")
Back to Journals » OncoTargets and Therapy » Volume 12
NCAPG Promotes The Proliferation Of Hepatocellular Carcinoma Through PI3K/AKT Signaling
Authors Gong C, Ai J, Fan Y, Gao J, Liu W , Feng Q, Liao W, Wu L
Received 31 May 2019
Accepted for publication 26 September 2019
Published 16 October 2019 Volume 2019:12 Pages 8537—8552
DOI https://doi.org/10.2147/OTT.S217916
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Chengwu Gong,1,* Jiyuan Ai,1,* Yun Fan,2 Jun Gao,1 Weiwei Liu,1 Qian Feng,3 Wenjun Liao,1 Linquan Wu1
1Department of General Surgery, The Second Affiliated Hospital of Nanchang University, Nanchang 330006, People’s Republic of China; 2Department of Neurology, Tongji Hospital, Huazhong University of Science and Technology, Wuhan 430000, People’s Republic of China; 3Department of Emergency Medicine, The Second Affiliated Hospital of Nanchang University, Nanchang 330006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Linquan Wu; Wenjun Liao
Department of General Surgery, The Second Affiliated Hospital of Nanchang University, No. 1, Minde Road, Nanchang 330006, People’s Republic of China
Tel +86 791 86311529
Fax +86 791 86262262
Email [email protected]; [email protected]
Purpose: Studies show that high expression of non-SMC condensin I complex subunit G (NCAPG) is associated with many tumors. In this study, we explore the mechanism by which NCAPG promotes proliferation in hepatocellular carcinoma (HCC).
Patients and methods: Liver cancer and paracancerous tissue specimens of 90 HCC patients were collected, and expression levels of NCAPG in these tissues and cell lines were evaluated by Western blotting and immunohistochemistry. HCC cells were transfected with siRNAs and plasmids, and pathway activators or inhibitors were added. The 5-ethynyl-2ʹ-deoxyuridine (EdU) proliferation assay was used to measure cell proliferation. Flow cytometry was used to evaluate cell apoptosis. Western blot assays were performed as a standard procedure to detect total protein expression. Treated HCC cells were subcutaneously injected into nude mice.
Results: Analysis using the Oncomine database showed that NCAPG was upregulated in HCC and immunohistochemistry and Western blot assays showed it was upregulated in both HCC tissues and HCC cell lines. The overexpression of NCAPG could promote HCC cell proliferation and reduce HCC cell apoptosis. More importantly, RNA-sequencing analysis predicted that NCAPG plays a role in the HCC via PI3K-AKT signaling pathway. The PI3K/AKT/FOXO4 pathway was aberrantly activated, and the expressions of apoptosis-related protein were altered when NCAPG was overexpressed or silenced both in vitro and in vivo. LY294002, a PI3K inhibitor, could eliminate the NCAPG role of promoting HCC cell proliferation and reducing HCC cell apoptosis, while 740Y-P, a PI3K activator, contributed to the opposite effect.
Conclusion: NCAPG functions as an oncogene in HCC and plays a role in promoting cell proliferation and antiapoptosis through activating the PI3K/AKT/FOXO4 pathway.
Keywords: NCAPG, hepatocellular carcinoma, PI3K/AKT, FOXO4, proliferation
Introduction
Hepatocellular carcinoma (HCC) is one of the most common and malignant tumors worldwide, which has a high degree of malignancy, poor prognosis, and a high recurrence rate.1,2 Most patients are diagnosed at advanced stages and miss the optimal time for surgical treatment.3 Although multiple genes and environmental factors are involved in the pathogenesis and progression of HCC,4 its underlying molecular mechanisms remain unclear. Thus, exploring the mechanisms that promote HCC growth is critical for early diagnosis and treatment.
Non-SMC condensin I complex subunit G (NCAPG), a mitotic associated chromosomal condensing protein,5 is a polypeptide composed of 1015 amino acids with a relative molecular weight of 114.1 kDa.6 NCAPG is encoded by the NY-MEL-3 gene, which is located on human chromosome 4p15.32.7 Studies show that high expression of NCAPG was associated with poor prognosis of prostate cancer,8 and knockdown of NCAPG combined with temozolomide treatment resulted in a combined suppressive effect on advanced pediatric glioma cell.9 Proteins encoded by NCAPG were hub proteins with high degrees in the protein–protein interaction (PPI) network of HCC.10 Preliminary results of our previous study found that NCAPG could promote cell proliferation in HCC.11 However, its mechanism by which NCAPG promotes proliferation in HCC remains unknown.
PI3K/AKT signaling is one of the most critical pathways for HCC development.12 Dysregulation of this pathway leads to decreased cell growth and enhanced apoptosis.13 PI3Ks and AKT are the core of this pathway, mediating downstream biological effects via various factors, such as NF-κB, VEGF, and FOXO.12 Forkhead Box transcription factor O (FOXO) family, consisting of FOXO1, O3, O4, and O6, regulates many biological processes, including oxidative stress, metabolism, immunity, and apoptosis.14 Investigations have found that PI3K/AKT/FOXO pathway plays a key role in various solid tumors, including breast,15 colorectal, and pancreatic cancer.16,17 Furthermore, Sheng et al found that oncoprotein BCR-ABL suppresses autophagy through PI3K/AKT/FOXO4 pathway in chronic myeloid leukemia.18 Whether PI3K/AKT/FOXO4 signaling is involved in the cell proliferation promoted by NCAPG in HCC still remains unclear and deserves further investigation.
In this study, we identified the abnormal upregulation of NCAPG expression in both HCC tissues and cell lines. We further confirmed that NCAPG functions as an oncogene in HCC and plays a roles in promoting cell proliferation and antiapoptosis. Moreover, we illuminated the involvement of NCAPG/PI3K/AKT/FOXO4 signaling pathway in the pathogenesis of HCC for the first time.
Materials And Methods
Study Population (Tissue Specimens)
In total, 90 HCC patients diagnosed between 2012 and 2017 were enrolled in this study. Each patient had undergone hepatectomy and did not receive any treatment before surgery, including radiotherapy or chemotherapy. Liver cancer and paracancerous tissue specimens were immediately collected, placed in liquid nitrogen, and stored at −80°C. This study was approved by the Ethical Review Committee of the Second Affiliated Hospital of Nanchang University. The procedures followed the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008. Written informed consent was obtained from all patients prior to their inclusion.
Cell Culture
The immortalized liver cell line (LO2) and four HCC cell lines (SMMC7721, MHCC97H, HCCLM3, and Huh-7) were used in this study, and all were procured from the Shanghai Institute of Cell Biology (Shanghai, China). All cell lines were cultured in high-glucose DMEM (Solarbio, Beijing, China) supplemented with 10% FBS (Biological Industries, Beit-Haemek, Israel), 100 µg/mL streptomycin and 100 U/mL penicillin at 37°C, with 5% CO2 in a humidified incubator. Cells in logarithmic growth were used for all experiments.
Immunohistochemistry (IHC)
IHC was performed as previously described.11 Briefly, paraffin-embedded samples were cut into 4-µm sections on a glass slide. The sections were then deparaffinized, hydrated, incubated with 0.3% hydrogen peroxide for 15 mins to block endogenous peroxidase, microwave heated at 100°C for 10 mins in sodium citrate buffer (10 mmol/L, pH 6.0) for antigen retrieval, and blocked with goat serum. Sections were then incubated with anti-NCAPG mouse monoclonal antibody (1:200; ab56382, Abcam, Danvers, MA, USA) at 4°C overnight, and then washed with PBS three times for 5 mins each. Sections were then incubated with secondary biotinylated goat antimouse IgG for 30 mins at 37°C. Diaminobenzidine and hematoxylin were used to stain the sections, which were subsequently sealed with neutral resins. Two pathologists blindly and randomly evaluated and semiquantitatively scored the staining intensity and percentage of positive cells. The previous study described the methods that we used to grade the staining intensity and score the percentage stained cells. The overall staining index was calculated by multiplying the grades and scores to reach a value from 0 to 9, which was finally designated as: 0–1, NCAPG nonoverexpression and 2–9, NCAPG overexpression.
Transfection
NCAPG siRNA, FOXO4 siRNA, and negative control (NC) siRNA were purchased from GenePharma (Shanghai, China). The NCAPG overexpression plasmid was purchased from GeneChem (Shanghai, China). HCC-LM3 and Huh-7 cells were used for siRNA studies, and SMMC7721 and MHCC97H cells were used for NCAPG overexpression studies. Lipofectamine 3000 (Invitrogen Life Technologies, Carlsbad, CA, USA) was used to transiently transfect cells with siRNAs and plasmids. Two different siRNA sequences were used to silence NCAPG: NCAPG_s1 sense: 5′-GGAGUUCAUUCAUUACCUUTT-3′ and antisense: 5′-AAGGUAAUGAAUGAACUCCTT-3′; NCAPG_s2 sense: 5′-GCUGAAACAUUGCAGAAAUTT-3′ and antisense: 5′-AUUUCUGCAAUGUUUCAGCTT-3′. Three different siRNA sequences were used to silence FOXO4: FOXO4_s1 sense: 5′-CGCGAUCAUAGACCUAGAUTT-3′ and antisense: 5′-AUCUAGGUCUAUGAUCGCGTT-3′; FOXO4_s2 sense: 5′-CAGCUUCAGUCAGCAGUUATT-3′ and antisense: 5′-UAACUGCUGACUGAAGCUGTT-3′; and FOXO4_s3 sense: 5′-GUGACAUGGAUAACAUCAUTT-3′ and antisense: 5′-AUGAUGUUAUCCAUGUCACTT-3′. Cells were seeded in 6-well plates, transfected for 48 hrs, and then harvested for further assays. LY294002 and 740Y-P were obtained from MedChemExpress (Monmouth Junction, NJ, USA). Unless otherwise stated, LY294002 and 740Y-P were used at the final concentrations of 25 and 10 µM, respectively. Cells were treated with drug(s) for 24 hrs prior to proliferation, apoptosis, or Western blot analysis.
Western Blotting
Western blot assays were performed as a standard procedure to detect total protein expression in tissues and cells after 48-hr transfection. Tissues and cells were lysed in RIPA buffer with 1% phenylmethanesulfonyl fluoride; extracted proteins were separated by 10% SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes. Then, the membranes were incubated with primary antibody at 4°C overnight. Tris-HCl solution + Tween-20 (TBST) was used to wash the membranes three times for 10 mins. Subsequently, they were incubated with horseradish peroxidase-conjugated secondary antibody for 1 hr at room temperature. Finally, the blots were detected with enhanced chemiluminescence, and band intensities were measured with Quantity-One software (Bio-Rad, Hercules, CA, USA). Mouse anti-NCAPG and -GAPDH, rabbit anti-PI3K, -AKT, -FOXO4, -p-FOXO4, -Bcl-2, and -Bax were purchased from Abcam (Cambridge, UK). Rabbit anti-p-PI3K was purchased from Wanleibio (Shenyang, China). Rabbit anticleaved caspase-3 was purchased from Proteintech (Wuhan, China). Rabbit anti-p-AKT (S473) was purchased from Cell Signaling Technology.
Cell Proliferation Analysis
The 5-ethynyl-2ʹ-deoxyuridine (EdU) proliferation assay was used to measure cell proliferation. We added 0.1 mL of 50 µM EdU (RiboBio Biotechnology, Guangzhou, China) into each well of 500 mL medium for 2 hrs. Then, cells were fixed with 4% polyformaldehyde in PBS at room temperature for 30 mins and subsequently incubated with Apollo staining solution and Hoechst 33342 for 30 mins. Fluorescence microscopy was performed in five randomly selected fields with an Olympus (Tokyo, Japan) microscope to analyze proliferation rates. Blue, Hoechst 33342 staining of nuclei with all cells; red, Apollo staining of EdU with proliferating cells; overlay, the percentage of proliferating cells. The stained cells were examined with a fluorescence microscope and photographed with camera. Image J was used to count the number of all cells and proliferating cells.
The Cell Counting Kit-8 (CCK-8) assay was also used to measure cell proliferation. The HCC cells were seeded into 96-well plates at a density of 1x104 cells/well and incubated for 24 hrs at 37°C in 5% CO2. After 0, 1, 2, 3, and 4 days of cultivation, 10 µL CCK-8 reagent (CCK-8; Dojindo Laboratories, Kumamoto, Japan) were added to each well followed by cultured for 2 hrs at 37°C in 5% CO2. The absorbance at 450 nm was measured using a microplate reader (Bio-Rad, Berkeley, CA, USA).
Apoptosis Analysis
Cells were stained with Annexin V-FITC and propidium iodide (BD Biosciences, Franklin Lakes, NJ, USA) to evaluate apoptosis by flow cytometry. Briefly, 1×106 cells were washed twice with PBS and stained with 5 µL Annexin V-FITC and 5 µL propidium iodide solution in 1× binding buffer for 15 mins at room temperature in the dark. Then, an additional 400 µL of 1× binding buffer was added to the cell suspension. Finally, apoptosis rates were determined by flow cytometry (BD Biosciences).
Cell Migration Assays
Cell migration was evaluated using Transwell inserts. The specific steps were performed as previously described.11
In Vivo Proliferation Assays
For in vivo proliferation assays, 1×107 cells in 100 µL PBS were subcutaneously injected into the flanks of male BALB/c-nu/nu mice (6–8 weeks old; n=6 per group) (Hunan SJA Laboratory Animal Co., Ltd., Hunan, China). Mice were sacrificed after 6 weeks, and tumor volumes were measured. All animal work was approved by the Ethics Committee for Animal Experiments of the Second Affiliated Hospital of Nanchang University and was performed in accordance with the guidelines by the UK Animals (Scientific Procedures) Act, 1986, and EU Directive 2010/63/EU.
Oncomine Data Analysis
Oncomine (http://www.oncomine.com) is an integrated cancer microarray database that contains unified bioinformatics resources from 715 datasets (version 4.4.4.3 after Q2 update 2013).19 We compared the mRNA expression of NCAPG from liver cancer datasets that contain data from both HCC tissues and normal liver tissues. Three datasets were included in our study: Wurmbach et al20 and Roessler et al (including Roessler Liver 1 and 2 datasets).21 The differential expression of NCAPG between HCC and normal liver tissues was analyzed by Unpaired t-test. According to the dataset Guichard Liver,22 which contains >90 patients, we also assessed the relationship between NCAPG expression and overall survival by the log-rank (Mantel–Cox) test.
RNA-Sequencing (RNA-Seq) Analysis
RNA of HCC-LM3 cells with NCAPG knockdown siRNA or NCAPG overexpression was extracted for RNA-Seq analysis. RNA degradation and contamination were monitored on 1% agarose gels. RNA purity was checked using the NanoPhotometer®spectrophotometer (IMPLEN, CA, USA). RNA concentration was measured using Qubit® RNA Assay Kit in Qubit®2.0 Fluorometer (Life Technologies). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). A total amount of 3 µg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq platform and 125 bp/150 bp paired-end reads were generated. Reference genome and gene model annotation files were downloaded from the genome website directly. Index of the reference genome was built using Hisat2 v2.0.5 and paired-end clean reads were aligned to the reference genome using Hisat2 v2.0.5. FeatureCounts v1.5.0-p3 was used to count the reads numbers mapped to each gene. And then, FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. Differential expression analysis of two conditions was performed using the DESeq2 R package (1.16.1). KEGG is a database resource for understanding high-level functions and utilities of the biological system, such as the cell, the organism, and the ecosystem, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies. We used clusterProfiler R package to test the statistical enrichment of differential expression genes in KEGG pathways.
Statistical Analysis
All statistical analyses were performed with SPSS 17.0 (IBM, Armonk, NY, USA). Kaplan–Meier plots and log-rank tests were used for survival analyses. Differences between groups were analyzed by Student’s t-test for two groups and one-way ANOVA for more than two groups. Data are presented as mean ± SD of at least three independent experiments. P values <0.05 were considered statistically significant.
Results
NCAPG Was Overexpressed In HCC Tissues And Cell Lines And Was Associated With Poor Prognoses
To assess the relationship between NCAPG and HCC, we first analyzed three microarray datasets from the Oncomine database. The results showed that NCAPG was significantly overexpressed in the majority of HCC tissues compared with adjacent non-neoplastic controls (Figure 1A). The median rank of NCAPG among upregulated genes in HCC was 179.0, based on a meta-analysis across three datasets using the Oncomine algorithms23 (563 samples, P=8.68E−65; Figure 1B). The result provided a clue that NCAPG was overexpressed in transcriptional level in HCC.
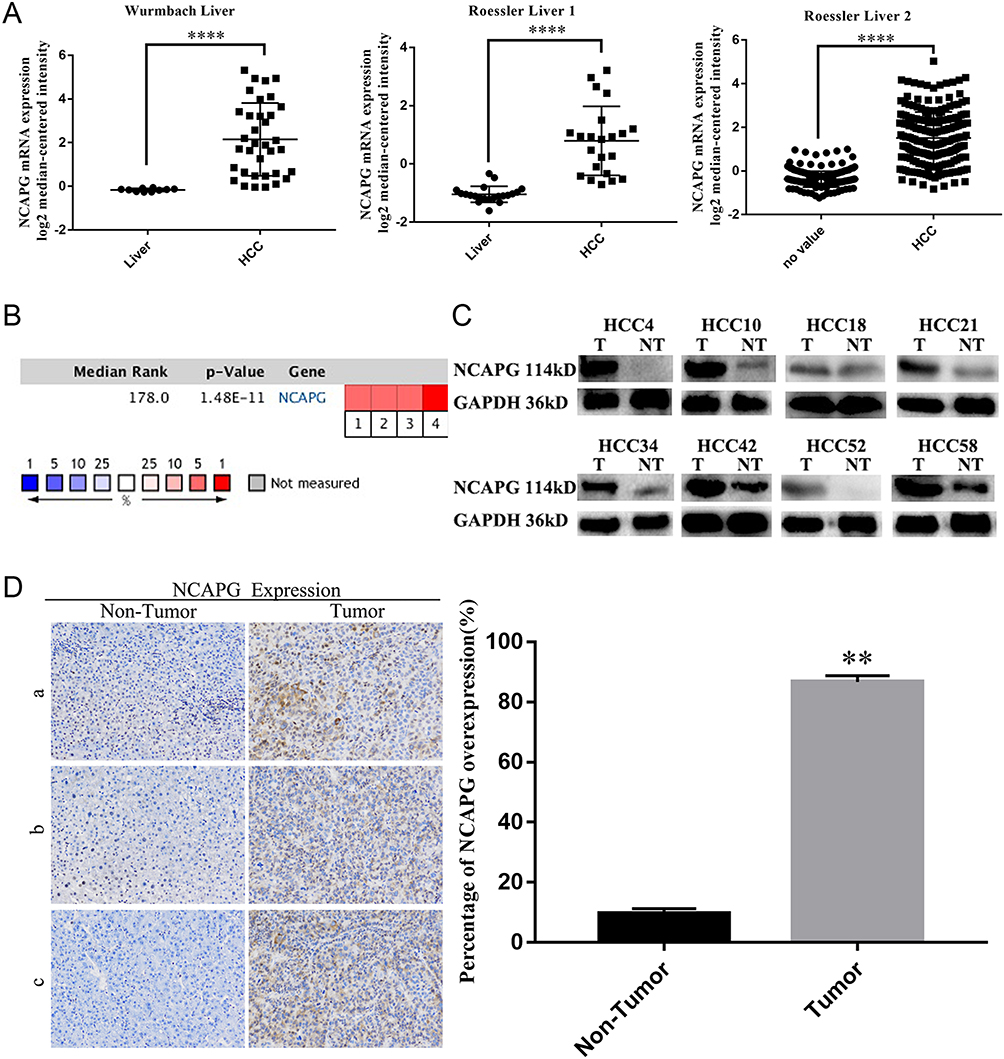 |
Figure 1 NCAPG was overexpressed in HCC tissues. Notes: (A) Representative image showed that NCAPG mRNA was overexpressed in HCC tissues compared with adjacent non-neoplastic controls three selected datasets from Oncomine database (Dataset 1, Wurmbach Liver; Dataset 2, Roessler Liver 1; Dataset 3, Roessler Liver 2, ****P<0.0001, independent Student’s t-test). (B) Cell color and the number above the cell in the lower panel indicate the best gene rank percentile for the analysis. Red, upregulated; blue, downregulated. (C) Western blot analysis of NCAPG in HCC tissues (T, tumor; NT, nontumor). (D) Immunohistochemical staining of NCAPG protein expression in 90 HCCs and their corresponding nontumor tissues. Representative cases (a/b/c) were shown (200×). The values indicate the mean ± SD of three independent experiments (**P<0.01, independent Student’s t-test).Abbreviations: NCAPG, non-SMC condensin I complex subunit G; HCC, hepatocellular carcinoma. |
To identify whether NCAPG is overexpressed in translational level in HCC, we next detected NCAPG protein levels in 90 paired primary HCC tissues and the corresponding adjacent normal tissues by Western blot. The results showed that NCAPG was significantly overexpressed in 83/90 (92.2%) primary liver cancer tissues compared with adjacent nontumor tissues (Figure 1C). In addition, NCAPG protein levels were assayed by IHC in the same 90 pairs of HCC and the corresponding nontumor tissues. Consistently, the results demonstrated the higher expression of NCAPG in 85/90 (94.4%) HCC tissues compared with their nontumor tissues (Figure 1D).
NCAPG expression was upregulated in both transcriptional and translational levels in HCC tissues, which may hint its involvement in the pathogenesis of HCC. And DisGenET enrichment analysis demonstrated that NCAPG plays a more important role in liver carcinoma than in other diseases (Figure 2A).
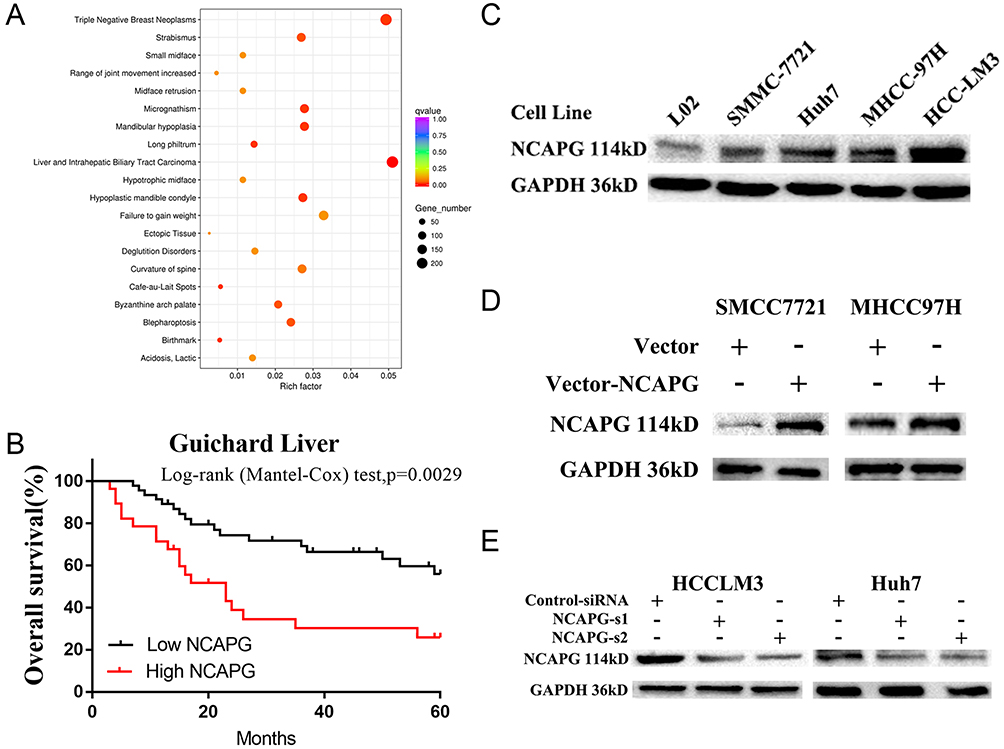 |
Figure 2 NCAPG plays a more important role in liver carcinoma than in other diseases and was associated with a poor overall survival. Notes: (A) DisGeNET Enrichment analysis of differentially expressed genes when NCAPG was overexpressed in HCC-LM3 cells. GeneRatio: ratio of the number of differential genes enriched in a certain disease to the total number of differential genes. (B) Kaplan-Meier overall survival curve of two HCC groups: low NCAPG and high NCAPG controls one selected datasets from the Oncomine database (Guichard Liver). (C) Western blot analysis was used to detect expressions of NCAPG in one immortalized liver cell line (LO2) and four HCC cell lines. (D) Western blot analysis was used to verify the effect of NCAPG overexpression. (E) Western blot analysis was used to verify the effect of NCAPG knockdown with siRNAs.Abbreviations: NCAPG, non-SMC condensin I complex subunit G; HCC, hepatocellular carcinoma. |
To assess the relationship between NCAPG expression and overall survival in HCC, we analyzed one microarray dataset (Guichard Liver) from the Oncomine database, and the results showed that NCAPG expression was associated with a poor overall survival (Figure 2B), similar to our previous study.11
NCAPG Promoted HCC Proliferation In Vitro And In Vivo
We further verified the expression of NCAPG in four HCC cell lines and one immortalized liver cell line by Western blot. Consistent with clinical HCC specimen, the results showed that NCAPG was upregulated in the four HCC cell lines compared with the immortalized liver cell line LO2 (Figure 2C). To explore the role of NCAPG in HCC proliferation, NCAPG was overexpressed in SMMC-7721 and MHCC-97H cells and silenced in HCC-LM3 and Huh-7 cells using two distinct siRNA duplexes. NCAPG expression was evaluated by Western blot (Figure 2D and E). EdU proliferation assays and CCK-8 assay showed that proliferation was significantly increased in NCAPG-overexpressing SMMC-7721 and MHCC-97H cells compared with their corresponding control cells (Figure 3A, B, E, and F). Conversely, proliferation was significantly decreased in siNCAPG-transfected HCC-LM3 and Huh-7 cells (Figure 3C, D, G, and H).
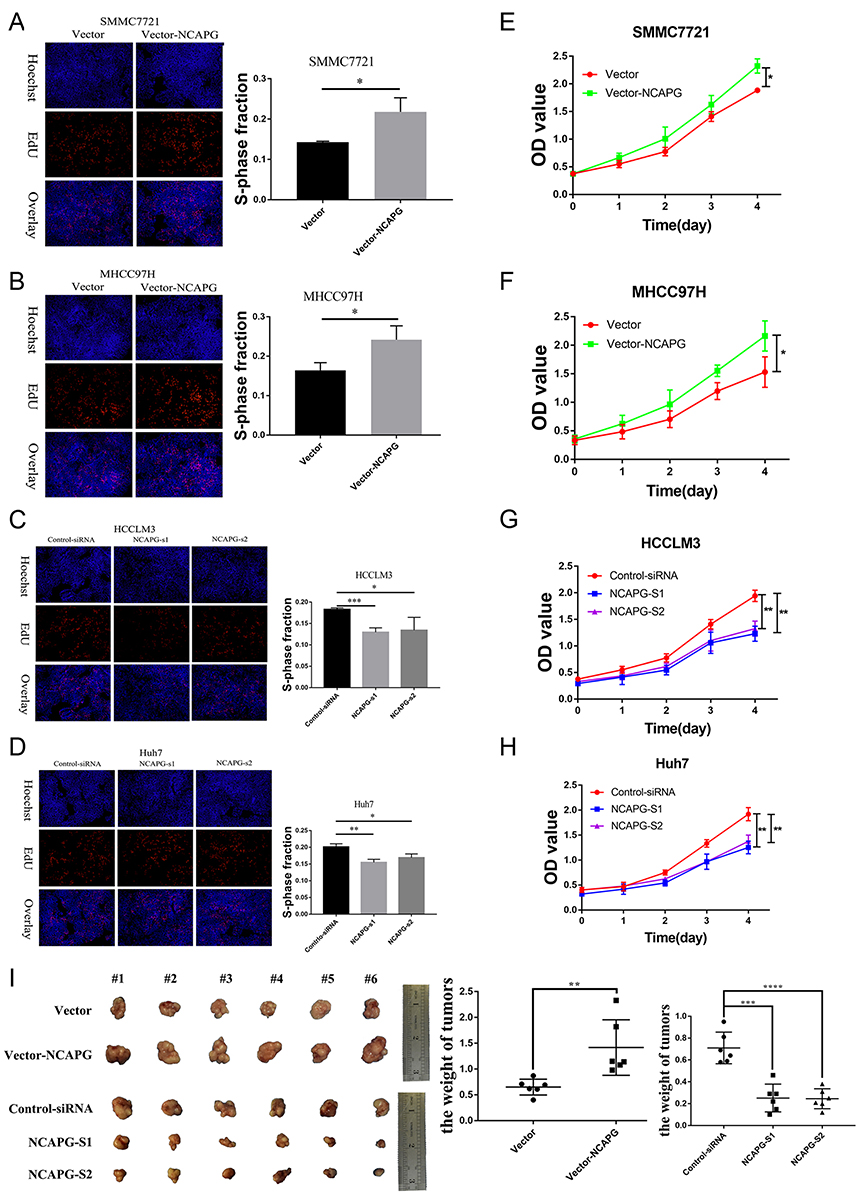 |
Figure 3 NCAPG promoted HCC proliferation in vitro and in vivo. Notes: (A, B, E, and F) Representative images of cell proliferation abilities in SMMC-7721 and MHCC-97H cells with NCAPG overexpression detected by EdU and CCK-8. Blue, Hoechst33342 staining of nuclei with all cells; red, Apollo staining of EdU with proliferating cells; overlay, the percentage of proliferating cells. The values indicate the mean ± SD of three independent experiments (*P<0.05, independent Student’s t-test). (C, D, G, and H) Representative images of cell proliferation abilities in HCC-LM3 and Huh-7 cells with NCAPG knockdown siRNA detected by EdU and CCK-8. The values indicate the mean ± SD of three independent experiments (*P<0.05, **P<0.01, ***P<0.001, independent Student’s t-test). (I) Cells were subcutaneously injected into the flanks of nude mice to detect proliferative ability. Tumor weight (g) was measured at 6 weeks after injection and expressed as the mean ± SD, n = 6 (**P<0.01, ***P<0.001, ****P<0.0001, independent Student’s t-test).Abbreviations: NCAPG, non-SMC condensin I complex subunit G; HCC, hepatocellular carcinoma; EdU, 5-ethynyl-2ʹ-deoxyuridine; CCK-8, Cell Counting Kit-8. |
We next examined the effects of altering NCAPG expression on proliferation in vivo. Tumor formation was observed in 6/6 and 6/6 mice injected with SMMC-7721-Vector and SMMC-7721-NCAPG cells, respectively, and the average tumor volumes of SMMC-7721-NCAPG cells was significantly larger than tumors induced by control cells. Conversely, tumor formation was observed in 6/6, 6/6, and 6/6 mice injected with Huh7-siRNA-control, Huh7-siRNA-S1, and Huh7-siRNA-S2, respectively. Moreover, the average volume of tumors induced by Huh7-siRNA-S1 and Huh7-siRNA-S2 cells was significantly reduced compared with controls (P<0.05; Figure 3I). Taken together, these data indicated that NCAPG promoted HCC cell proliferation.
NCAPG Reduced Apoptosis In HCC Cells
To explore the role of NCAPG in HCC apoptosis, the detection of apoptosis-related proteins, including Bcl-2, Bax, and cleaved caspase-3, in NCAPG-overexpressing and silencing HCC cells were performed. Western blot analysis showed that Bcl-2 was upregulated, while Bax and cleaved caspase-3 were downregulated in NCAPG-overexpressing cells compared with controls. Conversely, after the silencing of NCAPG, the expression of Bcl-2 was downregulated, while Bax and cleaved caspase-3 were upregulated compared with controls (Figure 4A and B).
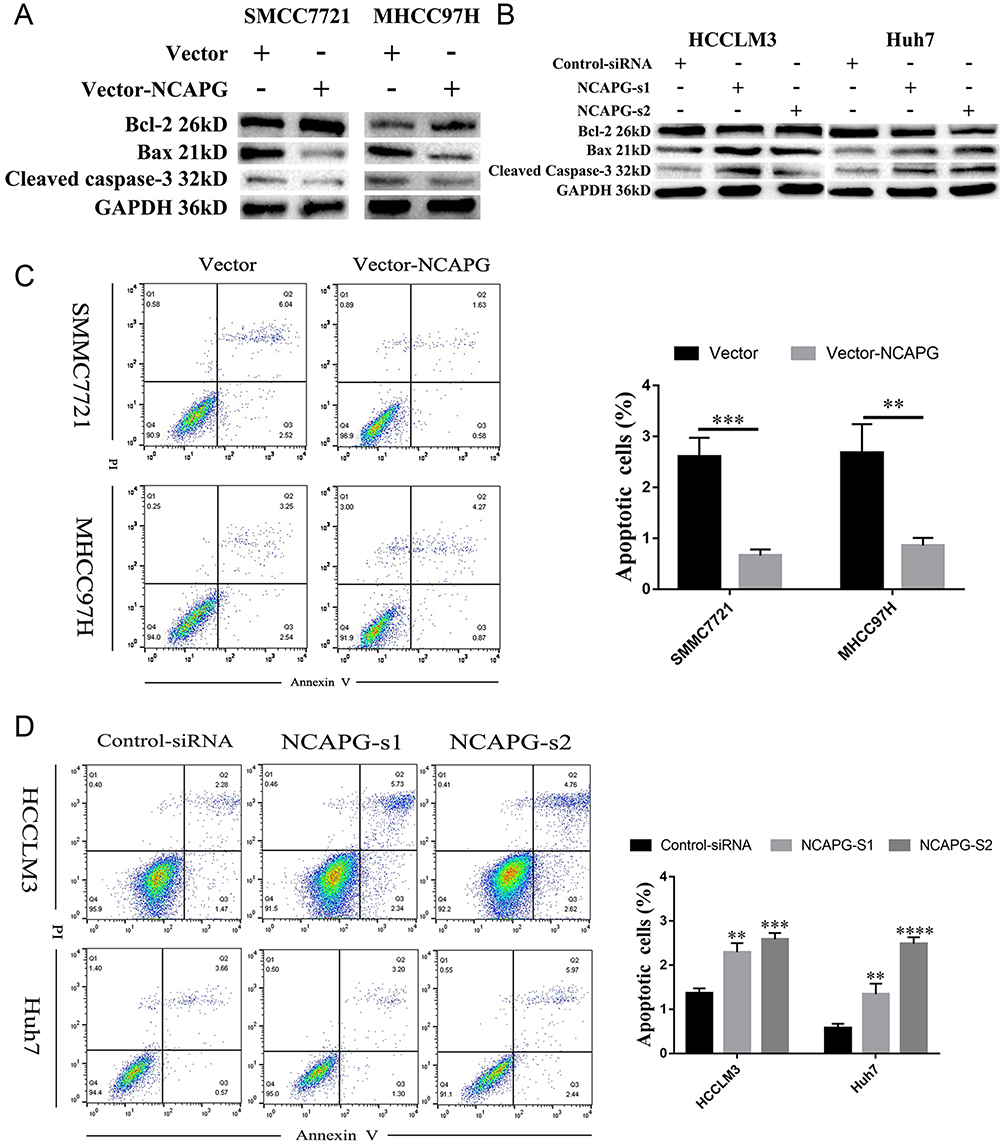 |
Figure 4 NCAPG reduced apoptosis in HCC cells. Notes: (A) Western blot analysis of SMMC-7721 and MHCC-97H cells transfected with NCAPG-overexpression plasmid. (B) Western blot analysis of HCC-LM3 and Huh-7 cells transfected with two different NCAPG siRNA. (C) Representative images (left) and summary bar chart (right) of cell apoptosis abilities in SMMC-7721 and MHCC-97H cells with NCAPG overexpression detected by cell apoptosis analysis. The values indicate the mean ± SD of three independent experiments (**P<0.01, ****P<0.0001, independent Student’s t-test). (D) Representative images (left) and summary bar chart (right) of cell apoptosis abilities in HCC-LM3 and Huh-7 cells with NCAPG knockdown siRNA detected by cell apoptosis analysis. The values indicate the mean ± SD of three independent experiments (**P<0.01, ***P<0.001, ****P<0.0001, independent Student’s t-test).Abbreviations: NCAPG, non-SMC condensin I complex subunit G; HCC, hepatocellular carcinoma. |
In addition, we evaluated the effect of NCAPG on the apoptosis of HCC-related cell lines, including SMMC-7721 and MHCC-97H cells by flow cytometry. Results showed that NCAPG-overexpression groups presented a lower percentage of early apoptosis compared with the control group (Figure 4C). Contrary to this, the early apoptosis rate was significantly increased in both HCC-LM3 and Huh-7 cells when NCAPG was silenced (Figure 4D). These data suggest that NCAPG reduced apoptosis of HCC and its molecular mechanism deserves further investigations.
NCAPG Promoted HCC Migration In HCC Cells
To explore the role of NCAPG in HCC migration, NCAPG was overexpressed in SMMC-7721 and MHCC-97H cells and silenced in HCC-LM3 and Huh-7 cells using two distinct siRNA duplexes. Transwell inserts were used to evaluate cell migration. The results showed that migration was significantly increased in NCAPG-overexpressing SMMC-7721 and MHCC-97H cells compared with their corresponding control cells (Figure 5A). Conversely, migration was significantly decreased in siNCAPG-transfected HCC-LM3 and Huh-7 cells (Figure 5B).
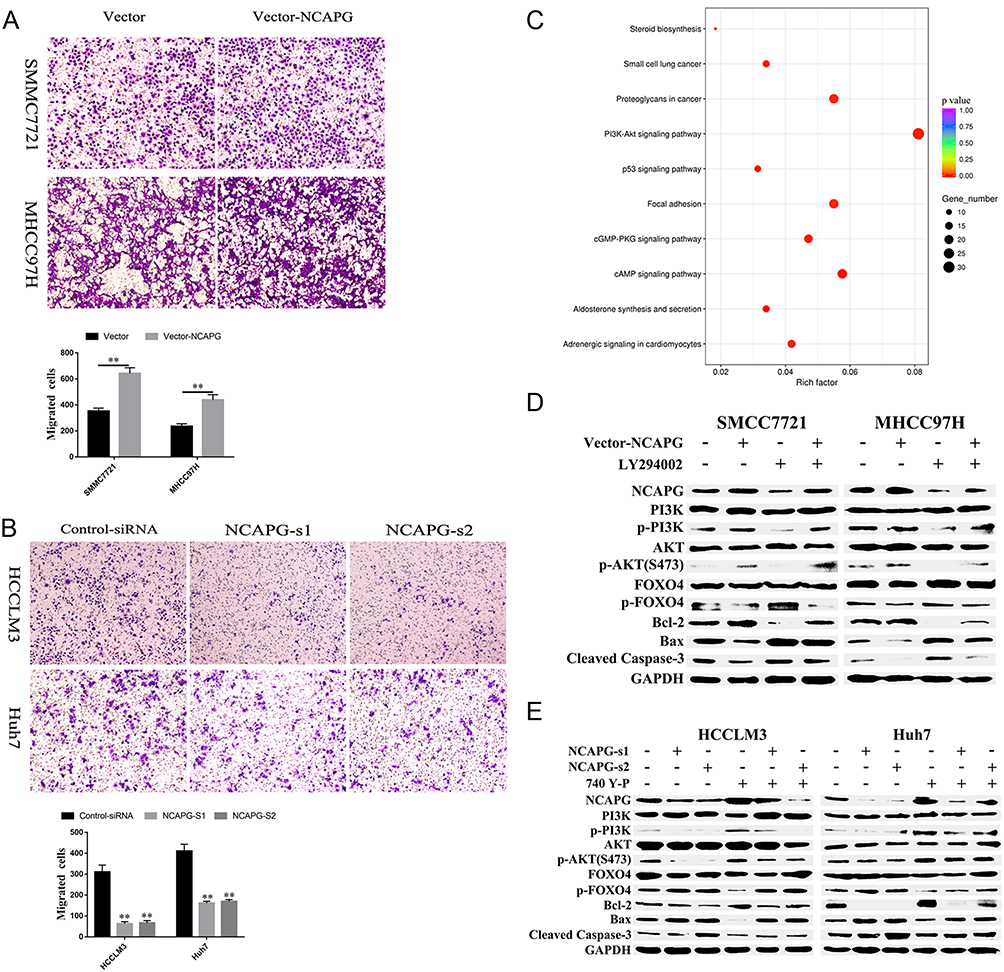 |
Figure 5 NCAPG plays a biological role in HCC by activating the PI3K/AKT/FOXO4 pathway. Notes: (A) Representative images of cell migration abilities in SMMC-7721 and MHCC-97H cells with NCAPG overexpression detected by Transwell migration assays. The values indicate the mean ± SD of three independent experiments (**P<0.01, independent Student’s t-test). (B) Representative images of cell proliferation abilities in HCC-LM3 and Huh-7 cells with NCAPG knockdown siRNA detected by Transwell migration assays. The values indicate the mean ± SD of three independent experiments (**P<0.01, independent Student’s t-test). (C) KEGG pathway analysis of differentially expressed genes when NCAPG was silenced in HCC-LM3 cells. GeneRatio: ratio of the number of differential genes enriched in a certain pathway to the total number of differential genes. (D) Western blot of SMMC-7721 and MHCC-97H cells transfected with NCAPG-overexpression plasmid and treated for 24 hrs with 25 μM LY294002. (E) Western blot of HCC-LM3 and Huh-7 cells transfected with NCAPG siRNA and treated for 24 hrs with 10 μM 740Y-P.Abbreviations: NCAPG, non-SMC condensin I complex subunit G; HCC, hepatocellular carcinoma. |
NCAPG Promoted Proliferation And Reduced Apoptosis By Activating The PI3K/AKT/FOXO4 Pathway
KEGG enrichment analysis of differentially expressed genes when NCAPG was silenced in HCC-LM3 cells showed that NCAPG may be involved in the regulation of the PI3K-AKT signaling pathway (Figure 5C). Therefore, we hypothesized that NCAPG mainly contribute to cell survival by regulating the PI3K/AKT pathway. Western blotting showed that the levels of p-PI3K, p-AKT (S473), and Bcl-2 were upregulated, while p-FOXO4, Bax, and cleaved caspase-3 were downregulated in the NCAPG-overexpressing cells. Furthermore, p-PI3K, p-AKT (S473), and Bcl-2 were downregulated, while p-FOXO4, Bax, and cleaved caspase-3 were upregulated when NCAPG was silenced (Figure 5D and E).
To further confirm the crucial role of PI3K/AKT activation in the oncogenic effect of NCAPG, the PI3K inhibitor LY294002 and PI3K activator 740Y-P were applied to identify whether they could attenuate or promote oncogenic phenotypes. Similar to the NCAPG-overexpression experiments, proliferation, apoptosis, and Western blot analyses of SMMC-7721 and MHCC-97H cells with the treatment of 25 µM LY294002 for 24 hrs were performed. In addition, similar to the NCAPG-silencing study, proliferation, apoptosis, and Western blot analyses of Huh-7 and HCC-LM3 cells treated with 10 µM 740Y-P for 24 hrs were conducted. Interestingly, Western blot analysis showed that LY294002 decreased the expression of p-PI3K, p-AKT(S473), and Bcl-2 and increased the expression of p-FOXO4, Bax, and cleaved caspase-3 in NCAPG-overexpressing cells, while 740Y-P played an opposite effect in NCAPG-silencing cells (Figure 5D and E). Furthermore, LY294002 attenuated cell proliferation and antiapoptosis induced by NCAPG overexpression (Figures 6A, B, E, F, and 7A). Contrary to this, 740Y-P rescued cell proliferation and antiapoptosis that resulted from NCAPG silencing (Figures 6C, D, G, H, and 7B). Together, these data highlighted that the oncogenic effect of NCAPG on HCC cell proliferation and its inhibitory effect on apoptosis were via modulating PI3K/AKT/FOXO4 signaling.
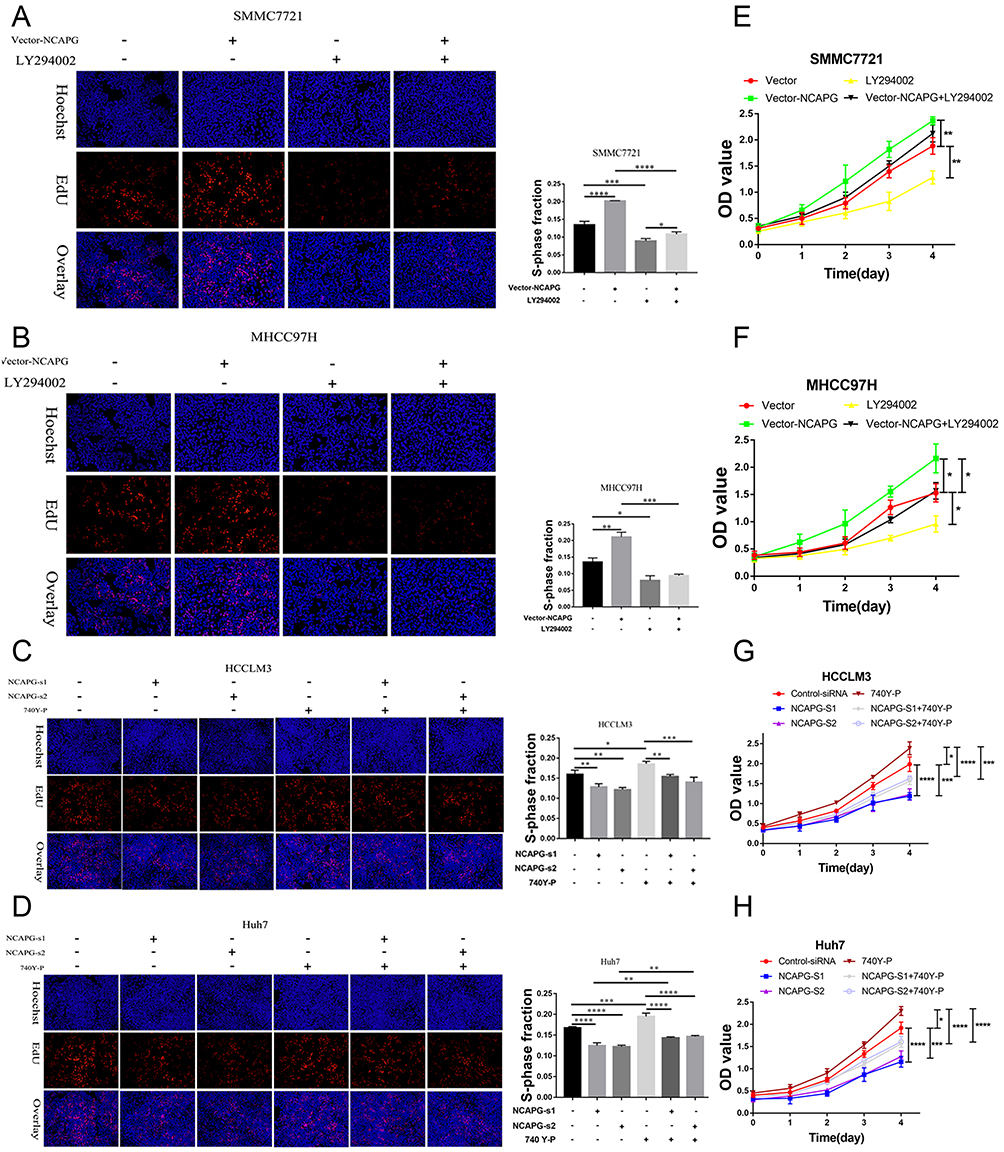 |
Figure 6 NCAPG promoted proliferation by activating the PI3K/AKT/FOXO4 pathway. Notes: (A, B, E, and F) Representative images of cell proliferation abilities in SMMC-7721 and MHCC-97H cells transfected with NCAPG-overexpression plasmid and treated for 24 hrs with 25 μM LY294002 detected by EdU and CCK-8. The values indicate the mean ± SD of three independent experiments (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, one-way ANOVA). (C, D, G, and H) Representative images of cell proliferation abilities in HCC-LM3 and Huh-7 cells transfected with NCAPG siRNA and treated for 24 hrs with 10 μM 740Y-P detected by EdU and CCK-8. The values indicate the mean ± SD of three independent experiments (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, one-way ANOVA).Abbreviations: NCAPG, non-SMC condensin I complex subunit G; EdU, 5-ethynyl-2ʹ-deoxyuridine; CCK-8, Cell Counting Kit-8. |
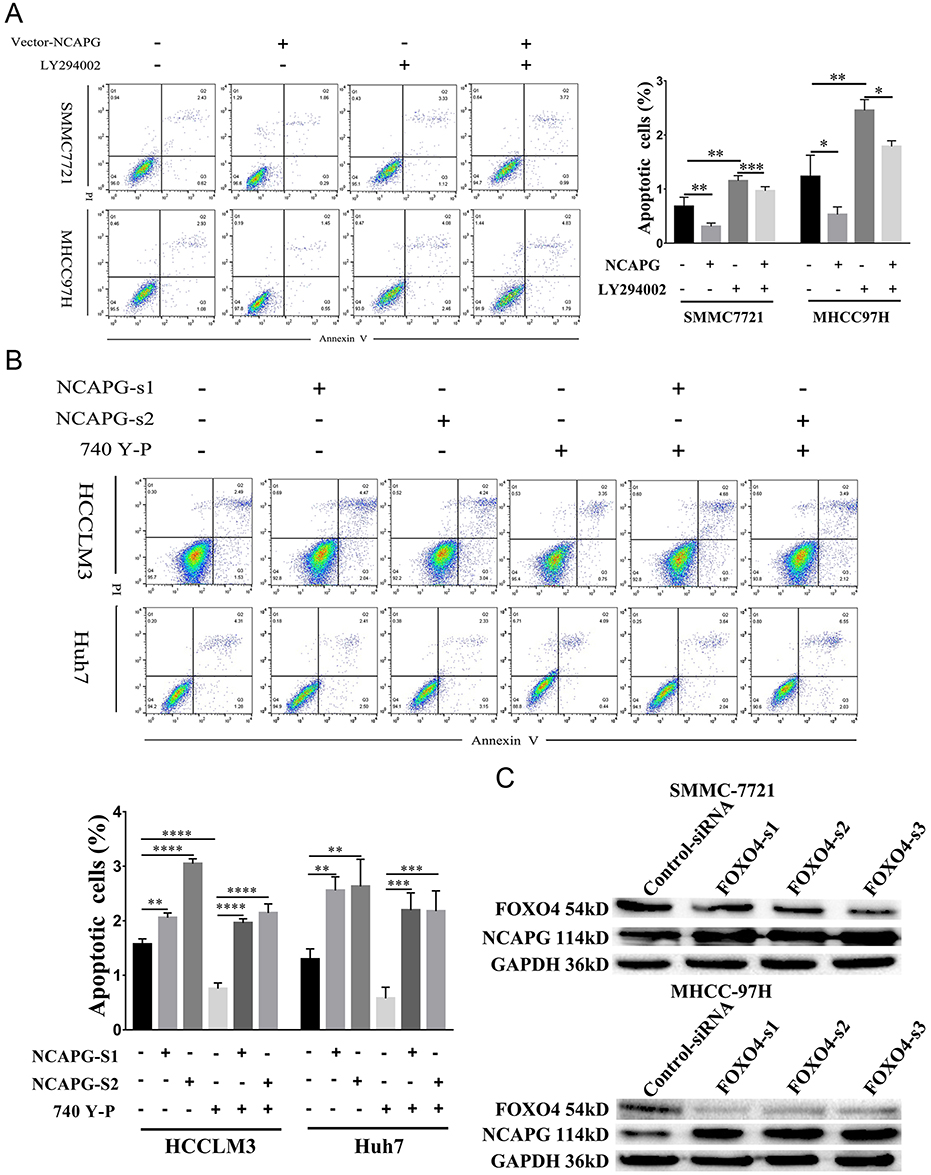 |
Figure 7 NCAPG reduced apoptosis by activating the PI3K/AKT/FOXO4 pathway. Notes: (A) Representative images (left) and summary bar chart (right) of cell apoptosis abilities in SMMC-7721 and MHCC-97H cells transfected with NCAPG-overexpression plasmid and treated for 24 hrs with 25μM LY294002 detected by cell apoptosis analysis. The values indicate the mean ± SD of three independent experiments (*P<0.05, **P<0.01, ***P<0.001, one-way ANOVA). (B) Representative images and summary bar chart of cell apoptosis abilities in HCC-LM3 and Huh-7 cells transfected with NCAPG siRNA and treated for 24 hrs 10μM 740Y-P detected by cell apoptosis analysis. The values indicate the mean ± SD of three independent experiments (**P<0.01, ***P<0.001, ****P<0.0001, one-way ANOVA). (C) Western blot analysis was used to detect the expression of FOXO4 and NCAPG in SMMC-7721 and MHCC-97H cells transfected with three different FOXO4 siRNA.Abbreviations: NCAPG, non-SMC condensin I complex subunit G; FOXO4, Forkhead Box transcription factor O (FOXO) family 4. |
NCAPG Promoted Migration By Activating The PI3K/AKT/FOXO4 Pathway
LY294002 attenuated cell migration induced by NCAPG overexpression (Figure 8A). Contrary to this, 740Y-P rescued cell migration that resulted from NCAPG silencing (Figure 8B). Together, these data highlighted that the oncogenic effect of NCAPG on HCC cell migration was via modulating PI3K/AKT/FOXO4 signaling.
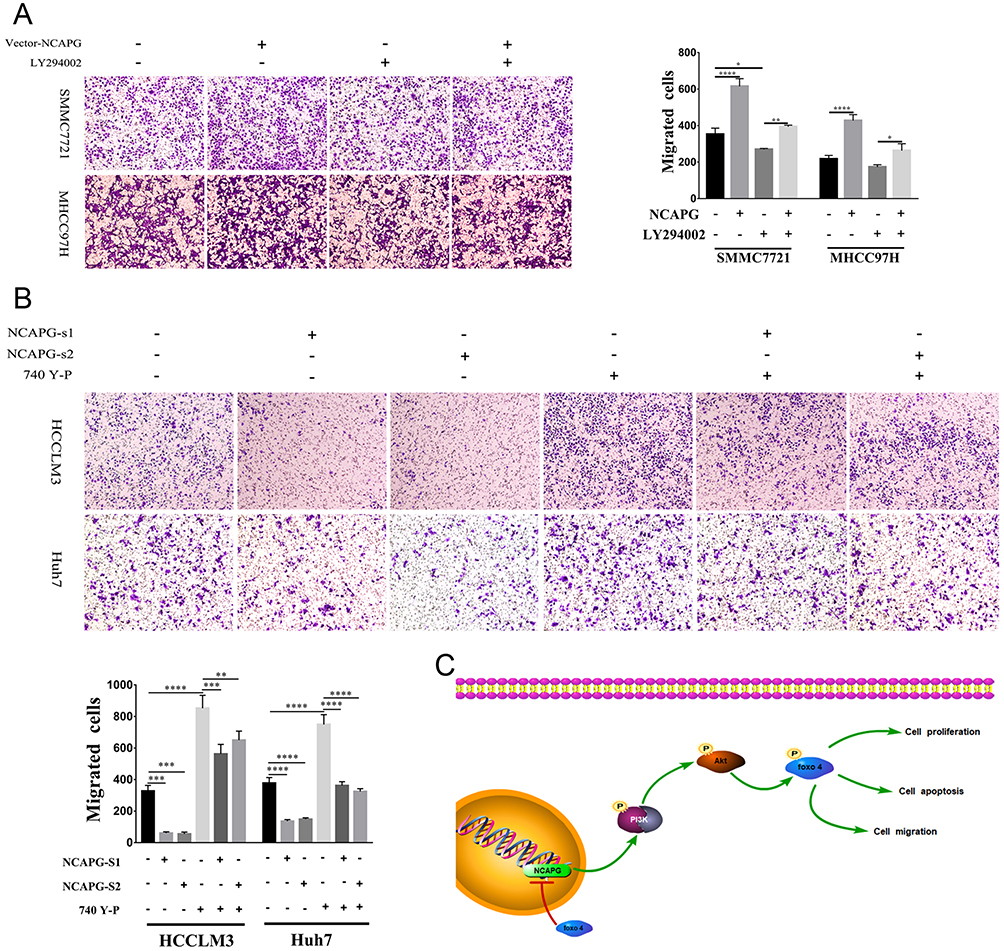 |
Figure 8 NCAPG promoted migration by activating the PI3K/AKT/FOXO4 pathway. Notes: (A) Representative images of cell proliferation abilities in SMMC-7721 and MHCC-97H cells transfected with NCAPG-overexpression plasmid and treated for 24 hrs with 25 μM LY294002 detected by Transwell migration assays. *P<0.05, **P<0.01, ****P<0.0001, one-way ANOVA. (B) Representative images of cell proliferation abilities in HCC-LM3 and Huh-7 cells transfected with NCAPG siRNA and treated for 24 hrs with 10 μM 740Y-P detected by Transwell migration assays. **P<0.01, ***P<0.001, ****P<0.0001, one-way ANOVA. (C) A final summary figure which summarizes our findings.Abbreviation: NCAPG, non-SMC condensin I complex subunit G. |
Silencing FOXO4 Increased NCAPG Expression
Negative feedback loops had been proven to exist in tumors; for example, inhibiting mTOR can block feedback inhibition of the pathway, resulting in persistent AKT activation in cancer cell lines.24 Therefore, we examined whether FOXO4 expression was associated with NCAPG. We found the overexpression of NCAPG contributed to the significant downregulation of FOXO4 in the study. To further examine whether FOXO4 could affect the expression of NCAPG reversely, we knocked down the expression of FOXO4 in SMMC-7721 and MHCC-97H by transfecting three different FOXO4-siRNA sequences. The expression of NCAPG was detected by Western blot analysis. Results showed that NCAPG expression was increased in FOXO4-siRNA groups compared with control-siRNA groups (Figure 7C). These data suggest that a positive feedback loop exists between NCAPG and FOXO4 in the PI3K/AKT/FOXO4 pathway. However, the underlying mechanism of how FOXO4 influences NCAPG expression remains to be elucidated in future studies.
Discussion
Tissue homeostasis critically depends on the balance between proliferation and cell death.25 Cancer is a disease characterized as dysregulated proliferation. Mutations in various proliferative pathways have a profound impact on aberrant cell division and the inhibition of differentiation.25 Apoptosis, a distinct, intrinsic cell death program, is a genetically regulated cellular suicide mechanism that occurs in various physiological and pathological situations.26 Thus, learning about the mechanisms of HCC proliferation and migration is useful to develop newer and better therapeutic strategies to improve the survival rate of HCC patients.
NCAPG encodes a subunit of the condensin complex, which is responsible for chromosome condensation and stabilization during mitosis and meiosis. Researches have demonstrated that NCAPG was involved in the pathogenesis of many kinds of neoplasms, including prostate cancer,8 pediatric glioma cell,9 renal cell carcinoma,27 multiple myeloma,28 and melanoma.29 Using bioinformatics analysis, studies found that NCAPG is a key gene in liver cancerigenesis and development.30 Here, we found the abnormal upregulation of NCAPG in HCC by analyzing three microarray datasets from the Oncomine database and further confirmed it in 90 paired primary HCC tissues and the corresponding adjacent normal tissues by IHC and Western blot, which indicate its putative tumor-promoting function in HCC. In addition, we found that NCAPG expression was associated with poor clinical outcomes. Furthermore, our data indicated that NCAPG promoted the proliferation and migration of HCC cells and inhibited the apoptosis of HCC cells in vitro and altered the size of xenograft tumors in nude mice in vivo in both NCAPG-overexpressing and NCAPG-silencing experiments, which is in favor of our previous research11 and demonstrated that NCAPG functions as oncogene in HCC. Similar to our results, researches have reported the overexpression of NCAPG and its effects on cell proliferation and apoptosis in HCC only by NCAPG-silencing experiments.31,32 However, up to now, the mechanism of NCAPG roles of promoting proliferation, migration, and antiapoptosis in HCC has not been investigated.
PI3Ks are heterodimeric lipid kinases that are composed of a regulatory and catalytic subunit, which are encoded by different genes.33,34 AKT, a downstream PI3K effector, regulates many biological processes, including proliferation, apoptosis, and growth. The PI3K/AKT pathway is dysregulated in many cancers, including breast, colon, ovarian, and pancreatic cancers.35–37 Abnormal activation of the PI3K/AKT pathway has also been reported in HCC.38,39 Interesting, our results found the excessive activation of PI3K/AKT signaling in NCAPG-overexpressing experiments and its inhibition in NCAPG-silencing experiments in HCC. Although a recent study has reported that the mRNA expressions of AKT were downregulated after NCAPG silencing in BEL-7404 cells, an HCC cell line,32 the study on the cause and effect between NCAPG and PI3K/AKT signaling in HCC pathogenesis is lacking up to now. Therefore, we further uncover whether NCAPG plays its role in promoting cell proliferation, migration, and inhibiting apoptosis through the PI3K/AKT signaling pathway in HCC. Results found that the addition of 740Y-P (PI3K activator) resulted in increased proliferation, migration, and decreased apoptosis in NCAPG-silencing cells, while the LY294002 (PI3K inhibitor) contributed to the opposite effect in NCAPG-overexpressing cells. Together, our data suggest that the oncogenic function of NCAPG in HCC is dependent on PI3K/AKT signaling.
FOXO4, which is considered to be at the outset of tumorigenesis,40 has been extensively identified to be a crucial transcription factor involved in the regulation of several essential types of cancer, such as leukemia, gastric cancer,41 colorectal cancer,42 prostate cancer,43 breast cancer,44 lung cancer,45 and neuroblastoma.46 It has been reported that AKT/ERK/FOXO4/ATF5 pathway was involved in the process of hepatitis B virus X protein and promoted tumorigenesis.47 Additionally, a recent study has reported the key role of Ras/Foxo4 signaling in CCDC50S-mediated cell proliferation and metastasis in HCC.48 To identify whether FOXO4 is involved in the NCAPG oncogenic phenotype, in the study, we detected the expression of FOXO4 and p-FOXO4 in both NCAPG-overexpressing and NCAPG-silencing cells. Results showed that NCAPG inhibited the expression of p-FOXO4. More importantly, our data demonstrated that LY294002 (PI3K inhibitor) rescued the decreased expression of p-FOXO4 in NCAPG-overexpressing cells, while 740Y-P (PI3K activator) showed the opposite result in NCAPG-silencing cells. These indicate that FOXO4 plays roles in NCAPG oncogenic functions in HCC and may be the downstream of the PI3K/AKT pathway. Interestingly, we found that silencing FOXO4 increased NCAPG expression, which reveals that there may be negative feedback loops between NCAPG and FOXO4 in the tumorigenesis of HCC. Further studies will be needed to expand our knowledge of the underlying mechanisms of the regulatory loop between them.
In summary, we further confirmed the overexpression of NCAPG in both HCC tissues and cell lines and its roles in promoting proliferation, migration, and antiapoptosis in HCC (Figure 8C). Notably, we illuminated that NCAPG plays its oncogenic roles in the pathogenesis of HCC through PI3K/AKT/FOXO4 pathway for the first time, which suggests that NCAPG may act as a prognostic therapeutic target in HCC.
Acknowledgments
We thank James P. Mahaffey, PhD, from Edanz Group for editing a draft of this manuscript. This study was supported by the National Natural Science Foundation of China, China [grant number 81860431], the Natural Science Foundation of Jiangxi Province, China [grant number 20171BAB205063], and the Young Science Foundation of Jiangxi Province, China [grant number 20171BAB215037].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LAT, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal AJD. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
3. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314.
4. Schulze K, Nault JC, Villanueva A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J Hepatol. 2016;65(5):1031–1042.
5. Eberlein A, Takasuga A, Setoguchi K, et al. Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC condensin I complex, subunit G (NCAPG) gene. Genetics. 2009;183(3):951.
6. Kimura K, Cuvier O, Hirano T. Chromosome condensation by a human condensin complex in Xenopus egg extracts. J Biol Chem. 2001;276(8):5417–5420.
7. Jager D, Stockert E, Jager E, et al. Serological cloning of a melanocyte rab guanosine 5ʹ-triphosphate-binding protein and a chromosome condensation protein from a melanoma complementary DNA library. Cancer Res. 2000;60(13):3584–3591.
8. Goto Y, Kurozumi A, Arai T, et al. Impact of novel miR-145-3p regulatory networks on survival in patients with castration-resistant prostate cancer. Br J Cancer. 2017;117(3):409–420.
9. Liang ML, Hsieh TH, Ng KH, et al. Downregulation of miR-137 and miR-6500-3p promotes cell proliferation in pediatric high-grade gliomas. Oncotarget. 2016;7(15):19723–19737.
10. Yan H, Li Z, Shen Q, et al. Aberrant expression of cell cycle and material metabolism related genes contributes to hepatocellular carcinoma occurrence. Pathol Res Pract. 2017;213(4):316–321. doi:10.1016/j.prp.2017.01.019
11. Liu W, Liang B, Liu H, et al. Overexpression of nonSMC condensin I complex subunit G serves as a promising prognostic marker and therapeutic target for hepatocellular carcinoma. Int J Mol Med. 2017;40(3):731–738. doi:10.3892/ijmm.2017.3079
12. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170(4):605–635. doi:10.1016/j.cell.2017.07.029
13. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. doi:10.1038/nrd1902
14. Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14(2):83–97. doi:10.1038/nrm3507
15. Smit L, Berns K, Spence K, et al. An integrated genomic approach identifies that the PI3K/AKT/FOXO pathway is involved in breast cancer tumor initiation. Oncotarget. 2016;7(3):2596–2610. doi:10.18632/oncotarget.6354
16. Sun Y, Tian H, Wang L. Effects of PTEN on the proliferation and apoptosis of colorectal cancer cells via the phosphoinositol-3-kinase/Akt pathway. Oncol Rep. 2015;33(4):1828–1836. doi:10.3892/or.2015.3804
17. Boreddy SR, Pramanik KC, Srivastava SK. Pancreatic tumor suppression by benzyl isothiocyanate is associated with inhibition of PI3K/AKT/FOXO pathway. Clin Cancer Res Off J Am Assoc Cancer Res. 2011;17(7):1784. doi:10.1158/1078-0432.CCR-10-1891
18. Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. 2011;118(10):2840–2848. doi:10.1182/blood-2010-12-322537
19. Rhodes DR, Yu J, Shanker K, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. doi:10.1016/s1476-5586(04)80047-2
20. Wurmbach E, Chen YB, Khitrov G, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007;45(4):938–947. doi:10.1002/hep.21622
21. Roessler S, Jia HL, Budhu A, et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010;70(24):10202–10212. doi:10.1158/0008-5472.CAN-10-2607
22. Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–698. doi:10.1038/ng.2256
23. Rhodes DR, Yu J, Shanker K, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A. 2004;101(25):9309–9314. doi:10.1073/pnas.0401994101
24. O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–1508. doi:10.1158/0008-5472.CAN-05-2925
25. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–348. doi:10.1038/35077213
26. Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776. doi:10.1038/35037710
27. Yamada Y, Arai T, Kojima S, et al. Regulation of antitumor miR-144-5p targets oncogenes: direct regulation of syndecan-3 and its clinical significance. Cancer Sci. 2018;109(9):2919–2936. doi:10.1111/cas.13722
28. Cohen Y, Gutwein O, Garach-Jehoshua O, Bar-Haim A, Kornberg A. The proliferation arrest of primary tumor cells out-of-niche is associated with widespread downregulation of mitotic and transcriptional genes. Hematology. 2014;19(5):286–292.
29. Ryu B, Kim DS, Deluca AM, Alani RM. Comprehensive expression profiling of tumor cell lines identifies molecular signatures of melanoma progression. PLoS One. 2007;2(7):e594. doi:10.1371/journal.pone.0000594
30. Zhang L, Huang Y, Ling J, et al. Screening and function analysis of hub genes and pathways in hepatocellular carcinoma via bioinformatics approaches. Cancer Biomark. 2018;22(3):511–521. doi:10.3233/CBM-171160
31. Zhang Q, Su R, Shan C, Gao C, Wu P. Non-SMC condensin I complex, subunit G (NCAPG) is a novel mitotic gene required for hepatocellular cancer cell proliferation and migration. Oncol Res. 2018;26(2):269–276. doi:10.3727/096504017X15075967560980
32. Liu K, Li Y, Yu B, Wang F, Mi T, Zhao Y. Silencing non-SMC chromosome-associated polypeptide G inhibits proliferation and induces apoptosis in hepatocellular carcinoma cells. Can J Physiol Pharmacol. 2018;96:1246–1254. doi:10.1139/cjpp-2018-0195
33. Guba M, von Breitenbuch BP, Steinbauer M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8(2):128–135. doi:10.1038/nm0202-128
34. Mayo LD, Dixon JE, Durden DL, Tonks NK, Donner DB. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J Biol Chem. 2002;277(7):5484–5489. doi:10.1074/jbc.M108302200
35. Bellacosa A, Feo DD, Godwin AK, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64(4):280–285. doi:10.1002/ijc.2910640412
36. Cheng JQ, Ruggeri B, Klein WM, et al. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci U S A. 1996;93(8):3636–3641. doi:10.1073/pnas.93.8.3636
37. Philp AJ, Campbell IG, Leet C, et al. The phosphatidylinositol 3ʹ-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61(20):7426–7429.
38. Pellegrino R, Calvisi DF, Neumann O, et al. EEF1A2 inactivates p53 via PI3K/AKT/mTOR-dependent stabilization of MDM4 in hepatocellular carcinoma. Hepatology. 2013;59(5):1886. doi:10.1002/hep.26639
39. Zhou Q, Lui VW, Yeo W. Targeting the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Future Oncol. 2011;7(10):1149–1167. doi:10.2217/fon.11.95
40. Jiang S, Yang Z, Di S, et al. Novel role of forkhead box O 4 transcription factor in cancer: bringing out the good or the bad. Semin Cancer Biol. 2018;50:1–12. doi:10.1016/j.semcancer.2018.04.007
41. Zhou L, Shang Y, Jin Z, et al. UHRF1 promotes proliferation of gastric cancer via mediating tumor suppressor gene hypermethylation. Cancer Biol Ther. 2015;16(8):1241–1251. doi:10.1080/15384047.2015.1056411
42. Kwon IK, Wang R, Thangaraju M, et al. PKG inhibits TCF signaling in colon cancer cells by blocking beta-catenin expression and activating FOXO4. Oncogene. 2010;29(23):3423–3434. doi:10.1038/onc.2010.91
43. Su B, Gao L, Baranowski C, et al. A genome-wide RNAi screen identifies FOXO4 as a metastasis-suppressor through counteracting PI3K/AKT signal pathway in prostate cancer. PLoS One. 2014;9(7):e101411. doi:10.1371/journal.pone.0101411
44. Kim SH, Miller FR, Tait L, Zheng J, Novak RF. Proteomic and phosphoproteomic alterations in benign, premalignant and tumor human breast epithelial cells and xenograft lesions: biomarkers of progression. Int J Cancer. 2009;124(12):2813–2828. doi:10.1002/ijc.24278
45. Xu MM, Mao GX, Liu J, et al. Low expression of the FoxO4 gene may contribute to the phenomenon of EMT in non-small cell lung cancer. Asian Pac J Cancer Prev. 2014;15(9):4013–4018. doi:10.7314/apjcp.2014.15.9.4013
46. Mei Y, Wang Z, Zhang L, et al. Regulation of neuroblastoma differentiation by forkhead transcription factors FOXO1/3/4 through the receptor tyrosine kinase PDGFRA. Proc Natl Acad Sci U S A. 2012;109(13):4898–4903. doi:10.1073/pnas.1119535109
47. Xu X, Fan Z, Kang L, et al. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J Clin Invest. 2013;123(2):630–645. doi:10.1172/JCI64265
48. Hong W, Zhang CZ, Lu SX, et al. A CCDC50 splice variant is modulated by SRSF3 and promotes hepatocellular carcinoma via the Ras signaling pathway. Hepatology. 2018;69:179–195.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.