Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Natural Compound α-PGG and Its Synthetic Derivative 6Cl-TGQ Alter Insulin Secretion: Evidence for Diminishing Glucose Uptake as a Mechanism
Authors Chen X, Daniels NA, Cottrill D, Cao Y, Wang X, Li Y, Shriwas P, Qian Y, Archer MW, Whitticar NB, Jahan I, Nunemaker CS, Guo A ![]()
Received 6 October 2020
Accepted for publication 24 December 2020
Published 24 February 2021 Volume 2021:14 Pages 759—772
DOI https://doi.org/10.2147/DMSO.S284295
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ming-Hui Zou
Xiaozhuo Chen,1– 7 Nigel A Daniels,1,4,5,8 David Cottrill,2,3 Yanyang Cao,2,3 Xuan Wang,2,3 Yunsheng Li,2 Pratik Shriwas,2,3 Yanrong Qian,2 Michael W Archer,1,4 Nicholas B Whitticar,4,9 Ishrat Jahan,1,4 Craig S Nunemaker,1,3– 5 Aili Guo10
1The Diabetes Institute at Ohio University, Athens, OH, 45701, USA; 2The Edison Biotechnology Institute, Athens, OH, 45701, USA; 3Department of Biological Sciences, Athens, OH, 45701, USA; 4Department of Biomedical Sciences, Athens, OH, 45701, USA; 5Heritage College of Osteopathic Medicine, Athens, OH, 45701, USA; 6Interdisciplinary Graduate Program in Molecular and Cellular Biology, Athens, OH, 45701, USA; 7Department of Chemistry and Biochemistry, Athens, OH, 45701, USA; 8Department of Specialty Medicine, Athens, OH, 45701, USA; 9Translational Biomedical Sciences Program, Ohio University, Athens, OH, 45701, USA; 10Department of Internal Medicine, Division of Endocrinology, Diabetes and Metabolism, University of California at Davis (UC Davis) School of Medicine, UC Davis Health Science, Sacramento, CA, 95817, USA
Correspondence: Aili Guo
Department of Internal Medicine, Division of Endocrinology, Diabetes and Metabolism, University of California at Davis (UC Davis) School of Medicine, UC Davis Health Science, PSSB, G400, 4150 V St., Sacramento, CA, 95817, USA
Tel +1 916-734-3730
Fax +1 916-734-2292
Email [email protected]
Craig S Nunemaker
Department of Biomedical Sciences, 1 Ohio University, Athens, OH, 45701, USA
Tel +1 740-593-2387
Fax +1 740-593-4795
Email [email protected]
Purpose: Previously we showed that natural compound α-penta-galloyl-glucose (α-PGG) and its synthetic derivative 6-chloro-6-deoxy-1,2,3,4-tetra-O-galloyl-α-D-glucopyranose (6Cl-TGQ) act to improve insulin signaling in adipocytes by increasing glucose transport. In this study, we investigated the mechanism of actions of α-PGG and 6Cl-TGQ on insulin secretion.
Methods: Mouse islets and/or INS-1832/13 beta-cells were used to test the effects of our compounds on glucose-stimulated insulin secretion (GSIS), intracellular calcium [Ca2+]i using fura-2AM, glucose transport activity via a radioactive glucose uptake assay, intracellular ATP/ADP, and extracellular acidification (ECAR) and mitochondrial oxygen consumption rates (OCAR) using Seahorse metabolic analysis.
Results: Both compounds reduced GSIS in beta-cells without negatively affecting cell viability. The compounds primarily diminished glucose uptake into islets and beta-cells. Despite insulin-like effects in the peripheral tissues, these compounds do not act through the insulin receptor in islets. Further interrogation of the stimulus-secretion pathway showed that all the key metabolic factors involved in GSIS including ECAR, OCAR, ATP/ADP ratios, and [Ca2+]i of INS-1832/13 cells were diminished after the compound treatment.
Conclusion: The compounds suppress glucose uptake of the beta-cells, which consequently slows down the rates of glycolysis and ATP synthesis, leading to decrease in [Ca2+]i and GSIS. The difference between adipocytes and beta-cells in effects on glucose uptake is of great interest. Further structural and functional modifications could produce new compounds with optimized therapeutic potentials for different target cells. The higher potency of synthetic 6Cl-TGQ in enhancing insulin signaling in adipocytes but lower potency in reducing glucose uptake in beta-cells compared to α-PGG suggests the feasibility of such an approach.
Keywords: glucose-stimulated insulin secretion, calcium influx, glucose transporter, α-PGG, 6Cl-TGQ
Introduction
Insulin is one of the most important hormones for energy homeostasis. It is produced by pancreatic beta-cells and secreted into blood in response to increased blood glucose concentrations.1,2 Insulin then binds to insulin receptor (IR) located on the surface of fat and muscle cells, triggering the IR-mediated signaling pathway, glucose transporter 4 (Glut4)-mediated glucose uptake, and consequently reducing blood glucose levels.3,4 The mechanism of insulin secretion involves sensing extracellular glucose concentration via glucose transporters, followed by increased glucose metabolism, increased ATP production, membrane depolarization, calcium influx, and granule exocytosis.5,6 Adipocytes sense increases in circulating levels of insulin to recruit more Glut4 transporter to the cell surface to take up glucose, whereas in contrast, pancreatic islets use transporter 2 (Glut2)-mediated glucose transport (Glut1 and Glut 3 in humans) as the primary sensor of circulating levels of blood glucose.7,8 The relationship between glucose and insulin thus defines the biological actions of peripheral adipocytes and pancreatic beta-cells in very different ways, but both making important contributions to the systemic regulation of glucose.
Insulin and its analogs have been widely used for treating patients with diabetes mellitus, mainly as injectable drugs given their polypeptide nature. Efforts have been made to develop non-injectable forms of insulin.9–17 Various small molecule compounds with insulin-like actions have been studied.18–30 Coupling a glucose uptake assay with HPLC fractionation, we identified that a group of polyphenolic compounds called hydrolysable tannin (or tannic acid) from a plant Lagerstroemia speciosa possess glucose transport stimulatory activity in adipocytes.31,32 Further compound fractionation and functional assays of tannic acid identified α-penta-galloyl-glucose (α-PGG) being the most potent compound.33 α-PGG functions the same way as insulin in adipocytes, ie, it binds to the α-subunit of IR and activates the IR-mediated PI3K-Akt signaling pathway, inducing Glut4-mediated glucose uptake.33 In addition, α-PGG reduces blood levels of glucose, triglycerides, and insulin in healthy and diabetic mice.33 Through a structure activity relationship (SAR) study, the structural components in α-PGG responsible for its glucose-lowering activity were identified.34 Among several dozen of compounds synthesized and tested, 6-chloro-6-deoxy-1,2,3,4-tetra-O-galloyl-α-D-glucopyranose (6Cl-TGQ), an analog of α-PGG, was developed, displaying the strongest IR-inducing activity in adipocytes and a highest potency in inducing IR-mediated signaling and blood-glucose-lowering activity in animals.35
The focus of this study is to examine the potential effects of α-PGG and 6Cl-TGQ on glucose-stimulated insulin secretion (GSIS) in beta-cells provided glucose is the primary driver of insulin secretion. However, since pancreatic beta-cells/islets also express insulin receptors, we were interested in determining whether α-PGG and 6Cl-TGQ could alter IR-signaling and/or glucose transport of beta-cells/islets to affect insulin secretion function. Using mouse islets and a modified rat pancreatic INS-1 cell line (INS-1832/13 cells), we examined the effects of our compounds on beta-cell function at multiple points along the stimulus-secretion coupling pathway, ie, glucose uptake, glycolytic activity, mitochondrial oxidative phosphorylation (OXPHOS), ATP/ADP ratios, intracellular calcium ([Ca2+]i), and glucose-stimulated insulin secretion (GSIS). We found that both compounds reduced insulin secretion, mostly by affecting glucose transport and metabolism of beta-cells, independently from any effects on insulin receptor signaling. The current study sheds light on how these compounds work in the pancreas and contributes to future development of optimized small molecules with therapeutic benefits.
Materials and Methods
Compounds and Cells
The compound α-PGG and 6Cl-TGQ were synthesized, as described previously.33–35 Compound solutions were prepared in sterile ddH2O. Bovine insulin was purchased from Sigma–Aldrich. Rat pancreatic INS-1832/13 cells were a generous gift from Dr. Newgard’s lab and maintained as described.36 The rat INS-1 cell line has been widely used in beta-cell functional studies; however, the magnitude of the response is far less than that seen in freshly isolated animal islets. The INS-1832/13 cell line, derived from the rat INS-1 cell line, exhibits markedly enhanced and stable responsiveness to glucose and several of its known stimuli.36 Compounds were studied in vitro at 40 µM unless indicated otherwise, chosen based on previous studies on these compounds.31–33 This dose selection is also supported by the pilot studies in INS-1832/13 cells that indicated that the GSIS IC50 values were in the range of 20–30 μM for α-PGG and ~40–50 μM for 6Cl-TGQ, respectively. The insulin receptor antagonist S961 (Sigma-Aldrich, St. Louis, MO) was used at 10 or 100nM in a subset of studies since the lower concentration had no effect (data were not included).
Animals
Studies were conducted using outbred female and male CD-1 mice (Charles River Laboratories, MA) or C57Bl/6 mice at 9–16 months of age from a colony housed at Ohio University. Mice were housed with a 12h light/dark cycle and free access to food and water.
Pancreatic Islet Isolation
Pancreatic islets were isolated as previously described in detail.37 Briefly, to isolate pancreas and islets, mice were euthanized under anesthesia. The common bile duct (CBD) was cannulated and a Liberase TL (Roche) solution was injected into CBD. Pancreas was removed and placed in a 50 mL conical tube for further pancreas digestion and islet purification. Digestion was carried out at 37°C for 12 minutes, after which ice cold RPMI-1640 plus 10% fetal bovine serum (v/v; GE Healthcare Life Sciences) was added to stop digestion. Islets were then selected by pipetting and washed with ice cold RPMI-1640 plus 10% fetal bovine serum. For the experiments examining [Ca2+]i and the effect of co-treatment with S961 on GSIS, islets were isolated by another method described in detail previously.38
Culture of Rat INS-1832/13 Cells
Cells were seeded at a density of 0.5x106/well in 1mL RPMI-1640 plus 10% fetal bovine serum at 37°C for a 24-well plate. At 48 hours after the initial seeding, the cell culture media were changed and GSIS assay was performed.
Glucose-Stimulated Insulin Secretion Assay (GSIS)
Islets isolated from C57BL/6J mice were incubated for 48 hours in RPMI-1640 plus 10% fetal bovine serum at 37°C to allow recovery from isolation. Then, lots of 10 islets were incubated for 30 min in 0.5 mL Krebs-Ringer buffer (KRB) that contains 2.5 mM glucose before serial incubations in 2.5 mM glucose and 16 mM glucose in the absence or presence of compounds, respectively, for GSIS assay. Different compound concentrations (0, 2.5, 5, 10, 20, 40, 60, and 80 µM) were used to produce a GSIS dose-response curve for α-PGG or 6Cl-TGQ. The half-maximal inhibitory concentration (IC50) values were calculated using software GraphPad. Separate GSIS experiments were conducted in both cultured INS-1832/13 cells and isolated islets at compound concentrations 40 µM. For each condition, 10 islets in triplicate were used, and experiments were repeated at least three times. The cells were incubated for 30 min in 0.5 mL Krebs-Ringer buffer (KRB) that contains 0.5 mM glucose before serial incubations in 2.5 mM glucose and 16 mM glucose, respectively, for GSIS assay. After treatment, conditioned media collected from each experiment of isolated pancreatic islets or cultured INS-1832/13 cells were analyzed for insulin by insulin (mouse) ultrasensitive ELISA (ALPCO, Salem, NH, USA) and normalized to islet total protein quantified using Pierce BCA assay (Life Technologies Corporation, Grand Island, New York, USA).
In the experiment investigating the effectiveness of the compounds on insulin secretion after inhibiting the insulin receptor, islets from male CD-1 mice were isolated, followed by incubation in RPMI-1640 plus 10% fetal bovine serum at 37°C for 24 hours to allow for recovery. Then, islets were pooled and size-matched to account for inherent differences in insulin secretion.39 Sets of 20 islets were placed in 1mL of KRB with no glucose, and then moved to 2.5 mM glucose, and finally 16mM glucose for 1 hour each. Treatment groups were performed in triplicate and included KRB with no compounds as a control, 40 µM α-PGG, or 40 µM 6Cl-TGQ, and each compound concomitantly with 100 nM S961. Islets were removed from the solution and the supernatant was collected and spun at 10,000 rpm for 10 minutes. The top 500 µL was collected and stored at −80°C. The following day, the samples were analyzed using an ultrasensitive mouse insulin ELISA kit (ALPCO, Salem, NH, USA). Data were collected using the FLUOstar Optima microplate reader (BMG Labtech Inc., Cary, NC) and analyzed with Microsoft Excel (Microsoft, Redmond, WA).
Intracellular Calcium [Ca2+]i Measurements
Intracellular calcium [Ca2+]i was measured using the ratiometric [Ca2+]i indicator fura-2 AM using methods described previously.40 Islets were loaded with 1 µM fura-2AM for 30 min in a modified KRB solution containing the following: 16 mM glucose (high glucose), 128 mM NaCl, 3 mM CaCl2, 5 mM KCl, 2 mM MgCl2, and 10 mM HEPES, pH 7.3 or 2.5 mM glucose (low glucose), 135 mM NaCl, 3 mM CaCl2, 5 mM KCl, 2 mM MgCl2, and 10 mM HEPES, pH 7.3. Islets were washed of excess surface fura-2AM by perifusing islets with KRB solution containing 2.5 mM or 16 mM glucose (depending on starting glucose concentration for the experiment) for ~15-min at 500 µL/min using a peristaltic pump (Minipuls 2, Gilson, Middleton, WI). Note that the lag time in this perifusion system for drugs to reach the islets is ~45–60 sec. Islets were recorded with a Hamamatsu ORCA-FLASH4.0 camera (Hamamatsu Photonics, Japan) attached to an Olympus BX51WIF fluorescence microscope (Olympus, Center Valley, PA) using 340 and 380 nm excitation light and 510 nm emission. Data were recorded with CellSens software (Olympus) and analyzed with Microsoft Excel (Microsoft, Redmond, WA).
Glucose Uptake Assay
The glucose transport stimulatory activity of compounds was analyzed by measuring the uptake of 2-deoxy-D-(3H)-glucose (PerkinElmer Life Sciences, Waltham, MA, USA) in isolated mouse pancreatic islets or INS-1832/13 cells as described previously with minor modifications.33,34 Briefly, 30 pancreatic islets in a 1 mL microfuge tube or INS-1832/13 cells grown in 24-well plates were washed twice with serum-free DMEM and incubated with 0.5 mL of the same medium in 10% CO2 at 37°C for 2 hours. The cells were washed three times with Krebs-Ringer-HEPES (KRP) buffer (136 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 1.25 mM MgSO4, and 10 mM sodium phosphate buffer at pH 7.4) and then incubated with 0.45 mL KRP buffer at 37°C for 30 minutes. Compounds were individually added to the islets or cells at predetermined concentrations and each condition was in triplicate, and then the islets or INS-1832/13 cells were incubated at 37°C for 15 minutes. Glucose uptake assay was initiated by the addition of 0.1 mL of KPR buffer supplemented with 1 μCi/mL [3H] 2-deoxy-D-glucose and 1 mM cold glucose as the final concentration to the cells. After 10 min, the medium was aspirated, and the plates were washed with ice-cold PBS to terminate the glucose uptake process. The cells were lysed with 0.45 mL of 1% triton X-100 and the radioactivity taken up was measured with a scintillation counter (Beckman Instruments).
Metabolic Rates and Mitochondrial Stress Measurements by Seahorse Assay
Glycolytic or mitochondrial oxidative phosphorylation (OXPHOS) rates of INS-1832/13 cells, 30,000 cells per well, treated with or without 40 µM of α-PGG or 6Cl-TGQ, were measured continuously with an XFe 24 Extracellular Flux Analyzer (Seahorse Bioscience) in the assay medium containing DMEM, 143 mM NaCl, 10 mM Glucose, 1 mM Pyruvate and 2 mM Glutamax. These rates were reported as the extracellular acidification rate (ECAR) for glycolysis and oxygen consumption rate (OCR) for mitochondrial OXPHOS.41,42 Respiratory control ratio (RCR), defined as the ratio of FCCP-rotenone to oligomycin-rotenone treatments, was also calculated where the initial OCR (time zero) was normalized to a constant value for all wells.
ATP/ADP Ratio
The ATP/ADP ratios of INS-1832/13 cells were measured with a commercial assay kit (Sigma-Aldrich) by following the assay instructions. Briefly, cells were seeded at a density of 2x104/well in 100μL RPMI-1640 medium plus 10% fetal bovine serum and grown at 37°C in a 96-well clear-bottom plate. Then, cells were treated with or without 500 μM phloretin (a known Glut2 inhibitor), 30 μM α-PGG, or 50 μM 6Cl-TGQ for 30 min. After treatment, media was removed, and ATP reagent was added to lyse the cells and provide luciferase and D-luciferin. Then, intracellular ATP levels were measured (RLUA) using a Veritas Microplate Luminometer (Turner BioSystems, Sunnyvale, CA). After ATP measurement, the plate was incubated for 10 min and residual ATP was measured (RLUB). ADP reagent was then added to convert ADP to ATP. Then, ATP levels were measured again (RLUC). ATP/ADP ratios were calculated using the formula: ATP/ADP ratio = RLUA/(RLUC – RLUB).
MTT Cell Viability Assay
Cell viability and proliferation (MTT) assays of α-PGG- or 6Cl-TGQ-treated INS-1832/13 cells were performed as previously described.43,44
Statistics Analysis
All data were analyzed with one-way ANOVA unless otherwise stated in the Results. In all figures, values are means ± standard errors (SEM) of samples. Each experiment was conducted in either duplicate or triplicate trials unless indicated otherwise in the Figure Legend. P<0.05 was set as the level of statistical significance. *p<0.05; **p<0.01; and ***p<0.001.
Results
α-PGG and 6Cl-TGQ Reduce GSIS in Isolated Mouse Pancreatic Islets and INS-1832/13 Cells
To determine if the two compounds impact pancreatic insulin secretion, GSIS assays were performed using isolated mouse pancreatic islets and INS-1832/13 cells. Isolated islets were treated with various concentrations of either α-PGG (Figure 1A) or 6Cl-TGQ (Figure 1B) in low glucose (2.5 mM) or high glucose (16 mM) GSIS assays. These compounds reduced GSIS from mouse pancreatic islets in a concentration-dependent manner with estimated IC50 of 20.7±1.2 μM for α-PGG and 32.9±1.1 μM for 6Cl-TGQ. This inhibitory effect was also observed in INS-1832/13 cells using 40μM of each compound (Figure 1C).
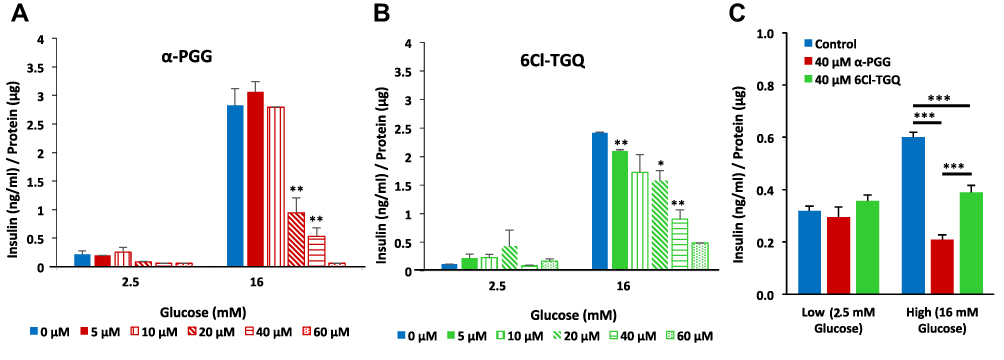 |
Figure 1 α-PGG and 6Cl-TGQ reduced glucose-stimulated insulin secretion (GSIS) in a concentration-dependent manner. Isolated mouse islets were treated with either α-PGG (A) or 6Cl-TGQ (B) at 0, 5, 10, 20, 40, and 60µM in low glucose (2.5mM) or high glucose (16mM) GSIS assays. Thirty minutes after the treatment, assay media were collected and measured for the secreted insulin, then normalized by cell protein contents. For each condition, 10 islets in triplicate were used. Data are expressed as means ± SEM. (C) Studies were repeated on INS-1832/13 cells using a dose of 40 µM for each compound. Data are expressed as means ± SEM. *p<0.05, **p<0.01, ***p<0.001. |
Effects on Insulin Secretion Do Not Appear to Require Insulin Receptor Activation in Beta-Cells
Since the compounds were previously shown to act through the insulin receptor in adipose tissue, we need to determine if insulin receptor (IR) binding activity was involved in the decrease in GSIS. We used insulin receptor antagonist S961 at 100nM to prevent insulin receptor signaling to re-examine the effects of our compounds. Performing a GSIS assay, α-PGG (Figure 2A) and 6Cl-TGQ (Figure 2B) both reduced insulin secretion in INS-1832/13 cells compared to untreated control in 2.5 mM and 16 mM glucose (p<0.001, n=3) as expected.45 When S961 was added to block insulin receptors along with each separate compound, no significant difference in insulin-lowering effect was observed for either compound (Figure 2A and B). S961 by itself also appeared to inhibit insulin secretion but to a lesser extent (P<0.05). Similar results were obtained using mouse islets (Figure 2C and D). In knowing the high IR binding affinity of S961, these results may suggest that the compounds are exhibiting their inhibitory actions on the beta-cell through an IR-independent mechanism. However, the observed weak inhibitory effect on GSIS of S961 itself leaves open the possibility that α-PGG and 6Cl-TGQ could be interacting with insulin receptors in some way.46
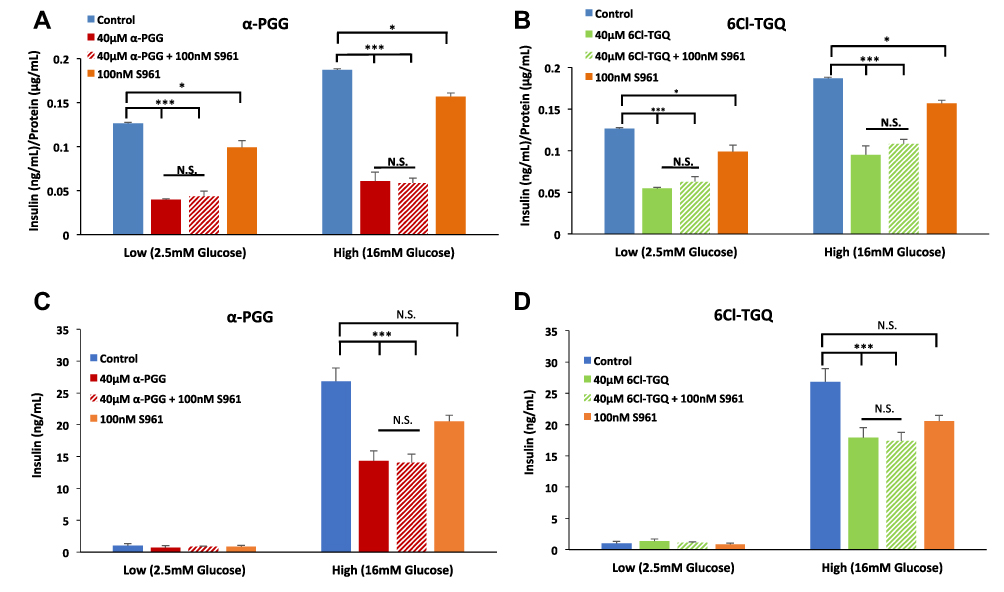 |
Figure 2 The insulin receptor antagonist S961 failed to block the inhibitory effects of the compounds on glucose-stimulated insulin secretion (GSIS). INS-1832/13 cells were exposed to low (2.5mM) and high (16mM) glucose along with either (A) 40µM α-PGG, 40µM α-PGG ± 100nM S961, or S961 alone or (B) 6CL-TGQ, 6CL-TGQ + 100nM S961, or S961 alone. (C and D) Islets isolated from CD-1 mice were exposed to the same conditions described for α-PGG in (A) and for 6Cl-TGQ in (B), respectively. For each condition, 20 islets in triplicate were used. Data are expressed as means ± SEM, N. S. = not significant *P<0.05, ***p<0.001. |
Compound Treatment Led to Reduction of Glucose-Stimulated Intracellular Calcium [Ca2+]i
To further examine the inhibitory effect of α-PGG and 6Cl-TGQ on islet function, we measured [Ca2+]i as a proximal marker of insulin secretion.47 Acute treatment with 40 µM α-PGG decreased [Ca2+]i levels in stimulatory (16 mM) glucose. As shown in Figure 3, the kinetics of this effect involves a rapid decline in [Ca2+]i levels, and a rapid washout during 5-min exposure to α-PGG (Figure 3A). The net effect of acute α-PGG exposure was a reduction in [Ca2+]i observed consistently for the 15 islets tested (Figure 3B). Similar results were obtained using 40 µM 6Cl-TGQ under the same experimental conditions (Figure 3C and D). These data indicate that α-PGG and 6Cl-TGQ act through the consensus pathway, also known as the K(ATP)-channel-dependent pathway, of insulin secretion.48
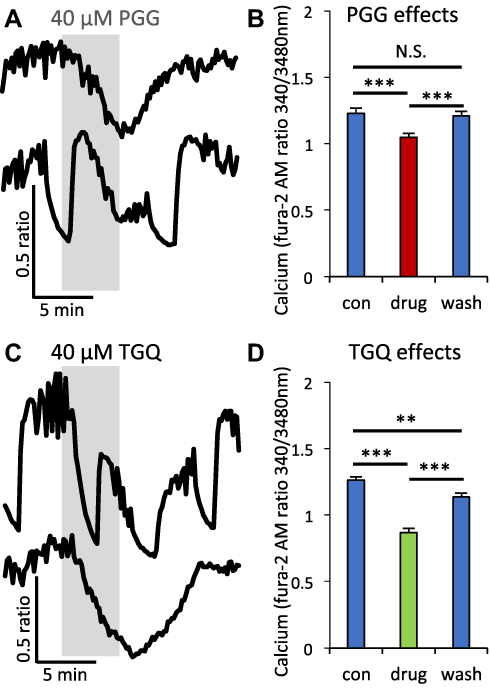 |
Figure 3 α-PGG and 6Cl-TGQ reduced glucose-stimulated intracellular calcium [Ca2+]i response. (A and C) [Ca2+]i traces showing effects of 5-min exposure to α-PGG (A) and 6CI-TGQ (C) in 16mM glucose. (B and D) Mean intracellular calcium ± SEM before (con), during (drug), and after (wash) exposure to α-PGG (B) and 6CI-TGQ (D). N=10–15 islets were used for testing each compound. N.S. = not significant, **P<0.01. ***P<0.001. |
We also examined whether these effects on calcium influx [Ca2+]i are linked to insulin signaling in β-cells since we showed previously that α-PGG and 6Cl-TGQ bind the insulin receptor in other cell types.26–28 First, we treated islets acutely with 100nM insulin and observed no significant change in [Ca2+]i (Figure 4A and B). Next, we blocked the insulin receptor acutely with S961 and again observed no significant change in [Ca2+]i (Figure 4C and D). These data suggest that neither insulin receptor activation by insulin or blockade by S961 has the type of effect on [Ca2+]i as our compounds. To provide additional evidence for separate pathways, we treated islets with α-PGG and 6Cl-TGQ in the presence of S961 to prevent any action through the insulin receptor. Despite this blockade, [Ca2+]i was significantly inhibited during acute treatment with α-PGG (Figure 4E and F) and 6Cl-TGQ (Figure 4G and H). These data strongly suggest that the primary effect of these compounds is not through insulin receptor activation although to affirm whether S961 blocks all receptor-mediated effects of α-PGG/6Cl-TGQ, it may be further investigated in adipocytes in the future. The studies that follow seek to identify the source of the actions of α-PGG and 6Cl-TGQ on islets.
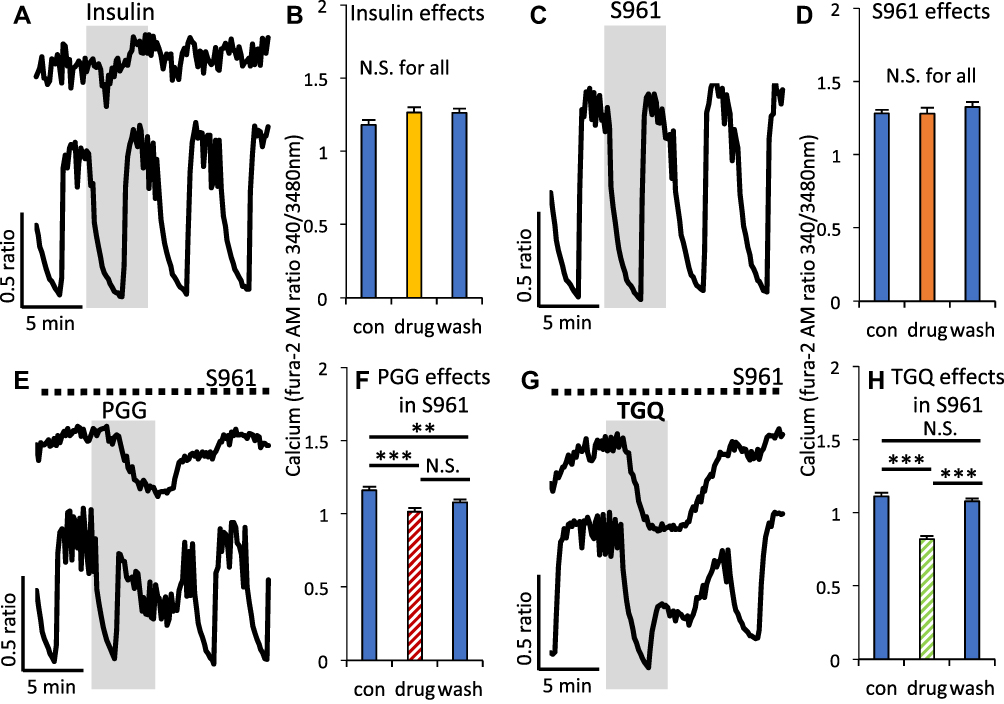 |
Figure 4 Modulating insulin receptor signaling had no effect on [Ca2+]i or on the inhibitory effects of α-PGG and 6Cl-TGQ. (A) [Ca2+]i traces showing effects of 5-min exposure to 100nM insulin in 16mM glucose. (B) Mean intracellular calcium ± SEM before (con), during (drug), and after (wash) exposure to insulin. Studies as described in (A and B) are shown for the insulin receptor antagonist S961 (C and D), α-PGG in the presence of S961 (E and F), and 6Cl-TGQ (G and H). N=10–15 islets were used for testing each compound. N.S. = not significant, **P<0.01. ***P<0.001. |
Reduction of Glycolysis, OXPHOS, and ATP/ADP Ratios
We first examined effects of our compounds on several aspects of glucose metabolism. The glycolytic rate is primarily determined by the glucose influx whilst the OXPHOS is partially dependent on the glycolytic rate. As expected, Seahorse metabolic rate analysis revealed that acute treatment with α-PGG led to significant reductions in the extracellular acidification rate (ECAR, Figure 5A) and in oxygen consumption rate (OCR, Figure 5C) in INS-1832/13 cells, while 6Cl-TGQ displayed similar but smaller reductions in both rates at the same tested concentration (Figure 5B and D). Further mitochondrial stress assay showed reduced mitochondrial ATP synthesis rates following α-PGG or 6Cl-TGQ treatment while α-PGG was more potent than 6Cl-TGQ (Figure 5E). The ATP/ADP ratios after α-PGG or 6Cl-TGQ treatment were significantly lower than vehicle-treated controls, but not as low as the known glucose transport inhibitor phloretin (Figure 5F). However, the respiratory control ratios (RCR), as an indicator of the capacities of mitochondrial oxidative phosphorylation, were not significantly different at 2.8, 2.9, and 2.9 for INS-1832/13 cells treated by either mock (as controls), α-PGG, or 6Cl-TGQ, respectively. Taken together, the overall reduction in metabolism in the INS-1832/13 cells would be most likely due to less available intracellular glucose pool resultant from inhibition of glucose uptake after the compound treatment. Importantly, the compounds did not affect the capacities of mitochondrial oxidative phosphorylation.
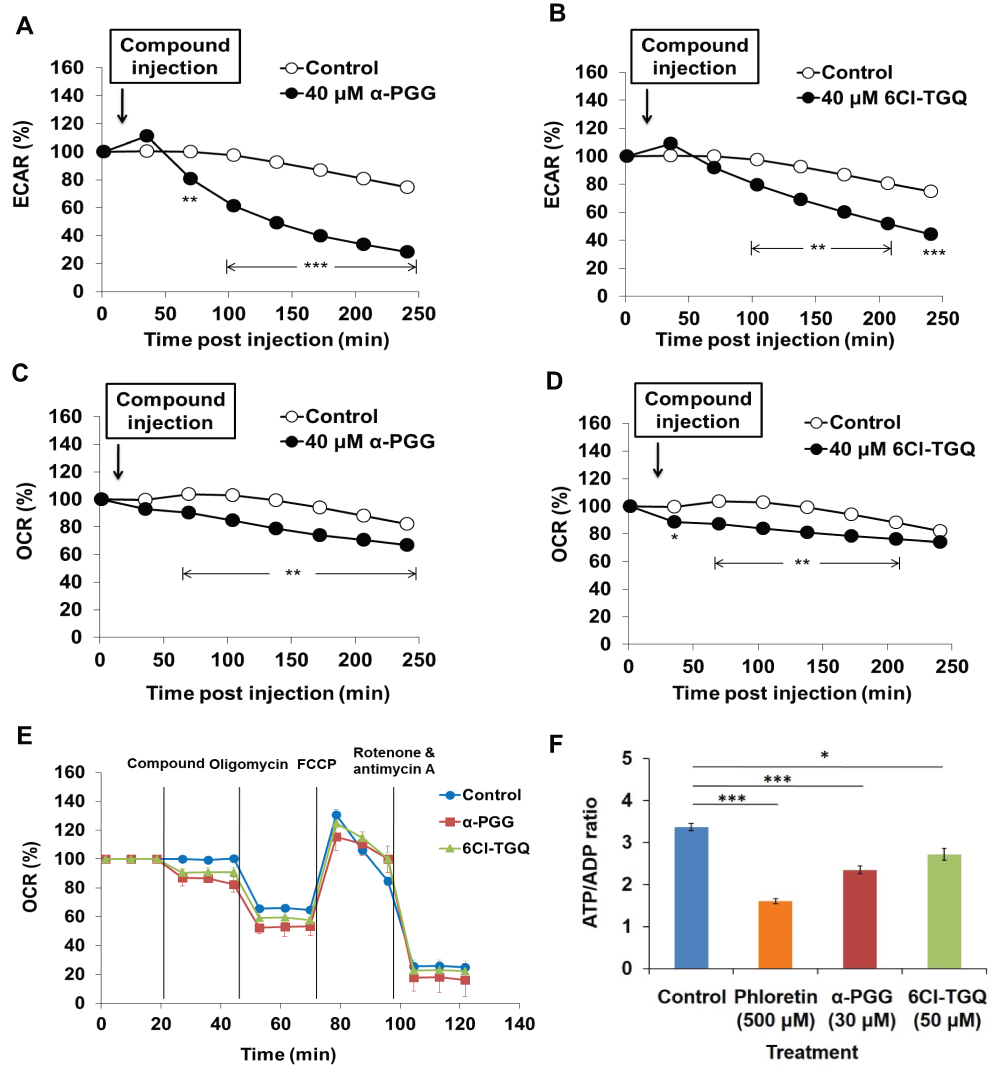 |
Figure 5 Effects of α-PGG and 6Cl-TGQ on rates of glycolysis, mitochondrial OXPHOS, and ATP/ADP ratio in rat INS-1832/13 cells. INS-1832/13 cells were treated with 40 µM of either α-PGG or 6Cl-TGQ and subjected to either the Seahorse metabolic analyses. (A and B) Effects of α-PGG and 6Cl-TGQ treatment on ECAR (glycolysis rate) in INS-1832/13 cells, respectively. (C and D) Effects of α-PGG and 6Cl-TGQ treatment on OCR (OXPHOS) in INS-1832/13 cells. (E) Mitochondrial stress test of α-PGG and 6Cl-TGQ. Vertical lines in the graph indicate times for addition of compound and mitochondrial inhibitors. (F) Effects of α-PGG and 6Cl-TGQ treatment on ATP/ADP ratio. Data are expressed as means ± SEM, N=3–6, *p<0.05, **p<0.01, ***p<0.001. |
Diminished Glucose Uptake in Isolated Pancreatic Islets and INS-1832/13 Cells
Since our compounds appear to reduce the rate of glucose metabolism, we next performed radioactive glucose uptake assays in isolated pancreatic islets (data not shown) and INS-1832/13 cells to determine if this effect is mediated by Glut2-dependent glucose transport. Indeed, both α-PGG (Figure 6A) and 6Cl-TGQ (Figure 6B) significantly diminished glucose uptake of INS-1832/13 cells in a dose-dependent manner. Further, 6Cl-TGQ was less potent in inhibiting glucose uptake than α-PGG. Because glucose uptake is the first step in the stimulus-secretion pathway, these results were consistent with the inhibitory activity of the compounds on glucose metabolism, [Ca2+]i, and insulin secretion that we observed. We also made direct comparisons between our compounds and insulin for effects on glucose uptake. As shown in Figure 6C, insulin had no inhibitory effects on glucose uptake, whereas 30μM α-PGG and 40μM 6Cl-TGQ reduced glucose uptake to 40% of control rates. These data indicate that our compounds have unique effects on beta-cells that are independent of any actions of insulin itself.
 |
Figure 6 α-PGG and 6Cl-TGQ diminished glucose transport in rat INS-1832/13 cells. Pancreatic INS-1832/13 cells treated with α-PGG or 6Cl-TGQ were subjected to a glucose uptake assay. After addition of compound and [3H] 2-deoxy-D-glucose, treated cell lines were lysed and were measured for their respective retained radioactivity. (A) α-PGG inhibited glucose uptake in INS-1832/13 cells. (B) 6Cl-TGQ inhibited glucose uptake in INS-1832/13 cells. (C) Insulin had no inhibitory effects on glucose uptake, whereas 30μM α-PGG and 40μM 6Cl-TGQ reduced glucose uptake to ~40% of control rates. Data are expressed as means ± SEM, N=3–6, *p<0.05. N.S. = not significant. |
α-PGG and 6Cl-TGQ Did Not Show Toxicity Towards Pancreatic Beta Cells
MTT assays on the compound-treated INS-1832/13 cells demonstrated that the treatment of 24 or 72 hours with either compound at 10–50 µM concentrations tested did not alter viability. The data of 24 hour-treatment were shown in (Figure 7). Phloretin, an inhibitor of glucose transport that induces apoptosis in cancer cells, reduced viability in INS-1832/13 cells following chronic exposure.49 Since it appears that these compounds do not show detectable toxicity or negative effect on cells’ growth or viability at the concentrations tested, it is very unlikely that the biological changes observed in other bioassays were nonspecific activity or toxicity of compounds to these cells.
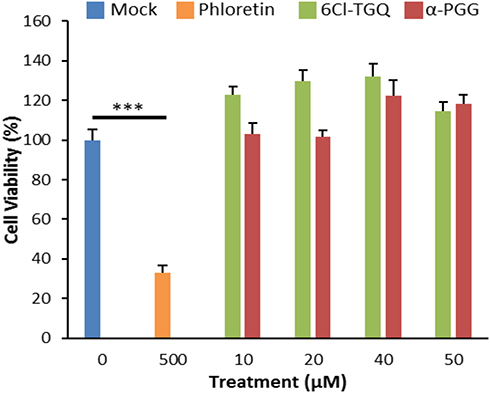 |
Figure 7 α-PGG and 6Cl-TGQ did not affect cell viability. Treatments of α-PGG or 6Cl-TGQ at various compound concentrations for 24h do not alter cell viability of INS-1832/13 cells. Phloretin used as a positive control for cell death. Data are expressed as means ± SEM, N=3–6, ***p<0.001. |
Discussion
We previously showed that herbal compound α-PGG is an orally deliverable small molecule that functions like insulin in adipocytes by binding to and activating the IR-mediated signaling pathways for glucose transport.33,50 Through a structure activity relation (SAR) study, we further identified an improved synthetic compound 6Cl-TGQ with more potent glucose-lowering effect.34,35 It is noteworthy that α-PGG and 6Cl-TGQ possess five and four galloyl groups, respectively. Some of the hydroxyl (OH) groups in each of the galloyl groups enable the compounds to form multiple hydrogen bonds with target proteins, leading to activation or inhibition of the protein activities. These OH groups also make the compounds relatively polar and hydrophilic, resulting in preferentially extracellular interactions with membrane-associated proteins such as the insulin receptor (IR) and glucose transporters (Gluts), rather than entering cells and interacting with intracellular proteins.33,35 These features may help to understand the results of present study, which demonstrated decrease in GSIS after compound treatment by diminishing glucose uptake and subsequent glucose metabolism in both isolated mouse pancreatic islets and cultured rat pancreatic INS-1832/13 cells.
α-PGG and 6Cl-TGQ Reduced GSIS in Beta-Cells
The feedback effects of insulin on insulin secretion are complex and controversial. The prevailing view is that insulin may stimulate insulin release from beta-cells, as reviewed in the literature.51 Other studies, however, have demonstrated inhibitory effects of insulin on the glucose-simulated insulin secretory pathway or both stimulatory and inhibitory effects depending on conditions.52–57
Based on these collective findings, we hypothesized that our compounds would bind with insulin receptors on beta-cells to affect insulin secretion, acting in either a stimulatory or an inhibitory fashion. We found that α-PGG and 6Cl-TGQ substantially reduce insulin secretion from INS-1832/13 cells and primary cultured mouse islets; however, the effects of our compounds on insulin secretion do not appear to mimic insulin itself. First, our compounds continue to inhibit insulin secretion even in the presence of S961, an insulin receptor antagonist with high IR binding affinity,46 although it should be noted that treatment with S961 on its own reduced insulin release. Second, evidently insulin had no impact on glucose uptake, the initiating step in glucose-stimulated insulin secretion of beta-cells, whereas our compounds inhibited glucose uptake by 50–60%, which is consistent with the magnitude of GSIS inhibition. Third, exposure to α-PGG or 6Cl-TGQ causes a rapid and reversible reduction in intracellular calcium [Ca2+]i in islets, but similar exposure to insulin has no such effect nor does blocking the insulin receptor pathway with S961. Together, these observations strongly suggest that our compounds do not work through the insulin receptor on beta-cells to inhibit insulin secretion.
Glucose Transport as a Possible Mechanism
It is well established that insulin production and secretion from pancreatic beta-cells is a multi-step, vigorously regulated process that is initiated from the glucose transport-mediated by Glut2 and followed by consequent increases in glycolytic rate and ATP production.7,58,59 ATP/ADP ratios are critical regulators of glucose-stimulated insulin secretion. In our study, inhibition of Glut2-mediated glucose transport by our compounds was speculated and confirmed by subsequent glucose uptake assays in both mouse pancreatic islets and rat INS-1832/13 cells. In line with these findings, Seahorse metabolic analysis indicated significantly decreased ECAR and OCR rates in compounds-treated INS-1832/13 cells as well as reduced mitochondrial ATP synthesis rates as determined by the mitochondrial stress test. Since Glut2 predominantly controls the glucose transport and glycolysis rates in rodent pancreatic beta-cells,7,8,60 the pronounced reduction in ECAR and less pronounced reductions in OCR and OXPHOS rates by α-PGG and 6Cl-TGQ are consistent with Glut2-mediated glucose transport as the target for their suppression of GSIS. Further, the ATP/ADP ratios in α-PGG or 6Cl-TGQ-treated INS-1832/13 cells were significantly lower than those in untreated cells, suggesting that the compound treatment reduced intracellular ATP levels. However, importantly, the compounds did not affect the capacities of mitochondrial oxidative phosphorylation as indicated by an unchanged RCR. This is consistent with the hypothetical role of the compounds as Glut2 inhibitors. Thus, we have identified two protein targets of α-PGG and 6Cl-TGQ, ie, insulin receptors in adipocytes as previously described and Glut2 in pancreatic beta-cells.33,35
A Hypothetical Model for the PGG/TGQ GSIS Inhibitory Activity in Pancreatic Beta-Cells
Based on our previous studies,33,35 and particularly the present one, a hypothetical model is proposed as shown in Figure 8 to explain how PGG/TGQ may inhibit glucose stimulated insulin secretion (GSIS). According to this model, PGG/TGQ bind to Glut2 located on the plasma membrane of pancreatic beta-cells, the primary glucose transporter expressed in rodent beta-cells. Compound-Glut interaction results in inhibition of Glut2-mediated transport of glucose into beta-cells, reducing ATP synthesis and Ca2+ influx, and finally leading to reduction in GSIS. This mechanism is very different from the one we uncovered before in adipocytes where PGG/TGQ bind to the insulin receptor (IR) and activate IR-Glut4 mediated glucose uptake in adipocytes.33,35 As mentioned before, the rationale is based on that the IR in beta-cells would not induce Glut4-mediated glucose uptake as in adipocytes since glucose transport in rodent beta-cells is thought to be primarily mediated by Glut2.7,8,60 This model remains to be validated, particularly the Glut2 binding part.
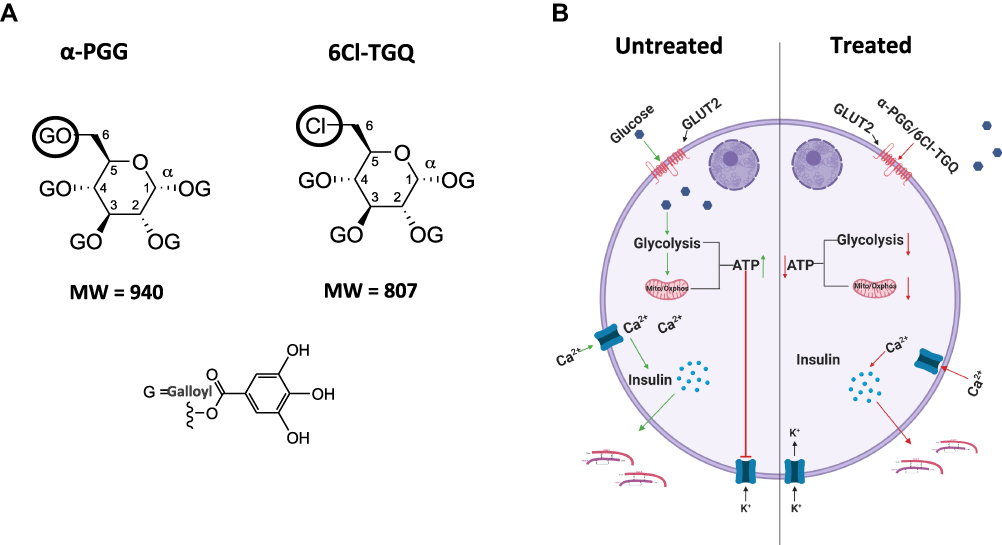 |
Figure 8 A hypothetical model explains the effects of PGG/TGQ on GSIS. (A) Structures of PGG and TGQ are included to show their relatively large molecular weights and their polarity with hydrophilic features and tendency of extracellularly functioning. (B) Model. Left, normal (untreated) GSIS in beta-cells. Right, beta-cells treated with α-PGG/6Cl-TGQ, resulting in reduced GSIS. See Discussion section for the detailed explanation of the mechanism underlying the action of the compounds. |
Two Targets Related to Glucose Metabolism: Peripheral Insulin Signaling and Insulin Secretion
We previously reported that our compounds had blood glucose-lowering effects in adipocytes and in a mouse model of both type 1 and type 2 diabetes.33,35 However, we noted that at higher concentrations, the blood glucose-lowering effects appeared to wane. Our data suggest a possible reason for this effect: a net effect of two competing activities of our compounds. At lower concentrations, the blood-glucose-lowering activity of the compounds at peripheral tissues (adipose and muscle) exceeded their pancreatic insulin secretion-inhibitory activity, resulting in an overall glucose-lowering activity. However, at higher concentrations, the insulin secretion-inhibitory activity in pancreas surpasses the blood-glucose-lowering activity of the compounds at peripheral tissues. When the compound concentration is further increased, it might have caused more potent reduction in pancreatic insulin secretion, leading to the disappearance of blood glucose-lowering effect of the compounds (unpublished observation).
By elucidating the effects of these compounds on beta-cells, the present study has made an important step forward in understanding how these compounds may be therapeutically beneficial. Moreover, synthetic 6Cl-TGQ, in comparison to α-PGG, has demonstrated more potent insulin-like activity in adipocytes and mice but less inhibitory effects on insulin secretion.35 This finding is supported by the specifics of the EC50 and IC50 values of α-PGG and 6Cl-TGQ, where the EC50 value of α-PGG in adipocytes for inducing IR-mediated glucose transport was higher at ~15 μM comparing to that of 6Cl-TGQ at ~10 μM.33,35 Conversely, the GSIS IC50 value of α-PGG was lower at ~20–30 μM comparing to 6Cl-TGQ at ~40 μM in INS-1832/13 cells. Taken together, these results suggest that 6Cl-TGQ is more potent than α-PGG in adipocytes but less potent in pancreatic beta cells. The observation of insulin-like activity and the pancreatic-inhibitory activity moving in opposite directions in 6Cl-TGQ strongly suggests that these two activities are structurally separable, at least partially if not completely. By performing additional structure activity relationship (SAR) studies to further separate the pancreatic inhibitory activity from the glucose-lowering activity of the compounds, it would allow us to generate new compounds with optimized therapeutic potentials; for instance, future compounds with enhanced glucose-lowering activity in peripheral tissues and/or lessened inhibition in insulin secretion would improve both glucose handling and diet-associated insulin over-secretion, a phenomenon not uncommon seen in patients with pre-diabetes and early-stage diabetes mellitus.
Conclusion
Our key finding is that the two compounds have inhibitory effects on glucose-stimulated insulin secretion, affecting glucose transport as a mechanism. It is of great interest to recognize the different effects of these orally deliverable natural compounds on glucose transport between peripheral tissues and pancreatic beta-cells. Through further SAR optimization, to generate new compounds with therapeutic potentials in a more targeted manner would be worth to explore.
Abbreviations
IR, insulin receptor; INS-1832/13 cells, a modified rat pancreatic INS-1 cell line; ECAR, extracellular acidification rate; OCR, oxygen consumption rate; OXPHOS, mitochondrial oxidative phosphorylation; RCR, respiratory control ratio; SAR, structure activity relationship.
Ethics Approval
All animal studies were conducted in accordance with the Ohio University guidelines for the use and care of laboratory animals with approved protocol. The use of pancreatic INS-1832/13 cells was approved by the institutional research ethics committee of the Ohio University.
Acknowledgments
We thank Misako Hata and Kathryn Corbin for technical assistance. We thank Emily Kunkler for assistance with collection of [Ca2+]i data.
Funding
Research awards from Ohio University supported the project for Chen and Guo. NIH R15 DK121247 supported the project for Nunemaker.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Fu Z, Gilbert ER, Liu D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr Diabetes Rev. 2013;9(1):25–53. doi:10.2174/157339913804143225
2. Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol. 2013;75(1):155–179. doi:10.1146/annurev-physiol-030212-183754
3. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi:10.1038/414799a
4. Saltiel AR. Insulin signaling in the control of glucose and lipid homeostasis. Handb Exp Pharmacol. 2016;233:51–71.
5. MacDonald PE, Joseph JW, Rorsman P. Glucose-sensing mechanisms in pancreatic beta-cells. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2211–2225. doi:10.1098/rstb.2005.1762
6. Jensen MV, Joseph JW, Ronnebaum SM, Burgess SC, Sherry AD, Newgard CB. Metabolic cycling in control of glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab. 2008;295(6):E1287–1297. doi:10.1152/ajpendo.90604.2008
7. Berger C, Zdzieblo D. Glucose transporters in pancreatic islets. Pflugers Arch. 2020;472(9):1249–1272. doi:10.1007/s00424-020-02383-4
8. Guillam MT, Hümmler E, Schaerer E, et al. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet. 1997;17(3):327–3230. doi:10.1038/ng1197-327
9. Leary AC, Dowling M, Cussen K, O’Brien J, Stote RM. Pharmacokinetics and pharmacodynamics of intranasal insulin spray (Nasulin) administered to healthy male volunteers: influence of the nasal cycle. J Diabetes Sci Technol. 2008;2(6):1054–1060. doi:10.1177/193229680800200613
10. Maltesen MJ, Bjerregaard S, Hovgaard L, Havelund S, van de Weert M. Quality by design - spray drying of insulin intended for inhalation. Eur J Pharm Biopharm. 2008;70(3):828–838. doi:10.1016/j.ejpb.2008.07.015
11. Arbit E, Kidron M. Oral insulin: the rationale for this approach and current developments. J Diabetes Sci Technol. 2009;3(3):562–567. doi:10.1177/193229680900300322
12. Stote R, Marbury T, Shi L, Miller M, Strange P. Comparison pharmacokinetics of two concentrations (0.7% and 1.0%) of Nasulin, an ultra-rapid-acting intranasal insulin formulation. J Diabetes Sci Technol. 2010;4(3):603–609. doi:10.1177/193229681000400314
13. He H, Ye J, Sheng J, Huang Y, Guanyi Chen WJ. Overcoming oral insulin delivery barriers: application of cell penetrating peptide and silica-based nanoporous composites. Front Chem Sci Eng. 2013;7(1):9–19. doi:10.1007/s11705-013-1306-9
14. Andreani T, de Souza ALR, Kiill CP, et al. Preparation and characterization of PEG-coated silica nanoparticles for oral insulin delivery. Int J Pharm. 2014;473(1–2):627–635. doi:10.1016/j.ijpharm.2014.07.049
15. Ramesan RM, Sharma CP. Recent advances in the oral delivery of insulin. Recent Pat Drug Deliv Formul. 2014;8(2):155–159. doi:10.2174/1872211308666140527143407
16. Razavi Rohani SS, Abnous K, Tafaghodi M. Preparation and characterization of spray-dried powders intended for pulmonary delivery of insulin with regard to the selection of excipients. Int J Pharm. 2014;465(1–2):464–478. doi:10.1016/j.ijpharm.2014.02.030
17. Zijlstra E, Heinemann L, Plum-Morschel L. Oral insulin reloaded: a structured approach. J Diabetes Sci Technol. 2014;8(3):458–465. doi:10.1177/1932296814529988
18. Zhang B, Salituro G, Szalkowski D, et al. Discovery of a small molecule insulin mimetic with antidiabetic activity in mice. Science. 1999;284(5416):974–977. doi:10.1126/science.284.5416.974
19. Air EL, Strowski MZ, Benoit SC, et al. Small molecule insulin mimetics reduce food intake and body weight and prevent development of obesity. Nat Med. 2002;8(2):179–183. doi:10.1038/nm0202-179
20. Strowski MZ, Li Z, Szalkowski D, et al. Small-molecule insulin mimetic reduces hyperglycemia and obesity in a nongenetic mouse model of type 2 diabetes. Endocrinology. 2004;145(11):5259–5268. doi:10.1210/en.2004-0610
21. Srivastava AK, Mehdi MZ. Insulino-mimetic and anti-diabetic effects of vanadium compounds. Diabet Med J Br Diabet Assoc. 2005;22(1):2–13. doi:10.1111/j.1464-5491.2004.01381.x
22. Garcia-Vicente S, Yraola F, Marti L, et al. Oral insulin-mimetic compounds that act independently of insulin. Diabetes. 2007;56(2):486–493. doi:10.2337/db06-0269
23. Lin B, Li Z, Park K, et al. Identification of novel orally available small molecule insulin mimetics. J Pharmacol Exp Ther. 2007;323(2):579–585. doi:10.1124/jpet.107.126102
24. He K, Chan C-B, Liu X, et al. Identification of a molecular activator for insulin receptor with potent anti-diabetic effects. J Biol Chem. 2011;286(43):37379–37388. doi:10.1074/jbc.M111.247387
25. Jung D-W, Ha -H-H, Zheng X, Chang Y-T, Williams DR. Novel use of fluorescent glucose analogues to identify a new class of triazine-based insulin mimetics possessing useful secondary effects. Mol Biosyst. 2011;7(2):346–358. doi:10.1039/C0MB00089B
26. Lee J, Jung D-W, Kim W-H, et al. Development of a highly visual, simple, and rapid test for the discovery of novel insulin mimetics in living vertebrates. ACS Chem Biol. 2013;8(8):1803–1814. doi:10.1021/cb4000162
27. Nankar RP, Doble M. Non-peptidyl insulin mimetics as a potential antidiabetic agent. Drug Discov Today. 2013;18(15–16):748–755. doi:10.1016/j.drudis.2013.04.005
28. Qiang G, Xue S, Yang JJ, et al. Identification of a small molecular insulin receptor agonist with potent antidiabetes activity. Diabetes. 2014;63(4):1394–1409. doi:10.2337/db13-0334
29. Alkhalidy H, Moore W, Zhang Y, et al. Small molecule kaempferol promotes insulin sensitivity and preserved pancreatic beta-cell mass in middle-aged obese diabetic mice. J Diabetes Res. 2015;2015:532984. doi:10.1155/2015/532984
30. Mukherjee S, Chattopadhyay M, Bhattacharya S, et al. Molecule also improves insulin sensitivity in diabetic mice. PLoS One. 2017;12(1):e0169809. doi:10.1371/journal.pone.0169809
31. Liu F, Kim J, Li Y, Chen X, Li J, Chen X. An extract of lagerstroemia speciosa L. has insulin-like glucose uptake-stimulatory and adipocyte differentiation-inhibitory activities in 3T3-L1 cells. J Nutr. 2001;131(9):2242–2247. doi:10.1093/jn/131.9.2242
32. Liu X, Kim J-K, Li Y, Li J, Liu F, Chen X. Tannic acid stimulates glucose transport and inhibits adipocyte differentiation in 3T3-L1 cells. J Nutr. 2005;135(2):165–171. doi:10.1093/jn/135.2.165
33. Li Y, Kim J, Li J, et al. Natural anti-diabetic compound 1,2,3,4,6-penta-O-galloyl-D-glucopyranose binds to insulin receptor and activates insulin-mediated glucose transport signaling pathway. Biochem Biophys Res Commun. 2005;336(2):430–437. doi:10.1016/j.bbrc.2005.08.103
34. Ren Y, Himmeldirk K, Chen X. Synthesis and structure−activity relationship study of antidiabetic penta-o-galloyl-d-glucopyranose and its analogues. J Med Chem. 2006;49(9):2829–2837. doi:10.1021/jm060087k
35. Cao Y, Li Y, Kim J, et al. Orally efficacious novel small molecule 6-chloro-6-deoxy-1,2,3,4-tetra-O-galloyl-α-D-glucopyranose selectively and potently stimulates insulin receptor and alleviates diabetes. J Mol Endocrinol. 2013;51(1):15–26. doi:10.1530/JME-12-0171
36. Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49(3):424–430. doi:10.2337/diabetes.49.3.424
37. Slepchenko KG, Daniels NA, Guo A, Li YV. Autocrine effect of Zn (2)(+) on the glucose-stimulated insulin secretion. Endocrine. 2015;50(1):110–122. doi:10.1007/s12020-015-0568-z
38. Carter JD, Dula SB, Corbin KL, Wu R, Nunemaker CS. A practical guide to rodent islet isolation and assessment. Biol Proced Online. 2009;11(1):3–31. doi:10.1007/s12575-009-9021-0
39. Slepchenko KG, Corbin KL, Nunemaker CS. Comparing methods to normalize insulin secretion shows the process may not be needed. J Endocrinol. 2019;241(2):149–159. doi:10.1530/JOE-18-0542
40. Whitticar NB, Strahler EW, Rajan P, Kaya S, Nunemaker CS. An automated perifusion system for modifying cell culture conditions over time. Biol Proced Online. 2016;18(1):19. doi:10.1186/s12575-016-0049-7
41. Wu M, Neilson A, Swift AL, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292(1):C125–136. doi:10.1152/ajpcell.00247.2006
42. Gohil VM, Sheth SA, Nilsson R, et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28(3):249–255. doi:10.1038/nbt.1606
43. Liu Y, Cao Y, Zhang W, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2012;11(8):1672–1682. doi:10.1158/1535-7163.MCT-12-0131
44. Qian Y, Wang X, Liu Y, et al. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett. 2014;351(2):242–251. doi:10.1016/j.canlet.2014.06.008
45. Cieniewicz AM, Kirchner T, Hinke SA, et al. Novel monoclonal antibody is an allosteric insulin receptor antagonist that induces insulin resistance. Diabetes. 2017;66(1):206–217. doi:10.2337/db16-0633
46. Schaffer L, Brand CL, Hansen BF, et al. A novel high-affinity peptide antagonist to the insulin receptor. Biochem Biophys Res Commun. 2008;376(2):380–383. doi:10.1016/j.bbrc.2008.08.151
47. Ramadan JW, Steiner SR, O’Neill CM, Nunemaker CS. The central role of calcium in the effects of cytokines on beta-cell function: implications for type 1 and type 2 diabetes. Cell Calcium. 2011;50(6):481–490. doi:10.1016/j.ceca.2011.08.005
48. Straub SG, James RF, Dunne MJ, Sharp GW. Glucose activates both K(ATP) channel-dependent and K(ATP) channel-independent signaling pathways in human islets. Diabetes. 1998;47(5):758–763. doi:10.2337/diabetes.47.5.758
49. Choi BY. Biochemical basis of anti-cancer-effects of phloretin-a natural dihydrochalcone. Mol Basel. 2019;24:278.
50. Cao Y, Himmeldirk KB, Qian Y, Ren Y, Malki A, Chen X. Biological and biomedical functions of penta-o-galloyl-d-glucose and its derivatives. J Nat Med. 2014;68(3):465–472. doi:10.1007/s11418-014-0823-2
51. Leibiger IB, Leibiger B, Berggren P-O. Insulin feedback action on pancreatic beta-cell function. FEBS Lett. 2002;532(1–2):1–6. doi:10.1016/S0014-5793(02)03627-X
52. Khan FA, Goforth PB, Zhang M, Satin LS. Insulin activates ATP-sensitive K(+) channels in pancreatic beta-cells through a phosphatidylinositol 3-kinase-dependent pathway. Diabetes. 2001;50(10):2192–2198. doi:10.2337/diabetes.50.10.2192
53. Persaud SJ, Asare-Anane H, Jones PM. Insulin receptor activation inhibits insulin secretion from human islets of Langerhans. FEBS Lett. 2002;510(3):225–228. doi:10.1016/S0014-5793(01)03268-9
54. Nunemaker CS, Zhang M, Satin LS. Insulin feedback alters mitochondrial activity through an ATP-sensitive K+ channel-dependent pathway in mouse islets and beta-cells. Diabetes. 2004;53(7):1765–1772. doi:10.2337/diabetes.53.7.1765
55. Hee-Park S, Lim B, Baek W-K, Bae J-H, Song D-K. Negative and positive feedback regulation of insulin in glucose-stimulated Ca2+ response in pancreatic beta cells. Diabetes Res Clin Pract. 2007;77(3):S143–149. doi:10.1016/j.diabres.2007.02.023
56. Ohsugi M, Cras-Meneur C, Zhou Y, et al. Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. J Biol Chem. 2005;280(6):4992–5003. doi:10.1074/jbc.M411727200
57. Jimenez-Feltstrom J, Lundquist I, Obermuller S, Salehi A. Insulin feedback actions: complex effects involving isoforms of islet nitric oxide synthase. Regul Pept. 2004;122(2):109–118. doi:10.1016/j.regpep.2004.06.004
58. Aspinwall CA, Lakey JR, Kennedy RT. Insulin-stimulated insulin secretion in single pancreatic beta cells. J Biol Chem. 1999;274(10):6360–6365. doi:10.1074/jbc.274.10.6360
59. Andrali SS, Sampley ML, Vanderford NL, Ozcan S. Glucose regulation of insulin gene expression in pancreatic beta-cells. Biochem J. 2008;415(1):1–10. doi:10.1042/BJ20081029
60. Pang K, Mukonoweshuro C, Wong GG. Beta cells arise from glucose transporter type 2 (Glut2)-expressing epithelial cells of the developing rat pancreas. Proc Natl Acad Sci U S A. 1994;91(20):9559–9563. doi:10.1073/pnas.91.20.9559
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.