")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 9
Natural and synthetic progestins enrich cancer stem cell-like cells in hormone-responsive human breast cancer cell populations in vitro
Authors Goyette S, Liang Y, Mafuvadze B , Cook MT, Munir M, Hyder SM
Received 22 February 2017
Accepted for publication 20 April 2017
Published 19 May 2017 Volume 2017:9 Pages 347—357
DOI https://doi.org/10.2147/BCTT.S135371
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Sandy Goyette,1,2 Yayun Liang,1,2 Benford Mafuvadze,1,2 Matthew T Cook,1,2 Moiz Munir,1,2 Salman M Hyder1,2
1Department of Biomedical Sciences, 2Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO, USA
Abstract: Clinical trials and studies have shown that combination estrogen/progestin hormone replacement therapy, but not estrogen therapy alone or placebo, increases breast cancer risk in postmenopausal women. Using animal models, we have previously shown that both natural and synthetic progestins (including medroxyprogesterone acetate [MPA], a synthetic progestin used widely in the clinical setting) accelerate the development of breast tumors in vivo and increase their metastasis to lymph nodes. Based on these observations, we have hypothesized that progestin-induced breast cancer tumor growth and metastasis may be mediated by an enrichment of the cancer stem cell (CSC) pool. In this study, we used T47-D and BT-474 hormone-responsive human breast cancer cells to examine the effects of progestin on phenotypic and functional markers of CSCs in vitro. Both natural and synthetic progestins (10 nM) significantly increased protein expression of CD44, an important CSC marker in tumor cells. MPA increased the levels of both CD44 variants v3 and v6 associated with stem cell functions. This induction of CD44 was blocked by the antiprogestin RU-486, suggesting that this process is progesterone receptor (PR) dependent. CD44 induction was chiefly progestin dependent. Because RU-486 can bind other steroid receptors, we treated PR-negative T47-DCO-Y cells with MPA and found that MPA failed to induce CD44 protein expression, confirming that PR is essential for progestin-mediated CD44 induction in T47-D cells. Further, MPA treatment of T47-D cells significantly increased the activity of aldehyde dehydrogenase (ALDH), another CSC marker. Finally, two synthetic progestins, MPA and norethindrone, significantly increased the ability of T47-D cells to form mammospheres, suggesting that enrichment of the CD44high, ALDHbright subpopulation of cancer cells induced by MPA exposure is of functional significance. Based on our observations, we contend that exposure of breast cancer cells to synthetic progestins leads to an enrichment of the CSC pool, supporting the development of progestin-accelerated tumors in vivo.
Keywords: breast cancer, progestins, medroxyprogesterone acetate, cancer stem cells, CD44, ALDH
Introduction
The past few decades have seen the successful development of highly effective breast cancer treatment and prevention options. However, an estimated 230,000 new cases of breast cancer are diagnosed every year in the United States, and approximately 40,000 US deaths are attributed to the disease annually.1 A subset of diagnosed breast cancer cases in postmenopausal women has been linked to the use of hormone replacement therapy (HRT) containing a combination of estrogen and progestin (P).2–5 In these combination HRT regimens, a synthetic P, such as medroxyprogesterone acetate (MPA), is included to prevent estrogen-induced endometrial cancer, which can arise as a consequence of unopposed estrogen action.6 Unfortunately, clinical trials in postmenopausal women show that, when compared with HRT therapies containing only estrogen, combination HRT is associated with an increased risk of invasive breast cancer.2–5 Furthermore, combination HRT has been linked to an increased risk of breast cancer recurrence and metastasis.7
Our laboratory is dedicated to identifying mechanisms responsible for increased breast cancer risk. Studies by both our group and others have shown that P induces potent angiogenic growth factors, such as vascular endothelial growth factor (VEGF).8–12 Increased VEGF provides developing tumors with a favorable tumor microenvironment, causing an increase in neovascularization and cell proliferation within the primary tumor. We have also shown that exposure of experimental animals to synthetic P reduces breast tumor latency and increases tumor growth.13,14 Most deaths from breast cancer occur following metastasis of the primary tumor to other tissues and organs such as the brain, lungs, and bone, a process that is highly dependent on increased angiogenesis. Studies by both our group and others have further shown that P induces cell transformation, increases cell motility, and enhances the metastatic potential of breast cancer.15,16
Breast cancer exhibits high phenotypic and functional heterogeneity and, therefore, a high degree of intratumoral variation.17 As a consequence, not all cells within a tumor can be targeted by traditional chemo- and endocrine therapy. Researchers have identified a small, highly tumorigenic cell population within breast tumors that demonstrates stem cell-like properties. In cancers of the breast, this cancer stem cell (CSC) subpopulation has been shown to possess the phenotypic signature of CD24low/-, CD44high, and ALDH+ (aldehyde dehydrogenase).18 Many studies have focused on identifying and characterizing CSCs, as their functions are linked to aggressive tumor growth, metastasis, and cancer recurrence.19,20 It is hypothesized that the bulk of a tumor is maintained by a small, self-renewing CSC population, which, in addition to creating an identical copy of itself, is able to generate multipotent progenitor cells.21 Such multi-potent progenitors generate committed progenitors, which in turn give rise to terminally differentiated cells of the myoepithelial, luminal epithelial, or alveolar subtype.22,23 Moreover, CSCs share important properties with normal stem cells. These include an ability to self-renew, initiate tumors, and generate heterogeneous and differentiated progeny.18 In the healthy breast, P plays an important role in expanding the mammary stem cell population during diestrus.24 Studies in mice have shown that P also plays a critical role in mammary gland development by expanding the mammary stem cell pool.25 Recent research suggests that synthetic hormones such as MPA may influence the CSC pool in established tumors.26,27
In this study, we show that exposure of breast cancer cells to P induces the expression of CD44, a cell surface glycoprotein that has been widely used as a diagnostic and prognostic marker in breast cancer.28 Furthermore, we show that exposure of T47-D breast cancer cells to MPA increases the activity of ALDH, an enzyme that is highly active in CSCs. Finally, we demonstrate that MPA treatment of T47-D breast cancer cells stimulates mammosphere formation, suggesting that the molecular changes occurring within CSCs in breast cancer cell populations in response to both natural and synthetic Ps are functionally significant.
Materials and methods
Cell lines and culture
All cell culture studies were approved by the University of Missouri Institutional Environmental Health and Safety Board. Hormone-responsive BT-474 and T47-D breast cancer cell lines were obtained from American Type Culture Collection (Manassas, VA, USA). T47-DCO-Y cells were kindly provided by Dr. Kate Horwitz, University of Colorado. These are derivatives of T47-D cells; briefly, the progesterone receptor (PR)-negative monoclonal T47-D cell line was created via cloning by limiting dilution and consequent flow cytometry analysis for PR-negative clones.29 All cells were maintained and grown at 37°C in phenol red-free Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich Co., St. Louis, MO, USA) in a humidified atmosphere of 5% CO2. Cells were harvested for various experiments with 0.05% trypsin–EDTA (Thermo Fisher Scientific). For all in vitro experiments, cells were first treated for 24 hours (for mammosphere formation, the treatment was for 48 hours) with DMEM/F12 supplemented with 5% dextran-coated charcoal (DCC)-treated FBS (Sigma-Aldrich Co.). Subsequently, cells were washed with phosphate-buffered saline (PBS) prior to treatment with specific ligands in fresh 5% DCC-treated FBS–DMEM/F12. Cells were treated with RU-486 (Sigma-Aldrich Co.) for 30 minutes prior to the addition of P to determine the specificity of P used. Because all ligands were diluted in ethanol, control cells were treated with ethanol vehicle.
Flow cytometry
After treatment, cells were washed once with PBS and harvested using Accutase (BD Biosciences, Franklin Lakes, NJ, USA). Cells were stained for 45 minutes on ice in 100 µL staining buffer (BD Biosciences) containing phycoerythrin (PE)-conjugated mouse antihuman CD24 (BD Biosciences) and allophycocyanin (APC)-conjugated mouse antihuman CD44 (BD Biosciences) antibodies. Samples were washed twice and resuspended in 1 mL staining buffer. A Beckman Coulter CyAn ADP flow cytometer and Summit 5.2 software were used for sample analysis. Unstained and single-staining controls were used to define gates, and an equal number of cells were evaluated for each sample.
ALDH activity was measured using the AldefluorTM kit (Stemcell Technologies, Vancouver, BC, Canada) and flow cytometry as per the manufacturer’s protocol.
RNA preparation and reverse transcription polymerase chain reaction (RT-PCR)
Total cellular RNA was isolated using RNAzol reagent (Molecular Research Center, Inc., Cincinnati, OH, USA). Briefly, samples were homogenized in RNAzol. RNase-free water was added to sediment DNA and proteins, after which RNA was precipitated in isopropanol. The resulting RNA pellet was washed with 75% ethanol. Integrity of RNA was determined by evaluating the 260/280 and 260/230 ratios using NanoDrop. RNA (1 µg) was subjected to RT-PCR utilizing the Invitrogen Superscript III One-Step RT-PCR amplification kit (Thermo Fisher Scientific) to assess CD44 transcript variant expression. RT-PCR conditions were as follows: 60°C for 30 minutes, 94°C for 2 minutes, followed by 35 cycles of 94°C for 15 seconds, 55°C for 30 seconds, 68°C for 60 seconds, and a final elongation step at 68°C for 5 minutes. RT-PCR products were electrophoresed on 1.5% agarose gels containing ethidium bromide in 0.5× Tris-buffer EDTA, pH 8.0, at 100 V, after which gels were analyzed using a BioRad Imager. RNA was also subjected to RT-PCR for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a housekeeping gene control. The primers used are listed in Table 1.
 | Table 1 Primers used in RT-PCR analysis Abbreviation: RT-PCR, reverse transcription polymerase chain reaction. |
Mammosphere formation assay
T47-D cells were grown in 100 mm dishes to 60% confluence. Cells were washed twice in PBS, then incubated with 5% DCC-treated FBS–DMEM/F12 medium (8 mL) for 48 hours. Cells were subsequently treated for 48 hours with either 1 or 5 nM of MPA or norethindrone (N-ONE) (Sigma-Aldrich Co.) in 5% DCC-treated FBS–DMEM/F12 medium. Following treatment, cells from each group were harvested separately and counted. Cells (5 × 103) in 0.1 mL complete Mammocult medium (Stemcell Technologies) were seeded onto ultra-low six-well adherent plates (Stemcell Technologies) for suspension cultures. Each well contained 2 mL of synthetic P diluted in complete Mammocult medium to the same final concentration as that used for the initial treatment. For controls, an equal volume of ethanol (synthetic P vehicle) was added to the medium. Incubations were carried out in triplicate. Every 48 hours, cells were retreated with 1 mL fresh P solution (or vehicle). Pictures of mammospheres were captured by EVOS light microscopy (10×). The number of mammospheres in each group was counted on day 5 by viewing 15–30 images per well using a size exclusion standard of 100 µm, and the number of mammospheres per 5000 cells was calculated. Representative pictures were taken on day 6.
Statistical analysis
Data were reported as mean ± standard error of the mean (SEM). Statistical significance was tested by one-way analysis of variance (ANOVA) using SigmaPlot software. Nonparametric measure based on ranks was used, as needed. When ANOVA indicated a significant effect (F-ratio, P < 0.05), the Student–Newman–Keuls multirange test was employed to compare the means of individual groups. When normality failed, significance was determined by Kruskal–Wallis test (one-way ANOVA by ranks) followed by the Student–Newman–Keuls test. For all comparisons, P < 0.05 was regarded as statistically significant.
Results
MPA induces CD44 protein expression in hormone-responsive human breast cancer cells in a dose- and time-dependent manner
We initially sought to determine whether P influenced the expression of CD44, an important CSC marker in breast cancer, in hormone-responsive breast cancer cells in vitro using two cell lines: T47-D and BT-474. T47-D and BT-474 breast cancer cells are of the luminal subtype and express both estrogen receptor and PR. In addition, BT-474 cells express elevated levels of Her2/neu.30 Flow cytometry analysis of CD44 density demonstrated that treatment of T47-D cells with MPA for 24 hours increased CD44 protein expression almost 10-fold, an effect that was attenuated by the PR antagonist RU-486 (Figure 1A), suggesting the involvement of classical nuclear PR in the increased CD44 expression observed with P stimulation. RU-486 treatment alone did not induce CD44 (Figure 1A). MPA treatment also produced a significant, but lesser, increase in CD44 expression in BT-474 cells (Figure 1B). The less robust increase in CD44 expression following MPA treatment in BT-474 cells compared with T47-D cells may be explained by the fact that T47-D cells possess higher levels of PR than those found in BT-474 cells. CD44 induction by MPA in T47-D cells was both dose and time dependent (Figure 1C and D). Exposure of T47-D cells to 10 nM MPA induced CD44 expression significantly after just 6 hours of treatment, and as little as 0.1 nM MPA significantly increased CD44 expression in T47-D cells, with 1 nM MPA saturating CD44 induction.
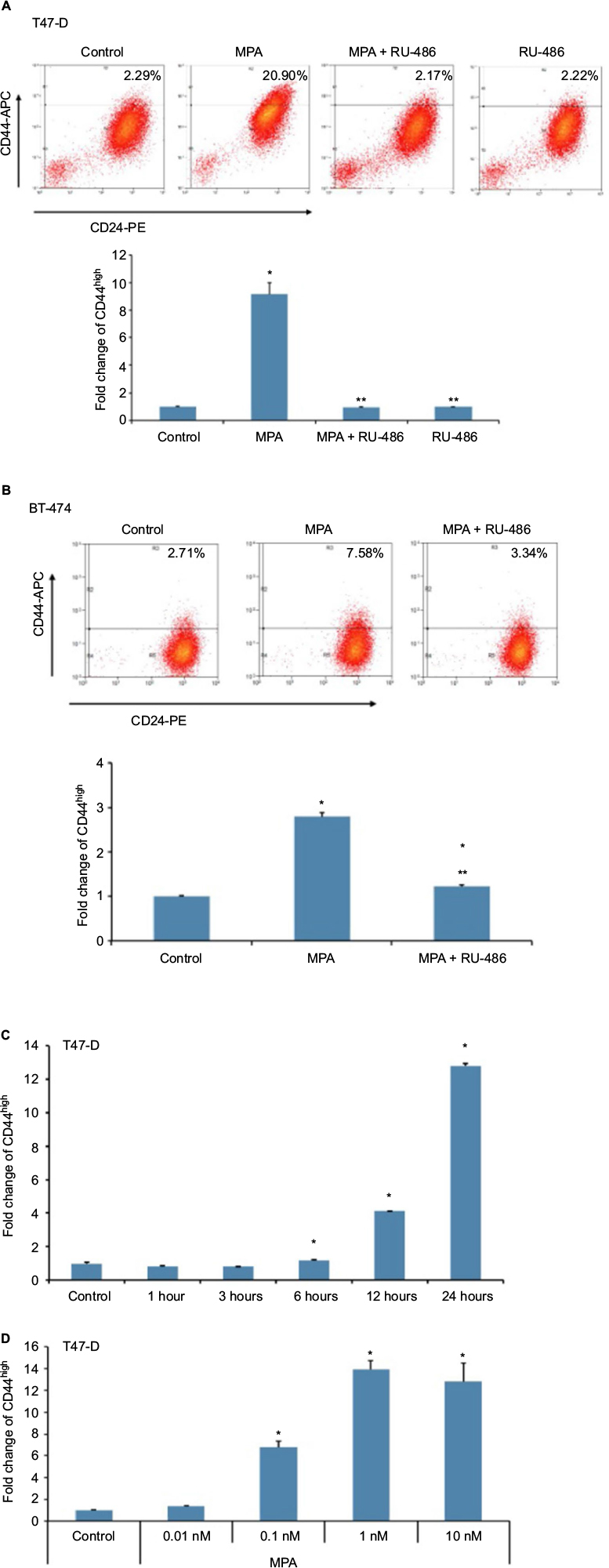 | Figure 1 Effect of MPA on CD44 protein expression in hormone-responsive human breast cancer cells. Notes: T47-D (A) and BT-474 (B) human breast cancer cells were treated at 37°C for 24 hours with 10 nM MPA, 10 nM MPA + 1 µM RU-486, or 1 µM RU-486 alone. Cells were labeled with CD44-APC and CD24-PE antibodies and analyzed using flow cytometry. (C) T47-D cells were treated at 37°C with 10 nM MPA for different periods of time, then CD44 expression was assessed by flow cytometry. (D) T47-D cells were treated with different concentrations of MPA for 24 hours, then CD44 expression was assessed by flow cytometry. Bar graphs quantitate changes in CD44 expression. Bars represent mean ± SEM (n = 3); fold change is presented compared with control value, which was set at 1. *Significantly different compared with controls; **significantly different compared with MPA-induced CD44 expression (ANOVA; P < 0.05). Abbreviations: ANOVA, analysis of variance; APC, allophycocyanin; MPA, medroxyprogesterone acetate; PE, phycoerythrin; SEM, standard error of the mean. |
Hormone induction of CD44 protein expression in T47-D cells is largely specific to Ps
Having established that a variety of Ps induce CD44 in hormone-responsive breast cancer cells, we next conducted studies to determine whether other steroid hormones, such as estrogens, androgens, and glucocorticoids, also induce CD44 protein expression in T47-D cells. Compared with both natural and synthetic Ps (such as MPA), dihydrotestosterone and dexamethasone did not significantly induce CD44 expression, whereas exposure to estradiol produced a small but significant increase in CD44 expression (Figure 2A). When we examined several synthetic Ps, including N-ONE and norgestrel (N-EL; both of which are widely used for both contraception and HRT), they all significantly induced CD44 expression in T47-D cells (Figure 2B). Notably, all P-mediated induction of CD44 expression in T47-D cells was blocked by the anti-progestin RU-486 (Figure 2B), implicating the involvement of PR in this process.
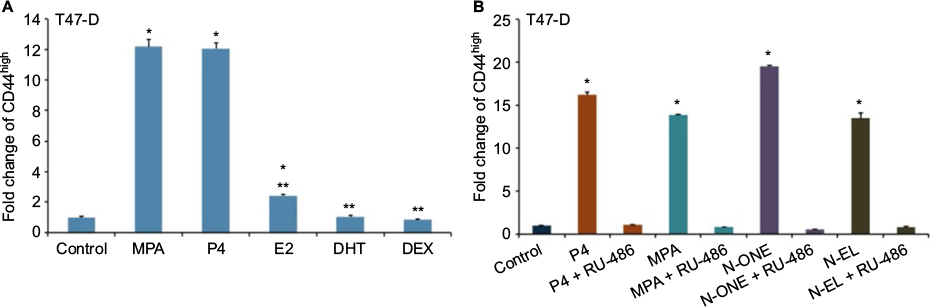 | Figure 2 Effects of steroid hormones on CD44 protein expression in T47-D cells. Notes: (A) T47-D cells were treated at 37°C for 24 hours with 10 nM ligand, then CD44 expression was assessed by flow cytometry. (B) T47-D cells were treated at 37°C for 24 hours with 10 nM ligand or 10 nM ligand + 1 µM RU-486, then CD44 expression was assessed by flow cytometry. Colors represent different progestins. Bars represent mean ± SEM (n = 3); fold change is presented compared with control value, which was set at 1. *Significantly different compared with controls, **significantly different compared with MPA-induced CD44 expression (ANOVA; P < 0.05). Abbreviations: ANOVA, analysis of variance; DEX, dexamethasone; DHT, dihydrotestosterone; E2, estradiol; MPA, medroxyprogesterone acetate; N-ONE, norethindrone; N-EL, norgestrel; P4, progesterone; SEM, standard error of the mean. |
Induction of CD44 protein expression in T47-D cells is PR dependent
Although our studies using RU-486 suggest that the induction of CD44 protein expression in hormone-responsive breast cancer cells by Ps is PR dependent, RU-486 is not a specific antiprogestin antagonist; that is, it is well established that it binds to other steroid receptors.31,32 Consequently, to confirm that the CD44 induction observed following P stimulation of these cells is indeed mediated through PR, we examined CD44 induction by MPA in T47-DCO-Y cells, a stable PR-negative monoclonal subline of the PR-positive T47-D cell line. Treatment of T47-DCO-Y cells with MPA failed to induce CD44 protein expression (Figure 3), confirming that PR is indeed essential for P-mediated CD44 induction in T47-D cells.
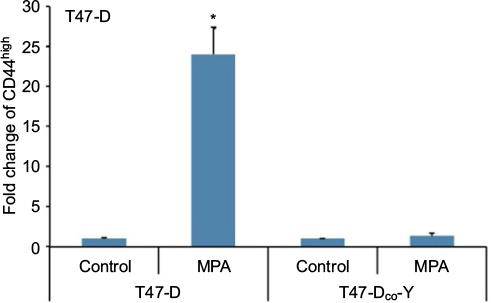 | Figure 3 Effect of MPA on CD44 protein expression in PR-negative T47-DCO-Y cells. Notes: T47-D (PR-positive) and T47-DCO-Y (PR-negative) cells were treated at 37°C for 24 hours with 10 nM MPA, then CD44 expression assessed by flow cytometry. Bars represent mean ± SEM (n = 3); fold change is presented compared with control value, which was set at 1. *Significantly different compared with controls (ANOVA; P < 0.05). Abbreviations: ANOVA, analysis of variance; MPA, medroxyprogesterone acetate; PR, progesterone receptor; SEM, standard error of the mean. |
MPA induces CD44v3 and CD44v6 transcript variant expression in T47-D cells
With 10 variant exons that are subject to alternative splicing, the CD44 gene gives rise to a myriad of different splice variants.33 To explore whether MPA induced specific CD44 splice variants in hormone-responsive breast cancer cells, we isolated RNA from T47-D cells treated with MPA ± RU-486 and then conducted RT-PCR using CD44 variant-specific primers. We found that MPA treatment significantly induced transcription of the CD44v3 and CD44v6 variants, and the induction was inhibited by RU-486 (Figure 4A) in a time-dependent manner (Figure 4B). Interestingly, previous research has suggested that CD44v3 may represent a CSC marker in head and neck cancers.34
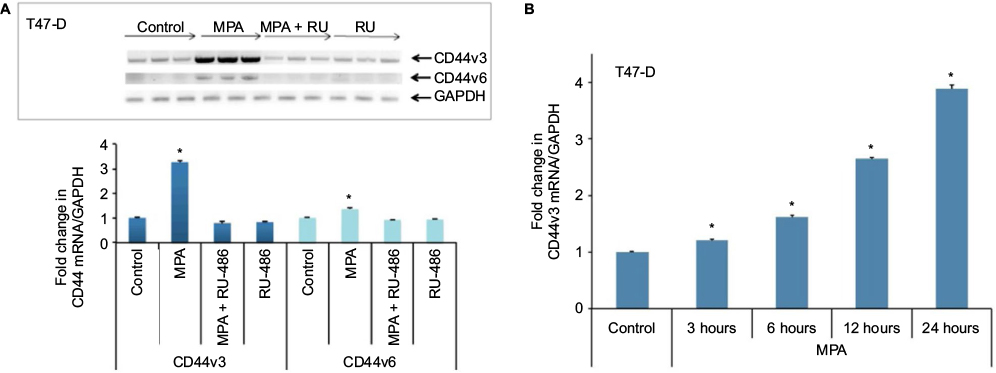 | Figure 4 Effect of MPA on the expression of CD44v3 and CD44v6 transcript variants in T47-D cells. Notes: (A) RNA was isolated from T47-D cells treated at 37°C for 24 hours with 10 nM MPA, 10 nM MPA + 1 µM RU-486, or 1 µM RU-486 alone, then analyzed by RT-PCR for CD44v3 and CD44v6 transcript variant expression. PCR products were electrophoresed on ethidium bromide–agarose gels, then photographed. Expression of GAPDH RNA was used for normalization. Quantification of CD44v3 and CD44v6 PCR product data presented in (A) normalized to GAPDH expression is shown in the panel. (B) RNA was isolated from T47-D cells treated at 37°C with 10 nM MPA for different periods of time, then analyzed by RT-PCR for CD44v3 transcript variant expression. Expression values were normalized to GAPDH expression. Bars represent mean ± SEM (n = 3); fold change is presented compared with control value, which was set at 1. *Significantly different compared with controls (ANOVA; P < 0.05). Abbreviations: ANOVA, analysis of variance; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MPA, medroxyprogesterone acetate; RT-PCR, reverse transcription polymerase chain reaction; SEM, standard error of the mean. |
MPA induces CD44v3 mRNA expression in T47-D cells at the transcriptional level
Actinomycin D, a cyclic polypeptide-containing antibiotic, inhibits RNA polymerase-mediated elongation of the newly synthesized RNA chain, and is therefore commonly used as a transcription inhibitor.35 To determine the mechanism by which MPA induces CD44v3 expression, we subjected T47-D cells treated with MPA ± actinomycin D for 6 hours to RT-PCR analysis of CD44v3 gene expression because it was the most abundant transcript measured. When actinomycin D was included in the incubation, the induction of CD44v3 mRNA expression by MPA was blocked (Figure 5), indicating that MPA most likely acts at the transcriptional level to induce CD44v3 mRNA expression.
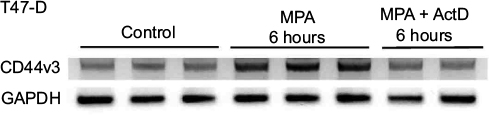 | Figure 5 Effect of ActD on MPA-induced increases in CD44v3 transcript expression in T47-D cells. Notes: RNA was isolated from T47-D cells treated at 37°C for 6 hours with 10 nM MPA or 10 nM MPA + ActD (1 µg/mL), then analyzed by RT-PCR for CD44v3 transcript variant and GAPDH expression. PCR products were electrophoresed on ethidium bromide–agarose gels, then photographed. Abbreviations: ActD, actinomycin D; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MPA, medroxyprogesterone acetate; RT-PCR, reverse transcription polymerase chain reaction. |
MPA increases ALDH enzyme activity in T47-D cells
ALDH has been established as a marker used to identify CSCs in breast cancer.18 We therefore conducted studies to ascertain the effect of MPA on ALDH enzyme activity in T47-D cells. MPA treatment of T47-D cells doubled the ALDHbright population, and this induction was blocked by RU-486 (Figure 6A). Because the ALDHbright and CD44high populations of cells are heterogeneous (containing not only CSCs or progenitor cells but also terminally differentiated cells), we further dissected these heterogeneous populations using a combination of well-characterized CSC markers (CD24, CD44, and ALDH). Treatment of T47-D cells with MPA increased the number of cells with the CD24-/low, CD44high, and ALDHbright phenotype (Figure 6B), strengthening further our hypothesis that P increases CSCs.
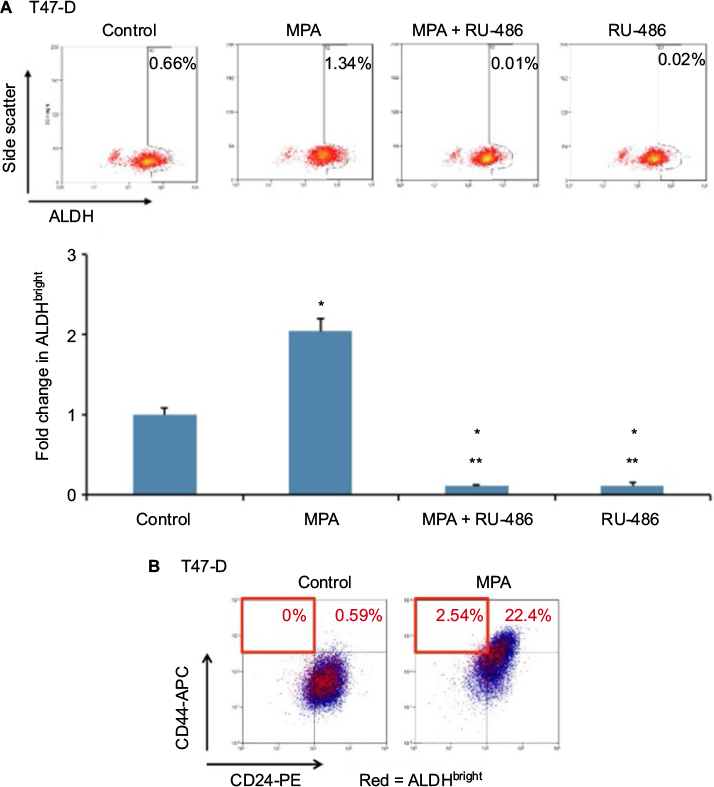 | Figure 6 Effect of MPA on ALDH enzyme activity in T47-D cells. Notes: (A) T47-D cells were treated at 37°C for 24 hours with 10 nM MPA, 10 nM MPA + 1 µM RU-486, or 1 µM RU-486 alone, then subjected to ALDEFLUOR assays. Cells were analyzed by flow cytometry to assess ALDH enzyme activity, with the ALDHbright gate set using cells treated with diethylaminobenzaldehyde (an inhibitor of the ALDH enzyme) and kept constant for all samples. Bars represent mean ± SEM (n = 3); fold change is presented compared with control value, which was set at 1. *Significantly different compared with controls; **significantly different compared with MPA-induced ALDH enzyme activity (ANOVA; P < 0.05). (B) T47-D cells were treated at 37°C for 24 hours with 10 nM MPA, then stained for CD24, CD44, and ALDH. Cells were analyzed by flow cytometry, with the CD24 gate and ALDH gate set based on negative-staining controls. The CD44 gate was set based on control levels observed. The highlighted red quadrant represents the CD24low, CD44high, ALDHbright population. Abbreviations: ALDH, aldehyde dehydrogenase; ANOVA, analysis of variance; MPA, medroxyprogesterone acetate; SEM, standard error of the mean. |
MPA increases the mammosphere-forming ability of T47-D cells
Studies have shown that breast CSCs and progenitor cells are enriched in anchorage-independent, non-adherent mammospheres.36 Assays to gage mammosphere formation are therefore excellent tools for evaluating CSC enrichment in breast cancer cell populations. When we subjected T47-D cells incubated with synthetic P (MPA or N-ONE) to mammosphere formation assays, we found an approximately two- to fourfold increase in the number of mammospheres formed compared with control-treated cells (Figure 7), suggesting that enrichment of the CD44high, ALDHbright subpopulation of cancer cells induced by MPA exposure is of functional significance.
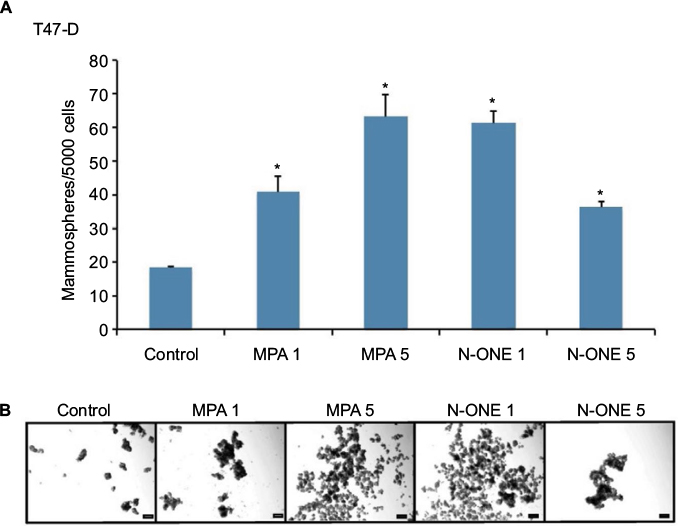 | Figure 7 Effect of MPA on mammosphere-forming ability of T47-D cells. Notes: T47-D cells were treated with either 1 nM or 5 nM MPA or N-ONE in 5% DCC-treated FBS–DMEM/F12 medium for 48 hours, and then subjected to mammosphere formation assays. (A) The number of mammospheres formed was counted after 5 days, and the number of mammospheres per 5000 cells was calculated. Bars represent mean ± SEM (n = 3). *Significantly different compared with controls (ANOVA; P < 0.05). (B) Representative micrographs at day 6. Scale bar= 100 µm. Abbreviations: ANOVA, analysis of variance; DCC, dextran-coated charcoal; DMEM, Dulbecco’s Modified Eagle’s Medium; FBS, fetal bovine serum; MPA, medroxyprogesterone acetate; N-ONE, norethindrone; SEM, standard error of the mean. |
Discussion
Clinical studies and trials in postmenopausal women have shown that combination HRT containing both estrogen and P is associated with an increased risk of breast cancer compared with the administration of estrogen or placebo alone.2–5 We have previously shown that both natural and synthetic Ps accelerate tumor development in in vivo animal models, a process that is blocked by antiprogestins. Furthermore, we found that P increases lymph node metastasis in these models,15 leading us to hypothesize that P-induced tumor growth and metastasis may be mediated by an enrichment of the CSC pool within tumors.
The CSC hypothesis rejects the notion that all cells within a tumor have the same tumorigenic and proliferative potential.37 Instead, it postulates that a rare subset of cancer cells, the CSCs, may be responsible for much of tumor initiation, maintenance, and metastasis. Clinicians trying to treat and cure breast cancer generally face two major obstacles – recurrence and metastasis – both of which are believed to be mediated by CSCs.19 During primary cancer treatment, CSCs activate cellular pathways that lead to increased survival and quiescence, allowing them to evade even highly aggressive cancer treatments.38 In addition, CSCs overexpress drug efflux pumps and have an increased capacity to activate antiapoptotic and pro-survival pathways.39 Further, after extended periods of quiescence, CSCs can initiate proliferation, reconstituting the tumor.40 Therefore, due to their various roles in tumor development, CSCs have become an attractive cancer therapeutic target in recent years.
In cancers of the breast, CSCs carry the phenotypic signature of being CD24-/low, CD44high, and ALDHhigh. CD44 is expressed in both normal stem cells and cancer cells, and is continuously used as an important stem cell marker in many different cancers.41,42 Furthermore, multiple studies have demonstrated that tumor cells that express high levels of CD44 exhibit CSC properties.34,43 In this study, we sought to better understand the mechanisms underlying P effects on breast cancer tumor growth and metastasis. We therefore expanded on our previous in vivo studies by examining whether Ps (both synthetic and natural) can enrich CSCs in hormone-responsive human breast cancer cells in vitro. We found that treatment of T47-D breast cancer cells with Ps induced CD44 protein expression through a PR-mediated mechanism, and that treatment of T47-D cells with MPA enriched CSC phenotype cells in a manner that was functionally significant. Further, we determined that treatment of T47-D cells with actinomycin D, which interferes with the elongation of the newly synthesized RNA strand by binding to the DNA strand near the transcription initiation complex, prevented induction of CD44 transcripts in the presence of MPA, suggesting that, by binding to PR, MPA affects CD44 transcription.
Both natural and synthetic Ps act hormonally through their traditional receptor to activate transcription of target genes. The two isoforms of PR (PR-A and PR-B) are generally coexpressed in mammalian cells.44 PR belongs to the family of ligand-regulated transcription factors. Both receptor isoforms contain a DNA-binding domain that binds to hormone response elements (HREs), which are short sequences of DNA that are typically found in the promoter region of target genes. Using a dual-reporter CD44 promoter clone, we investigated whether a 1316 bp fragment of the CD44 promoter (−1047/+268; GeneCopoeiaTM cat # HPRM10479-PG04) contains a PR-binding region that activates gene transcription. No promoter activation was observed (data not shown); consequently, we need to consider alternatives to promoter activation as the mechanism by which P induces CD44 expression.
Research has shown that HREs are not found exclusively in the promoter regions of a target gene but also in regions further up or downstream of the promoter. HREs have also been identified in untranslated and exonic regions.45 Further research is needed to identify and survey additional binding sites for PR that lie outside the promoter region on the CD44 gene. If PR does not actually bind to the CD44 gene to induce transcription, we may need to consider alternative methods of PR action on CD44 expression, including transcript stabilization or microRNA (miRNA) inactivation. Several miRNAs have been implicated to play a role in CSC regulation in P-dependent breast cancers. For example, failure of miR-27b to mediate repression of ectonucleotide pyrophosphatase/phosphodiesterase family member 1 has been shown to generate a subpopulation of cells that carry CSC markers and exhibit docetaxel resistance and high tumorigenicity.46 Alternatively, the miR-29 and miR-200 families have been linked to P-induced CSC enrichment.47
CD44 is a cell surface glycoprotein that is involved in a variety of important cellular functions, such as cell-to-cell communication, cell adhesion, and migration.28 Through extensive alternative splicing, cancer cells produce several isoforms of the mature CD44 protein, which mainly differ in the extracellular stem region of the cell surface glycoprotein. In addition to two constant regions, which give rise to the extracellular amino-terminal, transmembrane, and intracellular cytoplasmic tail domains of the mature CD44 protein, the CD44 gene consists of 10 variant exons, coding for the extracellular variant stem regions of the mature protein. We found that MPA treatment of T47-D cells specifically induced the CD44 transcript variants CD44v3 and CD44v6. CD44v6 has been shown to play an important role in extracellular matrix degradation and activation of invasive growth programs, both of which are closely linked to the process of metastasis.33 In addition, CD44v6 activates hyaluronic acid (HA) synthase 3, which synthesizes and secretes high-molecular-weight HA (HMW HA). HA is a principal substrate of the CD44 receptor, and HMW HA in particular has been shown to bind to CD44v3, causing increased expression of important CSC maintenance transcription factors, such as Oct4, Sox2, and Nanog.34 CD44v3 also interacts with other cell surface receptors, such as transforming growth factor-β receptor, whose downstream signaling pathways have been shown to activate Nanog expression.33,48 Nanog is a key stemness factor, and upregulation of Nanog is correlated with poor survival outcome of patients with various types of cancer.49 We have found that MPA treatment significantly upregulates Nanog transcripts in T47-D cells (data not shown). As a transcriptional regulator, Nanog activates and maintains gene programs that give CSCs unlimited self-renewal potential and pluripotency, both of which are key characteristics of CSCs. Chaffer and Weinberg20 speculate that CSCs possess some of the epithelial-to-mesenchymal transition-associated phenotypes, establishing a link between CSCs and metastasis. Immunohistochemical studies support this concept, showing that CD44v3 isoforms are preferentially expressed in metastatic lymph nodes, and that CD44v3 expression in primary tumors is associated with positive lymph nodes.50–52
Based on our observations, we contend that exposure of breast cancer cells to synthetic Ps, such as MPA, leads to an enrichment of CSCs, which would likely support the development of P-accelerated tumors in vivo. Due to the characteristics of CSCs, this enriched CSC pool greatly increases the likelihood for therapy resistance and the risk for metastasis. Our findings suggest that clinicians may be able to combat P-dependent tumor growth by blocking PR-mediated induction of CSC markers by immunotherapy, tissue-selective antiprogestins, or through a combination approach involving both immunotherapy against CD44 and small molecule targeting of PR.
Acknowledgments
This study was supported by generous gifts from donors of Ellis Fischel Cancer Center, University of Missouri, Columbia, Missouri, and by a faculty award from the College of Veterinary Medicine, University of Missouri. We also thank Dr Carolyn Henry for her invaluable support during the completion of this project. SMH is the Zalk Missouri Professor of Tumor Angiogenesis.
Disclosure
The authors report no conflicts of interest in this work.
References
American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015. | ||
Rossouw JE, Anderson GL, Prentice RL, et al; Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–333. | ||
Beral V. Million Women Study C. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362(9382):419–427. | ||
Chlebowski RT, Hendrix SL, Langer RD, et al; WHI Investigators. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women’s Health Initiative randomized trial. JAMA. 2003;289(24):3243–3253. | ||
Ross RK, Paganini-Hill A, Wan PC, Pike MC. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92(4):328–332. | ||
Kim JJ, Kurita T, Bulun SE. Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer. Endocr Rev. 2013;34(1):130–162. | ||
Holmberg L, Iversen OE, Rudenstam CM, et al; HABITS Study Group. Increased risk of recurrence after hormone replacement therapy in breast cancer survivors. J Natl Cancer Inst. 2008;100(7):475–482. | ||
Hyder SM, Chiappetta C, Stancel GM. Pharmacological and endogenous progestins induce vascular endothelial growth factor expression in human breast cancer cells. Int J Cancer. 2001;92(4):469–473. | ||
Hyder SM, Murthy L, Stancel GM. Progestin regulation of vascular endothelial growth factor in human breast cancer cells. Cancer Res. 1998;58(3):392–395. | ||
Jeng MH, Parker CJ, Jordan VC. Estrogenic potential of progestins in oral contraceptives to stimulate human breast cancer cell proliferation. Cancer Res. 1992;52(23):6539–6546. | ||
Liang Y, Hyder SM. Proliferation of endothelial and tumor epithelial cells by progestin-induced vascular endothelial growth factor from human breast cancer cells: paracrine and autocrine effects. Endocrinology. 2005;146(8):3632–3641. | ||
Mirkin S, Wong BC, Archer DF. Effect of 17 beta-estradiol, progesterone, synthetic progestins, tibolone, and tibolone metabolites on vascular endothelial growth factor mRNA in breast cancer cells. Fertil Steril. 2005;84(2):485–491. | ||
Liang Y, Besch-Williford C, Brekken RA, Hyder SM. Progestin-dependent progression of human breast tumor xenografts: a novel model for evaluating antitumor therapeutics. Cancer Res. 2007;67(20):9929–9936. | ||
Benakanakere I, Besch-Williford C, Schnell J, et al. Natural and synthetic progestins accelerate 7,12-dimethylbenz[a]anthracene-initiated mammary tumors and increase angiogenesis in Sprague-Dawley rats. Clin Cancer Res. 2006;12(13):4062–4071. | ||
Liang Y, Benakanakere I, Besch-Williford C, Hyder RS, Ellersieck MR, Hyder SM. Synthetic progestins induce growth and metastasis of BT-474 human breast cancer xenografts in nude mice. Menopause. 2010;17(5):1040–1047. | ||
Fu XD, Giretti MS, Baldacci C, et al. Extra-nuclear signaling of progesterone receptor to breast cancer cell movement and invasion through the actin cytoskeleton. PLoS One. 2008;3(7):e2790. | ||
Polyak K. Heterogeneity in breast cancer. J Clin Invest. 2011;121(10):3786–3788. | ||
Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–567. | ||
Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34(6):732–740. | ||
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. | ||
O’Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16(12):3113–3120. | ||
Tan BT, Park CY, Ailles LE, Weissman IL. The cancer stem cell hypothesis: a work in progress. Lab Invest. 2006;86(12):1203–1207. | ||
Zheng S, Xin L, Liang A, Fu Y. Cancer stem cell hypothesis: a brief summary and two proposals. Cytotechnology. 2013;65(4):505–512. | ||
Axlund SD, Sartorius CA. Progesterone regulation of stem and progenitor cells in normal and malignant breast. Mol Cell Endocrinol. 2012;357(1–2):71–79. | ||
Joshi PA, Jackson HW, Beristain AG, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465(7299):803–807. | ||
Finlay-Schultz J, Sartorius CA. Steroid hormones, steroid receptors, and breast cancer stem cells. J Mammary Gland Biol Neoplasia. 2015;20(1–2):39–50. | ||
Horwitz KB, Sartorius CA. Progestins in hormone replacement therapies reactivate cancer stem cells in women with preexisting breast cancers: a hypothesis. J Clin Endocrinol Metab. 2008;93(9):3295–3298. | ||
Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4(1):33–45. | ||
Sartorius CA, Groshong SD, Miller LA, et al. New T47D breast cancer cell lines for the independent study of progesterone B- and A-receptors: only antiprogestin-occupied B-receptors are switched to transcriptional agonists by cAMP. Cancer Res. 1994;54(14):3868–3877. | ||
Subik K, Lee JF, Baxter L, et al. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer (Auckl). 2010;4:35–41. | ||
Sun Y, Fang M, Davies H, Hu Z. Mifepristone: a potential clinical agent based on its anti-progesterone and anti-glucocorticoid properties. Gynecol Endocrinol. 2014;30(3):169–173. | ||
Heikinheimo O, Kekkonen R, Lahteenmaki P. The pharmacokinetics of mifepristone in humans reveal insights into differential mechanisms of antiprogestin action. Contraception. 2003;68(6):421–426. | ||
Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11(4):254–267. | ||
Bourguignon LY, Wong G, Earle C, Chen L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J Biol Chem. 2012;287(39):32800–32824. | ||
Sobell HM. Actinomycin and DNA transcription. Proc Natl Acad Sci U S A. 1985;82(16):5328–5331. | ||
Wang R, Lv Q, Meng W, et al. Comparison of mammosphere formation from breast cancer cell lines and primary breast tumors. J Thorac Dis. 2014;6(6):829–837. | ||
Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells – what challenges do they pose? Nat Rev Drug Discov. 2014;13(7):497–512. | ||
Vidal SJ, Rodriguez-Bravo V, Galsky M, Cordon-Cardo C, Domingo-Domenech J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene. 2014;33(36):4451–4463. | ||
Li Y, Rogoff HA, Keates S, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci U S A. 2015;112(6):1839–1844. | ||
Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res. 2011;17(15):4936–4941. | ||
Yan Y, Zuo X, Wei D. Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem cells translational medicine. 2015;4(9):1033–1043. | ||
Williams K, Motiani K, Giridhar PV, Kasper S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp Biol Med. 2013;238(3):324–338. | ||
Bourguignon LY, Earle C, Wong G, Spevak CC, Krueger K. Stem cell marker (Nanog) and Stat-3 signaling promote MicroRNA-21 expression and chemoresistance in hyaluronan/CD44-activated head and neck squamous cell carcinoma cells. Oncogene. 2012;31(2):149–160. | ||
Graham JD, Clarke CL. Expression and transcriptional activity of progesterone receptor A and progesterone receptor B in mammalian cells. Breast Cancer Res. 2002;4(5):187–190. | ||
Hyder SM, Nawaz Z, Chiappetta C, Stancel GM. Identification of functional estrogen response elements in the gene coding for the potent angiogenic factor vascular endothelial growth factor. Cancer Res. 2000;60(12):3183–3190. | ||
Takahashi RU, Miyazaki H, Takeshita F, et al. Loss of microRNA-27b contributes to breast cancer stem cell generation by activating ENPP1. Nat Commun. 2015;6:7318. | ||
Cittelly DM, Finlay-Schultz J, Howe EN, et al. Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene. 2013;32(20):2555–2564. | ||
Bourguignon LY, Singleton PA, Zhu H, Zhou B. Hyaluronan promotes signaling interaction between CD44 and the transforming growth factor beta receptor I in metastatic breast tumor cells. J Biol Chem. 2002;277(42):39703–39712. | ||
Wang ML, Chiou SH, Wu CW. Targeting cancer stem cells: emerging role of Nanog transcription factor. Onco Targets Ther. 2013;6:1207–1220. | ||
Wang SJ, Bourguignon LY. Role of hyaluronan-mediated CD44 signaling in head and neck squamous cell carcinoma progression and chemoresistance. Am J Pathol. 2011;178(3):956–963. | ||
Wang SJ, Wong G, de Heer AM, Xia W, Bourguignon LY. CD44 variant isoforms in head and neck squamous cell carcinoma progression. Laryngoscope. 2009;119(8):1518–1530. | ||
Wang SJ, Wreesmann VB, Bourguignon LY. Association of CD44 V3-containing isoforms with tumor cell growth, migration, matrix metalloproteinase expression, and lymph node metastasis in head and neck cancer. Head Neck. 2007;29(6):550–558. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.