Back to Journals » International Journal of Nanomedicine » Volume 19
Nanoparticle/Engineered Bacteria Based Triple-Strategy Delivery System for Enhanced Hepatocellular Carcinoma Cancer Therapy
Authors Yang M, Chen W, Gupta D, Mei C, Yang Y, Zhao B, Qiu L, Chen J
Received 19 December 2023
Accepted for publication 5 March 2024
Published 29 April 2024 Volume 2024:19 Pages 3827—3846
DOI https://doi.org/10.2147/IJN.S453709
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr RDK Misra
Meiyang Yang,1,2,* Weijun Chen,1,* Dhanu Gupta,3 Congjin Mei,1 Yang Yang,1 Bingke Zhao,1 Lipeng Qiu,1 Jinghua Chen1
1School of Life Sciences and Health Engineering, Jiangnan University, Wuxi, People’s Republic of China; 2School of Chemical and Material Engineering, Jiangnan University, Wuxi, People’s Republic of China; 3Department of Paediatrics, University of Oxford, Oxford, UK
*These authors contributed equally to this work
Correspondence: Lipeng Qiu; Jinghua Chen, School of Life Sciences and Health Engineering, Jiangnan University, Wuxi, 214122, People’s Republic of China, Email [email protected]; [email protected]
Background: New treatment modalities for hepatocellular carcinoma (HCC) are desperately critically needed, given the lack of specificity, severe side effects, and drug resistance with single chemotherapy. Engineered bacteria can target and accumulate in tumor tissues, induce an immune response, and act as drug delivery vehicles. However, conventional bacterial therapy has limitations, such as drug loading capacity and difficult cargo release, resulting in inadequate therapeutic outcomes. Synthetic biotechnology can enhance the precision and efficacy of bacteria-based delivery systems. This enables the selective release of therapeutic payloads in vivo.
Methods: In this study, we constructed a non-pathogenic Escherichia coli (E. coli) with a synchronized lysis circuit as both a drug/gene delivery vehicle and an in-situ (hepatitis B surface antigen) Ag (ASEc) producer. Polyethylene glycol (CHO-PEG2000-CHO)-poly(ethyleneimine) (PEI25k)-citraconic anhydride (CA)-doxorubicin (DOX) nanoparticles loaded with plasmid encoded human sulfatase 1 (hsulf-1) enzyme (PNPs) were anchored on the surface of ASEc (ASEc@PNPs). The composites were synthesized and characterized. The in vitro and in vivo anti-tumor effect of ASEc@PNPs was tested in HepG2 cell lines and a mouse subcutaneous tumor model.
Results: The results demonstrated that upon intravenous injection into tumor-bearing mice, ASEc can actively target and colonise tumor sites. The lytic genes to achieve blast and concentrated release of Ag significantly increased cytokine secretion and the intratumoral infiltration of CD4/CD8+T cells, initiated a specific immune response. Simultaneously, the PNPs system releases hsulf-1 and DOX into the tumor cell resulting in rapid tumor regression and metastasis prevention.
Conclusion: The novel drug delivery system significantly suppressed HCC in vivo with reduced side effects, indicating a potential strategy for clinical HCC therapy.
Keywords: hepatocellular carcinoma, engineered bacteria, chemotherapy, immunotherapy, anti-angiogenesis
Graphical Abstract:

Introduction
Hepatocellular carcinoma (HCC) is the third most common cause of cancer-related deaths globally.1,2 The incidence and mortality rate of this disease have witnessed a rapid surge, primarily attributed to factors such as chronic hepatitis B and C virus infections, aflatoxin exposure, smoking, obesity, and diabetes.3,4 Unfortunately, most HCC cases are diagnosed at an advanced stage, limiting treatment options and a poor 5-year survival rate (<15%).5 Current clinical treatments for advanced HCC include liver transplantation, surgical resection, radiofrequency ablation, transarterial chemoembolization, and multi-kinase inhibitors.6–8 Recently, immunotherapies, such as those targeting PD-1/PD-L1, have been clinically approved for HCC.9–11 While liver transplantation is considered the best treatment for HCC, it remains infrequent due to limited donor availability, high rate of tumor recurrence and metastasis post-surgery.5,12 Additionally, some patients face challenges in tumor resection, and for those with severe cirrhosis, resection can compromise normal liver functions. Radiofrequency ablation is a local treatment option for small HCCs ranging from 3 to 7 cm in diameter, but complete ablation of larger HCC tumors is not possible.13 Transarterial chemoembolization has gained recognition since 2018 for reducing tumor mass and alleviating pain in HCC patients.14,15 However, its efficacy in eradicating cancer cells, especially in highly vascularized tumors, remains limited.7,16
Currently, multi-kinase inhibitors and immunotherapy are the primary systemic therapies used in HCC treatment. However, the response rate to monotherapy remains below 20%.11,17 Although existing treatments offer modest improvement in survival and quality of life, recent advancements have been made in the realm of combination therapies. For example, the combination of atezolizumab and bevacizumab exhibited higher efficacy in advanced liver cancer patients compared to either monotherapy resulting in median overall survival of 19.2 months in Phase 3 clinical trials by Zhu et al.10 Another study demonstrated that the combination of tyrosine kinase inhibitors, immune checkpoint inhibitors, and transarterial chemoembolization resulted in an objective remission rate (ORR) of 52.8% in HCC patients.17 These findings indicate the potential of combination therapy, involving two or more agents, as a promising alternative to extend survival and enhance the quality of life for HCC patients.
Live tumor-targeting bacteria exhibit intrinsic tumor-targeting ability, and inhibit tumor cell growth to a certain extent, with various strains employing distinct mechanisms.18–21 Facultative anaerobic bacteria such as E. coli can be eliminated by the immune system in normal tissues shortly after systemic administration. However, the immunosuppressive microenvironment of the tumor facilitates bacterial colonization.22,23 Moreover, individual bacteria exhibit distinct tumor-targeting mechanism. For example, Listeria can infect not only specialized antigen-presenting cells (APC) but also myeloid-derived suppressor cells (MDSCs), which are then transported to the tumor microenvironment via MDSCs.24–26 When bacteria enter tumor tissue, they serve as natural immune adjuvant, inducing both innate and adaptive immune responses and altering the tumor immune microenvironment. Natural immune cells recognize pathogen-associated molecular patterns on bacteria, engulf and destroy them, secrete cytokines, and trigger specific adaptive immunity. In addition to the well-known CD4+ and CD8+T cells, there are also “innate” lymphocytes such as γδT cells, iNKT cells, MAIT cells, B cells, and more.27,28 In addition, bacteria utilize diverse physical and chemical properties (such as fat, protein, and other natural components), through the cross-presentation of dendritic cells, to induce CD4+ and CD8+T cells to combine and kill tumors.27,29
Additionally, bacteria can be modified to enhance the therapeutic effect by expressing therapeutic proteins, cytokines, precursor drug convertases, and immunomodulators.20,30–34 However, a more considerable challenge lies in the limited secretion of expressed proteins and enzymes outside the bacteria. Therefore, we genetically engineered live bacteria synthetic biology by introducing lytic genes that controlled the explosive release of proteins at tumor sites through population density-responsive switches.33,35,36 This results in a rapid increase in protein concentration at tumor sites, thereby achieving therapeutic effects. While studies on bacterial cancer therapy have shown promising results in experimental models, it is not recommended for standalone use in clinical tumor immunotherapy.37–39 We found that the combination of expressed (heparin sulfatase 1) Hsulf-1 enzyme of E. coli with the chemotherapy drug doxorubicin (DOX) can produce better therapeutic outcomes in solid tumors and metastases based on the previous studies.40 As there are approved bacteriological agents in clinical use in both Japan and China, along with DOX being an approved immune-antitumor drug, designing a comprehensive immunotherapy model in clinical practice is feasible.
This study employed no-npathogeniclive E. coli BL21 expressed Ag (ASEc) in situ as drug delivery carriers to transport the chemotherapy drug DOX and plasmid-encoded human sulfatase 1 (hsulf-1) enzyme (ASEc@PNPs) directly to the surrounding tumor tissue. Simultaneously, in order to address the challenge of antigen release in bacteria, we introduced lytic genes to achieve blast and concentrated release of live bacteria in the tumor. The nanoparticle loaded with the chemotherapy drug DOX and plasmid-encoded hsulf-1 enzyme (PNPs) was prepared via Schiff base reactions, which allowed the release of hsulf-1 and DOX in the tumor microenvironment. The Ag released by the lysed E. coli activated a specific immune response, whereas the lysed bacteria exerted an additional immune adjuvant effect, further enhancing the immune response. Combining this immune activation with DOX for chemotherapy and hsulf-1 for anti-angiogenesis therapy, this method provides three anti-HCC treatments within a single delivery system. Therefore, the controllable switch-release bacterial delivery system fulfills the dual function of tumor immune enhancement and drug delivery. The goal of suppressing tumor proliferation and metastasis was achieved through multiple mechanisms.
Materials and Methods
Materials
N,N-Dimethylformamide (DMF) was purchased from Energy Chemical Reagent (Shanghai, China). DOX·HCL was purchased from Jinan Daigang Biomaterial Engineering Company, China. Poly (ethyleneimine) (PEI25k), Citraconic anhydride (CA), N-Hydroxysuccinimide (NHS), and 1-Ethyl-3-(3-Dimethylaminopropyl) Carbodiimide (EDC) were purchased from Macklin Chemical Reagent (Shanghai, China). Dimethyl sulfoxide (DMSO) was purchased from Sinopharm Chemical Reagent (China). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) Quick Preparation kit was acquired from Beyotime Biotechnology (Shanghai, China). Calreticulin (CRT) polyclonal antibody and Fluorescein (FITC)–conjugated affinipure Goat Anti-Rabbit IgG (H+L) were obtained from Proteintech (USA). Extracellular Matrix (ECM) was purchased from Shanghai Nova Pharmaceuticals (China). Recombinant Human Vascular Endothelial G (VEGF165) was purchased from Sangon Biotechnology Company (Shanghai, China). Enzyme-linked immunosorbent assay (ELISA) kits (HBsAg, TNF-α, IL-6, and IFN-γ) were purchased from Ruixin Biotechnology Company (China). No endotoxin plasmid extraction kit, and 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) and 1.1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyaine iodide (DiR) were purchased from Yeasen Biotechnology Company (Shanghai, China). 4’,6-diamidino-2-phenylindole (DAPI) was purchased from Beyotime Biotechnology (Shanghai, China).
Cell Lines and Culture Conditions
HepG2 and H22 cells were cultured in the Roswell Park Memorial Institute 1640 medium (RPMI 1640), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin solution. HUVECs were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% FBS and 1% penicillin–streptomycin solution. The cells were incubated at 37°C in a 5% CO2 atmosphere. All cells were purchased from Meisen CTCC, Zhejiang, China.
Animals and Tumor Model
All Balb/c mice (aged 4–6 weeks and weighing 16 ± 2 g) were purchased from SiPeiFu Animal Experiment Company, China. Balb/c mice were kept in a specific pathogen-free (SPF) environment with a temperature of 20–26°C, a relative humidity of 40%–70%, alternating light and dark lighting for 12h/12h, and adequate food and water. All animal care and protocols were performed in accordance with the Guidelines for Animal Experimentation and were approved by the Ethics Committee of Medicine Laboratory Animal Center of Jiangnan University. The number was JN. No 20230330b1320527[099].
Construction of pTac/Ag and pTet/x174E Plasmid
The pMAL-C5X plasmid was purchased from Wuhan Miaoling Bioscience & Technology Co. Ltd. Escherichia coli (E. coli, strain BL21) was provided by Professor Liu Liming. The bacteria were placed in LB and cultured at 37°C with shaking at 220 rpm. The pTac/Ag and pTet/x174E plasmids were codon optimized and synthesized by GENEWIZ, Inc. Bacteria transformed with pTac/Ag and pTet/x174E were plated on ampicillin and kanamycin LB agar plates and incubated overnight. Positive transformants were denoted as ASEc.
Synthesis and Characterization of PNPs
Citraconic anhydride (CA)-doxorubicin (DOX) (CAD) was prepared using an anhydride ammonolysis reaction.40 CAD was bonded to PEI to prepare the PEI-CAD polymer (PD). CAD was dissolved in DMSO, followed by drop-wise addition of NHS and EDC for 4 h. The solution was dropped wisely, added to the PEI solution, and stirred for 24 h at room temperature in the dark. Thereafter, the product was purified by dialysis against distilled water (MWCO 3500 Da) and lyophilized to obtain the PD polymer, which was stored at 4°C for later use. The PD and hsulf-1 were suspended in deionized water, and the PD/hsulf-1 complex was prepared at a 5/1 (w/w) ratio. After incubation at room temperature for 30 min, the PD/hsulf-1 complex was obtained. The (CHO-PEG-CHO)-PD/hsulf-1 nanoparticles (PNPs) were synthesized using a Schiff base reaction between PEI and PEG.41,42 PD/hsulf-1 and (CHO-PEG-CHO) (molar ratio of 1:10) were dissolved in ddH2O (pH 8.0) and stirred for 4h at room temperature in the dark, and then the product was centrifugated at 10,000 rpm for 15 min. The precipitate was collected as the PNPs.
The chemical structures of the PD and PNPs were characterized by infrared spectrometry (TENSOR II, Bruker, Germany). The morphology of the PNPs was investigated using transmission electron microscopy (TEM, JEM-2100, JEOL, Ltd. Japan). The size distribution and zeta potential of the PNPs were measured by dynamic light scattering (DLS, ZEN 3600 instrument, Malvern, England) at 25°C three times. The DOX loading in PNPs was determined by UV–vis spectroscopy (UV-2550, SHIMADZU, Japan).
Preparation and Characterisation of Nanoparticle-Loaded E. coli
The PNPs were loaded onto the surface of ASEc through electrostatic interactions. PNPs were added and co-incubated with ASEc for 30 min. The ASEc@PNPs complexes were collected by centrifugation (4°C, 5000 rpm, 5 min) and washed three times with sterile PBS.
The viability of nanoparticle-loaded ASEc was assessed using the plate colony-counting method. Briefly, ASEc@PNPs complexes were diluted with sterile PBS at 100:1 and coated onto LB agar plates, after which the plates were cultured at 37°C overnight. The number of colonies on the plates was counted to calculate the viability of PNPs-loaded ASEc.
The morphology of the ASEc@PNPs complexes was investigated using TEM. DLS measured the complex’s size distribution and zeta potential. The ASEc@PNPs pellet was collected and stained with DAPI for 20–30 min. Confocal fluorescence images (CLSM, Ti2-E+A1, Nikon, Japan) were used to observe the cell morphology.
Cytotoxicity Test
The cytotoxicity of ASEc@PNPs complexes was evaluated in a transwell system. HepG2 cells were seeded (5 × 104 per well) in a transwell 24-well plate and co-cultured with DOX, PNPs (no hsulf-1), PNPs, ASEc, and ASEc@PNPs for 24 h. The viability of cells was analyzed using the MTT assay. Further, the cytotoxicity of DOX, PNPs (no hsulf-1), PNPs, ASEc, and ASEc@PNPs were investigated using the live/dead staining method. The tumor cells were co-stained with calcein-AM and propidium iodide (PI), after which they were imaged using CLSM.
Cell Uptake Studies
The flow cytometry assay (FCM) was used to assess the intracellular uptake test of PNPs and ASEc@PNPs. HepG2 cells were seeded (4 × 106 per well) in a 12-well plate. After culturing with PNPs and ASEc@PNPs medium for 3, 12, and 24 h, HepG2 cells were collected and resuspended in PBS solution for analysis. In addition, HepG2 cells were stained with DAPI for 30 min, purified with PBS three times, and fixed with 4% paraformaldehyde for 15 min. Finally, CLSM was carried out after washing the cells with PBS three times.
In vitro Transfection
We evaluated the effectiveness of PEI, PNPs, and ASEc@PNPs transfection vectors in delivering genetic material into HepG2 cells. FCM and CLSM were used to measure transfection efficiency by observing the percentage of green fluorescent protein expression by the transfected cells. First, HepG2 cells were seeded at a density of 2 × 105 cells/well in a 12-well plate and incubated for 24 h. Thereafter, the culture medium was removed, and the cells were washed with PBS before being replaced with a serum-free medium containing transfection media. Next, pGFP, PEI/pGFP, PNPs/pGFP, and ASEc@PNPs/pGFP complexes (4 μg pGFP in each sample) were incubated with the cells for 8–10 h, respectively. After incubation, the culture medium was replaced with a serum medium, and the cells were incubated for another 48 h. Transfection efficiency was evaluated by measuring cells expressing green fluorescent protein using FCM and CLSM. Further, cells were observed under a CLSM to monitor morphological changes and the co-delivery of DOX and DNA.
Wound Healing and Angiogenesis Assays
Wound-healing assay is a commonly used method to study cell migration and wound repair. To create wounds, 200 μL pipette tips were used to scrape across the monolayer, creating a “scratch” or “wound” that disrupted the uniformity of the cell layer. Relative cell migration was then measured by taking photographs of the wounds at 0, 24, and 48 h after the scratch was created. By photographing the wounds using an inverted microscope (Leica, Germany), we determined the percentage of cells that had migrated to the wound area over time, providing insights into the ability of these cells to repair damage and migrate.
In vitro, angiogenesis assay was conducted in 96-well plates coated with 30 µL of extracellular matrix (ECM), following the manufacturer’s instructions. Human umbilical vein endothelial cell (HUVEC) cultures (3 × 104 cells/well) were treated with PNPs (no hsulf-1) or PNPs in RPMI 1640 supplemented with 10% FBS. Briefly, the cells were pretreated with PNPs (no hsulf-1) or PNPs for 8 h at 37°C under 5% CO2. HUVECs were then plated on Matrigel-coated plates and incubated at 37°C under 5% CO2 for 6 h to allow tube formation. Subsequently, the cells were stained with Calcein-AM, and tube formation was evaluated.
In vitro Assessment of Macrophage Polarization
Macrophages were seeded in 6-well Transwell plates at a density of 3 × 105 cells/well. PNPs, BL21, and ASEc@PNPs were separately added into the wells. After 48 h, the cells were washed with PBS and collected using centrifugation. CD86-FITC was added after treatment with a permeabilization wash buffer; the cells were incubated for 30 min in the dark, centrifuged to remove the antibodies, and resuspended in PBS for FCM analysis. The suspension of each group was collected to measure the concentration of TNF-α and IL-6 using enzyme-linked immunosorbent assay (ELISA), following the instructions of the ELISA kit.
In vitro Induction of Immunogenic Cell Death (ICD)
To evaluate ICD in HepG2 cells, the expression of the ICD marker calreticulin (CRT) was quantified. PNPs (no hsulf-1), PNPs, and ASEc@PNPs media were separately added to confocal dishes containing HepG2 cells (1 × 105 cells/well) at a final DOX concentration of 0.1 μg/mL. After 24 h, the cells were washed with PBS. After fixation, CRT rabbit monoclonal antibodies were added for 12 h of incubation at 4°C. The cells were co-incubated with secondary antibody (Fluorescein [FITC]–conjugated AffiniPure goat anti-Rabbit IgG (H+L)) for 1 h in an incubator. Then DAPI was added to stain the nuclei. The cells were observed using flow cytometry and CLSM.
In vivo Biodistribution Analysis and Targeting Effect of ASEc@PNPs in a Mouse Tumor Model
ASEc@PNPs was injected into tumor-bearing mice via the tail vein; the mice were then sacrificed on days 1, 5, and 10. The heart, liver, spleen, lung, kidney, and tumor tissues were harvested and homogenized. The homogenates were diluted in a 10- or 200-fold gradient and plated. The cells were incubated for 24 h, and the colonies formed were counted. DiR-labeled ASEc and DiR-labeled ASEc@PNPs were intravenously injected into the H22 tumor-bearing mice separately. Major organs were obtained from the mice at different time points to monitor biodistribution and analyzed using IVIS Spectrum equipment from PerkinElmer.
In vivo Anti-Tumor Assay
H22 tumor-bearing mice were divided into seven groups (n = 5) and then administered PBS, DOX (0.25 mg/kg), PNPs (no hsulf-1), PNPs, AEc, ASEc@PNPs, and DOX (5 mg/kg). After 5 days of subcutaneous injection of H22 cells into Balb/c mice, PBS (i.v., 200 μL), DOX (5 μg, 200 μL), DOX (100 μg, 200 μL), PNPs (no hsulf-1) (5 μg, 200 μL), PNPs (DOX 5 μg, hsulf-1 60 μg, 200 μL), AEc (AEc dose was equal to the ASEc@PNPs, 200 μL), and ASEc@PNPs (DOX 5 μg, hsulf-1 60 μg) were intravenously administered into mice on days 0 and 5. Mouse weight and tumor size were monitored throughout the treatment process. After 14 days, the mice were sacrificed, and the major organs (heart, liver, spleen, lung, tumor, and kidney) were obtained for further analysis. These analyses included hematoxylin and eosin (H&E) staining for pathological state evaluation of different tissues and immunofluorescence staining for TUNEL assay (a technique for detecting DNA fragmentation in apoptotic cells) and Ki67 (a protein that indicates cell proliferation).
In vivo Antitumor-Immunity Assay
Tumor tissues were fixed with paraformaldehyde solution, sectioned, and stained with CRT antibody and DAPI for immunohistochemistry assays. The tissue sections were then imaged using a digital scanner (Pannoramic MIDI, 3DHISTECH). Furthermore, the serum samples were assayed for TNF-α, IL-6, and IFN-γ using ELISA kits. In order to assess the enrichment of CD4+ and CD8+T cells, the tumor tissues of the mice in different groups were obtained, fixed with paraformaldehyde solution, embedded in paraffin, sectioned, and stained with CD4 and CD8 antibodies, respectively. Finally, the tissue slices were observed using CLSM after staining with DAPI.
Lung Metastasis Inhibition
Two hundred microliters of sterile PBS solution containing 6 × 106 H22 cells were intravenously injected into female Balb/c mice via the tail vein. After 24 h, the mice were randomly divided into six groups (n = 5) and treated with PBS, PNPs (no hsulf-1), PNPs, AEc, and ASEc@PNPs at 0, 5, 10, and 15 days. Healthy mice were set as the negative control. After 3 weeks, the mice were euthanized, and their lung tissues were collected for analysis. H&E staining was performed to evaluate the pathological changes in the lung tissues.
Statistical Analysis
All results are presented as mean ± SEM and plotted using GraphPad 8.0.1 software (San Diego, CA, USA). Differences between three or more treatment groups were evaluated for significance using one-way ANOVA; differences between the two treatment groups were assessed using Student’s two-tailed t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 were used to indicate the significance of the difference.
Results and Discussion
The Design and Verification of the Production-Lysis Circuit (SLC)
The key function of ASEc@PNPs relies on an SLC to release Ag sustainably and periodically. This circuit is based on a production-lysis oscillation in which the lysis gene E is activated using acyl homoserine lactone (AHL) quorum-sensing signaling, resulting in controlled lysis of some bacteria.43–45 The SLC-Ag system was designed to colonize tumors and undergo intratumoral quorum lysis, leading to the simultaneous local delivery of Ag, DOX, hsulf-1, and immunostimulatory lysed bacterial adjuvants to stimulate antitumor immunity and promote tumor regression (Figure 1A).
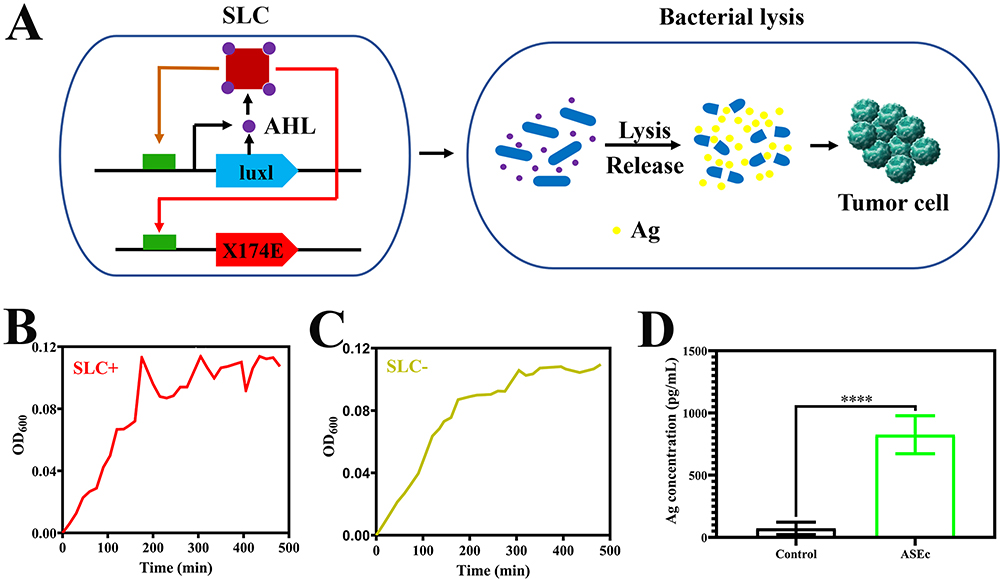 |
Figure 1 Quorum-induced release of Ag by engineered immunotherapeutic bacteria encoding a SLC. (A) E. coli with SLC reach a quorum and induce the phage-lysis protein X174E, leading to bacterial lysis and release of a constitutively produced. E. coli growth dynamics over time of SLC+ (B) and SLC− (C) E. coli in liquid culture (n = 3). (D) Ag concentration of the supernatant after Ag release E. coli cultured for 8 h. (****P < 0.0001). |
To confirm the expression and lysis-dependent release of Ag, non-pathogenic E. coli transformed with a single plasmid that encodes an SLC, and an additional plasmid that drives constitutive expression of Ag was used. The SLC+ strain produced AHL through the expression of LuxI and lysed due to the production of the X174E bacteriophage lysis protein, resulting in cycles of Ag accumulation and release. SLC+ and SLC− E. coli cultures were grown in lysogeny broth in a 48-well plate, and the OD600 was measured over time. SLC+-Ag-expressing E. coli exhibited multiple periodic dips between 200 min and 450 min, indicating rounds of synchronized lysis, whereas SLC− E. coli exhibited normal bacterial growth kinetics (Figure 1B and C). After verifying the synchronized lysis behavior, we evaluated the lysis-mediated release of Ag in batch cultures. The production and release of Ag in the SLC+ culture supernatant was detected by ELISA. The production and release of Ag was detected by ELISA (Figure 1D) from the culture supernatant of Ag release E. coli (containing pAg and pX174E). The control group was the strain with only pAg. The results showed that the protein release from the production-lysis circuit was 11.37-fold higher than that of the control group. The data obtained confirmed the ability of the production-lysis circuit to release Ag, which built up the basis for the application of production-lysis bacteria in vivo.
Preparation and Characterization of PNPs and ASEc@PNPs
Drug-loaded polymers were constructed by assembling PEI25k and CAD via amide reactions (Figure S1). Different molar ratios of PEI to CAD (1:1, 1:3, and 1:5) were tested in these experiments. From UV–Vis spectrophotometric analysis, DOX loading was the highest (7.28 ± 1.20) % when the molar ratio of PEI to CAD was 1:3 (Figure S2), and the zeta potential of the PD was +45.93±3.03 mV. Therefore, a molar ratio of PEI to CAD of 1:3 was used for synthesizing PDs. The hsulf-1 was loaded via electrostatic interactions. The optimal mass ratio of PD to hsulf-1 was found to be 5:1 based on nucleic acid glue results (Figure S3). To protect the plasmid, CHO-PEG-CHO was grafted to PD/hsulf-1 via Schiff base reactions (Figure S1). The PNPs were synthesized with different molar ratios of CHO-PEG-CHO to PD (5:1, 10:1, and 15:1). The average diameter was determined to be approximately 160 nm (Figure S4A). However, the zeta potential was −0.85 ± 0.55 mV when the molar ratio of CHO-PEG-CHO to PD was 15:1 (Figure S4B). Therefore, we chose a molar ratio of 10:1 to synthesize PNPs for further applications. The PNPs were confirmed using infrared spectrometry (Figure S5). The PNPs were spherical, with a hydrodynamic size of 162.16 ± 5.81 nm (Figure 2A), and a zeta potential of +3.12±0.69 mV. The loading percentage of DOX in the nanoparticle was (1.66±0.13) %, as determined by UV-Vis.
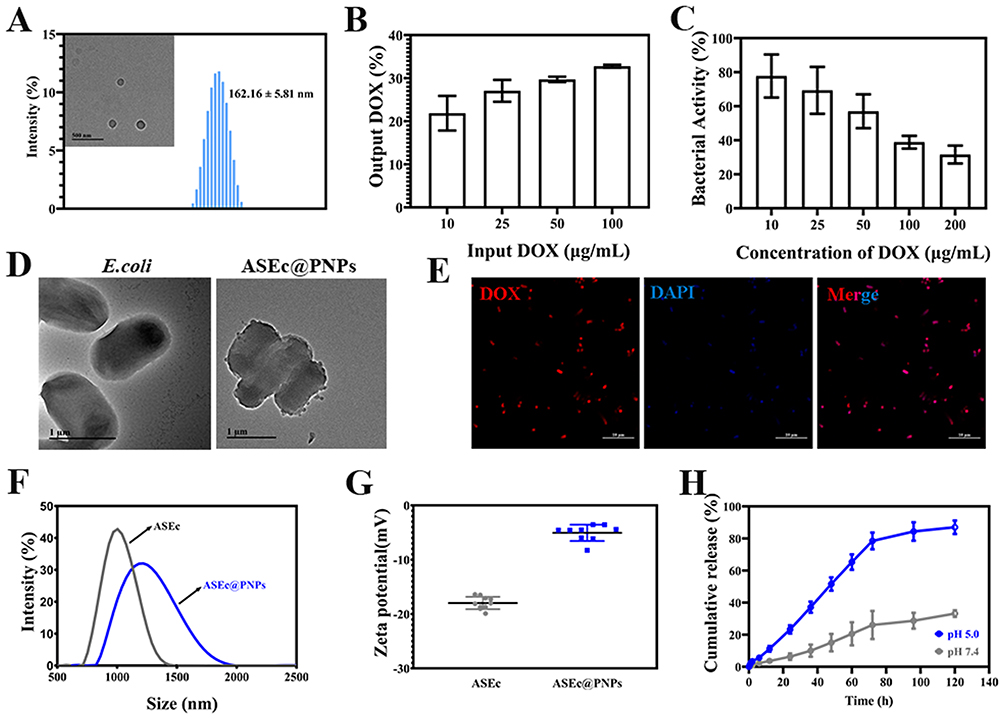 |
Figure 2 Characterization of ASEc@PNPs. (A) TEM image (inset) and size distribution of PNPs. (B) DOX relative loading of the ASEc. (C) Bacterial viability of ASEc@PNPs at different DOX concentrations. (D) TEM images of E. coli and PNPs-loaded ASEc. Scale bar: 1 μm. (E) CLSM image of PNPs-loaded ASEc. Red fluorescence represents PNPs, and blue fluorescence represents DAPI labeled ASEc. Scale bar: 10 μm. (F) Hydrodynamic diameter and (G) zeta potential of ASEc, and PNPs -loaded ASEc. (H) In vitro drug release curves of ASEc@PNPs. |
The PNPs could be loaded onto the negatively charged bacterial surface through electrostatic interactions due to the ζ-potential of ASEc of −18.26 ± 1.2 mV. The UV–Vis results showed that the DOX load of the ASEc increased slightly with increasing DOX concentrations (Figure 2B). To investigate the effect of the PNPs coating on the viability of the ASEc systems, the ASEc@PNPs were spread onto LB agar plates, and the total number of colonies was assessed after overnight incubation. The results showed that compared with that of untreated ASEc, increasing the amount of DOX induced a decrease in bacterial activity (Figure 2C). The DOX relative load of the ASEc reached 27.06% when the DOX concentration of 25 μg/mL. The bacterial activity was 69.33%. The attachment of particles to individual bacteria was characterized using TEM, which verified that the bacteria could carry PNPs (Figure 2D). In addition, CLSM images of ASEc incubated with DAPI and the PNPs clearly showed that the bacterium, which was stained blue, co-localized with the red-labeled complex, demonstrating that the bacteria can carry PNPs (Figure 2E). The hydrodynamic diameter and zeta potential of ASEc@PNPs were 1290.55 ± 188.89 nm and −5.05 ± 1.50 mV, respectively (Figure 2F and G). The change in the ASEc ζ-potential suggests that PNPs were successfully loaded onto ASEc. Additionally, SDS-PAGE analysis showed that the protein profile of ASEc@PNPs coincided with that of ASEc, suggesting that bacterial proteins were retained in ASEc@PNPs (Figure S6). As shown in Figure 2H, the release behaviors of DOX from ASEc@PNPs were both time- and pH-dependent. At pH 5.0, the cumulative release of DOX from ASEc@PNPs was 86.99% at 120 h. However, the release of DOX from both groups was 33.15% at pH 7.4. Drug release under acidic conditions was higher than that under physiological conditions (pH 7.4), which was due to the cleavage of the pH-responsive Schiff base bonds of ASEc@PNPs under acidic conditions.
In vitro Cytotoxicity Assay, Cellular Uptake and Transfection Studies
The antitumor effects of ASEc@PNPs were determined in HepG2 cells using an MTT assay to assess cell viability. As shown in Figure 3A, ASEc@PNPs c resulted in lower cell viability after 24 h than PNPs (no hsulf-1) and PNPs and cell survival rates were concentration-dependent. The cell survival rate of ASEc was 90.29%, while those of PNPs (no hsulf-1) and PNPs were 9.02% and 7.03%. Importantly, incubating HepG2 cells with ASEc@PNPs for 24 h led to more cell death than incubating them with pure PNPs (no hsulf-1) and PNPs at the DOX concentration of 10 μg/mL. The IC50 value of the ASEc@PNPs group was 0.22 μg/mL. A live/dead cell fluorescence assay was performed to confirm these results (Figure 3B), which indicated that ASEc@PNPs could efficiently kill cancer cells in vitro.
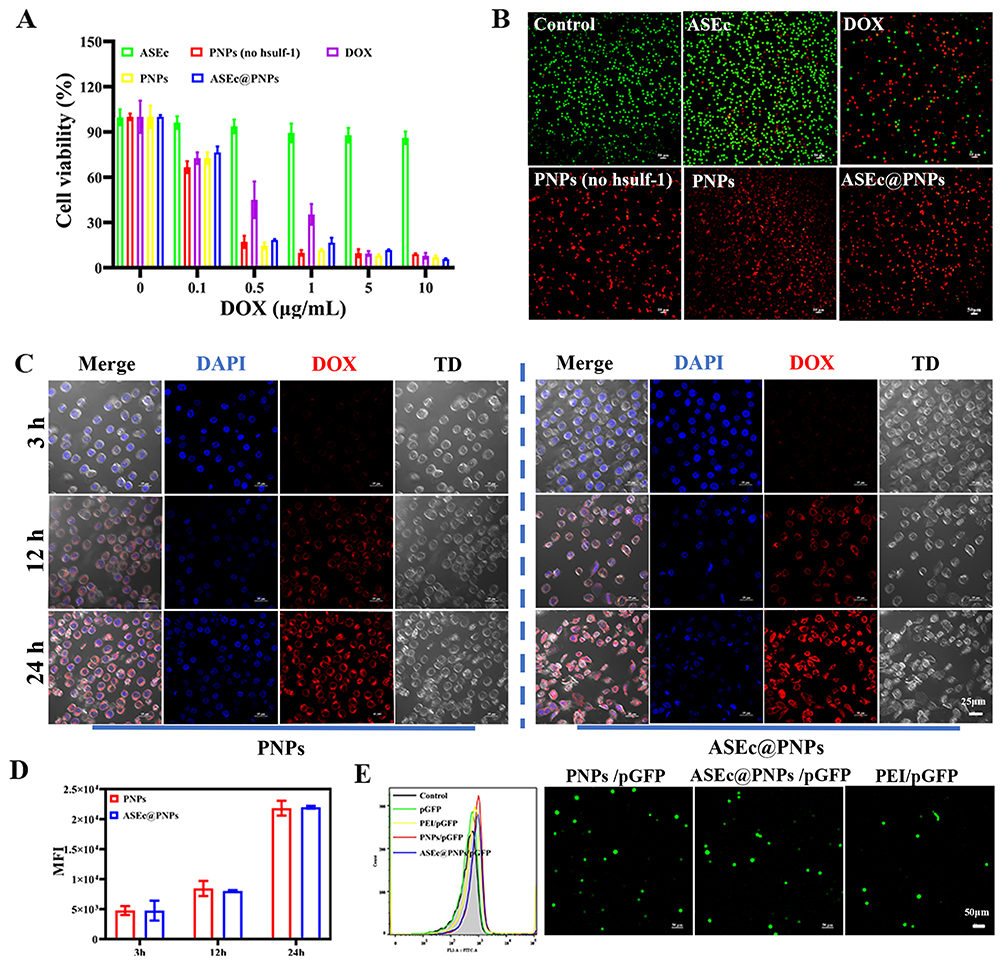 |
Figure 3 In vitro analysis of ASEc@PNPs. (A) Cytotoxicity of the ASEc, PNPs (no hsulf-1), PNPs and ASEc@PNPs. (B) CLSM images of HepG2 cells incubated with ASEc, PNPs (no hsulf-1), PNPs and ASEc@PNPs. Scale bar: 100 μm. Green and red represent live cells and dead cells, respectively. Cell uptake of PNPs and ASEc@PNPs through (C) CLSM images and (D) flow cytometry. Scale bar: 25 μm. Transfection of PNPs through (E) FCM and CLSM images. Scale bar: 50 μm. |
To assess the cellular uptake efficiency of PNPs and ASEc@PNPs, CLSM and flow cytometry experiments were carried out. Firstly, we analyzed the fluorescence intensity through detecting the red fluorescence intensities of DOX by flow cytometry after treating HepG2 cells at the same concentration of DOX for 3, 12, and 24 h (Figure 3C and D). The results showed that there was no significant difference between the PNPs group and the ASEc@PNPs group. Subsequently, CLSM images were applied to further investigate the uptake of HepG2 tumor cells. The results showed that the two groups entered HepG2 cell, whereas PNPs group and the ASEc@PNPs group internalized considerably more at 24 h (Figure 3C), indicating the presence of ASEc cannot affect the uptake of DOX by cells. Over time, PNPs on the surface of ASEc were released and uptaken by cells. Therefore, both the flow cytometry study and CLSM images revealed that PNPs onto the surface of the ASEc could be uptaken into cells effectively in vitro.
Furthermore, we investigated the DNA transfection of PNPs using a luciferase reporter plasmid. After incubating HepG2 cells with the PNPs/pGFP complex for 8–10 h, the culture medium was replaced with a serum-containing medium, and the cells were incubated for 48 h. The results were then examined using FCM (Figure 3E) and CLSM (Figure 3E). As shown in Figure 3E, both PNPs and ASEc@PNPs could successfully transfect HepG2 cells, with (10.62 ± 0.30) % and (7.37 ± 1.09) % of the cells exhibiting green fluorescence (generated by the pGFP plasmid), respectively. The total number of cells was determined using DAPI nuclear stain, as shown by the blue fluorescence signals in Figure 3E. These data indicate that the PNPs could be used as gene transfection vectors. Moreover, the co-delivery system could simultaneously deliver plasmid DNA and DOX into the same cells with high efficiency, as evidenced by both green and red fluorescent puncta in the HepG2 cells (Figure S7). These delivered agents could play distinct roles in antitumor therapies.
Wound Healing and Angiogenesis Assays
The up-regulation of hsulf-1 enzyme using gene transfection technology was considered as a potential therapeutic option for inhibiting tumor metastasis. Therefore, in this part of the assay, PNPs for the verification of gene function in vitro. Wound-healing was performed to evaluate the antimigration effect of PNPs. The results demonstrated that cell migration was inhibited in the PNPs group compared to the control, with a lower migration rate (15–20%) observed after 24 and 48 h of drug treatment (Figure 4A and B). This finding implies that therapy based on PNPs could improve the anti-migratory effect of cells. Furthermore, the traditional healing assay model for HCC cannot be used to evaluate the migration of HCC cells. Instead, the migration and invasion of endothelial cells were examined, which are essential for forming new blood vessels during neoangiogenesis, a critical event for tumor growth. Neovascularization depends on angiogenic stimuli to form and organize tubular networks. In vitro, models were used to promote the formation of a crosslinked network of HUVEC-lined tubes using ECM components as growth substrates. The results showed that VEGF promoted endothelial tubular morphogenesis, whereas hsulf-1 remarkably suppressed tube length compared to the control (control: 48,309.00 ± 10,313.92 μm; PNPs: 41,227.66 ±8826.99 μm) (Figure 4C and D). Tube formation in the PNPs group was also lower than that in the control and PNPs (no hsulf-1) groups, indicating an effective inhibitory function of hsulf-1 against VEGF-stimulated tubular structure formation in HUVECs.
 |
Figure 4 Hsulf-1 suppresses migration and angiogenesis. (A) Wound healing assays of HepG2 cells. Scale bar: 100 μm. Histograms show relative cell migration (B). Hsulf-1 inhibits VEGF-induced tube formation of HUVECs (C and D). Scale bar: 25 μm. |
Macrophage Polarization in vitro
Through in-depth studies on the relationship between some bacteria and cancer therapy, researchers have found that bacteria themselves can inhibit tumor growth by activating the body’s immunity. Moreover, numerous studies have shown that bacteria are able to induce different patterns of macrophage polarization. Lipopolysaccharides in the cell walls of E. coli and flagellin in the flagella can activate TLR4- and TLR5-related pathways, respectively, leading to the conversion of M2 macrophages to M1 macrophages.28,46 Macrophages were incubated with various E. coli formulations, and CD86+ cells were analyzed using FCM. The results showed that BL21 and ASEc@PNPs significantly increased the number of M1 macrophages compared with that of PNPs (Figure 5A). It is worth noting that after treating macrophages with BL21 and ASEc@PNPs (67.14 ± 1.15) % and (68.56 ± 0.63) % of the cells, respectively, were positive for the M1 phenotype. This result verified that the use of E. coli and ASEc@PNPs could convert M0 macrophages into M1 macrophages. To further investigate the macrophage polarization, TNF-α and IL-6 secretion by M1 macrophages was measured using an ELISA kit. The results demonstrated that BL21 and ASEc@PNPs upregulated the secretion of TNF-α and IL-6, and ASEc@PNPs exhibited a more remarkable polarization effect than the control (Figure 5B and C). These findings suggest that E. coli can effectively polarize macrophages into M1 macrophages, and ASEc@PNPs might be a potential candidate for promoting this conversion.
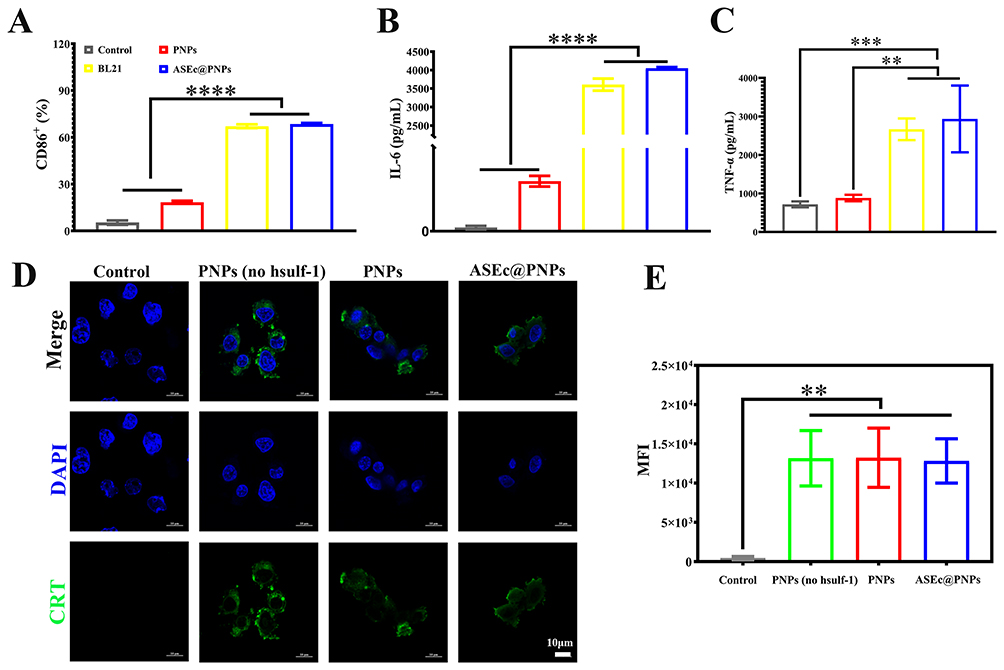 |
Figure 5 ASEc@PNPs induced the ICD of tumor cells and promoted macrophage polarization in vitro. (A) FCM analysis of the proportion of M1 macrophages (labeled with CD86+). Cytokine content of (B) TNF-α and (C) IL-6 tested by ELISA. (D) Immunofluorescence staining images of HepG2 cells. Scale bar: 10 μm. (E) Mean fluorescence intensity of CRT tested by FCM. (**p < 0.01, ***p < 0.001, ****p < 0.0001). Scale bar: 10 μm. |
Induction of ICD in vitro
Recent studies have demonstrated that DOX has the potential to induce ICD, leading to an antitumor immune response. Here, the cell surface expression of CRT, a crucial indicator of ICD, was detected by staining HepG2 cells with CRT polyclonal antibody. CLSM (Figure 5D) and FCM (Figure 5E) were used to determine the extent of CRT expression. Briefly, PNPs (no hsulf-1), PNPs, and ASEc@PNPs were incubated with HepG2 cells for 24 h in Transwell plates. FCM was used to detect the percentages of cells expressing CRT, which were similar among the PNPs (no hsulf-1), PNPs, and ASEc@PNPs groups, with no significant differences ((13.98 ± 3.5) %, (15.47 ± 0.83) %, and (11.68 ± 4.03) %, respectively (Figure S8)). Additionally, immunofluorescence staining showed green fluorescence in all groups, indicating CRT expression in the HepG2 cells (Figure 5D). Similar CRT expression levels were observed in HepG2 cells treated with the three formulations. Therefore, both FCM and immunofluorescence staining confirmed that PNPs (no hsulf-1), PNPs, and ASEc@PNPs trigger ICD in vitro.
In vivo Biodistribution and Tumor Retention of ASEc@PNPs
E. coli is a facultative anaerobe that preferentially colonizes the hypoxic and necrotic zones of a solid tumor after systemic administration. This tumor-targeting mechanism of bacteria is related to their hypoxia-targeting ability, chemotaxis, and motility, as well as the hypoxic, eutrophic, and immunosuppressive microenvironments, and chaotic vasculature in solid tumors.47,48 To investigate the distribution and retention behavior of ASEc@PNPs in vivo, colony counting assays and in vivo optical live imaging were performed. The ASEc@PNPs dispersions were intravenously injected into mice bearing H22 tumors on their flanks. At 1, 5, and 10 days after tail vein injection of ASEc@PNPs, the tumors and major organs were collected, homogenized, and the samples were spread onto LB agar plates. The distribution of ASEc@PNPs was analyzed by counting the E. coli colonies and normalizing them to the tissue weight. Due to the hypoxia-targeting ability of bacteria, ASEc accumulated more obviously at the tumor site than in other tissues. The results in Figure 6A and B show the time-dependent accumulation of ASEc@PNPs in the tumor over 5 days. ASEc were almost entirely concentrated in the tumor and liver in early periods, and the number of bacterial colonies in the tumor increased from days 0 to 5 and then decreased gradually.
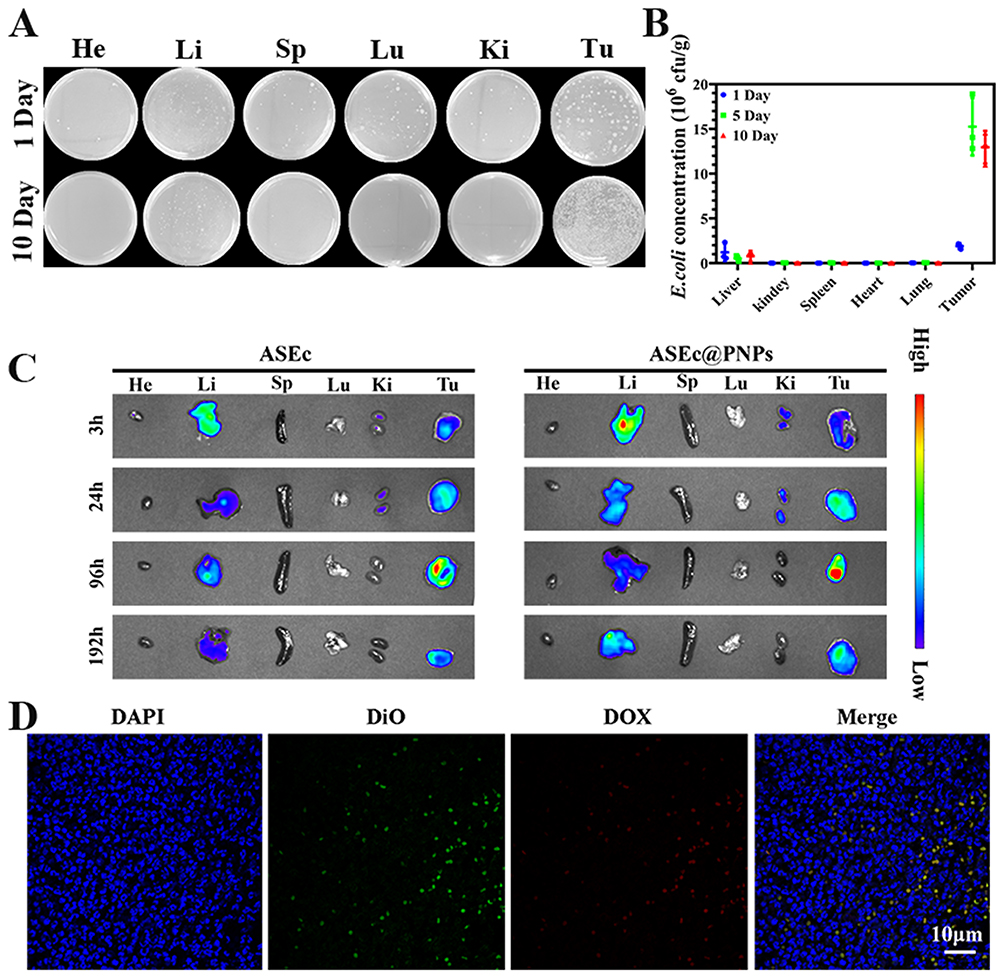 |
Figure 6 In vivo biodistribution of ASEc@PNPs. (A and B) E. coli colony numbers and pictures of plates of major organs and tumor tissues after being homogenized and incubated at 37 °C for 24 h (n = 3). (C) Ex vivo images of major organs and tumors at various time points after ASEc and ASEc@PNPs intravenous injection. (D) Immunofluorescence images of tumor sections intravenously injected with ASEc@PNPs at 24h. Scale bar: 10 μm. |
Based on the obtained results, ASEc was modified with DiR, and ASEc@PNPs was employed for long-term real-time fluorescence monitoring of the in vivo distribution. In tissues collected at various time points, the DiR fluorescence signal was observed in the liver and tumor 3 h post-injection, but the signal in the liver gradually weakened over time (Figure 6C). A strong fluorescence signal was observed at the tumor site 96 h following intravenous administration and was maintained until the end of the observation period (192 h). The fluorescence signal was significantly higher in the tumor than in other organs, indicating the excellent tumor-retention ability of ASEc@PNPs. Sections were stained with ASEc (DiO-membrane, green), DAPI (DNA, blue), and PNPs (red) to further investigate the localization of ASEc@PNPs in tumors using CLSM. The results showed that green fluorescence signals (ASEc) and red fluorescence signals (PNPs) accumulated in the tumor areas (Figure 6D), demonstrating that the active bacterial ASEc delivers PNPs to the tumor region. These results showed the superior targeting capacity of ASEc@PNPs to the tumor region owing to the hypoxic tropism of ASEc in vivo. They also demonstrated that ASEc@PNPs administered systemically could efficiently accumulate and be retained in tumors, whereas it was rapidly metabolized and eliminated in normal organs.
In vivo Anti-Tumor Study and Biosecurity Assay of ASEc@PNPs
The in vivo antitumor effects of ASEc@PNPs were assessed using H22 tumor-bearing murine models. The tumors were subcutaneously injected into the right flank of the mice, and the drug treatment procedure is illustrated in Figure 7A. Balb/c mice bearing large solid tumors (80–100 mm3) were injected with PBS, DOX (0.25 and 5 mg/kg), PNPs (no hsulf-1), PNPs, AEc, and ASEc@PNPs via the tail vein. The antitumor efficacy of each formulation was evaluated by determining tumor volume, survival rate, and body weight. Images of the excised tumors are shown in Figure 7B. The PBS group showed rapid tumor progression, with the tumor volume reaching 1135.69 ± 83.30 mm3 (13.26-fold higher than that in the ASEc@PNPs group). Although the administration of AEc alone initially slowed the tumor growth, likely due to the activation of innate immune cells by the bacterial adjuvant, the final tumor volume was 726.83 ± 81.11 mm3. In contrast, ASEc@PNPs treatment resulted in the slowest tumor growth, with a mean tumor volume of 85.62 mm3 (Figure S9A) and a tumor weight of 55.00 ± 29.33 mg (Figure 7D) on day 14. ASEc@PNPs led to a lower relative tumor volume (Figure 7C) and relative tumor growth rate (T/C), with a T/C rate of 3.62% (Figure S9B) that was attributed to the combination therapy with PNPs and ASEc could improve the anti-HCC efficacy. Accordingly, we hypothesized that continuous production and release of Ag by ASEc at the tumor site, in addition to the presence of DOX and the hsulf-1 enzyme, might activate CD8+ T cells, trigger cytokine release, and ultimately lead to HCC cell death. To investigate the in situ production of Ag, its levels in the serum and tumor tissues of mice were measured using ELISA kits. As shown in Figure 7F and G, ASEc could induce significantly higher Ag production than AEc, with Ag release occurring for at least 2 weeks (Figure S10A and B). Furthermore, the results of studying the tumor immune response in vivo confirmed the hypothesis. To further evaluate the in vivo antitumor activity, H&E, TUNEL, and anti-Ki67 staining were performed (Figure 7J). Nuclear damage and cytosolic degradation were most obvious in the ASEc@PNPs group. Moreover, the ASEc@PNPs group exhibited the largest apoptotic area, as observed using TUNEL staining, and the smallest proliferation area, as observed using anti-Ki67 staining, further demonstrating the enhanced antitumor function of the combination therapy.
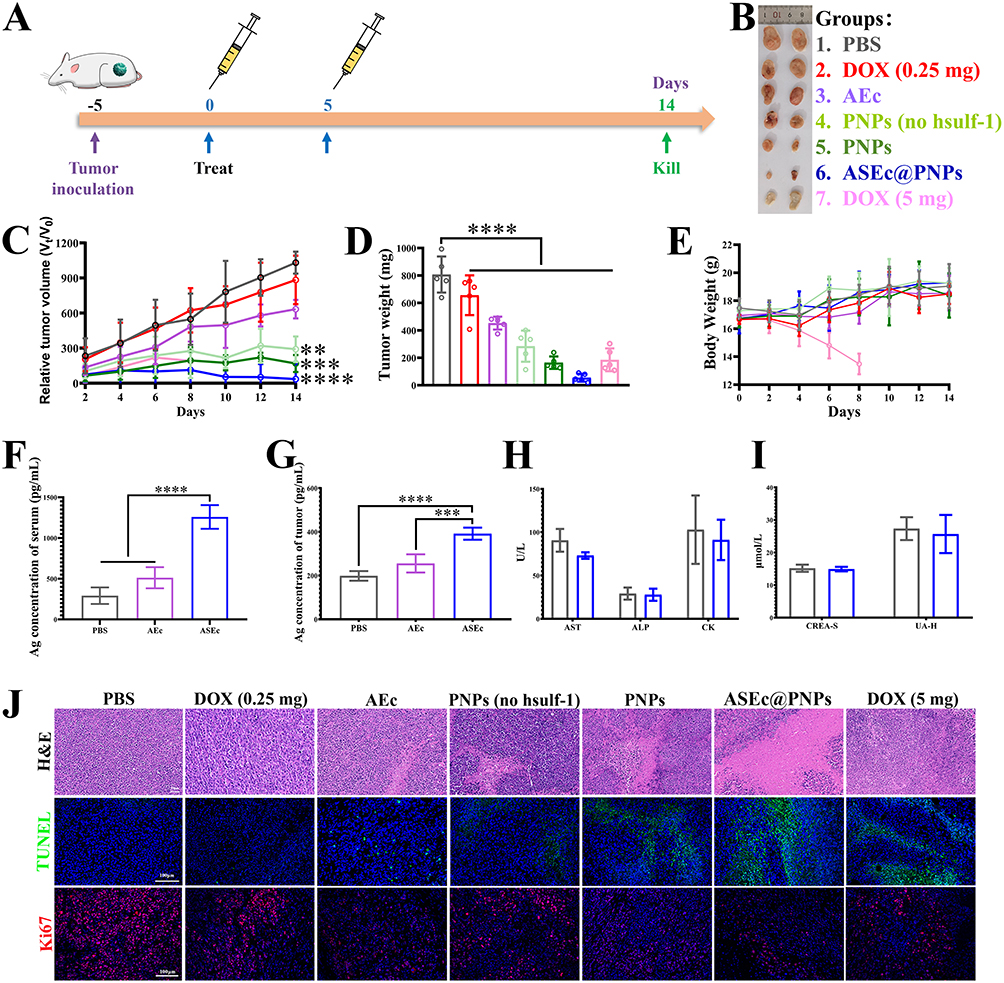 |
Figure 7 Anti-tumor effect in vivo. (A) The therapeutic schedule of ASEc@PNPs for inhibiting tumor growth (n = 5). (B) Images of isolated tumors on day 14. (C) Relative tumor volume variation of mice after different treatments. (D) Weight of the tumor after different treatments. (E) Average body weight of tumor-bearing mice during treatment. The serum (F) and tumor tissues (G) Ag levels of mice at 7 days. Serum levels of AST, ALP, CK (H), CREA-S, and UA-H (I). (J) H&E, TUNEL, and Ki67 staining images of the tumor tissues. Scale bar: 100 μm (**p <0.01, ***p <0.001, ****p < 0.0001). |
The safety profiles of drug delivery systems are crucial parameters, and, therefore, we conducted a series of biosafety experiments in mice using our formulations. In addition to the average body weight, H&E-stained images of the major organs and blood biochemistry indicators were analyzed to evaluate whether the developed formulations showed any detectable side effects. Body weights were measured as an indicator of systemic toxicity. Except for the 5 mg/kg free DOX group, the weight of the mice in all groups fluctuated around 18 g without significant changes during the treatment period, indicating that the materials were safe for the mice (Figure 7E). The weight of the mice in the 5 mg/kg free DOX group was the lightest, which was due to high cardiotoxicity of DOX and its reduction to semiquinone in the body. Additionally, as shown in Figure S11, the images of the H&E-stained major organs (heart, liver, spleen, and kidney) showed no obvious differences among the groups. Blood samples from the mice were obtained for in vivo biosecurity analysis. The values for liver function (including alkaline phosphatase [ALP] and aspartate aminotransferase [AST]), kidney function (including UREA-S and uric acid [UA-H]), and heart function (including creatine kinase [CK]) were maintained within the standard range (Figure 7H and I). This observation indicated that ASEc@PNPs is nontoxic and could be used in the delivery of antitumor drugs.
Anti-Tumor Immune Responses of ASEc@PNPs in vivo
Bacteria-based antitumor vaccines have attracted considerable interest.29,49,50 Bacteria are immunostimulatory agents that activate antitumor immunity. In this study, ASEc@PNPs was designed as an integrated nanosystem, in which Ag was chosen as the emodel antigen.51 Inspired by the promising in vitro polarization results, we further investigated the antitumor immune effect of ASEc@PNPs in vivo. An H22 tumor model was constructed by injecting H22 cells into the right hind legs of Balb/c mice. The tumor-bearing mice were intravenously administered PNPs (no hsulf-1), PNPs, AEc, or ASEc@PNPs. Recent studies have demonstrated the ability of DOX to induce ICD, thereby triggering an antitumor immune response. To evaluate DOX-induced ICD in tumor cells in vivo, immunofluorescence staining was performed to detect CRT expression. As shown in Figure 8A, pink fluorescence was observed in the PNPs (no hsulf-1), PNPs and ASEc@PNPs groups, indicating similar CRT expression in H22 cell-derived tumors treated with the three formulations. In addition, CD4+ and CD8+ T cell infiltration was assessed using immunofluorescence. The green fluorescence of CD4+ T cells and red fluorescence of CD8+ T cells were barely observable in the tissue sections of the PBS, PNPs (no hsulf-1) and PNPs groups. In contrast, relatively strong green and red fluorescence was observed in the ASEc@PNPs group (Figure 8E). This phenomenon indicated the effect of tumor-associated macrophage polarization and ICD, which weakens the suppression of T cells caused by the tumor microenvironment, thus activating CD4+ and CD8+ T cells, and improving their tumor infiltration. On day 14, the serum levels of the proinflammatory mediators IFN-γ, IL-6, and TNF-α remained higher in the ASEc@PNPs group than in the other groups. Specifically, the levels of IFN-γ, IL-6, and TNF-α were 2.07-, 4.08-, and 8.20-fold higher in the ASEc@PNPs group than in the PBS group (Figure 8B-D). The induction of cytokine secretion might play a role in the enhanced antitumor effect of combination therapy.
 |
Figure 8 In vivo CRT and anti-tumor immunity. (A) Immunofluorescence images of tumor sections after CRT staining (pink) assay. Scale bar: 2 mm. (B) IFN- γ, (D)TNF- α, and (C) IL-6 levels in the serum of mice treated as indicated 14 days after injection. (E) Immunofluorescence staining of CD4+ T cells (green) and CD8+ T cells (red) in the tumor tissues. Scale bar: 100 μm (*p <0.05, **p <0.01, ***p <0.001, ****p < 0.0001). |
Lung Metastasis Inhibition
Distant metastasis is one of the primary contributors to poor prognosis in patients with advanced HCC, and the lungs are the most common site of metastasis; approximately 55% of patients with HCC exhibit extrahepatic spread to the lungs.52 Heparan sulfate proteoglycans (HSPGs) modulate the binding of receptors and activation of signaling pathways involving specific growth factors in tumor cells, allowing the tumor to recur and metastasize.53 It was reported that hsulf-1, which hydrolyzes the 6-O sulfate-ester bonds of HSPGs, was underexpressed in HCC.54 Therefore, the inhibitory effect of ASEc@PNPs on lung metastasis was evaluated. A pulmonary metastasis mouse model was established by intravenous injection of H22 cells into the tail vein, as illustrated in Figure 9A. The tumor area in the lung tissue was further verified using H&E staining (Figure 9B). The lung tissues of the mice from the PBS, AEc, and PNPs (no hsulf-1) groups displayed evident tumor areas, whereas less profound tumor areas were observed in the lungs of the mice from the PNPs and ASEc@PNPs groups. This might be attributed to the tumor metastasis inhibition provided by upregulation of hsulf-1 expression in HCC cells.
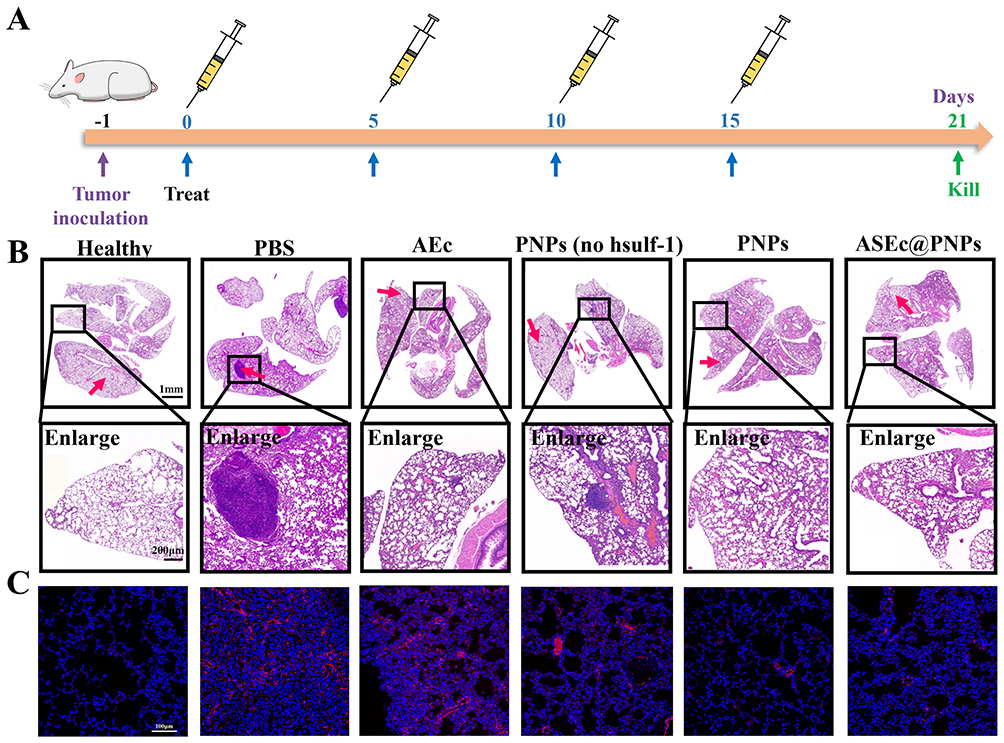 |
Figure 9 In vivo anti-pulmonary metastasis effect. (A) Schematic illustration of the schedule for drug treatment. (B) H&E analysis of the lung. Scale bars: 1mm, 200 μm. (C) Immunofluorescence assay of the α-SMA protein (red arrow). Scale bars:100 μm. |
Pulmonary fibrosis is the pathological basis of several fibroproliferative lung diseases.55,56 The invasion and residence of HCC cells in the lungs may activate normal fibroblasts to transition into myofibroblasts. To confirm pulmonary fibrosis, an immunofluorescence staining assay was performed to evaluate the α-smooth muscle actin (α-SMA) expression level in the lungs. As shown in Figure 9C, red signals were observed in the lung metastasis model but not in healthy lungs, indicating that metastatic HCC cells could activate quiescent fibroblasts in the lungs. Following drug treatment, α-SMA expression was downregulated in all treatment groups. Interestingly, almost no red fluorescent signal was observed in the PNPs and ASEc@PNPs groups, suggesting that PNPs and ASEc@PNPs effectively blocked fibroblast activation. This might be attributed to tumor metastasis inhibition resulting from the release of the hsulf-1 enzyme by PNPs. Therefore, ASEc@PNPs inhibited primary tumor growth and suppressed tumor metastasis. These results show that our combination formulation could achieve an ideal therapeutic effect. In summary, ASEc@PNPs exhibited a stronger antitumor effect than its individual components.
Conclusion
Chemotherapy is a universal method for HCC. However, the efficacy of a single chemotherapy agent in the clinic is limited owing to the biological characteristics of tumor sites. Combination therapy based on antitumor drugs with different mechanisms of action is the most effective method for treating HCC. To enhance the therapeutic effect, a strategy composed of enhanced tumor immunity-regulated bacteria and the loaded chemical-drugs/nucleic acids of PNPs was developed. In summary, we exploited functional and biohybrid drug delivery systems of ASEc@PNPs, which exhibited excellent tumor-targeting ability, and improved the tumor immunosuppressive microenvironment, robustly evoking systematic antitumor immune responses, and inhibiting HCC growth and metastasis, while reducing the destruction of normal tissues surrounding the tumor niche. The delivery system was constituted by bacteria and PNPs, engineered bacteria for enhancing anti-tumor immune response and PNPs to simultaneously kill tumor cells and inhibit tumor metastasis. In vivo assays indicated ASEc@PNPs could significantly accumulate in tumor tissues. The engineered bacteria could produce and release Ag for 2 weeks, activate CD4+ and CD8+ T cells in tumor tissues, and induce the secretion of cytokines. Moreover, PNPs could trigger ICD, which promotes the polarization of M0 macrophages to M1 and synergistically activates antitumor immunity and suppresses tumor growth. A biosafety assay showed that ASEc@PNPs would not cause obvious side effects. Overall, the drug delivery system is based on engineered bacteria and provides a new strategy to improve the immunotherapy efficiency for HCC treatment.
Abbreviations
HCC, hepatocellular carcinoma; DLS, dynamic light scattering; TEM, transmission electron microscopy; CLSM, confocal fluorescence images; FCM, flow cytometry assay; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; CHO-PEG2000-CHO, aldehyde group modified polyethylene glycol; PEI25k, poly(ethyleneimine); CA, citraconic anhydride; DOX, doxorubicin; AEc, engineering bacteria (pAg); ASEc, engineering bacteria (pAg+Px174E).
Acknowledgments
This work was supported by the Wuxi Science and Technology Development Fund Project (K20221018).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63(5):844–855. doi:10.1136/gutjnl-2013-306627
2. Okuda K. Hepatocellular carcinoma - history, current status and perspectives. Digest Liver Dis. 2002;34(9):613–616. doi:10.1016/S1590-8658(02)80200-67
3. Singh AK, Kumar R, Pandey AK. Hepatocellular carcinoma: causes, mechanism of progression and biomarkers. Curr Chem Genom Transl Med. 2018;12:9–26. doi:10.2174/2213988501812010009
4. Kong H, Ju E, Yi K, et al. Advanced nanotheranostics of CRISPR/Cas for viral hepatitis and hepatocellular carcinoma. Adv Sci. 2021;8(24):e2102051. doi:10.1002/advs.202102051
5. Karagiannakis DS. Systemic treatment in intermediate stage (Barcelona Clinic Liver Cancer-B) hepatocellular carcinoma. Cancers. 2023;16(1). doi:10.3390/cancers16010051
6. Zheng K, Wang X. Techniques and status of hepatic arterial infusion chemotherapy for primary hepatobiliary cancers. Ther Adv Med Oncol. 2024;16:1758–8359. doi:10.1177/17588359231225040
7. Yu YC, Mao YM, Chen CW, et al. CSH guidelines for the diagnosis and treatment of drug-induced liver injury. Hepatol Int. 2017;11(3):221–241. doi:10.1007/s12072-017-9793-2
8. Yi K, Kong H, Zheng C, et al. A LIGHTFUL nanomedicine overcomes EGFR-mediated drug resistance for enhanced tyrosine-kinase-inhibitor-based hepatocellular carcinoma therapy. Biomaterials. 2023;302:122349. doi:10.1016/j.biomaterials.2023.122349
9. Noori M, Jafari-Raddani F, Davoodi-Moghaddam Z, et al. Immune checkpoint inhibitors in gastrointestinal malignancies: an Umbrella review. Cancer Cell Int. 2024;24(1):10. doi:10.1186/s12935-023-03183-3
10. Zhu HD, Li HL, Huang MS, et al. Transarterial chemoembolization with PD-(L)1 inhibitors plus molecular targeted therapies for hepatocellular carcinoma (CHANCE001). Signal Transduct Target Ther. 2023;8(1):58. doi:10.1038/s41392-022-01235-0
11. Pinter M, Jain RK, Duda DG. The current landscape of immune checkpoint blockade in hepatocellular carcinoma. JAMA Oncol. 2021;7(1):113–123. doi:10.1001/jamaoncol.2020.3381
12. Escartin A, Sapisochin G, Bilbao I, et al. Recurrence of hepatocellular carcinoma after liver transplantation. Transplant Proc. 2007;39(7):2308–2310. doi:10.1016/j.transproceed.2007.06.042
13. Bi N, Shedden K, Zheng X, et al. Comparison of the effectiveness of radiofrequency ablation with stereotactic body radiation therapy in Inoperable stage I non-small cell lung cancer: a systemic review and pooled analysis. Int J Radiat Oncol Biol Phys. 2016;95(5):1378–1390. doi:10.1016/j.ijrobp.2016.04.016
14. Schonherr J, Seifert P, Guhne F, et al. Transarterial radioembolization (TARE) in patients with hepatocellular carcinoma: a comparison of palliative with bridging-to-transplant concepts. Cancers. 2024;16(1). doi:10.3390/cancers16010235
15. Xiang C, Shen X, Zeng X, et al. Effect of transarterial chemoembolization as postoperative adjuvant therapy for intermediate stage hepatocellular carcinoma with microvascular invasion: a multicenter cohort study. Int J Surg. 2023;110(1):315–323. doi:10.1097/JS9.0000000000000805
16. Zhao Y, Liu J, Xiong Z, et al. Exosome-derived miR-23a-5p inhibits HCC proliferation and angiogenesis by regulating PRDX2 expression: miR-23a-5p/PRDX2 axis in HCC progression. Heliyon. 2024;10(1):e23168. doi:10.1016/j.heliyon.2023.e23168
17. Yang F, Xu GL, Huang JT, et al. Transarterial chemoembolization combined with immune checkpoint inhibitors and tyrosine kinase inhibitors for unresectable hepatocellular carcinoma: efficacy and systemic immune response. Front Immunol. 2022;13:847601. doi:10.3389/fimmu.2022.847601
18. Zhou S, Gravekamp C, Bermudes D, et al. Tumour-targeting bacteria engineered to fight cancer. Nat Rev Cancer. 2018;18(12):727–743. doi:10.1038/s41568-018-0070-z
19. Wang S, Cheng M, Chen CC, et al. Salmonella immunotherapy engineered with highly efficient tumor antigen coating establishes antigen-specific CD8+ T cell immunity and increases in antitumor efficacy with type I interferon combination therapy. Oncoimmunology. 2024;13(1):2298444. doi:10.1080/2162402X.2023.2298444
20. Han ZY, Zhang C, An JX, et al. Metabolic regulation of tumor microenvironment with biohybrid bacterial bioreactor for enhanced cancer chemo‐immunotherapy. Adv Funct Mater. 2023;33:35. doi:10.1002/adfm.202302728
21. Zhou T, Wu J, Tang H, et al. Enhancing tumor-specific recognition of programmable synthetic bacterial consortium for precision therapy of colorectal cancer. NPJ Biofilms Microbiomes. 2024;10(1):6. doi:10.1038/s41522-024-00479-8
22. Yu X, Lin C, Yu J, et al. Bioengineered Escherichia coli nissle 1917 for tumour-targeting therapy. Microb Biotechnol. 2020;13(3):629–636. doi:10.1111/1751-7915.13523
23. Siddiqui NA, Ventrola AJ, Hartman AR, et al. An Engineered probiotic platform for cancer epitope-independent targeted radionuclide therapy of solid tumors. Adv Healthc Mater. 2023:e2202870. doi:10.1002/adhm.202202870
24. Liu Y, Lu Y, Ning B. Intravenous delivery of living Listeria monocytogenes elicits gasdmermin-dependent tumor pyroptosis and motivates anti-tumor immune response. ACS Nano. 2022;16(3):4102–4115. doi:10.1021/acsnano.1c09818
25. Curran CS, Rasooly A, He M, et al. Report on the NCI microbial-based cancer therapy conference. Cancer Immunol Res. 2018;6(2):122–126. doi:10.1158/2326-6066.CIR-17-0748
26. Lin Z, Meng F, Ma Y, et al. In situ immunomodulation of tumors with biosynthetic bacteria promote anti-tumor immunity. Bioact Mater. 2024;32:12–27. doi:10.1016/j.bioactmat.2023.09.007
27. Song S, Vuai MS, Zhong M. The role of bacteria in cancer therapy - enemies in the past, but allies at present. Infect Agent Cancer. 2018;13:9. doi:10.1186/s13027-018-0180-y
28. Yang X, Komatsu S, Reghu S, et al. Optically activatable photosynthetic bacteria-based highly tumor specific immunotheranostics. Nano Today. 2021;37:101100. doi:10.1016/j.nantod.2021.101100
29. Zhu J, Ke Y, Liu Q, et al. Engineered Lactococcus lactis secreting Flt3L and OX40 ligand for in situ vaccination-based cancer immunotherapy. Nat Commun. 2022;13(1):7466. doi:10.1038/s41467-022-35130-7
30. Savage TM, V. RL, Rae SS, et al. Chemokines expressed by engineered bacteria recruit and orchestrate antitumor immunity. Sci Adv. 2023;9:eadc9436. doi:10.1126/sciadv.adc94
31. Pan P, Dong X, Chen Y, et al. Engineered bacteria for enhanced radiotherapy against breast carcinoma. ACS Nano. 2022;16:801–812. doi:10.1021/acsnano.1c08350
32. Fan JX, Li ZH, Liu XH, et al. Bacteria-mediated tumor therapy utilizing photothermally-controlled TNF-alpha expression via oral administration. Nano Lett. 2018;18(4):2373–2380. doi:10.1021/acs.nanolett.7b05323
33. Canale FP, Basso C, Antonini G, et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature. 2021;598:662–666. doi:10.1038/s41586-021-04003-2
34. Jia HR, Zhu YX, Liu Y, et al. Direct chemical editing of gram-positive bacterial cell walls via an enzyme-catalyzed oxidative coupling reaction. Exploration. 2022;2(5):20220010. doi:10.1002/EXP.20220010
35. Chowdhury S, Castro S, Coker C, et al. Programmable bacteria induce durable tumor regression and systemic antitumor immunity. Nat Med. 2019;25(7):1057–1063. doi:10.1038/s41591-019-0498-z
36. Din MO, Danino T, Prindle A, et al. Synchronized cycles of bacterial lysis for in vivo delivery. Nature. 2016;536(7614):81–85. doi:10.1038/nature18930
37. Zhan Y, Zhang Y, Mao C, et al. Improved tumor infiltration and immunomodulation for tumor therapy: a pathway based on tetrahedral framework nucleic acids coupled bacterial nanocells. Nano Lett. 2023;23(1):353–362. doi:10.1021/acs.nanolett.2c03943
38. Guo H, Cao Z, Li J, et al. Integrating bacteria with a ternary combination of photosensitizers for monochromatic irradiation-mediated photoacoustic imaging-guided synergistic photothermal therapy. ACS Nano. 2023;17(5):5059–5071. doi:10.1021/acsnano.3c00032
39. Chen B, Zhang X, Cheng L, et al. Surface programmed bacteria as photo-controlled NO generator for tumor immunological and gas therapy. J Control Release. 2023;353:889–902. doi:10.1016/j.jconrel.2022.12.030
40. Yang M, Chen W, Yang F, et al. Engineered bacteria combined with doxorubicin nanoparticles suppress angiogenesis and metastasis in murine melanoma models. Acta Biomater. 2023;158(1):734–746. doi:10.1016/j.actbio.2022.12.027
41. Guan X, Guo Z, Wang T, et al. A pH-responsive detachable PEG shielding strategy for gene delivery system in cancer therapy. Biomacromolecules. 2017;18(4):1342–1349. doi:10.1021/acs.biomac.7b00080
42. Xu C, Wang P, Zhang J. Pulmonary codelivery of doxorubicin and siRNA by pH-sensitive nanoparticles for therapy of metastatic lung cancer. Small. 2015;11(34):4321–4333. doi:10.1002/smll.201501034
43. Gupta A, Reizman IM, Reisch CR, et al. Dynamic regulation of metabolic flux in engineered bacteria using a pathway-independent quorum-sensing circuit. Nat Biotechnol. 2017;35(3):273–279. doi:10.1038/nbt.3796
44. Stricker J, Cookson S, Bennett MR, et al. A fast, robust and tunable synthetic gene oscillator. Nature. 2008;456(7221):516–519. doi:10.1038/nature07389
45. Wu S, Liu J, Liu C, et al. Quorum sensing for population-level control of bacteria and potential therapeutic applications. Cellular Mol Life Sci. 2019;77(7):1319–1343. doi:10.1007/s00018-019-03326-8
46. Baicheng Wei JP, Yuan R, et al. Polarization of tumor-associated macrophages by nanoparticle loaded Escherichia coli combined with immunogenic cell death for cancer immunotherapy. Nano Lett. 2021;21(10):4231–4240. doi:10.1021/acs.nanolett.1c00209
47. Lou X, Chen Z, He Z, et al. Bacteria-mediated synergistic cancer therapy: small microbiome has a big hope. Nanomicro Lett. 2021;13(1):37. doi:10.1007/s40820-020-00560-9
48. Chen X, Li P, Luo B, et al. Surface Mineralization of engineered bacterial outer membrane vesicles to enhance tumor photothermal/immunotherapy. ACS Nano. 2024;18(2):1357–1370. doi:10.1021/acsnano.3c05714
49. Hu Q, Wu M, Fang C, et al. Engineering nanoparticle-coated bacteria as oral DNA vaccines for cancer immunotherapy. Nano Lett. 2015;15(4):2732–2739. doi:10.1021/acs.nanolett.5b00570
50. Vincent RL, G. CR, Fangda L, et al. Probiotic-guided CAR-T cells for solid tumor targeting. Science. 2023;382:211–218. doi:10.1126/science.add7034
51. Wang W, Xu H, Ye Q, et al. Systemic immune responses to irradiated tumours via the transport of antigens to the tumour periphery by injected flagellate bacteria. Nat Biomed Eng. 2022;6(1):44–53. doi:10.1038/s41551-021-00834-6
52. Chandradas S, Skillin CB, Frenette CT. Hepatocellular carcinoma: metastatic disease. Int Cancer Metastasis. 2017;163–175. doi:10.1016/B978-0-12-804003-4.00009-8
53. Liu P, Gou M, Yi T, et al. Efficient inhibition of an intraperitoneal xenograft model of human ovarian cancer by HSulf-1 gene delivered by biodegradable cationic heparin-polyethyleneimine nanogels. Oncol Rep. 2012;27(2):363–370. doi:10.3892/or.2011.1550
54. Li J, Mo ML, Chen Z, et al. HSulf-1 inhibits cell proliferation and invasion in human gastric cancer. Cancer Sci. 2011;102(10):1815–1821. doi:10.1111/j.1349-7006.2011.02024.x
55. Liu S-S, Liu C, Lv X-X, et al. The chemokine CCL1 triggers an AMFR-SPRY1 pathway that promotes differentiation of lung fibroblasts into myofibroblasts and drives pulmonary fibrosis. Immunity. 2021;54(9):2042–2056.e2048. doi:10.1016/j.immuni.2021.06.008
56. Gong X, Wu J, Wen J, et al. Dual-Ligand-Modified Nanoscale Liposomes Loaded with Curcumin and Metformin Inhibit Drug Resistance and Metastasis of Hepatocellular Carcinoma. ACS Appl Nano Mater. 2022;5(5):7063–7077. doi:10.1021/acsanm.2c01027
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.