Back to Journals » OncoTargets and Therapy » Volume 14
Natural Polyphyllins (I, II, D, VI, VII) Reverses Cancer Through Apoptosis, Autophagy, Mitophagy, Inflammation, and Necroptosis
Authors Ahmad B ![]() , Gamallat Y
, Gamallat Y ![]() , Khan MF
, Khan MF ![]() , Din SR, Israr M, Ahmad M, Tahir N, Azam N, Rahman KU, Xin W, Zexu W, Linjie P, Su P, Liang W
, Din SR, Israr M, Ahmad M, Tahir N, Azam N, Rahman KU, Xin W, Zexu W, Linjie P, Su P, Liang W
Received 3 December 2020
Accepted for publication 19 February 2021
Published 10 March 2021 Volume 2021:14 Pages 1821—1841
DOI https://doi.org/10.2147/OTT.S287354
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Bashir Ahmad,1,2 Yaser Gamallat,3 Muhammad Fiaz Khan,4 Syed Riaz Din,2 Muhammad Israr,1,5 Manzoor Ahmad,6 Naeem Tahir,2 Nasir Azam,2 Khalil Ur Rahman,2 Wang Xin,7 Wang Zexu,7 Peng Linjie,7 Pengyu Su,2 Wang Liang8
1Department of Biology, University of Haripur, KPK, I. R. Pakistan; 2College of Basic Medical Sciences, Dalian Medical University, Dalian, 116044, People’s Republic of China; 3Guangzhou Institute of Pediatrics, Guangzhou Women and Children’s Medical Center, Guangzhou, People’s Republic of China; 4Zoology Department, Hazara University, Mansehra, I. R. Pakistan; 5Biochemistry and Molecular Biology, College of Life Science, Hebei Normal University, Hebei, People’s Republic of China; 6Department of Chemistry, Malakand University, Chakdara, KPK, I. R. Pakistan; 7Department of Biotechnology, College of Basic Medical Sciences, Dalian Medical University, Dalian, 116044, People’s Republic of China; 8Stem Cell Clinical Research Center, National Joint Engineering Laboratory, Regenerative Medicine Center, The First Affiliated Hospital of Dalian Medical, Dalian City, Liaoning Province, 116011, People’s Republic of China
Correspondence: Wang Liang; Bashir Ahmad Email [email protected]; [email protected]
Abstract: Cancer is the second leading cause of mortality worldwide. Conventional therapies, including surgery, radiation, and chemotherapy, have limited success because of secondary resistance. Therefore, safe, non-resistant, less toxic, and convenient drugs are urgently required. Natural products (NPs), primarily sourced from medicinal plants, are ideal for cancer treatment because of their low toxicity and high success. NPs cure cancer by regulating different pathways, such as PI3K/AKT/mTOR, ER stress, JNK, Wnt, STAT3, MAPKs, NF-kB, MEK-ERK, inflammation, oxidative stress, apoptosis, autophagy, mitophagy, and necroptosis. Among the NPs, steroid saponins, including polyphyllins (I, II, D, VI, and VII), have potent pharmacological, analgesic, and anticancer activities for the induction of cytotoxicity. Recent research has demonstrated that polyphyllins (PPs) possess potent effects against different cancers through apoptosis, autophagy, inflammation, and necroptosis. This review summarizes the available studies on PPs against cancer to provide a basis for future research.
Keywords: natural products, saponins, polyphyllins, apoptosis, autophagy, inflammation, necroptosis
Introduction
Cancer is the second leading cause of mortality worldwide.1–4 A WHO report shows that cancer causes more deaths in the world compared with stroke and coronary heart diseases.5 Factors that increase cancer incidence include overpopulation and aging.6 Global epidemiologic and demographic transition signals indicate that cancer will increase in the next decade, especially in countries with low or middle income.7 A recent report has estimated that 18.1 million new cancer cases excluding non-melanoma skin cancer (17.0 million cases) and 9.6 million cancer-related deaths excluding non-melanoma skin cancer (9.5 million cases) were recorded in 2018.8 In both genders combined, lung cancer (11.6% of total reported cases) was more common, followed closely by female breast (11.6%), prostate (7.1%), and colorectal (6.1%) cancers. Lung cancer was also the leading cause of death (18.4% of the total reported cases), followed by colorectal cancer (9.2%), stomach (8.2%), and liver (8.2%) cancers.8
Available clinical treatment for cancer includes surgery, chemotherapy, and radiotherapy.9 Conventional therapies, including surgery, radiation, and chemotherapy, have limited success because of secondary resistance.1,10 Therefore, safe, non-resistant, less toxic, and convenient drugs are urgently required.1,10 NPs, primarily sourced from medicinal plants, are ideal for cancer treatment because of their low toxicity and high success.1,11,12 These NPs cure cancer by regulating different pathways, such as PI3K/AKT/mTOR, NF-kB, autophagy, MEK-ERK, inflammation, oxidative stress, and apoptosis.11
Paris (Melanthiaceae) consists of 29 species,13 many of which are used as traditional herbal medicines14 and distributed in Eastern Asia and Europe. Among 29 species of Paris,13 22 are found in China and its diversity center in China is South-west China.15 In China, extracts from Paris species are extensively used as herbal medicine.16 Paridis, the dry rhizome of Paris, is used as a main raw material in patent Chinese drugs, including “GongXue Ning,” “Jidesheng snake tablet,” and “Yunnan Baiyo”.17 For a thousand years, this herb has been used in traditional Chinese medicine for the treatment of various diseases, such as snakebite, parotitis, hemostasis, abscess, and fractures.17 Modern research explored its new pharmacological activities, including cytotoxic, hemolytic, antibiotic, spermicidal, styptovit analgesic, calming, immunoregulatory, anthelmintic, anti-inflammatory, and anticancer activities.18–22
Photochemical research found several active ingredients in the dried rhizome of Paris, including flavonoids, endophytic fungi, fatty acid ester, and steroid saponins, such as Polyphyllin I (PPI), Polyphyllin II (PPII), Polyphyllin VI (PPVI), and Polyphyllin VII (PPVII).17,21,23 Modern research revealed that the PPs have potential pharmacological, analgesic, and anticancer activities for the induction of cytotoxicity through apoptosis.24–26
PPI,15,27–31 PPII,15,27–31 PPVI,15,27–31 and PPVII15,27–32 have been isolated from 13 Paris species, including, P. polyphylla (ChongLou),32 P. caobangensis,15,27,28 P. cronquistii,15,27,28 P. cronquistii var. xichouensis,15,27,28 P. delavayi var. petiolata,15,27,28 P. fargesii,15,27,28 P. mairei,15,27,28 P. polyphylla,15,27–29 P. polyphylla var. alba,15,27,28 P. polyphylla var. chinensis,15,27,28 P. polyphylla var. younaninsis,15,27,28,30,31 P. vitenaminsis,15,27,28 P. axialis pseudothi,15,27,28 and P. polyphylla var.15,27,28 These studies reported that the amount of isolated compounds from the same species of different regions varies possibly because of climate changes.15,27 PPD,23,33–37 Paris-VII23,33,34 are derived from Rhizoma paridis,23,33,34 Paris polyphylla,37 P. forrestii.35,36 Rhizoma Paridis extracts also containing PPI, PPII, PPVI and PPVII.38
Among these PPs, PPI has a significant therapeutic effect on hepatocellular carcinoma, lung adenocarcinoma, and gastric cancer.39–41 PPII has different pharmacological activities, including hemolytic effect42 and anticancer (eg, against ovarian cancer).43 PPVI inhibits colon cancer by inducing apoptosis, PPVII is a potent compound for the treatment of cervical cancer by inhibiting the growth of Hela cells.44 PPD exerts a potent antiproliferative effect against different types of cancer cells, including HepG-2, MCF-7 and MDA-MB-231.25,45,46 These PPs exhibit an anticancer effect against different cancers through different mechanisms. This review summarizes the anticancer mechanisms of these PPs to provide basic knowledge for further studies.
Non-Mechanistic Studies of PPs in Different Cancers
This section discusses those studies in which PP mechanisms are not reported. PPD inhibits the growth of HMEC-1 cells and decreases angiogenesis by suppressing cell proliferation, tube formation, and migration in vitro.47 PPD induces apoptosis in HL-60, SMMC-7721, A-549, MCF-7, and SW480 cell lines.35 PPD inhibits the proliferation of ovarian cancer (OVCA) cell lines, including TYKNU, M41-R, TYKNU-R, M41-R, PE01, A2780S, Skov3, OVCAR8, MCAS, A2780CP, HEYA8, and OVCAR5. In all cell lines, PPD treatment can significantly decrease cisplatin IC50.48 Paris polyphylla mixtures containing PPD inhibit the migration of LA795 cells in vitro and inhibit the tumor growth in vitro.37 PPVII induces death in different cell lines, including gastric cancer SNU-5, lung cancer A-549, skin cancer carcinoma A431, oral cancer OECM-1, breast MCF-7, pancreas MiaPaca-2, colon HTB-39, human normal fibroblasts (FR2).
Mechanistic Studies of PPs in Different Cancers
This section discusses those studies in which PP mechanisms are reported.
Apoptosis: PPs Induce Apoptosis in Different Cancers Through the Following Mechanisms
Oxidative Stress
Oxidative stress is the disturbance in redox signaling and regulation or physiological imbalance in the production of reactive oxygen species (ROS), such as oxygen (O2) or hydrogen peroxide (H2O2), and the body’s ability to remove them.49 ROS are generated throughout the body as by-products of cellular aerobic metabolism, exposure to X-rays or ultraviolet light, and on-going stress.50 ROS play pivotal roles in cell signaling and the regulation of growth factors, transcription, cytokines, hormones, neuromodulation, apoptosis, and immunomodulation.50,51 ROS also function in different cell processes, including cell survival, proliferation, differentiation, gene expression, elimination of pathogens or foreign particles, and enzyme regulation.52,53 The high oxidative stress in cancer cells increases cell survival, proliferation, angiogenesis, and metastasis; disrupts cell death signaling; and causes drug resistance.54–56 Although ROS increase cell proliferation, they have been recently deemed useful in cancer treatment. Plant-derived compounds induce apoptosis in cancer cells by promoting ROS generation in these cells above the threshold level.54,56–58
Several PPs induce apoptosis in cancer cells through oxidative stress, which promotes the generation of ROS and the dissipation of mitochondrial membrane potential (MMP). In these PPs, PPI generates ROS and dissipates MMP in HCT 116 and MDA-MB-231 cells,59–61 PPII in HepG2,62 PPVI in HepaRG,63 PPG or PPVII in HepG-2 cells.64 Furthermore, PPD or PSI and PPG or PPVII cause dissipation of MMP in K562/A02 human leukemia drug-resistant and K562 cells65 and human NPC cells, respectively.66 ROS generation and MMP dissipation are reversed by the ROS inhibitor N-acetyl-l-cysteine (NAC) treatment.62–64 Oxidative stress is further summarized in Figure 1A.
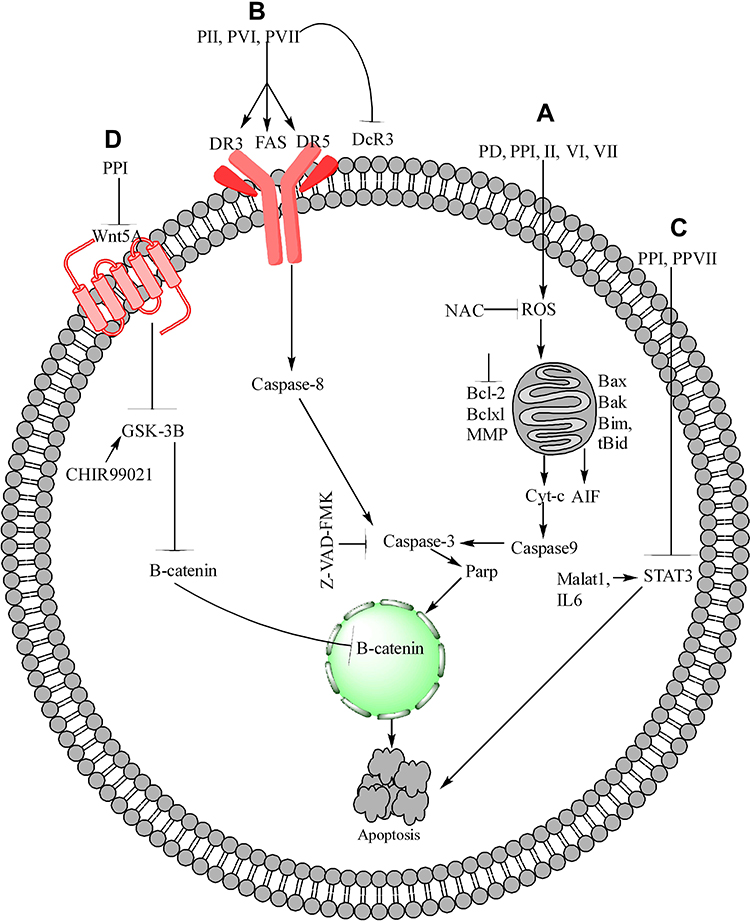 |
Figure 1 Molecular anticancer mechanisms of PPs. (A) In cancer cells, PPD, PPI, II, VI, and VII induce ROS generation, inhibit MMP, upregulate Bax, Bak, Bim, and tBid, and downregulate Bcl-2 and Bcl-xl, resulting in mitochondrial membrane permeability, allowing Cyt-c and AIF to enter the cytoplasm from the mitochondria. When Cyt-c and AIF accumulate in the cytoplasm, they cause the activation of caspase-3, caspase-9, and PARTP, leading to cell apoptosis. (B) In mitochondrial-independent pathway, PPII, VI, and VII upregulate FAS, DR3, and DR5 and downregulate DcR3, which further activate caspase-8, caspase-3, and PARO and cause cancer cell apoptosis. (C) In the STAT3 pathway, PPI and PPVII downregulate the Malat1 and IL-6 activation of STAT3 and cause apoptosis. (D) In the Wnt/β-catenin pathway, PPI inhibits Wnt5A, GSk-3B, and β-catenin and its translocation into the nucleus, leading to cell apoptosis. |
Mitochondrial-Dependent Pathway
The mitochondrial-dependent pathway is important for apoptosis induction, and any disturbance in this pathway prevents apoptosis. This pathway is regulated by B cell lymphoma-2 (Bcl-2) family proteins through changes in the permeability of the mitochondrial membrane for the release of different apoptotic proteins, including cytochrome-c (Cyt-c).67 Anti-apoptotic proteins, such as B cell lymphoma extra-large (BclxL), Bcl-2, Bcl-2-related protein A1, and myeloid cell leukemia 1, increase cell survival. Pro-apoptotic proteins, including Bcl-2 homologous killer, Bcl-2 associated X (BAX), and Bcl-2 homology domain 3 (BH3)-only proteins act as receptor mediators and cause apoptosis by inducing mitochondrial stress.68
BH3-only proteins have two subclasses: activators and depressors/sensitizers. Activators include total BH3 interacting domain death antagonist and Bcl-2 like protein-11. This subclass activates BAX/BAK directly and causes MMP depolarization. Depressor/sensitizers include Bcl-2-associated death promoter, Bcl-2-interacting killer, phorbol-12-myristate-13-acetate-induced protein-1, hara-kiri, and upregulated modulator of apoptosis p53. Instead of BAX/BAK activation, this subclass counterbalances anti-apoptotic proteins.69,70 Meanwhile, anti-apoptotic proteins block the death signaling pathway through the direct inhibition of activator BH3-only proteins or BAX/BAK activation.71 Anti-apoptotic proteins (Bcl-2 and Bcl-xl) cause cancer progression72 and therefore increase the resistance of tumor cells to different types of cell death stimuli, including anticancer drugs.70
In hepatocarcinoma cells, PPI and formosanin C synergistically downregulate BCL2 inhibition and BAX upregulation and release of cytochrome from the mitochondria. Cytochrome c release from the mitochondria activates caspase-3,9 and induces hepatocarcinoma cell apoptosis.73 In 143-B, MG-63, U-2 OS, non-small cell lung cancer (NSCLC), Saos-2, A549, SK-MES-1, H460, MDA-MB-231, MCF-7, U251, HepG-2, RPMI8226, SMC7721, and HO-8910M cells, PPI induces apoptosis through the mitochondrial-dependent pathway (MDP).74–83 In the MDP, PPI inhibits Bcl-274–79 and Bclxl75 while activating Bax74,75,78 and Bak,75 triggering Cyt-c release from the mitochondria to the cytosol.61,78,79 The PPI-induced release of Cyt-c in the cytosol activates caspase-9,61,82,83 caspase-3,74–77,80,82 and PARP.61,74–76,80,82 The activated PARP enters the nucleus and causes DNA damage, leading to cell apoptosis.77
PPII induces apoptosis in HepG2 cells through modulation of the mitochondrial pathway via downregulation of Bcl-2 and upregulation of Bax, triggering cytochrome c release from the mitochondria and caspase-9 activation. Activated caspase-9 further activates caspase-3 and PARP, respectively, resulting in apoptosis.62
In MCF-7 and MDA-MB-231 cells, PPD induces mitochondrial-dependent apoptosis through MMP dissipation,24 Bcl2 downregulation,24 and Bax upregulation,24 which further activate caspase-9,24 leading to cell apoptosis.24
In HepG2,25 R-HepG2,25 NB-69,84 K562/A02,65 U87 glioma,85 SGC790187, MCF-7,24 MDA-MB-231,24 and NSCLC cells, PPD or PSI induces apoptosis by modulating the mitochondrial-dependent pathway. In the mitochondrial-dependent pathway, PPD or PSI downregulates Bcl224,65,85–87 and upregulates Bax.24,65,85–87 As a result, the mitochondrial membrane becomes permeable, and cytochrome c65,86 and AIF25 are released from the mitochondria to the cytosol, causing the activation of caspase-9,84,86 caspase-3,25,65,84–87 and PARP24,25 and the apoptosis of cells.24,25 PPD-induced apoptosis was reversed by pretreatment with the inhibitor SP60012585 through modulation of Bcl-2,85 Bax,85 caspase-385 and with the caspase inhibitor z-DEVD-fmk through downregulation of caspase-3 in HepG2 and R-HepG2 cells.25
PPD or PSI also increases the apoptotic activity of other compounds, including camptothecin,88 10-hydroxycamptothecin,88 cisplatin,89 and hyperthermia.90
In H460,88 H446,88 and SGC-790189 cells, PSI sensitizes camptothecin-, 10-hydroxycamptothecin-, and cisplatin-89 induced apoptosis through modulation of the mitochondrial pathway. In the mitochondrial pathway, PSI inhibits Bcl288,89 and Bclxl88 while upregulating Bax,88,89 causing mitochondrial membrane permeability and cytochrome-C88 release from the mitochondria into the cytosol. As result, PSI increases caspase-988 and caspase-3 expression.88,89 Furthermore, PSI and hyperthermia at 43°C induce apoptosis in NSCLC cell lines through inhibition of Bcl2, Bax, and caspase-3 protein expression.90
PPVI in HepaRG cells induces MDP apoptosis through Bcl-2 downregulation and Bax upregulation, triggering cytochrome c release from the mitochondria and upregulating caspase-9,3 and PARP.63
PPVII66 or PPG64,91 induces apoptosis in SAS,91 OECM-192, HepG-2,64 and NPC66 cells through downregulation of Bclxl66,91 and Bcl264,66,91 and upregulation of Bak,91 Bad,64 Bax,64,66 Bim,91 and tBid91 levels. This phenomenon causes cytochrome c64 release from the mitochondria to the cytosol, which further increases the cleavage of caspase-364,66,91 and −9.64,66,91 z-VAD-FMK (a broad-spectrum caspase inhibitor) was used in the following experiments to clarify the relevance of PPG-induced cell death. PG combined with Z-VAD-FMK substantially increases the viability and decreases the apoptosis of HONE-1 and NPC-039 cells.66 Formosanin C and PPVII increase apoptosis in NCI‐H460 cells by downregulating Bcl-2 and upregulating Bax, caspase-3, −8, and 9.92 The summarized form of the mitochondrial-dependent pathway is depicted in Figure 1A.
Extrinsic Apoptotic Pathway
Tumor-necrosis-factor (TNF) family proteins, such as Fas or TNF receptor (TNFR-1), activate the extrinsic apoptotic pathway.93 Fas or TNFR-1 upregulates caspase-8 through Fas-associated death domain, generating a death signaling complex that activates caspase-3 and results in cell death.94,95 In different cancers, NPs regulate the extrinsic apoptotic pathway.96,97
PPII induces the apoptosis of HepG2 cells by modulating the extrinsic apoptotic pathway via upregulation of FAS, caspase-8, and caspase-3.62 PPVI induces apoptosis in a dose-dependent manner in HepaRG,63 A549,98 and NCI-H129998 cells by upregulating the expression of death receptor-3 (DR3),98 DR5,98 Fas,63,98 caspase-8,63 caspase-3,63,98 and cleaved PARP63,98 but downregulating the expression of decoy receptor-3 (DcR3).98 Pretreatment with Z-VAD-FMK (caspase inhibitor) on HepaRG cells increases the percentage of viable cells, indicating that PPVI induces cell apoptosis through the mitochondrial pathway and the Fas death-dependent pathway.63 PPVII or PPG induces apoptosis in a dose-dependent manner in human NPC,66 A549,98 NCI-H1299,98 HepG-2,64 SAS,91 and OECM-192 cells by upregulating the expression of DR3,98 DR5,98 Fas,98 caspase-8,64,66,91 cleaved PARP,98 and cleaved caspase-364,66,91,98 but downregulating the expression of DcR3.98 The summarized form of the extrinsic apoptotic pathway is depicted in Figure 1B.
Signal Transducer and Activator of Transcription-3 Pathway
Signal transducer and activator of transcription-3 (STAT3) regulates different cellular processes, including proliferation, differentiation, immune function, and survival.99 STAT3 is activated through its phosphorylation at tyrosine-705 (Y705) or serine-727 (S727).100,101 STAT3 can be activated by growth factor receptors, cytokine receptors, Janus activated kinases, sarcoma family kinases, and Abelson murine leukemia family kinases.102,103 STAT3 is expressed in different cancers 104,105 and leads to transformation, resistance to chemotherapy, and tumorigenesis.106 These findings show that STAT3 is a good therapeutic target in the development of new drugs against cancer.
In NSCLC cells, the overexpression of lncRNA-metastasis-associated lung adenocarcinoma transcript-1 (MALAT1) increases STAT3 expression, which causes resistance to gefitinib, whereas PPI reverses the MALAT1-induced overexpression of STAT3 and causes cell apoptosis.76 Another study demonstrated that PPI reverses the IL6/STAT3-induced resistance to erlotinib in HCC827 cells 108. PP7- or PPVII-induced apoptosis in HepG2 cells by downregulating STAT3.64 The STAT3 pathway is further summarized in Figure 1C.
Wnt/β-Catenin Pathway
Wnt/β-catenin pathway is an oncogenic pathway that plays a crucial role in cancer progression.108 β-catenin is an intracellular signal transducer that plays an important role in cadherin protein complex and activates the Wnt/β-catenin pathway during embryonic development and tumorigenesis.109–111 Following activation of this pathway, wnt binds to membrane proteins, including serpentine receptors, frizzled family, and low-density lipoprotein receptor-related protein-5/6, which is necessary for disheveled (Dsh/Dvl) phosphorylation. Activated Dsh/Dvl works at intermediate and transfer signals from the Wnt/β-catenin-receptor complex to axis and glycogen-synthase-kinase-3β (GSK-3β) to downregulate the phosphorylation of β-catenin.112–114 As a result of Wnt binding to its receptors, the unphosphorylated β-catenin accumulates in the cytoplasm. The accumulated β-catenin enters the nucleus, where its downstream target genes, such as c-myc, are activated.115 Furthermore, the Wnt-signaling pathway controls different cellular functions, including apoptosis, cell proliferation, migration, and invasion, which enhance the Wnt-dependent carcinogenesis.116,117
PPI inhibits cell growth, proliferation, and metastasis and induces apoptosis in 143-B, HOS, RPMI8226, and HO-8910M cells through the Wnt/β-catenin pathway. In the Wnt/β-catenin pathway, PPI inhibits Wnt5A and p-GSK-3β, further inhibiting β-catenin and its nuclear translocation.74,80,81,83 The PPI-reduced cell viability is abolished by GSK-3β specific inhibitor CHIR99021 while potentiated by β-catenin silencing.74 Furthermore, PPI inhibits the downstream regulator of b-catenin, such as surviving,80 as depicted in Figure 1D.
Cell Cycle
Cell cycle regulates cell growth at different checkpoints through the interaction of cyclin among exact cyclin-dependent kinases (CDKs), forming active complexes. The process ends before entering the new phase of the cell cycle.118 Then, CDKs are negatively regulated through different CDK inhibitors. CDK p21 controls the cell cycle at various checkpoints.119,120 Failed regulation of these checkpoints causes genomic rearrangement and mutation, resulting in genetic disturbance and ultimately cancer.120 Meanwhile, p53 is a key component that plays a crucial role in cell cycle regulation. A wide spectrum of damages and stresses activates p53.121,122 When p53 is activated through genotoxic stress, it controls the p21WAF1/CIP1/SDI1 genes encoding CDKs universal inhibitors, resulting in cell cycle inhibition.123 A plethora of research demonstrated that the anticancer compound causes death in cancer cells through induction of cell cycle arrest.124
In hepatocarcinoma, HCT 116, osteosarcoma 143-B, HOS, MG-63, U-2 OS, Saos-2, human myeloma RPMI8226, U266 cells, Du145, and PC3 cells, PPI causes cell cycle arrest in different phases, including G1, G2/M, S-phase, and G0/G1.59,73–75,78,80,125 In hepatocarcinoma cells, PPI and formosanin C synergistically upregulate p53, p21, and p27 and downregulate PCNA, CDK2, and Cyclin E, which lead to G1 phase cell cycle arrest.73 PPI in human myeloma RPMI8226 and U266 cells, U251 human glioma cells, HCT 116 cells 143-B and HOS cells induced ROS mediated G2/M phase cell cycle arrest through p21 upregulation which results in downregulations of cyclin B1, CD1 and c-myc.59,74,78,80 The ROS-mediated G2/M phase cell cycle arrest is reversed by NAC treatment.59 One study reported that PPI can cause S-phase cell cycle arrest in MG-63, U-2 OS, and Saos-2 osteosarcoma cells through downregulation of c-myc, Cyclin B1, Cyclin D1, and CDK1 time-dependently; this result suggests that the cell cycle arrest is due to cyclin/CD1 complex disturbance.75 Another study reported that PPI causes the G0/G1 phase cell cycle arrest in Du145 and PC3 cells with an unknown molecular mechanism.125
PPD or PSI induces G2/M phase cell cycle arrest in SGC7901,86 NSCLC,87 K562,65 and K562/A0265 through upregulation of CD1465 and p2165 and downregulation of cyclin B1,65,86 cyclin-dependent protein kinase,65 and Cdk1.86 PSI increases the cisplatin-induced G2/M phase cell cycle arrest in SGC-7901 cells through P21waf1/cip1 activation.89 PSI and hyperthermia at 43°C induce G2/M phase cell cycle arrest in NSCLC cell lines.90
PPVI induces apoptosis and G2/M phase cell cycle arrest in a dose-dependent manner in A549 and NCI-H1299 cells by upregulating the expression P53 and p21 Waf1/Cip1, which may inhibit cyclin B1 in NCI-H1299 cells.98 PPVI induces S phase cell cycle arrest in HepaRG cells by decreasing the expression of cyclin A2 and CDK2 and upregulating the expression of p21.63
In oral cancer OECM-1,91,126 SAS,91 NPC,66 A549,98 and NCI-H129998 cells, PPVII or PG induces cell cycle arrest at different stages, including G2/M,98,126 S-phase,91 and sub-G167 phase. PPVII induces G2/M phase cell cycle arrest by upregulating P53,98 p27,126 p21 Waf1/Cip1,98,126 Cdc251C,126 checkpoint kinases 1/2 (Chk1/2),126 and ataxia telangiectasia mutated126 and by downregulating cyclin B198,126 with no effect on p-Cdc21 (Tyr15).126 The G2/M phase cell cycle arrest due to p21and p27 has been confirmed through siRNA treatment.126 PPG or PPVII induces S-phase cell cycle arrest through the downregulation of Cyclin A and CDK2 expression in a dose-dependent manner. In addition, p21 and p27, inhibitors of the G1-to-S-promoting complex cyclin E-CDK2, significantly increase the protein level.91 However, the molecular mechanisms for sub-G1 phase cell cycle are unexplored.66 In gefitinib-sensitive PC-9 cells and acquired gefitinib-resistant H1975 cells, PPVII increases the sensitivity of gefitinib and induces G1 phase cell cycle arrest by upregulating p21 and downregulating CDK2, CDK4, Cyclin E, and Cyclin D1.127 The cell cycle is further summarized in Figure 2A–D.
 |
Figure 2 Anticancer molecular mechanisms of PPs. (A) In the PI3K/AKT/mTOR pathway, PI3K, PI3Kc2b, Akt, and mTOR are downregulated while Bax and caspase-3,9 and upregulated, leading to cell apoptosis. (B) In the ER stress pathway, PPI upregulates UPR, p-ERK, Bip, elF2α, ATF-4, and CHOP. PPD upregulates Bip/GRP78, PDI, and CHOP. Once CHOP becomes activated, it enters the nucleus and regulates UPR target genes, resulting in apoptosis. (C) In the JNK signaling pathway, PPI and PPD activate JNK and c-jun expression and result in cell apoptosis, while JNK inhibitor (SP600125) reverses the apoptosis. |
Phosphatidyl-Inositol 3-Kinase/Protein Kinase-B/Mammalian Target of Rapamycin Signaling Pathway
The phosphatidyl-inositol 3-kinase/protein kinase-B/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway promotes cell survival and growth through different molecular mechanisms.128,129 This pathway is activated in different types of cancer through multiple mechanisms.130–133 For example, AKT becomes activated as the phosphorylation of two residues including serine 473 (Ser 473) and threonine (Thr 308) of AKT occurs.134 After activation, AKT enters the nucleus, where they change the activities of transcription regulating factor. PI3K/AKT signaling elevates mTOR expression, which is associated with poor prognosis. Different NPs cure cancer through inhibition of the PI3K/AKT/mTOR pathway.135
PPI inhibits the proliferation and metastasis in ovarian cancer HO-8910M cells by downregulating PI3Kc2b.83 PP II increases the sensitivity of drug-resistant PC-9/ZD cells to gefitinib through the PI3K/Akt/mTOR pathway, in which it inhibits PI3K, AKT, and mTOR, which further activate Bax, caspase-9, and caspase-3 and trigger cell apoptosis.136 The PI3K/AKT/mTOR pathway is further summarized in Figure 3A.
 |
Figure 3 PPs induce cell cycle arrest in cancer cells. (A) PPI increases ROS generation and p21 expression while downregulating CB1, CG1 and c-myc expression, causing G2/M phase cell cycle arrest. (B) PPD, PPVI, and PPVII upregulate the expression of ATM, which increases p53, p21Waf1/Cip1, and p27 expression while activating chk1/2 and Cdc25C. The activated p27 and Cdc25C further inhibit cyclinB1 and CDK1 and cause G2/M phase cell cycle arrest. (C) PPI+FC upregulates the expression of p53, p21, and p27, which further inhibit CDK1, Cyclin E, CD1, and CDk4, resulting in G1 phase cell cycle arrest. PPI+FC also causes the G1 phase cell cycle arrest by inhibiting PCNA. (D) PPI, VI, and VII activate p53, p21, and p27, which further inhibit the expression of cmy-c, cyclin-B1, CDK1, Cyclin D1, Cyclin A, and CDK2 and lead to S phase cell cycle arrest. |
Endoplasmic Reticulum Stress
The endoplasmic reticulum (ER) plays a role in the synthesis, signaling, and sensing of eukaryotic cells. In performing these functions, the ER must regulate oxidizing and the Ca2+-rich folding environment. In the ER, Ca2+ buffering and protein folding are regulated by a number of chaperons, including calnexin, calreticulin, glucose-regulated protein GRP78 (BiP), and protein-disulfide isomerase. A number of pathophysiological conditions, including ER-Ca2+ depletion, viral infection, hypoglycemia, oxidative injury, and hypoxia, affect the ER homeostasis and cause ER stress. The ER responds to these path-physiological changes by activating integrated signal transduction pathways through unfolded protein response (UPR).137 The UPR controls the ER homeostasis by activating ER folding machinery components, controlling ER quality, and coordinating gene transcription and ER-associated degradation (ERAD) pathway. When ER stress increases, the UPR changes from pro-survival to pro-death response, which leads to the activation of intrinsic apoptosis.138 Mammals have various ER stress transducers, including activating transcription factor 6 (ATF6), inositol requiring enzyme 1, and protein kinase RNA like endoplasmic reticulum (p-ERK). The UPR has pro-survival and pro-apoptotic responses; pro-survival response activates ER chaperons, translation attenuation, and ERAD, whereas pro-apoptotic response activates C/EBP homologous protein-10 (CHOP)/GADD153 and caspase-12.139
PPI in MG-63, U-2 OS, and Saos-2 osteosarcoma cells dose-dependently activates the PERK branch of UPR, as determined by the increase in Bip expression, robust in eIF2α phosphorylation, and upregulation of ATF4 and GADD153 (CHOP) transcriptional factors.75
PPD induces the ER stress in human NSCLC NCI-H460 cell line by upregulating glucose-regulated protein 78 (Bip/GRP78), protein disulfide isomerase (PDI), and C/EBP homologous transcription factor (chop), triggering cell apoptosis.26 The ER pathway is further summarized in Figure 3B.
c-Jun N-Terminal Kinase Signaling Pathway
The c-Jun N-terminal kinase (JNK) pathway regulates various physiological processes, such as cell differentiation, death, survival, inflammation, protein expression, and proliferation. Any failure in this pathway causes a spectrum of diseases, including cardiac hyper-therapy, diabetes, cancer, asthma, and auto-immune diseases.140 JNK plays a major role in oncogenic changes. The JNK pathways eliminate apoptosis by downregulating Ras transformation.141 Different NPs induce apoptosis in cancer cells by regulating the JNK pathway.142–144
In human glioma U251 and ovarian cancer HO-8910M cells, PPI induces apoptosis through the JNK pathway by upregulating JNK and c-Jun.78,81,83 Furthermore, PPI inhibits tumor growth by upregulating c-Jun.81 PPD induces apoptosis in U87 glioma cells by upregulating NH2-terminal kinase (JNK) phosphorylation, which is reversed by JNK inhibitor SP600125; this result suggests that the apoptosis is due to JNK pathway regulation.85 The JNK pathway is further summarized and depicted in Figure 3C.
Nuclear Factor Kappa B Pathway
The nuclear factor kappa B (NF-kB) pathway is a complex pathway consisting of five homo- and hetero-dimers of the reticuloendotheliosis oncogene cellular homolog (Rel) family, such as RelA (p-65), RelB, c-Rel, NF-kB1 (p-50/p-65), and NF-kB2 (p50/p65).145 In cancers, the NF-kB pathway becomes dysregulated.146 Active NF-kB has been reported in different cancers, including breast, prostate, colon, liver, leukemia, lymphoma, and ovarian cancer.147–149 As the DNA becomes damaged, the NF-kB pathway becomes activated, which results in the activation of NF-kB targeted genes, including Cyclo-oxygenase-2 (COX-2)150 and inducible nitric oxide synthase (iNOS).151
These genes play pivotal roles in pro-survival anti-apoptosis. Therefore, NF-kB is a candidate for therapeutic resistance in different cancers. Different NPs have potential therapeutic efficacy against cancer by inhibiting NF-kB pathway activation in cancer cells.152
PPI alone or in combination with formosanin C or cisplatin inhibits cancer through the NF-kB pathway in different cancer cell lines, including hepatocarcinoma cells, osteosarcoma 143-B, and HOS cells.73,75,79 In the NF-kB pathway, PPI alone or in combination downregulates NF-kB p-6573 and IKBα. It also inhibits p65 translocation from the cytoplasm into the nucleus.73,75 Furthermore, this inhibition causes the downregulation of vascular endothelial growth factor (VEGF) and matrix metalloproteinase (MMP-9), leading to cell inhibition.73,79 One study demonstrated that the PPI in Du145 and PC3 cells inhibits NF-kB p65 in the cytoplasm, which further inhibits MUCIN1 directly and through HOTAIR. Furthermore, PPI inhibits p65 in the nucleus and HOT transcript antisense RNA (HOTAIR), which inhibits MUCIN1 expression and decreases Du145 and PC3 cell proliferation.125
PPI inhibits the migration, invasion, and proliferation of PC3 and DU145 cells by inhibiting HOTAIR, which further inhibits DNA methyl-transferase 1 (DNMT1) and enhancer of zeste homolog 2 (EZH2) expression. Inhibition of EHZ2 expression also inhibits cell proliferation.153 The NF-kB pathway is further summarized and illustrated in Figure 4A.
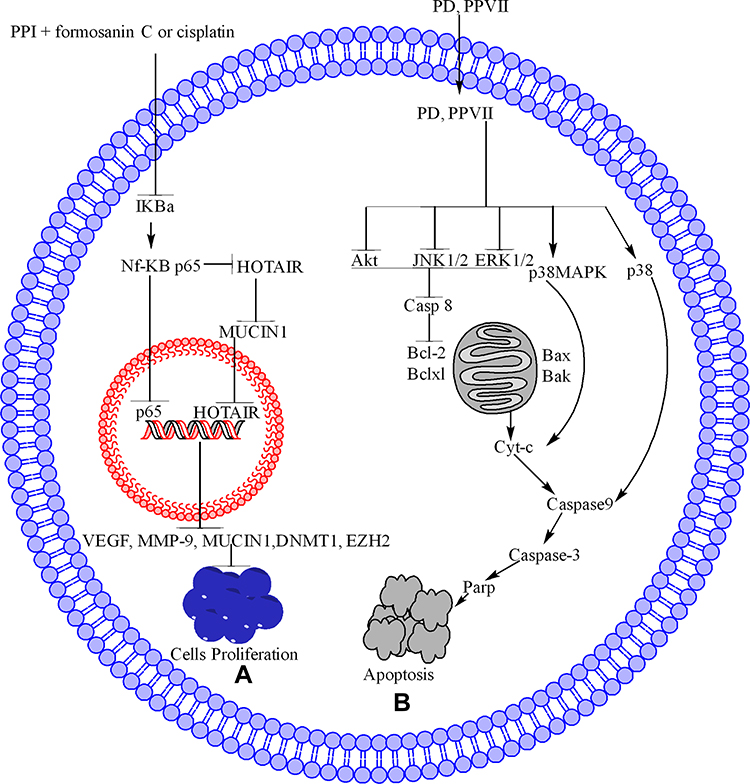 |
Figure 4 PPs inhibit cancer cell proliferation and induce apoptosis in cancer cells through the NF-kB and MAPK pathways. (A) In the NF-kB pathway, PPI+FC or Cisplatin inhibit the IKBα, which further inhibit the NF-kB p65, on one side its translocation to nucleus while on other side they inhibit the HOTAIR, MUCIN1 which further inhibit the HOTAIR translocation into nucleus. Once they inhibit the p65 and HOTAIR translocation into nucleus, they further inhibit VEGE, MMP-9, MUCIN1, DNMT1 and EZH2 which lead to inhibition of cell proliferation. (B) In the MAPK pathway, PPD and PPVII inhibit AKT, JNK1/2, and ERK1/2 expression, which further inhibit caspase-8, Bcl-2, and Bcl-xl while upregulating Bak and Bax, resulting in mitochondrial membrane permeability, allowing Cyt-c to translocate from the mitochondria into the cytoplasm. PPD and PPVII activate p38MAPK, thereby increasing Cyt-c expression; as a result, caspase-9 also becomes activated. PPD and PPVII also activate p38 and caspase-9, respectively. Upon activation, caspase-9 further activates caspase-3 and PARP, triggering the apoptosis of cancer cells. |
Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated-Kinase Pathways
The mitogen activated-protein kinase/extracellular signal-regulated kinase (MAPK/ERK) or Ras-Raf-MERK-ERK pathway possesses different cascades, but Ras-Raf-MERK-ERK 1 and 2 (ERK1/2) are mostly dysregulated in human cancer.154 This pathway regulates different cellular functions, including cell growth, differentiation, apoptosis, proliferation, migration, and senescence.155 The protein molecules of the MAPK/ERK pathway are activated through phosphorylation. Following activation, ERK enters the nucleus, where different transcription factors are activated. When these transcription factors are activated, they attach to the promoter region with different genes, including apoptosis inhibitory genes, cytokines, growth factors, and genes that increase cell proliferation.156 Dysregulation of this pathway causes drug resistance, tumorigenesis, and senescence,155,157,158 as detected in many human cancers.159,160 The MAPK pathway consists of ERK, JNK, and p-38MAK,161 which play a critical role in normal and cancerous cell proliferation and cause drug resistance.162,163 Small molecules targeting kinases in the MAPK pathway are not only useful in patients with NSCLC but also in patients with SCLC.164 NPs have a potential effect against cancer through MAPK pathways. Recently reported NPs, including PSI or PPD, PPVII or PPG have a potential antitumor effect through MAPK pathways.64,88,91
PSI or PPD sensitizes the CPT/HCPT-mediated inhibition of p38 MAPK and activation of phosphorylation of p38 MAPK in H1299 cells and the suppression of AKT and ERK pathway activation in H460 cells.88
PPVII or PPG induces apoptosis in HepG-2,64 OECM-192, and SAS91 cells by upregulating AKT,91 JNK,64,91 ERK,64,91 and p38.64,91 The involvement of the MAPK64,91 was evaluated using Akt inhibitor (LY294002),91 JNK inhibitor SP600125,64,91 ERK inhibitor PD98059,64,91 (5 μM), and p38 inhibitor SB20358064,91 to pre-treat HepG2,64 OECM-192, and SAS91 cells before the treatment of PP7. The MAPK64 (AKT, ERK1/2, p38 and JNK1/2)91 inhibitors could significantly reduce the expression of apoptosis-related proteins in HepG2, OECM-1,91 and SAS91 cells and decrease the apoptosis64,91 and necrosis64 of HepG2 cells treated with PP7.64
PPG-induced apoptosis in NPC cells through ERK1/2 and JNK1/2 and confirmed through the use of ERK1/2 inhibitor (U0126) increases the apoptosis and JNK1/2 inhibitor (SP600125), which inhibit the apoptosis caused by JNK upregulation.66 The PP anticancer effect through MAPK pathways is further summarized in Figure 4B.
AUTOPHAGY: PPs Induce Autophagy in Different Cancers Through the Following Mechanisms
PPs Induces Autophagy Through the PI3K/AKT/mTOR and AMPK/mTOR Pathways
A new target for cancer treatment is autophagy, which is activated through chemotherapy, nutrient deprivation, and oxidative stress and causes the degradation of damaged cytoplasmic organelles and proteins in response to external stress.165,166 Different signaling pathways are involved in autophagy, including adenosine mono-phosphate activated protein kinase (AMPK) and PI3K/AKT/mTOR pathways.167,168 The PI3K/AKT pathway serves as a positive regulator of the mTOR pathway and a negative regulator of autophagy in cancer cells169 [11], but the disruption of PI3K/AKT/mTOR pathways through anticancer agents results in autophagy. AMPK regulates energy homeostasis and causes autophagy through inhibition of mTOR complex 1.168,170 Furthermore, the JNK pathway is involved in the autophagy of cancer cells in response to pharmacological stress.171,172 A number of studies revealed that autophagy is triggered by inhibition of the PI3K/AKT/mTOR pathway through a link with activation of the JNK pathway.173,174 Autophagy protein Beclin-1 interacts with Bcl-2, forms Beclin-1/Bcl-2 complex, and inhibits autophagy.175 Activated JNK causes Bcl-2 phosphorylation, results in Bcl-2 degradation, dissociates Beclin-1 from the Beclin-1/Bcl-2 complex, and causes autophagy.176–179 Furthermore, JNK activation is essential for anticancer agent-induced autophagy cell death.176,180
One of the hallmarks of autophagy is the conversion of light chain 3I (LC3I) into its lapidated form LC3II, which is essential for autophagosome formation in autophagy.181 Another marker of autophagic flux is P62, which has an inverse relation to autophagy activity. It binds to LC3 and degrades in autophagy.182 Moreover, in the study of autophagy, a commonly used approach is the examination of autophagic flux and autophagosome formation. A critical crosstalk between apoptosis and autophagy is Bcl-2, which inhibits autophagy through binding with Beclin-1 that initiates autophagosome formation during autophagy.175
PPI induces protective autophagy through the PI3K/AKT/mTOR pathway by inhibiting mTOR (S2448), AKT (S473), 70-kDa ribosomal protein S6 kinase (p70S6K (T389)), and 4E (eIF4E)-binding protein 1(4EBP1 (T37/46)) and increasing LC3I conversion into LC3II. In SMMC7721 and HepG2 cells, EGF markedly increases the phosphorylation of Akt and p70S6K and reverses LC3II, which suggests that the autophagy is due to the PI3K/AKT/mTOR pathway.82
PPI induces ROS-mediated autophagy in HCT 116 cells by inhibiting mTOR (s2448) and AKT (S473), p62 expression and increasing LC3II expression.59 The increase in LC3II is reversed by NAC pre-treatment.59
PPVII or PPG treatment induces autophagy in HepG-2,183 HONE-1, and NPC-03966 cells via the PI3K/AKT/mTOR and AMPK/mTOR pathways. In the PI3K/AKT/mTOR pathway, PPVII inhibits the expression of PI3K,66,183 Akt183 and mTOR,66,183 activated mTOR (ser2448),66 total mTOR,66 Raptor,66 Rictor,66 and GβL66 in a dose-dependent manner.66,183 PPG-induced autophagic death is confirmed when Baf A1 and wortmannin (autophagy inhibitors) increase cell viability and PPG treatment alone exerts no effect on the apoptosis of HONE-1 and NPC-039 cells.66 PPVII also increases AMPK phosphorylation, which further inhibits mTOR and induces autophagy in HepG-2 cells through modulation of the AMPK/mTOR pathway.183 These mechanisms are further summarized in Figure 5.
 |
Figure 5 PPI induces protective autophagy through the PI3K/AKT/mTOR pathway by inhibiting AKT (S473), mTOR (S2448), p70S6K (T389), and 4EBP1 (T37/46) and increasing the conversion of LC3I conversion into LC3II. EGF markedly increases the phosphorylation of Akt and p70S6K and reverses LC3II, suggesting the autophagy is due to the PI3K/AKT/mTOR pathway. In the PI3K/AKT/mTOR pathway, PPVII inhibits PI3K Akt and mTOR activated mTOR (ser2448), total mTOR Raptor, Rictor and GβL in a dose-dependent manner. PPVII also increases AMPK phosphorylation, which further inhibits mTOR and induces autophagy in HepG-2 cells by modulating the AMPK/mTOR pathway. PPG or PPVII induces autophagy through the Akt, p38MAPK, ERK1/2, and JNK pathways. PPG or PPVII activates JNK1/2 and inhibits Akt, p38 MAPK, and ERK1/2, which further increase the conversion of LC3I to LC3II, P62 degradation, and formation of LC3-positive structures or LC3 puncta. Furthermore, PPVII treatment decreases the level of total Bcl-2 decreases but increases those of p-Bcl-2 and Beclin. The AKT inhibitor (LY294002) and JNK1/2 inhibitor (SP600125) increase, suggesting that PPG induces autophagy via the Akt, p38 MAPK, and JNK1/2 pathways. Moreover, LY294002, U0126, and SP600125 significantly attenuate PG-induced LC3-II activation, suggesting that the activation of ERK1/2 and JNK1/2 is involved in PG-induced autophagy. |
PPs Induces Autophagy Through the Akt, p38MAPK, ERK1/2, and JNK Signaling Pathways
A new target for cancer treatment is autophagy, which is activated through chemotherapy, nutrient deprivation, and oxidative stress and causes the degradation of damaged cytoplasmic organelles and proteins in response to external stress.165,166 Previous research demonstrated that the AKT and MAPK pathways are involved in autophagy and apoptosis.172,184–186
PPG or PPVII induces autophagy through the Akt, p38MAPK, ERK1/2, and JNK pathways in HepG-2,183 NPC-039,66 OECM-1,91 and SAS91 cells. PPG or PPVII activates JNK1/266,91,183 and inhibits AKT,66 p38 MAPK,66 and ERK1/2,91 which further increases the conversion of LC3I to LC3II,66,183 P62 degradation,66,183 and formation of LC3-positive structures or LC3 puncta.66,183 Furthermore, PPVII treatment decreases total Bcl-2183 while increases p-Bcl-2183 and Beclin.66,183 The AKT inhibitor (LY294002)66 and JNK1/2 inhibitor (SP600125)66,183 decrease PPG-induced autophagy, whereas p38 MAPK inhibitor (SB203580)66 increases this process, suggesting that the PPG-induced autophagy is due to the Akt, p38 MAPK, and JNK1/2 pathways.66,183 Moreover, LY294002, U0126, and SP600125 significantly attenuate PG-induced LC3-II activation, suggesting that the activation of ERK1/2 and JNK1/2 is involved in PG-induced autophagy.91 These mechanisms of autophagy are further summarized in Figure 5.
Inflammation Pathway
Inflammation is a physiological response for the protection of the body from tissue injury or infection and plays a critical role in different types of human cancers and other chronic diseases.187 In innate immune system, macrophages are the immune cells that play a central role in inflammation and protect the body from harmful stimuli. Active macrophages play important roles in host defenses against pathogens. They exert phagocytic activities and enhance inflammatory responses by producing different inflammatory factors, such as nitric oxide (NO) and prostaglandin-E2; proinflammatory mediators, such as iNOS, nitrogen species, metalloproteinases, and COX-2; and pro-inflammatory cytokines, such as TNF-α, interleukin-6 (IL-6), and IL-1β, which trigger other immune cells to the infection site.188–190 The overexpression of inflammation-related cytokines and mediators by activated macrophages is associated with the pathophysiology of different inflammatory and autoimmune diseases.191–193 Lipopolysaccharide (LPS) is a potent macrophage activator and endotoxin derived from the cell wall of Gram-negative bacteria.194 When LPS stimulates the macrophages, the expression of cytokines and mediator is regulated by NF-kB through MAPKs, including JNKs, ERKs, and p38-MAPK.195,196 Macrophages secrete MMP-9, which controls leukocyte migration in inflammatory diseases.197 In macrophages, MMP-9 is regulated by LPS via the NF-kB and MAPK pathways.198–200 Therefore, targeting MAPKs, NF-kB, and MMP-9 through NPs may have a potential effect on the treatment of inflammatory diseases.
In RAW264.7 cells, PPVII inhibits the LPS-induced phosphorylation of p38, JNK, and ERK, which indicate the involvement of MAPKs in the suppression of cytokines and mediator by PPVII.199 Macrophage produces pro-inflammatory cytokines (major components of inflammation), including interleukin-6 (IL-6), IL-1β, and TNF-α, and the PPVII-inhibited productions of these cytokines are markedly increased in LPS-treated cells.199 Furthermore, PPVII inhibits IkB-α phosphorylation, increases the p65 level in the cytoplasm that is inhibited in the nucleus, and downregulates PGE-2, NO, iNOS, COX-2, and MMP-9 at the mRNA and protein levels in LPS-activated cells.199 The above discussion suggests that PPVII inhibits inflammation through the MAPK and NF-kB pathways, as illustrated in Figure 6A.
 |
Figure 6 PPs inhibit inflammation and induce necroptosis and mitophagy in cancer cells. (A) PPVII inhibits the inflammation by inhibiting the LPS-induced expression of TNF-α, IL-1B, and IL-6. PPVII also inhibits the inflammation through the NF-kB and MAPK pathways. In the NF-kB pathway, PPVII inhibit the IKB-α, p65 and its translocation to the nucleus which further inhibit iNOS, COX-2, and MMP-9 and inhibit inflammation. In the MAPK pathway, PPVII inhibits p38-MAPK, ERK1/2, and JNK1/2, resulting in inhibition of inflammation. (B) PPD induces necroptosis in cancer cells, whereas necrosulfonamide reverses this inhibition. (C) PPI induces the translocation of DRP1 to the mitochondria by dephosphorylating DRP1 at ser-637, causing mitochondrial fission. PPI increases the stabilization of full-length PINK1 at the surface of the mitochondria, leading to P62, PARK2, ubiquitin, and LC3B-II recruitment to the mitochondria and then to mitophagy. PPI-induced mitophagy is suppressed markedly by PINK1 knockdown. DRP1 suppression through shRNA or mdivi-1 inhibits the knockdown of PINK1 and PPI-induced mitochondrial fragmentation. These results suggest that PINK1 depletion leads to fission and mitochondrial fragmentation. |
Necroptosis
Cell death usually occurs through two main pathways, namely, programmed cell death or apoptosis200 and direct cell damage or necrosis. Programmed necrosis or necroptosis is a new type of necrosis that has been recently reported.201 Necroptosis mechanisms involve the initial formation of a complex that contains receptor-interacting protein kinase-1 (RIPK1) induced by LPS and TNF. Furthermore, a complex is formed among RIPK3, RIPK1, Fas bound death domain protein, and caspase-8.202 By contrast, when the caspase-8 is inactive, RIPK3 and RIPK1 become activated and a RIP3 substrate known as mixed lineage kinase domain-like (MLKL) is phosphorylated, which attach to the cell membrane and form a hole in the cell membrane that causes the destruction of the cell membrane via necroptosis.203–205
PPD-induced necroptosis in LA-N-2 and IMR-32 cells, which is confirmed when the cells are co-treated with necrosulfonamide (a specific inhibitor of RIPK3 associated with necroptosis), through inhibition of PPD-induced cell death,84 as illustrated in Figure 6B.
Mitophagy
Mitochondrial autophagy or mitophagy is a cellular pathway that helps in the removal of damaged mitochondria.206,207 Mitophagy plays a crucial role in the control of cancer microenvironment, cancer cell survival, and death. The study of molecular mechanisms of mitophagy might be important for the development of new cancer therapies.208 The PIKK1/PINK2 pathway regulates mitophagy. PARK-2 is a RING domain-containing E3 ubiquitin ligase, which is activated via autoubiquitination.209 In depolarized state, mitochondria use the uncoupling reagents, including carbonyl cyanide m-chlorophenyl-hydrazone (CCCP), and PARK2 enters the mitochondria and promotes its degradation.210 Furthermore, PARK2 overexpression induces the degradation of depolarized mitochondria via mitophagy.211 Given that PARK2 also selectively binds only to damaged mitochondria, it might help ensure the specificity of mitophagy.212 PTEN-induced kinase 1 (PINK1) has a targeting sequence located in the mitochondria.213 PINK1 protects the mitochondria from neurotoxin-induced injury, whereas loss of PINK1 function or mutation causes ROS-mediated mitochondrial injury.214 The full-length expression of PIK1 increases CCCP-mediated mitophagy or autophagy.215 Under stress conditions, the depolarization of mitochondrial membranes prevents the mitochondrial uptake and processing of PINK1; therefore, the unprocessed PINK1 on the outer membrane of the mitochondria recruits PARK2 and eliminates the damaged mitochondria through mitophagy.210 Dynamin-related protein 1 (DRP1) inhibitor mdivi-1 prevents mitophagy by inhibiting the fusion–fission cycle, which shows the importance of mitochondrial fission in mitophagy.216 The mitochondrial fission mediated by DRP-1 causes LC3B lipidation and mitophagy, for which PINK1 and PARK2 are required.217 A recent study has demonstrated that the LC3B-II autophagosome targets the mitochondrial membrane through interaction with C18-ceramide-LC3B-II, increases lethal mitophagy, and inhibits tumor growth.218 The above description demonstrates that mitophagy might be helpful in the identification of new therapies for cancer treatment.
In MDA-MB-231 cells, PPI induces DRP1 translocation to the mitochondria through dephosphorylating of DRP1 at ser-637, causing mitochondrial fission. PPI increases the stabilization of full-length PINK1 at the surface of mitochondria, leading to P62, PARK2, ubiquitin, and LC3B-II recruitment to the mitochondria and mitophagy. PPI-induced mitophagy is suppressed markedly with PINK1 knockdown. DRP1 suppression through shRNA or mdivi-1 inhibits the knockdown of PINK1 and PPI-induced mitochondrial fragmentation. These results suggest that PINK1 depletion leads to fission and mitochondrial fragmentation,61 as shown in Figure 6C.
In vivo Anticancer Effect and Toxicity of PPs
PPI inhibits tumor growth in lung cancer tumor xenograft77 and xenograft orthotopic mouse model74 without any side effects.74 PPI inhibits ovarian tumor in nude mice by downregulating Wnt5a,81 in PC-9-ZD xenograft by inhibiting MALAT1 and STAT3 expression,76 and in prostate cancer xenograft mouse model by inhibiting HOTAIR, DNMT1, and EZH2 expression.153 Furthermore, in MDA-MB-231 xenografts, PPI inhibits tumor growth that is enhanced by PINK1 knockdown. These findings show that PPI might be a good therapy for cancer.61 PPI inhibits the vasculogenic mimicry and microvessel density in PLC and PLC/Twist1 cells. Furthermore, immunohistochemically staining showed that PPI inhibits the expression of Twist1, VE-cadherin, vimentin, VEGFR1, and VEGFR2 and increases E-cadherin expression in tumor xenograft.219 PPI also overcomes the erlotinib resistance in HCC827-tumor xenografts by inhibiting IL-6; decreasing mesenchymal markers FN1, VIM, and SNAIL; and upregulating epithelial marker E-cadherin 109. In the embryo of zebrafish, PPD decreases the formation of inter-segmental vessels.47 PPD markedly inhibits the tumor in MCF-7 bearing nude mice with no obvious toxicity to heart and liver tissue nor increases the level of plasma enzymes, including aspartate transaminase, alanine transaminase, lactase dehydrogenase, and creatine kinase.24 PPD-containing saponins inhibit tumor growth in H22 tumor-bearing mice.35 In A549 xenografts, PPVI inhibits the tumor by causing apoptosis and G/M phase cell cycle arrest through upregulation of DR3, DR5, and p53 and attenuation of cyclin B1.98 PPVII and gefitinib inhibit tumor growth in acquiring gefitinib-resistant xenograft compared with either treatment alone. Furthermore, in xenograft model, PPVII and gefitinib activate the expression of p21 while reducing the expression of CDK4, CDK2, Cyclin D1, and Cyclin E compared with either treatment alone.127 In the NPC orthotopic graft model, PPG inhibits tumor growth by downregulating Ki67.66
In vivo Anti-Inflammatory Effect and Toxicity of PPs
In Mice
PP7 dose-dependently suppresses the formation of xylene-induced ear edema in mice, suggesting that PP7 can potently inhibit acute inflammation in vivo. PP7 inhibits cotton pellet-induced granuloma formation in a dose-dependent manner in mice, indicating that it also inhibits chronic inflammation.199
In Zebrafish
Zebrafish (Danio rerio) is a freshwater fish used as a vertebrate model organism for different inflammation studies because its acquired and innate immunity is highly similar to that of mammals. The transparency of zebrafish embryo and larvae allows the dynamic and noninvasive imaging of in vivo inflammation.220–224 In adult zebrafish, the most abundant leukocytes are neutrophils; in BACmpx::GFP transgenic zebrafish, larvae possess green fluorescence in neutrophils, allowing the observation of neutrophil distribution in zebrafish.225,226 The numbers of neutrophils circulating near the lateral line neuromasts allow the measurement of inflammation. Copper sulfate and Lip+o-polysaccharide induce inflammation in zebrafish.
PPVII strongly inhibits NO generation and decreases the heartbeat and the size of yolk sac edema in LPS-stimulated inflammation in zebrafish embryos.199 Furthermore, pre-treatment with PPVII reduces the recruitment of neutrophils to the injured area, suggesting the potential effect of PVII against inflammation.199
Conclusion
PPs, including PPI, II, III, VI, VII, and PPD, are derived from different plants. These PPs exhibit potential effects against various cancers through different mechanisms in vitro and in vivo. PPs show their therapeutic effect on different cancers through apoptosis, autophagy, necroptosis, mitophagy, and inflammation. Among PPs, PPI targets apoptosis and autophagy through the PI3K/AKT/mTOR pathway. Thus, PPI is a candidate therapeutic drug for cancers activated by this pathway. In addition, although PPs target different pathways, a specific link among all pathways is lacking. Thus, researchers need to focus and find the link among these pathways. These PPs are ideal therapeutic drugs, and further research on their anticancer effect is highly encouraged.
Acknowledgment
The study was financially supported by the National Natural Science Foundation of China (81373875) and Liaoning Provincial Program for Top Discipline of Basic Medical Science (2012CB518803).
Disclosure
All authors declare that they have no competing interests in this work.
References
1. Pengyu SAB, Lijuan Z. Natural β-elemene: advances in targeting cancer through different molecular pathways. North Am J Aced Res. 2018;1(4):27.
2. Ahmad B, Khan S, Nabi G, et al. Natural gypenosides: targeting cancer through different molecular pathways. Cancer Manag Res. 2019;11:2287–2297.
3. Su P, Ahmad B, Ahmad M, et al. Targeting Cancer through PI3K/AKT/mTOR pathway with selected natural products (β-Elemene, Puerarin and Gypenosides). Am J Biomed Sci Res. 2020;8:4.
4. Pengyu SBA, Ullah H, Hussain B, Khan SZ, Mohammad SAJK, Ahmad M. Targeting cancer through autophagy with Β-Elemene and Puerarin. Am J Biomed Sci Res. 2020;8.
5. Organization WH. Global Health Observatory Data Repository. 2011. Number of Deaths (World) by Cause; 2015.
6. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
7. Bray F. Transitions in human development and the global cancer burden. World Cancer Report. 2014;54–68.
8. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
9. Qi F, Li A, Inagaki Y, et al. Chinese herbal medicines as adjuvant treatment during chemo- or radio-therapy for cancer. Biosci Trends. 2010;4(6):297–307.
10. Ahmad B, Khan S, Nabi G, et al. <p>Natural gypenosides: targeting cancer through different molecular pathways. Cancer Manag Res. 2019;11:2287–2297. doi:10.2147/CMAR.S185232
11. Ahmad B, Khan S, Liu Y, et al. Molecular mechanisms of anticancer activities of puerarin. Cancer Manag Res. 2020;12:79–90.
12. Jalal S, Ahmad B, Zhang T, Guo L, Huang L. SANTAMARINE: mechanistic studies on multiple diseases. Chem Biol Drug Des. 2020. doi:10.1111/cbdd.13666
13. Ji Y, Yang L, Chase MW, et al. Plastome phylogenomics, biogeography, and clade diversification of Paris (Melanthiaceae). BMC Plant Biol. 2019;19(1):543.
14. Wei JC, Gao WY, Yan XD, Wang Y, Jing SS, Xiao PG. Chemical constituents of plants from the genus Paris. Chem Biodivers. 2014;11(9):1277–1297.
15. Zhao Y, Zhang J, Yuan T, et al. Discrimination of wild Paris based on near infrared spectroscopy and high performance liquid chromatography combined with multivariate analysis. PLoS One. 2014;9(2):e89100.
16. Liu F, Meng Y, He K, et al. Comparative analysis of proteomic and metabolomic profiles of different species of Paris. J Proteomics. 2019;200:11–27.
17. Zhang J-Y, Wang Y, Zhao Y-L, et al. Phytochemicals and bioactivities of Paris species. 2011;13.
18. Li Y, Man S, Li J, et al. The antitumor effect of formosanin C on HepG2 cell as revealed by 1H-NMR based metabolic profiling. Chem Biol Interact. 2014;220:193–199.
19. Liu Z, Gao W, Jing S, et al. Correlation among cytotoxicity, hemolytic activity and the composition of steroidal saponins from Paris L. J Ethnopharmacol. 2013;149(2):422–430.
20. Zhao Y, Kang LP, Liu YX, et al. Steroidal saponins from the rhizome of Paris polyphylla and their cytotoxic activities. Planta Med. 2009;75(4):356–363.
21. Zhao J, Mou Y, Shan T, et al. Antimicrobial metabolites from the endophytic fungus Pichia guilliermondii isolated from Paris polyphylla var. yunnanensis. Molecules. 2010;15(11):7961–7970.
22. Wang GX, Han J, Zhao LW, Jiang DX, Liu YT, Liu XL. Anthelmintic activity of steroidal saponins from Paris polyphylla. Phytomedicine. 2010;17(14):1102–1105.
23. Man S, Gao W, Zhang Y, et al. Characterization of steroidal saponins in saponin extract from Paris polyphylla by liquid chromatography tandem multi-stage mass spectrometry. Anal Bioanal Chem. 2009;395(2):495–505.
24. Lee MS, Yuet-Wa JC, Kong SK, et al. Effects of polyphyllin D, a steroidal saponin in Paris polyphylla, in growth inhibition of human breast cancer cells and in xenograft. Cancer Biol Ther. 2005;4(11):1248–1254.
25. Ong RC, Lei J, Lee RK, et al. Polyphyllin D induces mitochondrial fragmentation and acts directly on the mitochondria to induce apoptosis in drug-resistant HepG2 cells. Cancer Lett. 2008;261(2):158–164.
26. Siu FM, Ma DL, Cheung YW, et al. Proteomic and transcriptomic study on the action of a cytotoxic saponin (Polyphyllin D): induction of endoplasmic reticulum stress and mitochondria-mediated apoptotic pathways. Proteomics. 2008;8(15):3105–3117.
27. Yang Y, Zhang J. Quantitative analysis in combination with fingerprint technology and chemometric analysis applied for evaluating six species of wild Paris using UHPLC-UV-MS. 2016;2016:3182796.
28. Xiao XH, Yuan ZQ, Li GK. Separation and purification of steroidal saponins from Paris polyphylla by microwave-assisted extraction coupled with countercurrent chromatography using evaporative light scattering detection. J Sep Sci. 2014;37(6):635–641.
29. Wang B, Ji S, Zhang H, et al. Liquid chromatography tandem mass spectrometry in study of the pharmacokinetics of six steroidal saponins in rats. Steroids. 2013;78(12–13):1164–1170.
30. Wu Z, Zhang J, Xu F, Wang Y, Zhang J. Rapid and simple determination of polyphyllin I, II, VI, and VII in different harvest times of cultivated Paris polyphylla Smith var. yunnanensis (Franch.) Hand.-Mazz by UPLC-MS/MS and FT-IR. J Nat Med. 2017;71(1):139–147.
31. Yang Y, Jin H, Zhang J, Zhang J, Wang Y. Quantitative evaluation and discrimination of wild Paris polyphylla var. yunnanensis (Franch.) Hand.-Mazz from three regions of Yunnan Province using UHPLC-UV-MS and UV spectroscopy couple with partial least squares discriminant analysis. J Nat Med. 2017;71(1):148–157.
32. Li ZH, Wan JY, Wang GQ, Zhao FG, Wen JH. Identification of compounds from Paris polyphylla (ChongLou) active against Dactylogyrus intermedius. Parasitology. 2013;140(8):952–958.
33. Liu Z, Wang J, Gao W, et al. Formulation and in vitro absorption analysis of Rhizoma paridis steroidal saponins. Int J Pharm. 2013;441(1–2):680–686.
34. Wu S, Gao W, Qiu F, Man S, Fu S, Liu C. Simultaneous quantification of Polyphyllin D and Paris H, two potential antitumor active components in Paris polyphylla by liquid chromatography-tandem mass spectrometry and the application to pharmacokinetics in rats. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;905:54–60.
35. Wang YH, Shi M, Niu HM, et al. Substituting one Paris for another? In vitro cytotoxic and in vivo antitumor activities of Paris forrestii, a substitute of Paris polyphylla var. yunnanensis. J Ethnopharmacol. 2018;218:45–50.
36. Wang Z, Zhou J, Ju Y, Zhang H, Liu M, Li X. Effects of two saponins extracted from the polygonatum Zanlanscianense pamp on the human leukemia (HL-60) cells. Biol Pharm Bull. 2001;24(2):159–162.
37. Shuli M, Wenyuan G, Yanjun Z, Chaoyi M, Liu Y, Yiwen L. Paridis saponins inhibiting carcinoma growth and metastasis in vitro and in vivo. Arch Pharm Res. 2011;34(1):43–50.
38. Yin X, Qu C, Li Z, et al. Simultaneous determination and pharmacokinetic study of polyphyllin I, polyphyllin II, polyphyllin VI and polyphyllin VII in beagle dog plasma after oral administration of Rhizoma Paridis extracts by LC-MS-MS. Biomed Chromatogr. 2013;27(3):343–348.
39. Jiang H, Zhao P, Feng J, Su D, Ma S. Effect of Paris saponin I on radiosensitivity in a gefitinib‑resistant lung adenocarcinoma cell line. Oncol Lett. 2014;7(6):2059–2064.
40. Shi Y-M, Yang L, Geng Y-D, Zhang C, Kong L-Y. Polyphyllin I induced-apoptosis is enhanced by inhibition of autophagy in human hepatocellular carcinoma cells. Phytomedicine. 2015;22(13):1139–1149.
41. Yue G, Wei J, Qian X, et al. Synergistic anticancer effects of polyphyllin I and evodiamine on freshly-removed human gastric tumors. PLoS One. 2013;8(6):e65164.
42. Ning L, Zhou B, Zhang Y, Li X. Study on hemolytic mechanism of polyphyllin II. Zhongguo Zhong Yao Za Zhi= Zhongguo Zhongyao Zazhi= China Journal of Chinese Materia Medica. 2015;40(18):3623–3629.
43. Yang M, Zou J, Zhu H, et al. Paris saponin II inhibits human ovarian cancer cell‑induced angiogenesis by modulating NF-κB signaling. Oncol Rep. 2015;33(5):2190–2198.
44. Zhang W, Zhang D, Ma X, Liu Z, Li F, Wu D. Paris saponin VII suppressed the growth of human cervical cancer Hela cells. Eur J Med Res. 2014;19(1):41.
45. Cheung JY-N, Ong RC-Y, Suen Y-K, et al. Polyphyllin D is a potent apoptosis inducer in drug-resistant HepG2 cells. Cancer Lett. 2005;217(2):203–211.
46. Lee M-S, Chan JY-W, Kong S-K, et al. Effects of polyphyllin D, a steroidal saponin in Paris polyphylla, in growth inhibition of human breast cancer cells and in xenograft. Cancer Biol Ther. 2005;4(11):1248–1254.
47. Chan JY, Koon JC, Liu X, et al. Polyphyllin D, a steroidal saponin from Paris polyphylla, inhibits endothelial cell functions in vitro and angiogenesis in zebrafish embryos in vivo. J Ethnopharmacol. 2011;137(1):64–69.
48. Al Sawah E, Marchion DC, Xiong Y, et al. The Chinese herb polyphyllin D sensitizes ovarian cancer cells to cisplatin-induced growth arrest. J Cancer Res Clin Oncol. 2015;141(2):237–242.
49. Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8(9–10):1865–1879.
50. Mittler R, Vanderauwera S, Suzuki N, et al. ROS signaling: the new wave? Trends Plant Sci. 2011;16(6):300–309.
51. Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72(11):1493–1505.
52. Gorlach A, Dimova EY, Petry A, et al. Reactive oxygen species, nutrition, hypoxia and diseases: problems solved? Redox Biol. 2015;6:372–385.
53. Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol. 2015;6:260–271.
54. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591.
55. Hong YH, Uddin MH, Jo U, et al. ROS accumulation by PEITC selectively kills ovarian cancer cells via UPR-mediated apoptosis. Front Oncol. 2015;5:167.
56. Zhu L, Ren L, Chen Y, Fang J, Ge Z, Li X. Redox status of high-mobility group box 1 performs a dual role in angiogenesis of colorectal carcinoma. J Cell Mol Med. 2015;19(9):2128–2135.
57. Wei C, Xiao Q, Kuang X, Zhang T, Yang Z, Wang L. Fucoidan inhibits proliferation of the SKM-1 acute myeloid leukaemia cell line via the activation of apoptotic pathways and production of reactive oxygen species. Mol Med Rep. 2015;12(5):6649–6655.
58. Seo KH, Ryu HW, Park MJ, et al. Mangosenone F, A furanoxanthone from Garciana mangostana, induces reactive oxygen species-mediated apoptosis in lung cancer cells and decreases xenograft tumor growth. Phytother Res. 2015;29(11):1753–1760.
59. Yu S, Wang L, Cao Z, et al. Anticancer effect of Polyphyllin Iota in colorectal cancer cells through ROS-dependent autophagy and G2/M arrest mechanisms. Nat Prod Res. 2018;32(12):1489–1492.
60. Cheng H, Su JJ, Hou HJ, Li QL. [Effect and mechanism of polyphyllin I on human cervical cancer cell HeLa in vitro]. Zhong Yao Cai. 2013;36(11):1815–1819. Chinese.
61. Li GB, Fu RQ, Shen HM, et al. Polyphyllin I induces mitophagic and apoptotic cell death in human breast cancer cells by increasing mitochondrial PINK1 levels. Oncotarget. 2017;8(6):10359–10374.
62. Wang W, Dong X, You L, et al. Apoptosis in HepaRG and HL-7702 cells inducted by polyphyllin II through caspases activation and cell-cycle arrest. 2019;234(5):7078–7089.
63. Liu Y, Dong X, Wang W, You L. Molecular mechanisms of apoptosis in HepaRG Cell Line Induced by Polyphyllin VI via the fas death pathway and mitochondrial-dependent pathway. 2018;10:5.
64. Zhang C, Jia X, Bao J, et al. Polyphyllin VII induces apoptosis in HepG2 cells through ROS-mediated mitochondrial dysfunction and MAPK pathways. BMC Complement Altern Med. 2016;16:58.
65. Wu L, Li Q, Liu Y. Polyphyllin D induces apoptosis in K562/A02 cells through G2/M phase arrest. J Pharm Pharmacol. 2014;66(5):713–721.
66. Chen JC, Hsieh MJ, Chen CJ, et al. Polyphyllin G induce apoptosis and autophagy in human nasopharyngeal cancer cells by modulation of AKT and mitogen-activated protein kinase pathways in vitro and in vivo. Oncotarget. 2016;7(43):70276–70289.
67. Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal. 2013;19(6):546–558.
68. Reed JC. Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006;13(8):1378–1386.
69. Letai A. BCL-2: found bound and drugged! Trends Mol Med. 2005;11(10):442–444.
70. Kim H, Rafiuddin-Shah M, Tu HC, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8(12):1348–1358.
71. Green DR. At the gates of death. Cancer Cell. 2006;9(5):328–330.
72. Zhu MY, Chen F, Niyazi M, Sui S, Gao DM. Variation in apoptotic gene expression in cervical cancer through oligonucleotide microarray profiling. J Low Genit Tract Dis. 2015;19(1):46–54.
73. Liu J, Man S, Liu Z, Ma L, Gao W. A synergistic antitumor effect of polyphyllin I and formosanin C on hepatocarcinoma cells. Bioorg Med Chem Lett. 2016;26(20):4970–4975.
74. Chang J, Li Y, Wang X, et al. Polyphyllin I suppresses human osteosarcoma growth by inactivation of Wnt/beta-catenin pathway in vitro and in vivo. Sci Rep. 2017;7(1):7605.
75. Chang J, Wang H, Wang X, et al. Molecular mechanisms of Polyphyllin I-induced apoptosis and reversal of the epithelial-mesenchymal transition in human osteosarcoma cells. J Ethnopharmacol. 2015;170:117–127.
76. Yang Q, Chen W, Xu Y, Lv X, Zhang M, Jiang H. Polyphyllin I modulates MALAT1/STAT3 signaling to induce apoptosis in gefitinib-resistant non-small cell lung cancer. Toxicol Appl Pharmacol. 2018;356:1–7.
77. Kong M, Fan J, Dong A, Cheng H, Xu R. Effects of polyphyllin I on growth inhibition of human non-small lung cancer cells and in xenograft. Acta Biochim Biophys Sin (Shanghai). 2010;42(11):827–833.
78. Liu J, Zhang Y, Chen L, et al. Polyphyllin I induces G2/M phase arrest and apoptosis in U251 human glioma cells via mitochondrial dysfunction and the JNK signaling pathway. Acta Biochim Biophys Sin (Shanghai). 2017;49(6):479–486.
79. Han W, Hou G, Liu L. Polyphyllin I (PPI) increased the sensitivity of hepatocellular carcinoma HepG2 cells to chemotherapy. Int J Clin Exp Med. 2015;8(11):20664–20669.
80. Liang Y, Li X, He X, et al. Polyphyllin I induces cell cycle arrest and apoptosis in human myeloma cells via modulating beta-catenin signaling pathway. Eur J Haematol. 2016;97(4):371–378.
81. Gu L, Feng J, Zheng Z, Xu H, Yu W. Polyphyllin I inhibits the growth of ovarian cancer cells in nude mice. Oncol Lett. 2016;12(6):4969–4974.
82. Shi YM, Yang L, Geng YD, Zhang C, Kong LY. Polyphyllin I induced-apoptosis is enhanced by inhibition of autophagy in human hepatocellular carcinoma cells. Phytomedicine. 2015;22(13):1139–1149.
83. Gu L, Feng J, Xu H, Luo M, Su D. Polyphyllin I inhibits proliferation and metastasis of ovarian cancer cell line HO-8910PM in vitro. J Tradit Chin Med. 2013;33(3):325–333.
84. Watanabe S, Suzuki T, Hara F, Yasui T, Uga N, Naoe A. Polyphyllin D, a steroidal saponin in Paris polyphylla, induces apoptosis and necroptosis cell death of neuroblastoma cells. Pediatr Surg Int. 2017;33(6):713–719.
85. Yu Q, Li Q, Lu P, Chen Q. Polyphyllin D induces apoptosis in U87 human glioma cells through the c-Jun NH2-terminal kinase pathway. J Med Food. 2014;17(9):1036–1042.
86. Xiao M, Dai X, He X, et al. Paris saponin I induces G(2)/M cell cycle arrest and apoptosis in human gastric carcinoma SGC7901 cells. J Huazhong Univ Sci Technolog Med Sci. 2011;31(6):768–772.
87. Jiang H, Zhao PJ, Su D, Feng J, Ma SL. Paris saponin I induces apoptosis via increasing the Bax/Bcl-2 ratio and caspase-3 expression in gefitinib-resistant non-small cell lung cancer in vitro and in vivo. Mol Med Rep. 2014;9(6):2265–2272.
88. Liu Z, Zheng Q, Chen W, et al. Chemosensitizing effect of Paris Saponin I on Camptothecin and 10-hydroxycamptothecin in lung cancer cells via p38 MAPK, ERK, and Akt signaling pathways. Eur J Med Chem. 2017;125:760–769.
89. Song S, Du L, Jiang H, Zhu X, Li J, Xu J. Paris Saponin I sensitizes gastric cancer cell lines to cisplatin via cell cycle arrest and apoptosis. Med Sci Monit. 2016;22:3798–3803.
90. Zhao P, Jiang H, Su D, Feng J, Ma S, Zhu X. Inhibition of cell proliferation by mild hyperthermia at 43 C with Paris Saponin I in the lung adenocarcinoma cell line PC-9. Mol Med Rep. 2015;11(1):327–332.
91. Hsieh MJ, Chien SY, Lin JT, Yang SF, Chen MK. Polyphyllin G induces apoptosis and autophagy cell death in human oral cancer cells. Phytomedicine. 2016;23(13):1545–1554.
92. Cui J, Man S. The synergistic anticancer effect of formosanin C and polyphyllin VII based on caspase-mediated cleavage of Beclin1 inhibiting autophagy and promoting apoptosis. 2019;52(1):e12520.
93. Locksley RM, Killeen N, Lenardo MJ, The TNF. TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104(4):487–501.
94. Ashkenazi A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 2008;19(3–4):325–331.
95. Fulda S. Targeting extrinsic apoptosis in cancer: challenges and opportunities. Semin Cell Dev Biol. 2015;39:20–25.
96. Fatehchand K, Santhanam R, Shen B, et al. Active hexose-correlated compound enhances extrinsic-pathway-mediated apoptosis of Acute Myeloid Leukemic cells. 2017;12(7):e0181729.
97. Kang TH, Yoon G, Kang IA, Oh HN, Chae JI, Shim JH. Natural Compound Licochalcone B induced extrinsic and intrinsic apoptosis in human skin melanoma (A375) and Squamous Cell Carcinoma (A431) cells. 2017;31(12):1858–1867.
98. Lin Z, Liu Y, Li F, et al. Anti-lung cancer effects of Polyphyllin VI and VII potentially correlate with apoptosis in vitro and in vivo. Phytother Res. 2015;29(10):1568–1576.
99. Siveen KS, Sikka S, Surana R, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845(2):136–154.
100. Qin HR, Kim HJ, Kim JY, et al. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008;68(19):7736–7741.
101. Huang G, Yan H, Ye S, Tong C, Ying QL. STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates. Stem Cells. 2014;32(5):1149–1160.
102. Harada D, Takigawa N, Kiura K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers (Basel). 2014;6(2):708–722.
103. Demaria M, Giorgi C, Lebiedzinska M, et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY). 2010;2(11):823–842.
104. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7(1):41–51.
105. Zhou J, Ong CN, Hur GM, Shen HM. Inhibition of the JAK-STAT3 pathway by andrographolide enhances chemosensitivity of cancer cells to doxorubicin. Biochem Pharmacol. 2010;79(9):1242–1250.
106. Zhao M, Gao FH, Wang JY, et al. JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non-small-cell lung cancer. Lung Cancer. 2011;73(3):366–374.
107. Lou W, Chen Y, Zhu KY, Deng H, Wu T, Polyphyllin WJ. I Overcomes EMT-Associated Resistance to Erlotinib in Lung Cancer Cells via IL-6/STAT3 Pathway Inhibition. Biol Pharm Bull. 2017;40(8):1306–1313.
108. Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat Rev Cancer. 2014;14(11):722–735.
109. Xu Q, Krause M, Samoylenko A, Vainio S. Wnt Signaling in Renal Cell Carcinoma. Cancers (Basel). 2016;8:6.
110. Bullions LC, Levine AJ. The role of beta-catenin in cell adhesion, signal transduction, and cancer. Curr Opin Oncol. 1998;10(1):81–87.
111. Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev. 1999;9(1):15–21.
112. Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281(32):22429–22433.
113. Bienz M. The subcellular destinations of APC proteins. Nat Rev Mol Cell Biol. 2002;3(5):328–338.
114. Stamos JL, Weis WI. The β-catenin destruction complex. Cold Spring Harb Perspect Biol. 2013;5(1):a007898.
115. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26.
116. Schambony A, Wedlich D. Wnt-5A/Ror2 regulate expression of XPAPC through an alternative noncanonical signaling pathway. Dev Cell. 2007;12(5):779–792.
117. Majid S, Saini S, Dahiya R. Wnt signaling pathways in urological cancers: past decades and still growing. Mol Cancer. 2012;11:7.
118. Khan M, Rasul A, Yi F, Zhong L, Ma T. Jaceosidin induces p53-dependent G2/M phase arrest in U87 glioblastoma cells. Asian Pac J Cancer Prev. 2011;12(12):3235–3238.
119. Lu MC, Yang SH, Hwang SL, et al. Induction of G2/M phase arrest by squamocin in chronic myeloid leukemia (K562) cells. Life Sci. 2006;78(20):2378–2383.
120. Yang G, Chang B, Yang F, et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin Cancer Res. 2010;16(12):3171–3181.
121. Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26(9):1306–1316.
122. Bourougaa K, Naski N, Boularan C, et al. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol Cell. 2010;38(1):78–88.
123. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310.
124. Khan M, Zheng B, Yi F, et al. Pseudolaric Acid B induces caspase-dependent and caspase-independent apoptosis in u87 glioblastoma cells. Evid Based Complement Alternat Med. 2012;2012:957568.
125. Xiang S, Zou P, Wu J, et al. Crosstalk of NF-kappaB/P65 and LncRNA HOTAIR-Mediated Repression of MUC1 Expression Contribute to Synergistic Inhibition of Castration-Resistant Prostate Cancer by Polyphyllin 1-Enzalutamide Combination Treatment. Cell Physiol Biochem. 2018;47(2):759–773.
126. Cai X, Guo L, Pei F, Chang X, Zhang R. Polyphyllin G exhibits antimicrobial activity and exerts anticancer effects on human oral cancer OECM-1 cells by triggering G2/M cell cycle arrest by inactivating cdc25C-cdc2. Arch Biochem Biophys. 2018;644:93–99.
127. Wang H, Fei Z, Polyphyllin JH. VII increases sensitivity to gefitinib by modulating the elevation of P21 in acquired gefitinib resistant non-small cell lung cancer. J Pharmacol Sci. 2017;134(3):190–196.
128. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28(6):1075–1083.
129. Steelman LS, Chappell WH, Abrams SL, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY). 2011;3(3):192–222.
130. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20(1):87–90.
131. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554.
132. Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3(10):1221–1224.
133. Kang S, Denley A, Vanhaesebroeck B, Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2006;103(5):1289–1294.
134. Vincent EE, Elder DJ, Thomas EC, et al. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br J Cancer. 2011;104(11):1755–1761.
135. Moselhy J, Srinivasan S, Ankem MK, Damodaran C. Natural Products That Target Cancer Stem Cells. Anticancer Res. 2015;35(11):5773–5788.
136. Zheng R, Jiang H, Li J, Liu X, Polyphyllin XH. II Restores Sensitization of the Resistance of PC-9/ZD Cells to Gefitinib by a Negative Regulation of the PI3K/Akt/mTOR Signaling Pathway. Curr Cancer Drug Targets. 2017;17(4):376–385.
137. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789.
138. Schroder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65(6):862–894.
139. Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7(4):335–345.
140. Kumar A, Singh UK, Kini SG, et al. JNK pathway signaling: a novel and smarter therapeutic targets for various biological diseases. Future Med Chem. 2015;7(15):2065–2086.
141. Kennedy NJ, Davis RJ. Role of JNK in tumor development. Cell Cycle. 2003;2(3):199–201.
142. Yan L, Liu X, Yin A, Wei Y, Yang Q, Kong B. Huaier aqueous extract inhibits cervical cancer cell proliferation via JNK/p38 pathway. Int J Oncol. 2015;47(3):1054–1060.
143. Li X, Lao Y, Zhang H, et al. The natural compound Guttiferone F sensitizes prostate cancer to starvation induced apoptosis via calcium and JNK elevation. BMC Cancer. 2015;15:254.
144. Zhai T, Hei Z, Ma Q, et al. Shikonin induces apoptosis and G0/G1 phase arrest of gallbladder cancer cells via the JNK signaling pathway. Oncol Rep. 2017;38(6):3473–3480.
145. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46(5):705–716.
146. Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8(1):49–62.
147. Arkan MC, Greten FR. IKK- and NF-kappaB-mediated functions in carcinogenesis. Curr Top Microbiol Immunol. 2011;349:159–169.
148. Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25(51):6817–6830.
149. Prasad S, Ravindran J, Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010;336(1–2):25–37.
150. Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J Biol Chem. 1995;270(52):31315–31320.
151. Park JH, Jeong YJ, Won HK, Choi SY, Park JH, Oh SM. Activation of TOPK by lipopolysaccharide promotes induction of inducible nitric oxide synthase through NF-kappaB activity in leukemia cells. Cell Signal. 2014;26(5):849–856.
152. Nabekura T, Hiroi T, Kawasaki T, Uwai Y. Effects of natural nuclear factor-kappa B inhibitors on anticancer drug efflux transporter human P-glycoprotein. Biomed Pharmacother. 2015;70:140–145.
153. Xiang S, Zou P, Tang Q, et al. HOTAIR-mediated reciprocal regulation of EZH2 and DNMT1 contribute to polyphyllin I-inhibited growth of castration-resistant prostate cancer cells in vitro and in vivo. Biochim Biophys Acta Gen Subj. 2018;1862(3):589–599.
154. Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):103–119.
155. Chang F, Steelman LS, Lee JT, et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17(7):1263–1293.
156. McCubrey JA, Steelman LS, Abrams SL, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22(4):708–722.
157. Martelli AM, Evangelisti C, Chiarini F, et al. The emerging role of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogenesis. Biochim Biophys Acta. 2010;1803(9):991–1002.
158. Martelli AM, Evangelisti C, Chiarini F, McCubrey JA. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1(2):89–103.
159. Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13(12):928–942.
160. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290.
161. Yip PY. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl Lung Cancer Res. 2015;4(2):165–176.
162. Baek SH, Ko JH, Lee JH, et al. Ginkgolic Acid Inhibits Invasion and Migration and TGF-β-Induced EMT of Lung Cancer Cells Through PI3K/Akt/mTOR Inactivation. J Cell Physiol. 2017;232(2):346–354.
163. Zhao RS, Wang W, Li JP, Liu CM, He L. Gelsolin Promotes Radioresistance in Non-Small Cell Lung Cancer Cells Through Activation of Phosphoinositide 3-Kinase/Akt Signaling. Technol Cancer Res Treat. 2017;16(4):512–518.
164. Cristea S, Sage J. Is the Canonical RAF/MEK/ERK Signaling Pathway a Therapeutic Target in SCLC? J Thorac Oncol. 2016;11(8):1233–1241.
165. Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24(1):69–79.
166. Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6(6):505–510.
167. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71.
168. Kim J, Kundu M, Viollet B, Guan KLAMPK. and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141.
169. Liu B, Bao JK, Yang JM, Cheng Y. Targeting autophagic pathways for cancer drug discovery. Chin J Cancer. 2013;32(3):113–120.
170. Kimura N, Tokunaga C, Dalal S, et al. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells. 2003;8(1):65–79.
171. Kim AD, Kang KA, Kim HS, et al. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis. 2013;4(8):e750.
172. Sui X, Kong N, Ye L, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344(2):174–179.
173. Kim A, Yim NH, Ma JY. Samsoeum, a traditional herbal medicine, elicits apoptotic and autophagic cell death by inhibiting Akt/mTOR and activating the JNK pathway in cancer cells. BMC Complement Altern Med. 2013;13:233.
174. Zhang L, Wang H, Xu J, Zhu J, Ding K. Inhibition of cathepsin S induces autophagy and apoptosis in human glioblastoma cell lines through ROS-mediated PI3K/AKT/mTOR/p70S6K and JNK signaling pathways. Toxicol Lett. 2014;228(3):248–259.
175. Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell Sep. 2005;122(6):927–939.
176. Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4(7):949–951.
177. Li DD, Wang LL, Deng R, et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene. 2009;28(6):886–898.
178. Park KJ, Lee SH, Lee CH, et al. Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem Biophys Res Commun. 2009;382(4):726–729.
179. Zhang YX, Kong CZ, Wang LH, et al. Ursolic acid overcomes Bcl-2-mediated resistance to apoptosis in prostate cancer cells involving activation of JNK-induced Bcl-2 phosphorylation and degradation. J Cell Biochem. 2010;109(4):764–773.
180. Shimizu S, Konishi A, Nishida Y, et al. Involvement of JNK in the regulation of autophagic cell death. Oncogene. 2010;29(14):2070–2082.
181. Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO j. 2000;19(21):5720–5728.
182. Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544.
183. Zhang C, Jia X, Wang K, et al. Polyphyllin VII Induces an Autophagic Cell Death by Activation of the JNK Pathway and Inhibition of PI3K/AKT/mTOR Pathway in HepG2 Cells. PLoS One. 2016;11(1):e0147405.
184. Sun H, Wang Z, Yakisich JS. Natural products targeting autophagy via the PI3K/Akt/mTOR pathway as anticancer agents. Anticancer Agents Med Chem. 2013;13(7):1048–1056.
185. Strozyk E, Kulms D. The role of AKT/mTOR pathway in stress response to UV-irradiation: implication in skin carcinogenesis by regulation of apoptosis, autophagy and senescence. Int J Mol Sci. 2013;14(8):15260–15285.
186. Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS j. 2010;277(1):2–21.
187. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. Dec. 2010;49(11):1603–1616.
188. Bruscia EM, Bonfield TL. Cystic Fibrosis Lung Immunity: the Role of the Macrophage. J Innate Immun. 2016;8(6):550–563.
189. Decano JL, Mattson PC, Aikawa M. Macrophages in Vascular Inflammation: origins and Functions. Curr Atheroscler Rep. 2016;18(6):34.
190. Gopinath VK, Musa M, Samsudin AR, Sosroseno W. Role of interleukin-1beta and tumour necrosis factor-alpha on hydroxyapatite-induced phagocytosis by murine macrophages (RAW264.7 cells). Br J Biomed Sci. 2006;63(4):176–178.
191. Moncada S. Nitric oxide: discovery and impact on clinical medicine. J R Soc Med. 1999;92(4):164–169.
192. Ahmad N, Chen LC, Gordon MA, Laskin JD, Laskin DL. Regulation of cyclooxygenase-2 by nitric oxide in activated hepatic macrophages during acute endotoxemia. J Leukoc Biol. 2002;71(6):1005–1011.
193. Bondeson J. The mechanisms of action of disease-modifying antirheumatic drugs: a review with emphasis on macrophage signal transduction and the induction of proinflammatory cytokines. Gen Pharmacol. 1997;29(2):127–150.
194. Matsuno R, Aramaki Y, Arima H, et al. Contribution of CR3 to nitric oxide production from macrophages stimulated with high-dose of LPS. Biochem Biophys Res Commun. 1998;244(1):115–119.
195. Buchanan MM, Hutchinson M, Watkins LR, Yin H. Toll-like receptor 4 in CNS pathologies. J Neurochem. 2010;114(1):13–27.
196. Huang GJ, Huang SS, Deng JS. Anti-inflammatory activities of inotilone from Phellinus linteus through the inhibition of MMP-9, NF-κB, and MAPK activation in vitro and in vivo. PLoS One. 2012;7(5):e35922.
197. Huang GJ, Yang CM, Chang YS, et al. Hispolon suppresses SK-Hep1 human hepatoma cell metastasis by inhibiting matrix metalloproteinase-2/9 and urokinase-plasminogen activator through the PI3K/Akt and ERK signaling pathways. J Agric Food Chem. 2010;58(17):9468–9475.
198. Lai WC, Zhou M, Shankavaram U, Peng G, Wahl LM. Differential regulation of lipopolysaccharide-induced monocyte matrix metalloproteinase (MMP)-1 and MMP-9 by p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J Immunol. 2003;170(12):6244–6249.
199. Zhang C, Li C, Jia X, et al. In Vitro and In Vivo Anti-Inflammatory Effects of Polyphyllin VII through Downregulating MAPK and NF-kappaB Pathways. 2019;24:5.
200. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257.
201. Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119.
202. Feoktistova M, Wallberg F, Tenev T, Geserick P, Leverkus M, Meier P. Techniques to Distinguish Apoptosis from Necroptosis. Cold Spring Harb Protoc. 2016;2016(4):
203. Galluzzi L, Kepp O, Kroemer G. MLKL regulates necrotic plasma membrane permeabilization. Cell Res. 2014;24(2):139–140.
204. Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16(6):329–344.
205. Wang H, Sun L, Su L, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–146.
206. Zhu J, Wang KZ, Chu CT. After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy. Nov. 2013;9(11):1663–1676.
207. Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011;7(3):297–300.
208. Lu H, Li G, Liu L, Feng L, Wang X, Jin H. Regulation and function of mitophagy in development and cancer. Autophagy. 2013;9(11):1720–1736.
209. Beasley SA, Hristova VA, Shaw GS. Structure of the Parkin in-between-ring domain provides insights for E3-ligase dysfunction in autosomal recessive Parkinson’s disease. Proc Natl Acad Sci U S A. 2007;104(9):3095–3100.
210. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803.
211. Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107(1):378–383.
212. Cai Q, Zakaria HM, Simone A, Sheng ZH. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012;22(6):545–552.
213. Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191(5):933–942.
214. Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008;105(32):11364–11369.
215. Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211–221.
216. Dagda RK, Cherra SJ
217. Lutz AK, Exner N, Fett ME, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem. 2009;284(34):22938–22951.
218. Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8(10):831–838.
219. Xiao T, Zhong W, Zhao J, et al. Polyphyllin I suppresses the formation of vasculogenic mimicry via Twist1/VE-cadherin pathway. Cell Death Dis. 2018;9(9):906.
220. Trede NS, Zapata A, Zon LI. Fishing for lymphoid genes. Trends Immunol. 2001;22(6):302–307.
221. Park KH, Cho KH. A zebrafish model for the rapid evaluation of pro-oxidative and inflammatory death by lipopolysaccharide, oxidized low-density lipoproteins, and glycated high-density lipoproteins. Fish Shellfish Immunol. 2011;31(6):904–910.
222. Sivamani S, Joseph B, Kar B. Anti-inflammatory activity of Withania somnifera leaf extract in stainless steel implant induced inflammation in adult zebrafish. J Genetic Eng Biotechnol. 2014;12(1):1–6.
223. Quezada M, Alvarez M, Peña OA, et al. Antiangiogenic, antimigratory and antiinflammatory effects of 2-methoxyestradiol in zebrafish larvae. Comp Biochem Physiol C Toxicol Pharmacol. 2013;157(2):141–149.
224. Chen YH, Wang WH, Wang YH, Lin ZY, Wen CC, Chern CY. Evaluation of the anti-inflammatory effect of chalcone and chalcone analogues in a zebrafish model. Molecules. 2013;18(2):2052–2060.
225. Lieschke GJ, Oates AC, Crowhurst MO, Ward AC, Layton JE. Morphologic and functional characterization of granulocytes and macrophages in embryonic and adult zebrafish. Blood. 2001;98(10):3087–3096.
226. Renshaw SA, Loynes CA, Trushell DM, Elworthy S, Ingham PW, Whyte MK. A transgenic zebrafish model of neutrophilic inflammation. Blood. 2006;108(13):3976–3978.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.