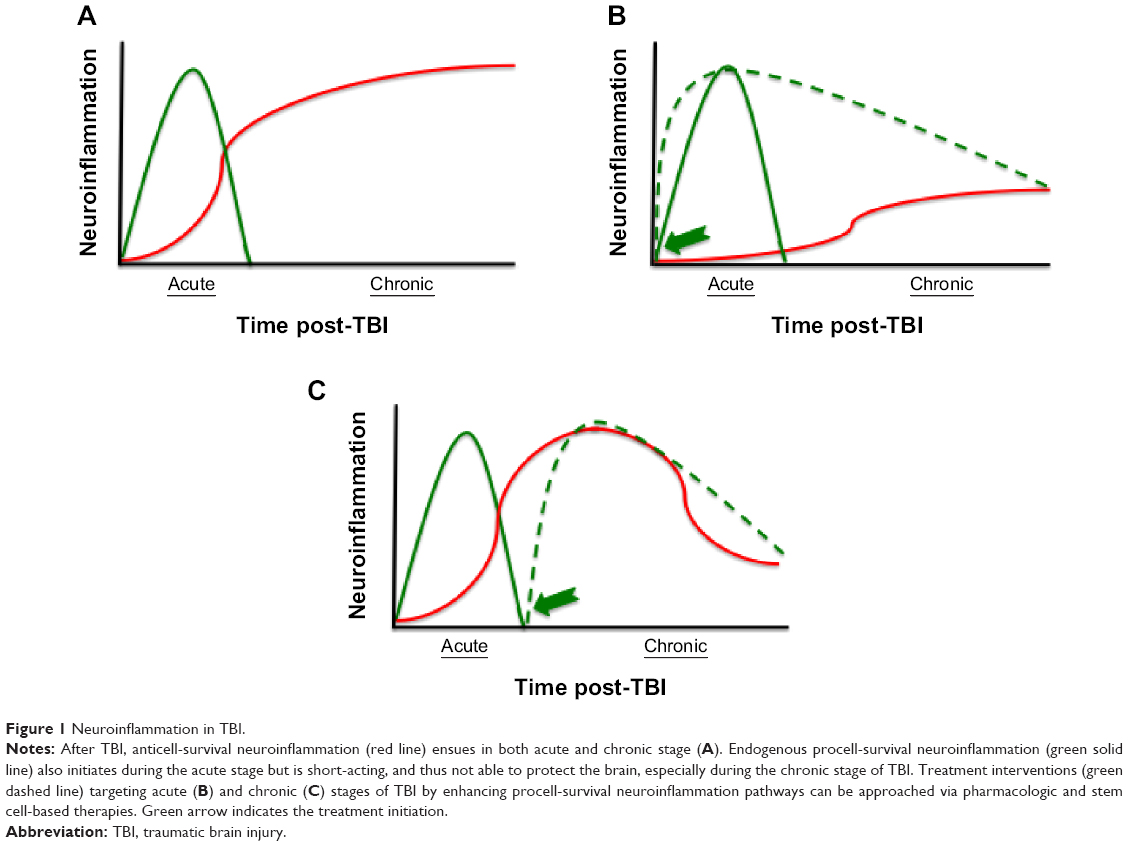 | Figure 1 Neuroinflammation in TBI. |
Clinical manifestations of TBI
After suffering TBI, around 30%–80% of patients experience postconcussive symptoms.27 Most patients fully recover in a couple hours or days, although it may take a couple of weeks. However, depending on the severity of the injury, there are some cases in which victims do not recover and the symptoms persist for years.28 Factors such as being female, being older, having a lower socioeconomic status, and having previous mental health problems make patients more prone to chronic postconcussive symptoms.27,29 Clinical manifestations of mild TBI consist of a combination of physical and neuropsychiatric symptoms, which include behavioral and cognitive disorders.
There are many forms of TBI other than mild TBI. Similar symptoms may develop from TBI (ie, contusions to the brain, or even diffuse axonal injuries), but they are often worsened (ie, coma). The clinical manifestations of these types of TBI, computed tomography scan findings, and management will differ. This review focuses mainly on symptoms found in mild TBI, as they are most prevalent.
Of the physical symptoms of TBI, headaches are the most common, with around 25%–90% of post–mild TBI patients reporting it.27 Dizziness and nausea are other common symptoms, along with fatigue, sleep disruption, hearing problems, and visual disturbances.28 As a result of damage to the frontal or temporal lobe, TBI patients are also prone to seizures, which may present a challenge for diagnosis and treatment (ie, differential diagnosis between TBI and epilepsy).27
Cognitive disorders after TBI primarily include attention deficit, memory problems, and executive dysfunction.28 Attention deficit is very common and interferes with other functions, making daily tasks harder than before.30 The extent of these cognitive impairments is directly correlated to the severity of injury.31 There are different explanations for the underlying mechanism of these symptoms. Injury to the frontal lobe and subcortical systems results in loss of function.30,31 White matter shearing and diffuse axonal injuries may result in interruption and inefficiencies in processing information, along with slower mental speed and stamina.30
Behavioral symptoms associated with TBI are recognized as changes in the personality of the patient. These include irritability, mood changes, aggression, impulsivity, self-centered behavior, and poor persistence.30,32 Other symptoms related to TBI are depression (sadness, low energy and motivation, not liking oneself, hopelessness), anxiety, and posttraumatic stress disorder.27,28,33 In addition, as noted earlier, TBI may increase the risk of developing Parkinson’s disease, Alzheimer’s disease, and other neurodegenerative diseases in the long term.16–18,34,35 These behavioral symptoms are more prevalent and are severely manifested in females, older adults, those of lower socioeconomic status, and those with previous mental health problems.29 The recognition of these comorbidity factors will facilitate the diagnosis and treatment of TBI, especially in the mild form.
As previously mentioned, TBI patients are prone to seizures, which makes it difficult to diagnose whether TBI or epilepsy is to blame for some symptoms. Regardless of the diagnoses, some latent forms of epileptiform processes disturb cognitive and emotional balance.27 This can lead to so-called complex partial seizure-like symptoms, which also effect inflammatory changes (ie, increased levels of interleukin 6 [IL-6]). Research that further expands knowledge on the mechanism of these processes may be of importance with respect to specific indications of anticonvulsants.
Secondary cell death and neuroinflammation
Once TBI occurs, a number of short- and long-term detrimental processes begin to affect the brain. The initial insult first leads to a primary injury caused by the mechanical damage from shearing, tearing, and/or stretching of neurons, axons, glia, and blood vessels.36 The primary injury can be diffuse, focal, or a combination of both, making the damage in each case heterogeneous. In most cases, any type of TBI-induced primary injury results in direct neural cell loss and necrotic cell death.37
The primary injury triggers a secondary wave of biochemical cascades, together with metabolic and cellular changes. This occurs within seconds to minutes after the traumatic insult and can last for days, months, or years. It often leads to progressive neurodegeneration and delayed cell death, exacerbating the damage from the primary injury.37,38 The secondary wave is mainly detected in the injury site and surrounding tissue, although neurodegeneration in brain areas located far from the primary impact has recently been recognized.3 The secondary wave consists of excitotoxicity, oxidative stress, mitochondrial dysfunction, blood–brain barrier (BBB) disruption, and inflammation.2,5,15,33,39–44 All these processes contribute to neurological deficits separately, but at the same time, these cell death processes interact, worsening the progressive outcome of TBI.39
There are several secondary cell death mechanisms that accompany TBI. Excitotoxicity, characterized by the injured nerve cells secreting large amounts of intracellular glutamate into the extracellular space,45 has been detected after TBI. This significantly increased concentration of neurotransmitter, glutamate, in the synaptic space46 overstimulates the AMPA and NMDA receptors of surrounding nerve cells. These receptors stay activated, allowing an influx of sodium and calcium ions into the cell.45 The high concentration of calcium ions in the cytosol leads to the activation of protein phosphatases, phospholipases, and endonucleases. Eventually, the DNA is fragmented, and structures and membranes of the cell are deteriorated. This results in cell death through a hybrid form of apoptosis and necrosis.47 The overstimulation of glutamate receptors also results in the increased production of nitric oxide, free radicals, and prodeath transcription factors.48
Another secondary cell death event that occurs soon after TBI is oxidative stress, characterized by aberrant accumulation of reactive oxygen species (ROS) and reactive nitrogen species (RON).49 Because of their highly reactive properties, free radical levels are normally kept low via regulation of the production and degradation rate by enzymes and antioxidants.50 After TBI, a significant increase in ROS and impairment of antioxidants that lower the levels is seen.51 When the generation of ROS/RON is too large, it leads to major cell dysfunction, as its oxidative capabilities damage all biomolecules.52 ROS cause lipoperoxidation of the cell membrane, which results in the dysfunction of many structures and organelles, such as the mitochondria and oxidizing proteins that affect membrane pores.50 It may also fragment DNA, causing mutations.50 ROS are also related to the infiltration of neutrophil, which induces an inflammatory response that, in turn, increases the generation of ROS.51 Overall, oxidative stress cascade results in large neuronal cell death.
Mitochondrial dysfunction has also been documented as a key secondary cell death mechanism in TBI.53 The mitochondria is a major source of ROS from the electron transport chain.54 After TBI, the stabilizing mechanisms of levels of ROS become impaired, resulting in increased concentrations.55 Lipid peroxidation-mediated oxidative damage to the mitochondrial membrane negatively affects its structure and function.56 The mitochondria also works as a calcium ion buffer, releasing and absorbing the ions as needed to maintain homeostasis.38 However, when the calcium ion load becomes too large from excitotoxicity, the function of the mitochondria becomes impaired. The mitochondrial permeability transition pore, associated with the mitochondrial inner membrane, is a calcium ion-dependent pore.57 With the excess calcium ions, the pore stays active, disrupting the mitochondrial membrane potential.57 Without a membrane potential, the mitochondria is unable to produce ATP, and the ATP synthase may actually consume ATP instead of producing it. With mitochondrial break down, toxins and apoptotic factors are released into the cell, activating the caspase-dependent apoptosis.58 This causes the cell to commit suicide.
An equally persistent secondary cell death in TBI is BBB breakdown.44,53,59 The BBB consists of tightly connected endothelial cells, but its normal function is contingent on interactions with closely located glia and astrocytes. It is highly selective, creating an environment for the brain parenchymal cells free of blood–borne factors and immune cells.60 BBB dysfunction is related to neuronal cell death and cognitive decline and limits the effectiveness of therapies.60,61 Its dysfunction triggers many other secondary injuries, including cell death, oxidative stress, and inflammation, causing the brain to swell, with higher intracranial pressure and ischemia.61 The primary injury disrupts the tight junctions, allowing an influx of peripheral immune cells and circulating factors (albumin, thrombin, and fibrinogen).60 These events affect the interaction between BBB endothelial cells and astrocytic glial cells, further contributing to the effects of BBB dysfunction by increasing its permeability.60 One of the underlying mechanisms regarding BBB dysfunction after TBI is the up-regulation of protein matrix metallopeptidase 9 (MMP-9).62 This digests the tight junctions, disrupting its proper function.62 BBB breakdown also allows an influx of larger molecules such as leukocytes that increase the osmotic force in the brain.63 This results in edema and higher intracranial pressure, which are directly related to ischemia and further cell death.63
Next, we focus on our research group’s long-standing interest in neuroinflammation, which is also responsible for major secondary cell death after TBI.2–4,15,19,33,43 Neuroinflammation initiates after impact and has been detected as lasting in humans for up to at least 17 years.64 Neuroinflammation involves immune cells, microglia, cytokines, chemotactic cytokines (chemokines), and other inflammatory mediators that interfere with the brain’s endogenous capacity to repair itself, exacerbating neuronal death.65 After the initial injury, an endogenous inflammatory response is triggered to defend the injury site from invading pathogens and to repair the damaged cells. The complement is activated to perform these functions and recruits inflammatory cells into the intrathecal compartment.39 The activation of the complement is also accompanied by the infiltration of neutrophils, monocytes, and lymphocytes across the BBB.66 These secrete prostaglandins, free radicals, proinflammatory cytokines, and other inflammatory mediators that, in turn, up-regulate the expression of chemokines and cell adhesion molecules.66,67 This results in immune cells and microglia mobilizing into the brain parenchyma.66
Peripheral and resident microglia cells, which are systemic macrophages, both mount a protective response against TBI, limiting the extent of the damage by separating the injured tissue from the healthy tissue.68 However, microglial activation in TBI is excessive, and proinflammatory cytokines such as tumor necrosis factor (TNF)-α, IL-1β, IL-6, IL-12, and interferon γ are released.4,39 The up-regulation of these cytokines increases the permeability of the BBB by higher expression of cell adhesion molecules in the endothelial cells and by an increased production of chemokines. This results in an increased inflammatory response.60 Sustained microglial activation also produces neurotoxic molecules and free radicals, which lead to other mechanisms of secondary cell death.4,37 Glial cells also play a major role in the leukocyte entry across the BBB. T cells produce cytokines that stimulate glial cells to greatly produce and express chemokines that ease the migration of leukocytes by up-regulating the expression of adhesion molecules. This leads to an amplification of the inflammatory reaction.69 In addition, activated microglial cells increase the expression of major histocompatibility complex class II (MHCII+), which is directly correlated to neurodegeneration.4
Although activated microglia often act as a major contributor in the inflammatory response, there are instances in which they do not. They can polarize into distinct phenotypes, depending on the microenvironment in which they are activated. An M1 phenotype, also referred as “classical” activation, is promoted in the presence of lipopolysaccharide and interferon γ.4,68 This subtype is characterized by the increased synthesis of proinflammatory cytokines and low levels of antiinflammatory cytokines such as IL-10.70 In contrast, when exposed to cytokines IL-4 or IL-13, microglia polarize into the M2 phenotype.70 This “alternative activation” dampens the inflammatory response by reducing the production of proinflammatory cytokines and increasing the production of IL-10, transforming growth factor 1β (both anti-inflammatory cytokines), and suppressor of cytokine signaling.4,68
Astrocytes also play a role in the injury site after TBI. Along with other neurotrophic factors, they up-regulate brain-derived neurotrophic factors that support and guide axons in their recovery, increase cell proliferation, and promote the long-term survival of neurons by inhibiting programmed cell death.68,71,73 Astrocytes also regulate extracellular glutamate levels, which consequently reduce glutamate excitotoxicity to neurons and other cells.68 Accordingly, astrocytes are thought to exert many beneficial effects post-TBI.68 However, when the presence of astrocytes is too large and they become overactivated, it can lead to detrimental effects in the brain. The astrocytes secrete inhibitory extracellular matrix, building a dense physical and chemical barrier surrounding the injury site (glial scar), which encapsulates and isolates the axons. This protects the remaining healthy brain from the neurotoxic environment of the injury site, but it also interferes and prevents the regeneration and repair of the damaged tissue.68,72
Neuroinflammation-based therapies
Damage resulting from the primary injury is almost impossible to treat and can only be prevented by using safety equipment (wearing helmets and/or seatbelts) to avoid accidents. As such, researchers and clinicians focus on secondary cell death when contemplating treatments for TBI. The delayed damage caused by secondary events allows a window of opportunity to treat the injury. Therefore, it is very convenient to target neuroinflammation for TBI therapy. It takes considerably more time for the inflammatory cells to reach the injured brain and contribute to the secondary cell death damage than it takes other secondary death mechanisms, such as glutamate excitotoxicity.74 This delayed onset provides an extended window of opportunity in which treatments can be administered, greatly increasing the chances of a successful intervention and preventing further damage. When considering treatments for neuroinflammation in TBI, it is important to note that inflammation has both beneficial and detrimental effects. Prior studies have shown that high doses of antiinflammatory agents actually lead to worse outcomes.75 In addition to inhibiting the detrimental effects of neuroinflammation, these robust treatments may also retard the beneficial ones.
Stand-alone drugs targeting inflammation have been explored for TBI. Minocycline, a tetracycline derivative with antiinflammatory properties, is one potential drug treatment for TBI.76–79 These properties, along with its excellent human safety profile, as deemed by the US Food and Drug Administration, and its ability to cross the BBB, make minocycline a superlative candidate for clinical trials.76,80 In studies using animal models, minocycline afforded improvement in tissue damage and in inflammation, leading to improved outcomes. The drug reduces the secretion of proinflammatory cytokines and chemokines, together with other mediators of inflammation, reducing nitric oxide by direct scavenging and inhibiting the overactivation and proliferation of microglia cells.80,81 Targeting microglia cells is crucial, as it is accompanied by the reduction of cytokines IL-1β, IL-6, and MMP-9, all of which mediate the proinflammatory response.75,81,82 However, data from other studies suggest that minocycline has no therapeutic effect on TBI. These conflicting results may be a result of variations such as dosage and interval of administration, but they still warrant additional studies.81,83
Another stand-alone drug being investigated is melatonin. A hormone from the pineal gland, melatonin has shown neuroprotective properties.84–94 As a lipophilic enzyme, it can easily cross cell membranes and carry out its functions. Its antiinflammatory capabilities are related to inhibition of microglial activation and lower secretion of proinflammation cytokines including IL-1β and TNF-α.91,92 These are directly related to an increased inflammatory response. Similar to minocycline, the effect of melatonin has varied. The results of successful trials include lower brain edema and reduced cortical neural degeneration, which would suggest improved cognitive deficits.93 However, experimental models have failed to show cognitive improvements, with the dosage of therapeutic administered suggested as the root cause.83 Therefore, melatonin may be a potential therapy to counter secondary damage caused by inflammation. Additional studies are needed to assess the long-term feasibility, safety, and efficacy of melatonin in multiple models of TBI.
Statins, drugs normally used to lower cholesterol, have also shown many neuroprotective properties, including antiinflammatory effects in a mouse model of subarachnoid hemorrhage.95 Statins suppress the activation of microglia by inhibiting the signaling pathways of toll-like receptor 4 and nuclear factor kB (NF-kB) activation, as well as some small G-proteins.40,96 It also suppresses the activation of astrocytes by inhibiting epidermal growth factor receptors, which play a central role in astrogliosis.97 Therefore, the suppressed microglia and astrocytes do not perform their proinflammatory effects, resulting in decreased expression of proinflammatory cytokines IL-1β and TNF-α, intracellular adhesion molecules, and intercellular adhesion molecules ameliorating the inflammatory response.40,95,97 Results from statin treatment in TBI models have shown improved neuronal survival, growth, and differentiation and reduction of functional impairment.40 In clinical trials of patients reporting TBI, administration of rosuvastatin over the course of 10 days resulted in minor improvements in amnesia and disorientation, as assessed by the Galveston Orientation Amnesia Test.98 Statins are well tolerated by patients, and their mild adverse effects are well-defined and can be easily monitored. Although statins have a record of safety and efficacy in preclinical models of other disease implications (eg, stroke, intracerebral hemorrhage), more rigorous laboratory studies in TBI models may improve clinical outcomes.
Preclinical studies using stem cells have shown promising results as a regenerative therapy for TBI.2,3,99 Stem cells are unspecialized cells that can regenerate damaged tissue because of their endogenous ability to differentiate into multiple cell lineages, their high proliferative capacity, and the trophic support they provide to surviving cells.2,100,101 They are especially valuable in the treatment of brain damage, as they can modulate inflammation, stimulate endogenous repair mechanisms, and potentially differentiate into specific cells, such as neurons, replacing the dead ones.3,102–104 Positive results from these therapies include decreased cognitive and motor dysfunction after a traumatic event, as well as functional and physiological improvements, less inflammation and neural cell death, and increased regeneration of damaged tissue.3,105 Stem cells can be sourced from embryo, fetus, and adult cells; the first two have higher plasticity, trophic support, and proliferation. However, unlike adult stem cells, they risk uncontrolled proliferation and tumor formation. In addition, adult stem cells avoid the ethical and moral issues that are related to embryonic and fetal cells.2,3,72
Mesenchymal stem cells (MSCs) have shown therapeutic potential in TBI. They are able to migrate to the injury site, modulate the inflammatory response, proliferate, and activate T lymphocytes, neutrophils, and microglia.106–108 The suppression of inflammation and immunomodulation by MSCs is attributed to the secretion of TNF-α-stimulated gene/protein 6 (TSG-6). TSG-6 is an antiinflammatory protein that is induced by TNF-α and IL-1. It interferes with the signaling pathway involved in the activation of toll-like receptors and NF-kB.106,109 The latter regulates many genes involved in the inflammatory response and is also correlated to the synthesis of proinflammatory cytokines. Therefore, with the secretion of TSG-6, the NF-kB pathway is inhibited together with the proinflammatory cascade that follows.109 At the same time, TSG-6 binds and inactivates fragments of hyaluronan, which are characterized by their proinflammatory role.109 It also shifts the behavior of T cells from producing proinflammatory cytokines (interferon γ) to producing antiinflammatory cytokines (IL-4).110
MSCs also reduce the production of chemokines CXCL2, CCL2, and RANTES. Chemokines increase the permeability of the BBB, allowing peripheral leukocytes to migrate into the damaged brain parenchyma. They are usually secreted by microglia and neurons, and may lead to an immune and inflammatory response.106 The chemokine CXCL2 participates in the infiltration of neutrophils. CCL2, a chemoattractant, stimulates the migration of immune cells. RANTES is able to activate T cells.106,111 MSCs down-regulate production of CCL2 by secreting transforming growth factor β. This factor activates Smad3, which suppresses the transcription of CCL2 in the cell.111 As a result of MSCs reducing the production of these chemokines, the detrimental effects caused by leukocytes, neutrophils, and microglia after TBI are ameliorated, reducing secondary damage.
One issue transplanted stem cells face is that they cannot thrive in the inhospitable environment of the injury site.112 Because of this, stem cells should be used in combination with other factors that improve the environment, can further enhance the regeneration of damaged tissue, and improve stem cell neuroprotection. This can be achieved by using granulocyte-colony stimulating factor (G-CSF). There is evidence supporting G-CSF’s capability to enhance endogenous stem cells and promote stemness maintenance.3 It demonstrates a moderate antiinflammatory effect, ameliorates motor dysfunction, reduces brain edema, and improves the control of glutamate levels. Likewise, treatment with human umbilical cord blood cells alone results in a moderate decrease of hippocampal cell death and enhanced neurogenesis. However, the neuroprotective effects of both therapies are synergized when administered as a combination, exhibiting a robust beneficial effect in endogenous neurogenesis and cell death reduction.3 Combining these therapies also leads to longer-lasting effects than those exhibited by the treatments administered alone.3
G-CSF, a drug approved by the US Food and Drug Administration for treatment, is a cytokine that acts through receptor-mediated transport by recruiting endogenous stem cells from the bone marrow into the peripheral blood. These stem cells can then promote neuroprotection in the injury site.3 Experimental data also suggest that G-CSF crosses the BBB and binds to the G-CSF receptor on neurons and microglia. As a result, inflammatory cytokines are down-regulated, angiogenesis is promoted in the brain, and antiapoptotic pathways are triggered.3 Stem cells mobilized from the bone marrow can also play an indirect role in the periphery, where they synthesize and release trophic growth factors, chemokines, and cytokines that contribute to the protection of the damaged tissue, improving brain repair.3
Future direction of antiinflammation as a therapy for TBI
The double-edged sword of neuroinflammation after TBI offers a venues for understanding disease mechanisms and treatments. Immune cells, astrocytes, cytokines, and chemokines are all necessary components for brain repair, and it is their excessive levels that contribute to the secondary cell death damage in TBI. Finding a treatment strategy that can modulate the influx of these cells to the injured brain in a time-dependent manner, allowing their relative activation and deactivation, is crucial to exploit their beneficial effects toward TBI treatment. Future studies are necessary to further advance our understanding of secondary cell death mechanisms (inflammation) in an effort to repurpose their outcome from being an exacerbating factor to serving as a therapeutic agent against TBI. A full recognition of disease etiology, their clinical manifestations, and their detrimental consequences will aid in the research and development of therapeutic regimens for TBI.
Acknowledgments
Dr Borlongan is supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke 1R01NS071956-01, Department of Defense W81XWH-11-1-0634, James and Esther King Foundation for Biomedical Research Program, SanBio Inc, KM Pharmaceuticals, NeuralStem Inc, and Karyopharm Inc.
Disclosure
The authors report no conflicts of interest in this work.
References
Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7(8):728–741. | ||
Tajiri N, Acosta SA, Shahaduzzaman M, et al. Intravenous transplants of human adipose-derived stem cell protect the brain from traumatic brain injury-induced neurodegeneration and motor and cognitive impairments: cell graft biodistribution and soluble factors in young and aged rats. J Neurosci. 2014;34(1):313–326. | ||
Acosta SA, Tajiri N, Shinozuka K, et al. Combination therapy of human umbilical cord blood cells and granulocyte colony stimulating factor reduces histopathological and motor impairments in an experimental model of chronic traumatic brain injury. PLoS ONE. 2014;9(3): e90953. | ||
Hernandez-Ontiveros DG, Tajiri N, Acosta S, Giunta B, Tan J, Borlongan CV. Microglia activation as a biomarker for traumatic brain injury. Front Neurol. 2013;4:30. | ||
Yu S, Kaneko Y, Bae E, et al. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. 2009;1287:157–163. | ||
Niyonkuru C, Wagner AK, Ozawa H, Amin K, Goyal A, Fabio A. Group-based trajectory analysis applications for prognostic biomarker model development in severe TBI: a practical example. J Neurotrauma. 2013; 30(11):938–945. | ||
Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2(7872):81–84. | ||
Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT; Workshop Scientific Team and Advisory Panel Members. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25(7):719–738. | ||
Bazarian JJ, Cernak I, Noble-Haeusslein L, Potolicchio S, Temkin N. Long-term neurologic outcomes after traumatic brain injury. J Head Trauma Rehabil. 2009;24(6):439–451. | ||
Cerecedo-López CD, Kim-Lee JH, Hernandez D, Acosta SA, Borlongan CV. Insulin-associated neuroinflammatory pathways as therapeutic targets for traumatic brain injury. Med Hypotheses. 2014;82(2): 171–174. | ||
Diamandis T, Gonzales-Portillo C, Gonzales-Portillo GS, et al. Diabetes insipidus contributes to traumatic brain injury pathology via CD36 neuroinflammation. Med Hypotheses. 2013;81(5):936–939. | ||
Parikh S, Koch M, Narayan RK. Traumatic brain injury. Int Anesthesiol Clin. 2007;45(3):119–135. | ||
Malkesman O, Tucker LB, Ozl J, McCabe JT. Traumatic brain injury – modeling neuropsychiatric symptoms in rodents. Front Neurol. 2013;4:157. | ||
Dikmen SS, Corrigan JD, Levin HS, Machamer J, Stiers W, Weisskopf MG. Cognitive outcome following traumatic brain injury. J Head Trauma Rehabil. 2009;24(6):430–438. | ||
Acosta SA, Tajiri N, de la Pena I, et al. Alpha-synuclein as a Pathological Link between Chronic Traumatic Brain Injury and Parkinson’s disease. J Cell Physiol. 2014;n/a. | ||
Stefanis L. α-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2(2):a009399. | ||
Su E, Bell MJ, Wisniewski SR, et al. α-Synuclein levels are elevated in cerebrospinal fluid following traumatic brain injury in infants and children: the effect of therapeutic hypothermia. Dev Neurosci. 2010;32(5–6): 385–395. | ||
Uryu K, Chen XH, Martinez D, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208(2):185–192. | ||
Tajiri N, Kellogg SL, Shimizu T, Arendash GW, Borlongan CV. Traumatic brain injury precipitates cognitive impairment and extracellular Aβ aggregation in Alzheimer’s disease transgenic mice. PLoS ONE. 2013;8(11):e78851. | ||
Corrigan JD, Selassie AW, Orman JA. The epidemiology of traumatic brain injury. J Head Trauma Rehabil. 2010;25(2):72–80. | ||
Langlois JA, Rutland-Brown W, Thomas KE. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths. Atlanta, GA: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2006. | ||
Machamer JE, Temkin NR, Dikmen SS. Neurobehavioral outcome in persons with violent or nonviolent traumatic brain injury. J Head Trauma Rehabil. 2003;18(5):387–397. | ||
Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: A public health perspective. J Head Trauma Rehabil. 1999;14(6):602–615. | ||
Duckworth JL, Grimes J, Ling GS. Pathophysiology of battlefield associated traumatic brain injury. Pathophysiology. 2013;20(1):23–30. | ||
Warden D. Military TBI during the Iraq and Afghanistan wars. J Head Trauma Rehabil. 2006;21(5):398–402. | ||
Dilley M, Avent C. Long-term neuropsychiatric disorders after traumatic brain injury. In: Uehara T, ed. Psychiatric Disorder – Worldwide Advances. London: InTech; 2011. http://dx.doi.org/10.5772/28548 | ||
Riggio S. Traumatic brain injury and its neurobehavioral sequelae. Psychiatr Clin North Am. 2010;33(4):807–819. | ||
Bryan CJ. Multiple traumatic brain injury and concussive symptoms among deployed military personnel. Brain Inj. 2013;27(12):1333–1337. | ||
Mott TF, McConnon ML, Rieger BP. Subacute to chronic mild traumatic brain injury. Am Fam Physician. 2012;86(11):1045–1051. | ||
Tramontana MG, Cowan RL, Zald D, Prokop JW, Guillamondegui O. Traumatic brain injury-related attention deficits: treatment outcomes with lisdexamfetamine dimesylate (Vyvanse). Brain Inj. 2014;28(11):1461–1472. | ||
Siopi E, Llufriu-Dabén G, Fanucchi F, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M. Evaluation of late cognitive impairment and anxiety states following traumatic brain injury in mice: the effect of minocycline. Neurosci Lett. 2012;511(2):110–115. | ||
Belanger HG, Kretzmer T, Vanderploeg RD, French LM. Symptom complaints following combat-related traumatic brain injury: relationship to traumatic brain injury severity and posttraumatic stress disorder. J Int Neuropsychol Soc. 2010;16(1):194–199. | ||
Acosta SA, Diamond DM, Wolfe S, et al. Influence of post-traumatic stress disorder on neuroinflammation and cell proliferation in a rat model of traumatic brain injury. PLoS ONE. 2013;8(12):e81585. | ||
Franzblau M, Gonzales-Portillo C, Gonzales-Portillo GS, et al. Vascular damage: a persisting pathology common to Alzheimer’s disease and traumatic brain injury. Med Hypotheses. 2013;81(5):842–845. | ||
Fleminger S. Long-term psychiatric disorders after traumatic brain injury. Eur J Anaesthesiol Suppl. 2008;42(suppl 42):123–130. | ||
Adams JH, Graham DI, Gennarelli TA. Head injury in man and experimental animals: neuropathology. Acta Neurochir Suppl (Wien). 1983;32:15–30. | ||
Kabadi SV, Faden AI. Neuroprotective strategies for traumatic brain injury: improving clinical translation. Int J Mol Sci. 2014;15(1):1216–1236. | ||
Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160(1):226–234. | ||
Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury – an inflammatory disease? Brain Res Brain Res Rev. 2005;48(2):388–399. | ||
Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31(12):596–604. | ||
Bramlett H, Dietrich WD III. Long-term consequences of traumatic brain injury: Current status of potential mechanisms of injury and neurologic outcomes. J Neurotrauma. Epub 2014 Aug 26. | ||
Shahaduzzaman M, Acosta S, Bickford PC, Borlongan CV. α-Synuclein is a pathological link and therapeutic target for Parkinson’s disease and traumatic brain injury. Med Hypotheses. 2013;81(4):675–680. | ||
Acosta SA, Tajiri N, Shinozuka K, et al. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS ONE. 2013;8(1):e53376. | ||
Glover LE, Tajiri N, Lau T, Kaneko Y, van Loveren H, Borlongan CV. Immediate, but not delayed, microsurgical skull reconstruction exacerbates brain damage in experimental traumatic brain injury model. PLoS ONE. 2012;7(3):e33646. | ||
Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol. 2013;698(1–3):6–18. | ||
Quintard H, Patet C, Suys T, Marques-Vidal P, Oddo M. Normobaric hyperoxia is associated with increased cerebral excitotoxicity after severe traumatic brain injury. Neurocrit Care. 2014. http://dx.doi.org/10.1007/s12028-014-0062-0 | ||
Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46(4):281–309. | ||
Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15(11):1382–1402. | ||
Dasuri K, Zhang L, Keller JN. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med. 2013;62:170–185. | ||
Mutinati M, Pantaleo M, Roncetti M, Piccinno M, Rizzo A, Sciorsci RL. Oxidative stress in neonatology: a review. Reprod Domest Anim. 2014;49(1):7–16. | ||
Wang JW, Wang HD, Cong ZX, et al. Puerarin ameliorates oxidative stress in a rodent model of traumatic brain injury. J Surg Res. 2014;186(1):328–337. | ||
Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842(8):1240–1247. | ||
Hayashi T, Kaneko Y, Yu S, et al. Quantitative analyses of matrix metalloproteinase activity after traumatic brain injury in adult rats. Brain Res. 2009;1280:172–177. | ||
Li Q, Zhang T, Wang J, et al. Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem Biophys Res Commun. 2014;444(2):182–188. | ||
Zhu KJ, Huang H, Chu H, Yu H, Zhang SM. Alterations in enterocyte mitochondrial respiratory function and enzyme activities in gastrointestinal dysfunction following brain injury. World J Gastroenterol. 2014;20(28):9585–9591. | ||
Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem. 2010;114(1):271–280. | ||
Mbye LH, Singh IN, Sullivan PG, Springer JE, Hall ED. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Exp Neurol. 2008;209(1):243–253. | ||
Cheng G, Kong RH, Zhang LM, Zhang JN. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br J Pharmacol. 2012;167(4):699–719. | ||
Shojo H, Kaneko Y, Mabuchi T, Kibayashi K, Adachi N, Borlongan CV. Genetic and histologic evidence implicates role of inflammation in traumatic brain injury-induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience. 2010;171(4):1273–1282. | ||
Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood–brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. 2011;2(4):492–516. | ||
Pop V, Sorensen DW, Kamper JE, et al. Early brain injury alters the blood–brain barrier phenotype in parallel with β-amyloid and cognitive changes in adulthood. J Cereb Blood Flow Metab. 2013;33(2):205–214. | ||
Yu F, Wang Z, Tanaka M, et al. Posttrauma cotreatment with lithium and valproate: reduction of lesion volume, attenuation of blood–brain barrier disruption, and improvement in motor coordination in mice with traumatic brain injury. J Neurosurg. 2013;119(3):766–773. | ||
Yang SH, Gustafson J, Gangidine M, et al. A murine model of mild traumatic brain injury exhibiting cognitive and motor deficits. J Surg Res. 2013;184(2):981–988. | ||
Giunta B, Obregon D, Velisetty R, Sanberg PR, Borlongan CV, Tan J. The immunology of traumatic brain injury: a prime target for Alzheimer’s disease prevention. J Neuroinflammation. 2012;9(1):185. | ||
Tajiri N, Hernandez D, Acosta S, et al. Suppressed cytokine expression immediatey following traumatic brain injury in neonatal rats indicates an expeditious endogenous anti-inflammatory response. Brain Res. 2014;1559:65–71. | ||
Fluiter K, Opperhuizen AL, Morgan BP, Baas F, Ramaglia V. Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J Immunol. 2014;192(5):2339–2348. | ||
Bellander BM, Singhrao SK, Ohlsson M, Mattsson P, Svensson M. Complement activation in the human brain after traumatic head injury. J Neurotrauma. 2001;18(12):1295–1311. | ||
Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26(8):1191–1201. | ||
Ransohoff RM. The chemokine system in neuroinflammation: an update. J Infect Dis. 2002;186(s2)(suppl 2):S152–S156. | ||
Chhor V, Le Charpentier T, Lebon S, et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav Immun. 2013;32:70–85. | ||
Zhao Z, Alam S, Oppenheim RW, Prevette DM, Evenson A, Parsadanian A. Overexpression of glial cell line-derived neurotrophic factor in the CNS rescues motoneurons from programmed cell death and promotes their long-term survival following axotomy. Exp Neurol. 2004;190(2):356–372. | ||
Rodrigues MC, Glover LE, Weinbren N, et al. Toward personalized cell therapies: autologous menstrual blood cells for stroke. J Biomed Biotechnol. 2011;2011:194720. | ||
Wu LY, Bao XQ, Sun H, Zhang D. Scavenger receptor on astrocytes and its relationship with neuroinflammation. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2014;36(3):330–335. | ||
Szmydynger-Chodobska J, Gandy JR, Varone A, Shan R, Chodobski A. Synergistic interactions between cytokines and AVP at the blood-CSF barrier result in increased chemokine production and augmented influx of leukocytes after brain injury. PLoS ONE. 2013;8(11):e79328. | ||
Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7(1):22–30. | ||
Xu L, Fagan SC, Waller JL, et al. Low dose intravenous minocycline is neuroprotective after middle cerebral artery occlusion-reperfusion in rats. BMC Neurol. 2004;4(1):7. | ||
Fagan SC, Edwards DJ, Borlongan CV, et al. Optimal delivery of minocycline to the brain: implication for human studies of acute neuroprotection. Exp Neurol. 2004;186(2):248–251. | ||
Machado LS, Kozak A, Ergul A, Hess DC, Borlongan CV, Fagan SC. Delayed minocycline inhibits ischemia-activated matrix metalloproteinases 2 and 9 after experimental stroke. BMC Neurosci. 2006;7(1):56. | ||
Matsukawa N, Yasuhara T, Hara K, et al. Therapeutic targets and limits of minocycline neuroprotection in experimental ischemic stroke. BMC Neurosci. 2009;10(1):126. | ||
Kovesdi E, Kamnaksh A, Wingo D, et al. Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front Neurol. 2012;3:111. | ||
Homsi S, Federico F, Croci N, et al. Minocycline effects on cerebral edema: relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res. 2009;1291:122–132. | ||
Guo ZD, Wu HT, Sun XC, Zhang XD, Zhang JH. Protection of minocycline on early brain injury after subarachnoid hemorrhage in rats. Acta Neurochir Suppl. 2011;110(Pt 1):71–74. | ||
Kelso ML, Scheff NN, Scheff SW, Pauly JR. Melatonin and minocycline for combinatorial therapy to improve functional and histopathological deficits following traumatic brain injury. Neurosci Lett. 2011;488(1):60–64. | ||
Campolo M, Ahmad A, Crupi R, et al. Combination therapy with melatonin and dexamethasone in a mouse model of traumatic brain injury. J Endocrinol. 2013;217(3):291–301. | ||
Senol N, Naziroğlu M. Melatonin reduces traumatic brain injury-induced oxidative stress in the cerebral cortex and blood of rats. Neural Regen Res. 2014;9(11):1112–1116. | ||
Gonzales-Portillo GS, Lozano D, Aguirre D, et al. An update on the use of melatonin as a stroke therapeutic. Minerva Med. 2014. | ||
Shinozuka K, Staples M, Borlongan CV. Melatonin-based therapeutics for neuroprotection in stroke. Int J Mol Sci. 2013;14(5):8924–8947. | ||
Borlongan CV, Sumaya I, Moss D, et al. Melatonin-secreting pineal gland: a novel tissue source for neural transplantation therapy in stroke. Cell Transplant. 2003;12(3):225–234. | ||
Borlongan CV, Yamamoto M, Takei N, et al. Glial cell survival is enhanced during melatonin-induced neuroprotection against cerebral ischemia. FASEB J. 2000;14(10):1307–1317. | ||
Poulos SG, Borlongan CV. Artificial lighting conditions and melatonin alter motor performance in adult rats. Neurosci Lett. 2000;280(1):33–36. | ||
Wang Z, Wu L, You W, Ji C, Chen G. Melatonin alleviates secondary brain damage and neurobehavioral dysfunction after experimental subarachnoid hemorrhage: possible involvement of TLR4-mediated inflammatory pathway. J Pineal Res. 2013;55(4):399–408. | ||
Ding K, Wang H, Xu J, Lu X, Zhang L, Zhu L. Melatonin reduced microglial activation and alleviated neuroinflammation induced neuron degeneration in experimental traumatic brain injury: Possible involvement of mTOR pathway. Neurochem Int. 2014;76:23–31. | ||
Ding K, Wang H, Xu J, et al. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. | ||
Yürüker V, Naziroğlu M, Senol N. Reduction in traumatic brain injury-induced oxidative stress, apoptosis, and calcium entry in rat hippocampus by melatonin: Possible involvement of TRPM2 channels. Metab Brain Dis. 2014. http://dx.doi.org/10.1007/s11011-014-9623-3 | ||
Uekawa K, Hasegawa Y, Ma M, et al. Rosuvastatin ameliorates early brain injury after subarachnoid hemorrhage via suppression of superoxide formation and nuclear factor-kappa B activation in rats. J Stroke Cerebrovasc Dis. 2014;23(6):1429–1439. | ||
Wang KW, Chen HJ, Lu K, et al. Simvastatin attenuates the cerebral vascular endothelial inflammatory response in a rat traumatic brain injury. Ann Clin Lab Sci. 2014;44(2):145–150. | ||
Wu H, Mahmood A, Lu D, et al. Attenuation of astrogliosis and modulation of endothelial growth factor receptor in lipid rafts by simvastatin after traumatic brain injury. J Neurosurg. 2010;113(3):591–597. | ||
Tapia-Perez J, Sanchez-Aguilar M, Torres-Corzo JG, et al. Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758). J Neurotrauma. 2008;25(8):1011–1017. | ||
Tajiri N, Duncan K, Antoine A, et al. Stem cell-paved biobridge facilitates neural repair in traumatic brain injury. Front Syst Neurosci. 2014;8:116. | ||
Tajiri N, Duncan K, Borlongan MC, et al. Adult stem cell transplantation: is gender a factor in stemness? Int J Mol Sci. 2014;15(9):15225–15243. | ||
Antonucci I, Di Pietro R, Alfonsi M, et al. Human second-trimester amniotic fluid cells are able to create embryoid body-like structures “in vitro” and to show typical expression profiles of embryonic and primordial germ cells. Cell Transplant. 2014. | ||
Liu R, Zhang Z, Lu Z, et al. Human umbilical cord stem cells ameliorate experimental autoimmune encephalomyelitis by regulating immunoinflammation and remyelination. Stem Cells Dev. 2013;22(7): 1053–1062. | ||
Antonucci I, Pantalone A, Tete S, et al. Amniotic fluid stem cells: a promising therapeutic resource for cell-based regenerative therapy. Curr Pharm Des. 2012;18(13):1846–1863. | ||
Rodrigues MC, Voltarelli J, Sanberg PR, et al. Recent progress in cell therapy for basal ganglia disorders with emphasis on menstrual blood transplantation in stroke. Neurosci Biobehav Rev. 2012;36(1):177–190. | ||
de la Peña I, Sanberg PR, Acosta S, Lin SZ, Borlongan CV. Umbilical cord blood cell and granulocyte-colony stimulating factor: combination therapy for traumatic brain injury. Regen Med. 2014;9(4):409–412. | ||
Zhang R, Liu Y, Yan K, et al. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J Neuroinflammation. 2013;10(1):106. | ||
Borlongan CV, Glover LE, Tajiri N, Kaneko Y, Freeman TB. The great migration of bone marrow-derived stem cells toward the ischemic brain: therapeutic implications for stroke and other neurological disorders. Prog Neurobiol. 2011;95(2):213–228. | ||
Borlongan CV. Bone marrow stem cell mobilization in stroke: a ‘bonehead’ may be good after all! Leukemia. 2011;25(11):1674–1686. | ||
Watanabe J, Shetty AK, Hattiangady B, et al. Administration of TSG-6 improves memory after traumatic brain injury in mice. Neurobiol Dis. 2013;59:86–99. | ||
Russo I, Barlati S, Bosetti F. Effects of neuroinflammation on the regenerative capacity of brain stem cells. J Neurochem. 2011;116(6):947–956. | ||
Yoo SW, Chang DY, Lee HS, et al. Immune following suppression mesenchymal stem cell transplantation in the ischemic brain is mediated by TGF-β. Neurobiol Dis. 2013;58:249–257. | ||
Dela Peña I, Sanberg PR, Acosta S, Tajiri N, Lin SZ, Borlongan CV. Stem cells and G-CSF for treating neuroinflammation in traumatic brain injury: aging as a comorbidity factor. J Neurosurg Sci. 2014;58(3):145–149. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.