Back to Journals » International Journal of Nanomedicine » Volume 19
Nanoparticle-Mediated Synergistic Chemoimmunotherapy for Cancer Treatment
Authors Lang X, Wang X, Han M, Guo Y
Received 16 December 2023
Accepted for publication 7 May 2024
Published 21 May 2024 Volume 2024:19 Pages 4533—4568
DOI https://doi.org/10.2147/IJN.S455213
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jie Huang
Xiaoxue Lang,1 Xiangtao Wang,1 Meihua Han,1 Yifei Guo1– 4
1Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, People’s Republic of China; 2Key Laboratory of Bioactive Substances and Resources Utilization of Chinese Herbal Medicine, Ministry of Education, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, People’s Republic of China; 3Key Laboratory of New Drug Discovery Based on Classic Chinese Medicine Prescription, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 4Beijing Key Laboratory of Innovative Drug Discovery of Traditional Chinese Medicine (Natural Medicine) and Translational Medicine, Beijing, People’s Republic of China
Correspondence: Yifei Guo, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, No. 151, Malianwa North Road, Haidian District, Beijing, 100193, People’s Republic of China, Tel +86 18810811910, Email [email protected]
Abstract: Until now, there has been a lack of effective strategies for cancer treatment. Immunotherapy has high potential in treating several cancers but its efficacy is limited as a monotherapy. Chemoimmunotherapy (CIT) holds promise to be widely used in cancer treatment. Therefore, identifying their involvement and potential synergy in CIT approaches is decisive. Nano-based drug delivery systems (NDDSs) are ideal delivery systems because they can simultaneously target immune cells and cancer cells, promoting drug accumulation, and reducing the toxicity of the drug. In this review, we first introduce five current immunotherapies, including immune checkpoint blocking (ICB), adoptive cell transfer therapy (ACT), cancer vaccines, oncolytic virus therapy (OVT) and cytokine therapy. Subsequently, the immunomodulatory effects of chemotherapy by inducing immunogenic cell death (ICD), promoting tumor killer cell infiltration, down-regulating immunosuppressive cells, and inhibiting immune checkpoints have been described. Finally, the NDDSs-mediated collaborative drug delivery systems have been introduced in detail, and the development of NDDSs-mediated CIT nanoparticles has been prospected.
Keywords: chemoimmunotherapy, chemotherapy, immunotherapy, NDDSs
Graphical Abstract:

Introduction
Cancer is a global health problem, for example, in the United States alone, 2,001,140 new cancer cases and 611,720 cancer-related deaths are expected to occur in 2024.1 For decades, the most widely used traditional treatments used by cancer patients have been surgery, chemotherapy and radiotherapy. Surgery and radiotherapy are effective for local and non-metastatic cancers but struggle to work when the cancer has spread. Emerging therapies, including gene therapy and hormone therapy, are also attracting researchers’ attention. Gene therapy manipulates cells by introducing genetic material into cells that do not function properly. Gene therapy is still experimental and there is no guarantee it will have a positive effect on everyone. After the infusion, the body’s immune system may respond differently to the virus carrying the altered gene, with a low fault tolerance rate. Hormone therapies work by interfering with hormone signals, and they may attack different parts of the signaling pathway to slow the growth of cancer. Hormone therapy is usually combined with surgery/radiation/chemotherapy. In these cases, hormone therapy is considered an adjunct therapy. Chemotherapy is a way of selectively destroying tumor cells or at least limiting their proliferation using low molecular weight drugs.2 Cancer drugs can reach all organs of the body through the bloodstream, so chemotherapy is currently the preferred method for metastatic cancers.3 The four most common mechanisms of action include alkylating agents, antimetabolites, antimicrotubule agents, and topoisomerase inhibitors.4 However, chemotherapy often fails in cancer treatment due to the physiological complexity of cancer, which results in poor drug pharmacokinetics, multiple drug resistance (MDR) in clinical practice.5 In addition, chemotherapeutic drugs generally have non-specific targets, which may cause systemic toxicity and severe side effects, including nausea, vomiting, fatigue, hair loss, gastrointestinal side effects, central and peripheral neurotoxicity, etc. Most side effects subside at the end of treatment, but some can cause permanent damage to the kidneys, heart, lungs or reproductive system.6,7 Therefore, the search for new treatment modalities and the development of combined treatment is imminent.
Immunotherapy takes advantage of the patient’s immune system to selectively target tumor cells and is currently considered the fifth treatment option for cancer therapy in addition to surgery, chemotherapy, molecular targeted therapy, and radiotherapy.8,9 In 1891, William B. Coley injected streptococcal organisms into an inoperable cancer patient to stimulate the immune system, and finally the tumor disappeared.10 This is one of the first examples of immunotherapy. Based on available clinical trials, chimeric antigen receptor T cells (CAR-T cells) immunotherapy is significantly superior to current standard chemotherapeutics in the treatment of multiple hematologic malignancies.11 Despite the expectations raised by immunotherapy as a promising cancer treatment modality, it has not achieved desirable results in the therapy of certain clinical tumor types and patients. In the tumor microenvironment (TME), some immune cell phenotypes completely deprive the immune effect, or that do not properly infiltrate the tumor, which are not responsive to immune checkpoint blockade (ICB) monotherapy.4 In addition, the complex tumor environment leads to immunotherapy resistance, low immunogenicity of vaccines, and significant immune-related adverse events (irAEs), which limit the therapeutic efficacy of single immunotherapy.12
Chemoimmunotherapy (CIT) is able to combine conventional chemotherapy with current immunotherapeutic to synergistically inhibit tumor progression, metastasis, and recurrence.13 Chemotherapy drugs have been considered immunosuppressive before, but recent experimental data show that they enable to improve the anti-tumor effectiveness of immunotherapy by regulating the immunosuppressive microenvironment, modulating the expression of tumor antigen or immune checkpoint molecules, enhancing the cross-presentation of tumor antigen, and overcoming partial immunosuppression.14 The effective time of chemotherapy and immunotherapy are complementary. Chemotherapeutic drugs work quickly but for a short duration, while immunotherapy shows strong long-term anti-tumor effects. In addition, immunotherapy can kill chemotherapy-resistant cells and cancer stem cells to make up for the deficiency of chemotherapy.12 Unfortunately, although CIT has great success in treating hematologic tumors, its therapeutic effect on solid tumors remains limited.15,16 Low drug delivery efficiency, different physicochemical properties of drugs, and differences in delivery targets are all obstacles to co-delivery. These factors cause the pharmacokinetics, in vivo distribution, and efficacy of combined delivery to be uncertain.13,17,18 Therefore, it is crucial to design a vector that can simultaneously and deliver these two drugs and enable effective synergistic combined treatment.
In recent years, nano-based drug delivery systems (NDDSs) have received increasing attention for their outstanding performance in disease treatment, especially in cancer treatment strategies.19 NDDS has been used in a variety of cancer treatment regimens. For example, in gene therapy, exogenous genetic material needs to enter the nucleus for expression, so the design of suitable drug delivery systems is necessary. Nanocarriers represented by liposomes and plasmids have the advantages of low cost, simple preparation, easy large-scale production, high safety, and unlimited length of foreign genes, which have attracted the attention of some researchers. NDDSs can improve drug solubility, stability, and targeting, and have great potential in CIT drug delivery. In this review, in addition to describing current immunotherapies, the immunomodulatory effects of chemotherapy are also discussed. Subsequently, nanotechnology-based co-delivery systems and multiple drug delivery modes are also described. Finally, we prospect the application prospect of nanoparticle-mediated anti-tumor chemotherapy immunotherapy.
Cancer Immunotherapy
The key to understanding cancer cell avoidance of immune killing is to figure out the concept of cancer immunoediting. It consists of three consecutive segments: elimination, equilibrium, and escape. The host’s innate and acquired immune systems jointly identify and respond to tumor-specific antigens (TSAs) during the elimination phase. Some mutated tumor cells survive and enter the equilibrium phase. In this stage, the acquired immune system suppresses complete tumor growth and at the same time applies selective pressure on some of the cells to make them further mutate, and is gradually resistant to immune elimination. Eventually, the mutated tumor cells enter the escape phase. Thus, the immune system plays a dual role in inhibiting and promoting tumor development.20 Cancer immunotherapy progresses depending on the study of tumor escape mechanism, which utilizes human’s own immune regulation to improve anti-tumor immune response by stimulating or inhibiting the components and activities of the immune system. In this section, several immunotherapies will be described, including immune checkpoint blockade (ICB), adoptive cellular immunotherapy (ACI), cancer vaccines, oncolytic virotherapy (OVT) and cytokine therapy.
Immune Checkpoint Blockade (ICB)
James Allison and Tasuku Honjo were awarded the 2018 Nobel Prize in Physiology or Medicine for their contribution to the discovery of negative immunomodulatory therapies for cancer treatment.21 Immune checkpoint inhibitors (ICIs) enhance anti-tumor immune responses by targeting immune-related receptors on the surface of T-lymphocytes.22,23 Over the past decade, the most widely used ICIs have been antibodies that inhibit cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed death-1 (PD-1), and programmed cell death-ligand 1 (PD-L1).
T-cell activation is based on two sequential signals. First, major histocompatibility complex (MHC) I or II on specific antigen-presenting cells (APCs) binds to T-cell receptors (TCRs). Next, a costimulatory signal is needed to amplify the response. The interaction of T-cell transmembrane protein CD28 with the B7 molecule on the APCs surface is the most important aspect of these co-stimulatory signals. CTLA-4 has a homologous conformation with CD28, and its affinity is significantly higher than that of CD28. Therefore, CTLA-4 weakens costimulatory signals by competing with CD28 for their common ligands B7-1 (CD80) and B7-2 (CD86).24,25 The use of antibodies and fusion proteins to target the CD28/CTLA-4 pathway holds considerable promise in cancer and therapy.26 Linsley et al first reported that anti-CTLA-4 antibodies have the ability to stimulate T-cell activation, and the Allison laboratory reported the tumor-suppressive effect of antibodies injected with CTLA-4 in tumor-bearing mice and led to immunity against secondary contact with tumor cells.27,28 In 2011, the US Food and Drug Administration (FDA) approved Ipilimumab for melanoma, which is the first ICI targeting CTLA-4. It was the first available ICI, and ushered in a new era of cancer immunotherapy.29,30 Subsequently, another human IgG2 monoclonal antibody Tremelimumab blocking CTLA-4 was developed by Pfizer and AstraZeneca, which was combined with durvalumab for unresectable hepatocellular carcinoma (uHCC). The antibody can also be used in combination with durvalumab and platinum-based chemotherapy, respectively, to treat metastatic non-small cell lung cancer (mNSCLC) without the sensitized epidermal growth factor receptor (EGFR) mutation or with anaplastic lymphoma kinase (ALK) genomic tumor aberrations. Both of these were approved in the US in 2022.31
Compared to blocking CTLA-4, blocking the PD-1/PD-L1 axis can reduce side effects.32 At present, antibodies that block the PD-1/PD-L1 are approved as a first or second-line therapy for varieties of cancers.33,34 PD-1 can interact with two ligands, namely PD-L1 (CD274 or B7-H1) and PD-L2 (CD273 or B7-DC).35 PD-L2 is mainly expressed on dendritic cells (DCs) and macrophages, while PD-L1 is widely expressed on epithelial cells, vascular endothelial cells, and mesenchymal stem cells (MSCs), and is induced by type I and type II interferons, tumor necrosis factor-α (TNF-α), and pro-inflammatory factors such as VEGF.36 In the TME, high levels of PD-1 are expressed by activated T cells, which induces the expression of PD-L1 locally by causing the release of cytokines such as interferon-gamma (IFN-γ), interleukins (ILs), and TNF-α. Upregulated PD-L1 plays a major part in blocking the immune response and facilitating self-tolerance by preventing the over-activation of T cells after interacting with PD-1 on T cells through protein-protein interactions.37 Overall, the PD-1/ PD-L1 axis has been more deeply developed and utilized than the PD-1/ PD-L2 axis.34 The currently developed antibodies against PD-1 include pembrolizumab, nivolumab, and cemiplimab, while antibodies against PD-L1 include atezolizumab, avelumab, and durvalumab, which have been used in squamous-cell non-small cell lung cancer (NSCLC), Hodgkin’s lymphoma, renal cell carcinoma, and other cancer immunotherapy regimens (Table 1).
 |
Table 1 Anti-PD-1/PD-L1 Cancer Immunotherapy Regimens |
In addition, new inhibition pathways are being studied, and several emerging immune checkpoint targets are currently in clinical trials (Figure 1), such as Lymphocyte activation gene-3 (Lag-3), V-domain Ig suppressor of T cell activation (VISTA), T cell immunoglobulin-3 (Tim-3), T cell immunoglobulin and ITIM domain (TIGIT), and so on.52,53
 |
Figure 1 Immune checkpoint inhibitors are used to treat cancer. CD8+ T cells express PD-1, CTLA-4, TIM3 and LAG3. CTLA-4 and TIM3 were highly expressed on Tregs. TIM3 was significantly expressed in TAMs and NK cells. Binding of PD-1 to PD-L1 expressed on tumor cells can promote apoptosis of CD8+ T cells. CTLA-4 inhibits T cell proliferation and induces Tregs activity by binding to CD80/86 in APCs. The interaction between TIM3 and the ligand galactin-9 on the surface of MDSCs also induced T cell apoptosis. Reprinted with permission from Chen Y, Hu H, Yuan X, Fan X, Zhang C. Advances in immune checkpoint inhibitors for advanced hepatocellular carcinoma. Front Immunol. 2022;13:896752. doi:10.3389/fimmu.2022.896752 . Copyright © 2022.54 |
Adoptive Cell Transfer Therapy (ACT)
The process of ACT involves taking the patient’s own immune cells, massively expanding them outside the body, and reinfusing them into the patient to kill the tumor.55 Here, we highlight several commonly used ACT therapies.
Tumor-infiltrating lymphocyte (TIL) treatment depends on the isolation and in vitro expansion of T cells in tumors and has successfully treated a significant proportion of patients with metastatic melanoma. In 1988, Rosenberg published the first report on the prosperous treatment of melanoma using TIL-ACT.56 TIL is primarily used as a second-line treatment, and melanoma remains the primary treatment type for TIL in most clinical trials.57 It is worth noting that TIL treatment is also applicable to some extent to other solid tumors and has shown initial efficacy in breast, cervical, and colorectal cancers.58–60
Gene editing and gene transfer techniques have extremely facilitated the development of the ACT. T cell receptor-engineered T cell (TCR-T) treatment utilizes genetic modifications of T lymphocytes to make them tumor antigen-specific.61 Insertion of high-functioning tumor-specific TCRs into patients’ T cells can modify the T cell genome and redirect T lymphocytes, which can subsequently be expanded in vitro (Figure 2).62 Despite the low density of target cell antigens, TCR-T cells can be activated in large numbers because these cells possess all the helper molecules of the TCR signaling pathway.63 However, TCR-T cells specifically recognize HLA-I/peptide complexes, so the use of TCR-T therapy is limited to patients expressing the appropriate HLA-I isotype, thus preventing the use of this approach in most cancer patients. In addition, immune escape due to loss or downregulation of HLA molecules is an ordinary mode in cancer and perhaps limits the application of the TCR-T cell approach.64
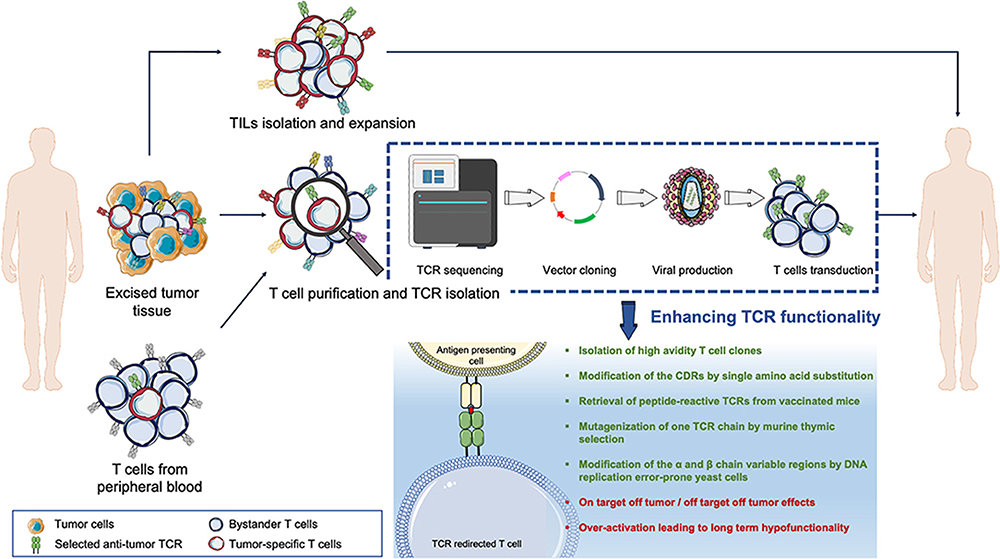 |
Figure 2 Overview of adoptive T cell therapy in TCR. Tumor-reactive lymphocytes can be isolated from the patient’s tumor mass or peripheral blood. T cells can be expanded in vitro and re-infused back into the patient, for example in TILs therapy. The tumor-reactive T cell receptor (TCR) gene can be isolated/sequenced via a vector and transferred into recipient T cells to redirect T cell specificity for tumor epitopes. Reprinted with permission from Manfredi F, Cianciotti BC, Potenza A, et al. TCR redirected T cells for cancer treatment: achievements, hurdles, and goals. Front Immunol. 2020;11:1689. doi:10.3389/fimmu.2020.01689. Copyright © 2020.62 |
Both TCRs and TIL treatment rely on external co-stimulation and the presentation of new epitopes targeted via the MHC-I complex, which is frequently down-regulated in tumor cells. Chimeric antigen receptors (CARs) are genetically engineered hybrid receptors composed of the extracellular structural domain single-chain variable fragment (scFv) as well as intracellular signaling structural domains. Theoretically, scFv recognizes all surface antigens on cells, so the targeting of tumor cells is not limited by MHC and is not reliant on the processing and expression of target epitopes.65 In the design of the first-generation CAR, the signal sequence was taken from CD3 complex ζ chain. However, subsequent studies found that the signal strength of the first-generation CAR-T was insufficient to effectively maintain the proliferation and activation of T cells, and did not achieve satisfactory results in clinical practice.66 To overcome the shortcomings of first-generation CAR-T, researchers have cascaded co-stimulatory molecular sequences based on first-generation CAR-T, generally derived from the CD28 family (including CD28 and inducible T Cell Costimulator (ICOS)) or the tumor necrosis factor receptor (TNFR) family (including 4–1BB, CD27, and OX40).67 Due to the different mechanisms of action of signal pathway sequences derived from the CD28 family and TNFR family, the final designed CAR-T product may have different mechanisms of action. Some studies have shown that CARs based on CD28 have stronger initial anti-tumor activity, while the 4–1BB signaling pathway improves CAR-T cell persistence.55 To further enhance the function and lifespan, third-generation CARs were manufactured by combining two co-stimulatory elements (mainly the aforementioned CD28 and TNFR signaling sites) in an intracellular region, but to date, no studies have been published in which the third-generation is significantly superior to the second generation.68,69 In addition to the first three generations of CAR-Ts described above, a fourth generation of CAR-Ts, “armored” CARs, whose full name is T cell redirected for antigen-unrestricted cytokine-initiated killing (TRUCKS),66 further improves effectiveness against solid tumors and at least antigen-negative tumors. So far, the successful clinical applications of CAR-T cells have focused on CD19 and CD22 molecules in B-cell Acute Lymphoblastic Leukemia (B-ALL) and B-cell lymphoma, as well as B-cell Material Antigen (BCMA) for multiple myeloma.70–72 At present, the FDA has approved five CAR-T protocols, examples include Abecma (idecabtagene vicleucel), Yescarta (axicabtagene ciloleucel), Kymriah (tisageneleucel), Tecartus (breucabtagene autoleucel), and Breyanzi (lisocabtagene maraleucel).73 Kymriah and Yescarta were launched in 2017 and 2018, respectively, and almost half a million patients worldwide have received the injections.74 However, the infiltration and transport of CAR-T cells, as well as the suppressive microenvironment and heterogeneity of solid tumors, remain barriers to solid tumor therapy.69
Cancer Vaccines
Vaccines currently under development fall into three main categories, namely cellular vaccines, viral vector vaccines, and molecular vaccines consisting of peptides, DNA, or RNA.
Cellular vaccines refer to those prepared using killed cancer cells or APCs loaded with tumor antigens. Tumor cell-derived vaccines use the entire tumor cells or subpopulation of tumor cells (e g tumor cell membranes) as antigen sources. These vaccines avoid recognizing immunogenic antigens meanwhile providing a large number of different tumor-associated antigens (TAAs) as immune targets. The main ones include whole-cell vaccines, tumor cell lysate (TCL) vaccines, tumor cell membrane-derived vaccines, etc. Whole tumor cell vaccines contain whole antigen profiles of tumor cells and can be used to activate both CD4+ T helper cells and CD8⁺ cytotoxic lymphocytes, but progress in clinical translation has been poor.75 One of the limiting factors is that patients may not have enough tumor cells to prepare sufficient doses of whole-cell vaccine. In recent years, researchers have transformed tumor cells into in-situ vaccines through ICDs induced by chemotherapy, photodynamic/photothermal, thereby avoiding complex cell preparations, which leaves infinite potential for whole-cell vaccine research. TCL vaccines effectively present a variety of antigens to activate T cell responses and also provide dendritic cells with immune regulatory cytokines.76 However, their inherent instability and complex composition often lead to poor dendritic cell uptake and inefficient antigen cross-presentation. Therefore, the development of suitable delivery carriers and purification methods is necessary. In contrast to patient-derived whole-cell vaccines and TCLs, tumor cell membranes are enriched with cancer antigens while avoiding the effects of excess non-tumor-associated antigenic material. Tumor cell membranes can be flexibly adjusted or modified, which opens up a variety of possibilities for membrane coating platforms.77 In 2010, the FDA approved the marketing of the first cancer vaccine, Provenge®, which was derived from patient-derived DC cells, co-cultured and activated in vitro which uses prostate acid phosphatase and granulocyte-macrophage colony-stimulating factor (GM-CSF) combined with proteins, and then injected back into the patient.78 This has raised concerns about the use of autoimmune cells for immunotherapy and is a good example of individualized medicine, but the complex process increases labor and costs and may limit its development.79
Virus-like particles (VLPs) are self-assembling viral protein complexes but are not infectious. VLPs can effectively target and activate DC cells, resulting in the presentation of viral antigens to MHC-I and MHC-II, which triggers B- and T-cell immunity (Figure 3).80 VLPs range in size from 20–200 nm and can be produced in more than 170 different expression host systems, including bacterial, insect, yeast, or mammalian cells.81 Viral vector vaccines can bind to a wide range of antigens from different proteins that can be functionalized devoid of affecting their antigenicity.80 VLPs can be an effective adjuvant even in the absence of antigen loading.82 The combination of modified human papillomavirus 16 (HPV16) VLP with a photoactivatable drug, IRDye-700DX, triggered an effective and durable antitumor response when activated by near-infrared light (NIR).83 As an effective preventive and therapeutic vaccine strategy, VLPs have played a crucial role in the field of prevention of cancers caused by human oncoviruses. Prophylactic vaccines based on VLPs against hepatitis B virus (HBV) and human papillomavirus (HPV) have been developed, such as Gardasil 9, CervarixⓇ, GardasilⓇ, and Heplisav-BⓇ.84,85
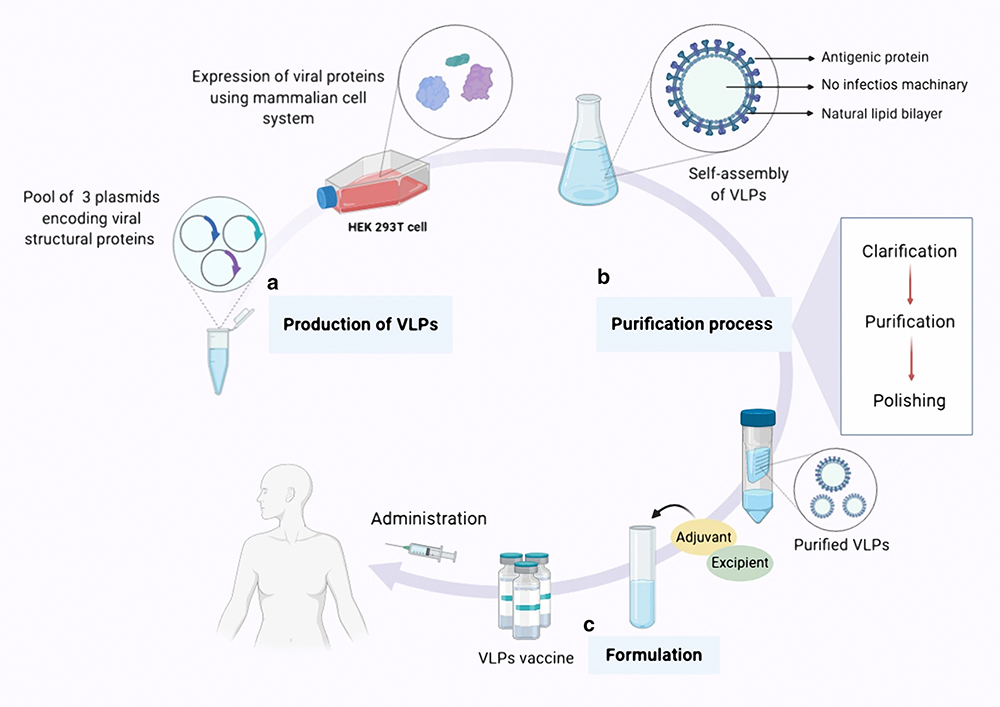 |
Figure 3 Overview of VLP-based vaccine expression, purification, and formulation. (a) Production stage: clone interested viral structural genes and express viral proteins with self-assembly capability in a suitable expression platform. (b) Purification stage: downstream processing, such as clarification and purification, and finally obtaining the purified complete VLP. (c) Formulation stage: adjuvants and other ingredients are added to the vaccine formulation to achieve safe and effective vaccination products. Reprinted with permission from Nooraei S, Bahrulolum H, Hoseini ZS, et al. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J Nanobiotechnol. 2021;19(1):59. doi:10.1186/s12951-021-00806-7. Copyright © 2021.86 |
Peptide vaccines have easily controlled structures with well-defined sequences. Short peptide vaccines are typically 9 amino acids in length and elicit cytotoxic TCD8 response by interacting with MHC I. As the smallest CD8 epitope, they can directly activate effector cells without the need for APCs to further process the antigen. Long peptide vaccines are 20–30 amino acids in length and can potentially activate responses to TCD8 and TCD4.87 Short peptide vaccines may undergo protein degradation and have a short duration of the immune response, while long peptide vaccines have disadvantages including susceptibility to enzymatic degradation, rapid clearance, and insufficient uptake at the injection site.88 Nucleic acid vaccines not only activate multiple epitopes simultaneously but are also simple to operate, which makes them an attractive anti-cancer immunotherapy.89 Studies have shown that mammalian cells transfected can express genes encoded on plasmid DNA. Recent advances in molecular biology and recombinant techniques have provided the basis for plasmid gene manipulation and the development of DNA vaccines.90 In addition, the safety of the DNA vaccine has been established in studies and therefore multiple injections are allowed at the time of administration.91 Two strategies can be used to improve DNA vaccines, one is to select optimal antigens to insert into plasmid DNA, and the other is to combine DNA vaccines with other complementary therapies.92 In 2020, the FDA urgently emergency approval for two mRNA vaccines manufactured by Pfizer-BioNTech/BNT162b2 and Moderna/mRNA-1273 for the prevention of coronavirus disease 2019 (COVID-19).93,94 The next year, the first COVID-19 vaccine manufactured by Pfizer-BioNTech received FDA approval (marketed as Comirnaty). Compared to other types of vaccines, mRNA carries antigenic genetic information that can be translated into proteins very rapidly once it enters the cell, making mRNA vaccines non-integrated, safer, and free of mutational risk.95 In addition, mRNA vaccines also act as self-adjuvants, with APCs recognizing mRNA and triggering pattern recognition receptors (PRRs). However, naked RNA is susceptible to degradation by nucleases. Effective mRNA delivery is therefore a key factor in the success of therapy.96
The search for a suitable antigen has been the focus of vaccine design for many years. Ideally, antigens should not be expressed in normal cells but are expected to be expressed specifically in cancer cells. It is present in all cancer cells and is highly immunogenic.97 Current antigens include mainly TAAs and TSAs. TAAs are not only overexpressed in tumor cells, but are also expressed in normal cells, including oncogenic viral antigens, tissue differentiation antigens, and overexpressed antigens.98 Up to now, most cancer vaccines have targeted TAAs, but the normal cellular damage associated with their off-target effects is a concern; secondly, recognizable high-affinity T cells are cleared by the immune system during development, while CD8 T cells targeting TAAs have low functional affinity and are not sufficient to achieve significant efficacy.99 TSAs include cancer germline antigens and tumor-specific mutated antigens, mutations that occur in tumor cells that create new autoantigenic epitopes, called neoepitopes or neoantigens. These antigens are not expressed in normal cells, preventing “off-target” damage to non-malignant tissues and potentially circumventing the central tolerance of T cells to their own epitopes.98
Although the development of cancer vaccines is ascending, they are not produced and applied in clinic broadly. The failure of cancer vaccines is perhaps due to inadequate tumor immunogenicity. Therefore, improving the immunogenicity of antigens, combating tumor immune escape mechanisms, and realizing effective delivery of tumor vaccines are urgent issues that researchers need to address today. More types of tumor antigens and more valid delivery routes will facilitate tumor vaccine research, and the study of maximizing tumor immunogenicity should be strengthened.100
Oncolytic Virotherapy (OVT)
Oncolytic viruses (OVs) are tumor-killing viruses that specifically infect tumor cells. In recent years, OVs have been widely considered an ideal anticancer method because of their unique tumor tendency and low toxicity to normal cells. The anti-tumor mechanism of OVs mainly includes the following three aspects (Figure 4): ① OVs selectively replicate in tumor cells and directly cause tumor cell lysis. Tumor debris will cause the body’s immune response to speed up the removal of tumors. ② OVs can mediate anti-tumor immune responses and stimulate innate and adaptive immune responses. They can promote ICD and release PAMPs and DAMPs, causing the release of a series of cytokines (IFNs, TNF-α, IL-6, IL-1) and activation of immune cells (T cells, NK cells, DC cells). ③ OVs can infect and lyse vascular endothelial cells (VECs), thereby disrupting vascular structure and affecting tumor blood supply.101 Four OVs drugs are approved worldwide: H101, Teserpaturev, ECHO-7 (discontinued), and Talimogene laherparepvec (T-VEC). T-VEC, developed by Amgen, is an improved herpes simplex virus type 1 (HSV1) containing the GM-CSF coding sequence. It is currently the only oncolytic viral therapy widely recognized worldwide and was approved by the FDA in 2015 for treating unresectable metastatic melanoma.102 In a randomized open phase 3 trial in patients with unresectable stage IIIB-IVM1c melanoma. 295 patients received T-VEC and 141 were assigned to GM-CSF (2:1 randomization). In a preliminary analysis of overall survival (OS), the median OS was 23.3 months for T-VEC and 18.9 months for GM-CSF. A complete response (CR) was achieved in 16.9% of patients treated with T-VEC, compared to 0.7% of patients treated with GM-CSF. 72% of patients had no recurrence at 3 years after CR, and an estimated 88.5% were alive at 5 years.103,104
 |
Figure 4 Mechanism of action of oncolytic virus. The virus replication in tumor cells triggers the ICD, which causes the release of a series of cytokines (IFNs, TNF-α, IL-6, IL-1) and the activation of immune cells (T cells, NK cells, DC cells). OVs can infect and dissolve vascular endothelial cells, affect tumor blood supply, and reshape TME. Reprinted with permission from Volovat SR, Scripcariu DV, Vasilache IA, et al. Oncolytic virotherapy: a new paradigm in cancer immunotherapy. Int J Mol Sci. 2024;25(2):1180. doi:10.3390/ijms25021180. Copyright © 2024.105 |
Over the past decade, rapid progress has been made in the OVT field, but only in the intratumoral way. Although intratumoral administration ensures that OVs act directly on tumor lesions, systemic delivery is more practical for hard-to-reach lesions and metastatic tumors. However, intravenous administration of OVs faces several problems, including neutralizing antibodies, complement activation, antiviral cytokines, and non-specific uptake by other tissues.106 Therefore, the virus must persist in circulation without depletion or degradation, while selectively infecting tumor cells. There have been many researches on the vectors of whole-body delivery of OVs, which can be divided into biological vectors and abiotic vectors. Biologic vectors include some living cells (such as T cells or mesenchymal stem cells), extracellular vesicles, and cell membrane vectors with intrinsic tumor-taxis. Abiotic carriers include liposomes, magnetic nanocarriers, and polymer-based nanocarriers.107,108 Chen et al prepared an oncolytic virus-T cell chimera (ONCOTECH), which encoded CRISPR-Cas9 targeting PD-L1 into oncolytic adenovirus (OAs) and shielded it with an engineered biofilm.109 Membrane-camouflaged OAs can physically attach to T cell surfaces by recognizing TCRs or CARs. The results showed that in vivo administration of ONCOTECH in the melanoma model led to a strong accumulation of OAs in tumor cells and a 50% reduction in PD-L1 expression. In addition, a single dose resulted in 80% survival in mice within 70 days. This suggests that combining viral therapy with other immunotherapies is a promising therapeutic platform.
Overall, OVT is gaining more attention as an emerging tool in cancer therapy, but transporting the virus to tumor cells remains a challenge. In addition, some OVs have limited efficacy and low persistence in target tissues. To overcome these limitations, further genetic engineering of OVs is needed. Moreover, combining OVT with other anti-cancer treatments may offer promising ideas.
Cytokine Therapy
Cytokines are a class of low molecular weight proteins or small molecular peptides that can transmit information between cells and have the function of immune regulation. Major cytokines involved in cell communication in homeostasis and disease include interleukins, interferons, some tumor necrosis factor (TNF) superfamily members, chemokines, and growth factors. There is strong evidence that some cytokines, particularly those involved in adaptive immune responses, contribute to anti-tumor immunity. IFN-α was the first cytokine approved in 1986 for treating Hairy cell leukemia (HCL). It can up-regulate MHC molecules, promote DC cell migration enhance cytotoxic T cell activity, and directly inhibit malignant cells.110 IL-2 is a key cytokine that regulates the adaptive/innate immune system and plays a crucial role in T cell development and expansion (Figure 5). It can promote the proliferation of CD4+ and CD8+ T cells, induce the differentiation of helper T cells, and enhance the cytolytic activity of NK and CD8+ T cells.111 Intravenous infusion of high-dose IL-2 was approved by the FDA in 1992 and 1998 for the treatment of metastatic renal cell carcinoma (RCC) and melanoma, respectively.112 In addition, other cytokines are also being evaluated, such as IL-7, IL-12, IL-10, IL-15 and IFN-γ.110
 |
Figure 5 Interaction of IL-2/IL-2R signal transduction in TME. Lower levels of IL-2 can promote the regulation of the microenvironment, which may promote tumor growth, while higher levels can act as a stimulant for immune cells and promote tumor elimination. On the one hand, it can stimulate Tregs to inhibit the anti-tumor response; on the other hand, it can induce the infiltration of CTL and NK cells, thus enhancing the anti-tumor immune response. The arrows of different sizes in the figure indicate the different intensities of IL-2 production and its effect on different cell types. Reprinted with permission from Muhammad S, Fan T, Hai Y, Gao Y, He J. Reigniting hope in cancer treatment: the promise and pitfalls of IL-2 and IL-2R targeting strategies. Mol Cancer. 2023;22(1):121. doi:10.1186/s12943-023-01826-7. Copyright © 2023.113 |
The short blood half-life, pleiotropy, and adverse tissue distribution of cytokines have caused a series of side effects and made cytokine monotherapy not meet people’s expectations.114 At present, the clinical research direction of cytokine-based therapies focuses on combining them with other immunotherapies, modifying cytokines to change their pharmacokinetics and binding affinity, and promoting the delivery of cytokines in vivo.115 Some researchers constructed NK-92 cells expressing NKG2D and IL-21, and the expression of IL-21 enhanced the cytotoxic function against A549 and H1975 cells, the expression of CD107a, and the production of IFN-γ of NKG2D CAR NK-92 cells. NKG2D-IL-21 CAR-NK-92 cells significantly reduced tumor growth and promoted higher levels of IFN-γ compared to NKG2D CAR-NK-92 cells.116 In recent years, protein engineering-based approaches to tailor the function of cytokines have attracted researchers’ attention. Recombinant cytokine therapies, including directed evolution and PEGylation, have made great progress.115 The toxicity of IL-2 is associated with activation of the high-affinity trimer IL-2Rαβγ complex. The IL-2Rαβγ complex is highly expressed in Treg cells and stimulates immunosuppressive cells, which antagonizes anticancer therapy. Using a directed evolution technique to create an IL-2 “superkine” to improve the affinity for the IL-2Rβγ complex, the mutant protein has a 200-fold greater affinity for IL-2Rβγ, eliminating the affinity difference between the dimeric IL-2Rβγ and trimeric IL-2Rαβγ complexes and leading to a greater anti-tumor response in vivo mouse model. Fusion of this IL-2 “superkine” with albumin (MDNA11) is currently underway in Phase I/II clinical trials (NCT05086692).117 In addition to binding properties, the circulating half-life of cytokines also determines the in vivo effects of the relevant therapeutic agents. Researchers have developed a PEGylated IL-10 (Pegilodecakin) that can activate tumor CD8 T cells in mice and induce an objective tumor response in patients. Increased INF-γ and granzyme B levels and expansion and activation of CD8 T cells in cancer patients treated with Pegilodecakin. PEGylated increases local IL-10 concentrations without causing serious side effects.118 Several techniques for the targeting and delivery of cytokines are also flourishing. Fusing cytokines to other proteins helps to promote tumor localization and overcome the poor pharmacokinetic properties and adverse biodistribution profiles of cytogenic subdrugs. For example, some researchers have fused IL-12 into the ED-B domain of fibronectin, significantly enhancing the targeting and anti-tumor activity of IL-12.119 In addition, some nanocarriers, including dextran, chitosan, liposomes, PLGA, gold, and magnetic particles have the potential to address many of the limitations of cytokine delivery.120 Some researchers prepared PcDNA3.1-dsNKG2D-IL-21 plasmid nanoparticles based on chitosan. The NKG2D-IL-21 fusion protein theoretically recognizes tumor cells through the NKG2D group, and the IL-21 group activates T cells. The nanoparticles accumulated in tumor tissue after intravenous injection ~ 4–24 h, and delayed tumor growth and extended the life span of tumor-bearing mice by activating NK and T cells in vivo.121
With rapid advances in artificial intelligence and machine learning-based protein structure prediction, some more complex de novo protein design cytokines will be developed. Such non-human proteins have completely different amino acid sequences, which may be more likely to cause immunogenicity. The same is true for DARPins and nanobodies, which largely limit the safety and effectiveness of this class of drugs. In addition, some modifications can affect the signaling activity of cytokines, such as steric hindrance that may arise from high molecular weight PEG. Therefore, as protein engineering strategies become more complex and multifaceted, it will be important to understand the mechanisms of how each modification affects both pharmacokinetics/pharmacodynamics and receptor activation.122
Chemotherapy-Induced Immunomodulation
Although immunotherapy has taken its place in the prevention and treatment of tumors, it has been ineffective in clinical practice. This is probably related to the tumor-suppressive microenvironment and “cold tumor”. Several inhibitory receptors in the TME, such as CTLA-4, PD-1, and Tim-3; immunosuppressive cytokines including IL-10, IL-6, transforming growth factor-β (TGF-β), vascular endothelial growth factor, etc. can block DC and T-cell function.123 The “cold tumor” means that the tumor tissue lacks TILs, and the TME is immunosuppressive.124 Therefore, it is necessary to develop collaborative therapeutic approaches to activate the tumor immune microenvironment (TIME) and improve the therapeutic effect. In the late 1960s, researchers found that the efficacy of antitumor drugs may be caused by a synergistic effect between the killing effect of the drug itself and the host immune system.125 The modulatory effects of chemotherapy on immunity include four main aspects (Table 2).
 |
Table 2 Immunological Effects of Conventional Antitumor Agents |
Inhibition of Immune Checkpoint (IC) Expression
In recent years, some chemotherapy drugs have been found to affect the function of IC (Figure 6). The research progress of interfering histone deacetylase 2 inhibitor (HDAC2i) in Hepatocellular carcinoma (HCC) has been reported. It can block PD-L1 acetylation-dependent nuclear translocations to reprogram the expression of immune response-related genes and regulate IFN-γ to affect PD-L1.144 SN-38 is an activated form of irinotecan. Researchers have found that SN-38 can reduce the expression of PD-L1 by inhibiting specific carcinogenic signaling pathways, such as c-Myc and Myc targets. In addition, SN-38 can promote the recruitment of NKs or CD8+ T cells and reduce the infiltration of Tregs or TAMs in TME.133 Lenvatinib is a multikinase inhibitor that has been approved by the FDA for first-line use in the treatment of unresectable HCC. Studies have shown that Lenvatinib leads to decreased expression of PD-L1 by blocking FGFR4. It can also limit the differentiation of Tregs to improve the efficacy of anti-PD-1.145 A phase Ib single-arm study showed that lenvatinib in combination with pembrolizumab resulted in an objective response rate (ORR) of 46.0% (95% CI, 36.0% to 56.3%) per mRECIST and 36.0% (95% CI, 26.6% to 46.2%) per RECIST v 1.1. A related double-blind randomized controlled phase III study has also been initiated to evaluate the safety and efficacy of the combination as a first-line treatment for uHCC (NCT03713593).146
 |
Figure 6 Potential mechanism of HDAC2i blocking PD-1/PD-L1. HDAC2i can affect PD-L1 by blocking PD-L1 acetylation-dependent nuclear translocations, reprogramming the expression of immune response-related genes, and regulating IFN-γ. Reprinted with permission from Han R, Ling C, Wang Y, Lu L. Enhancing HCC treatment: innovatively combining HDAC2 inhibitor with PD-1/PD-L1 inhibition. Cancer Cell Int. 2023;23(1):203. doi:10.1186/s12935-023-03051-0. Copyright © 2023.144 |
Induced Immunogenic Cell Death (ICD)
Certain chemotherapeutic agents, photodynamic therapy (PDT), photothermal therapy (PTT), oncolytic viruses, and radiotherapy (RT) are the main causes of ICD.147 Dying tumor cells expose tumor antigens to APC and produce damage-associated molecular patterns (DAMPs), including calreticulin (CRT), adenosine triphosphate (ATP), heat-shock proteins (HSP70 and HSP90), high-mobility group box-1 (HMGB1), type I IFNs and IL-1. Innate PRRs recognize these DAMPs, some examples include toll-like receptors (TLRs) and NOD-like receptors (NLRs), which activate tumor-specific immune responses (Figure 7A). Several chemotherapeutic drugs have been shown to have ICD effects, including doxorubicin (DOX), paclitaxel (PTX), and oxaliplatin (OXA).147,148 Ma et al prepared a DOX-based mannose nanogels (DM NGs), which can induce strong ICD, enhance CRT migration to the cell membrane, and release HMGB1.149 Ren et al designed a self-assembled cascade bioreactor FeS-GOx@PTX (FGP), which could effectively increase CRT and HMGB-1 expression. Subsequently, FGP combined with anti-CTLA-4 was shown to completely cure the primary tumor and actively inhibit distant tumors by enhancing cytotoxic T lymphocyte (CTL) infiltration.150 Teniposide upregulates ICD characteristics, promotes antigen presentation by tumor cells, and enhances T-cell recognition. In rodent colon cancer, teniposide induced strong anti-tumor CD8+ T cell immunity and significant tumor suppression.134 Wang et al constructed bovine serum albumin (BSA)/ferritin nano agonist combining Mn2+ and β-lapachone (Lap) (BSA-Man@Mn2+-Ft@Lap). Lap can induce ICD and release rich tumor-derived dsDNA into the TME, which will be delivered to DC via the Mn2+-stimulated cGAS-STING signaling pathway. Subsequently, the activation and infiltration of effector T cells were enhanced, ultimately promoting systemic anti-tumor immunity.135
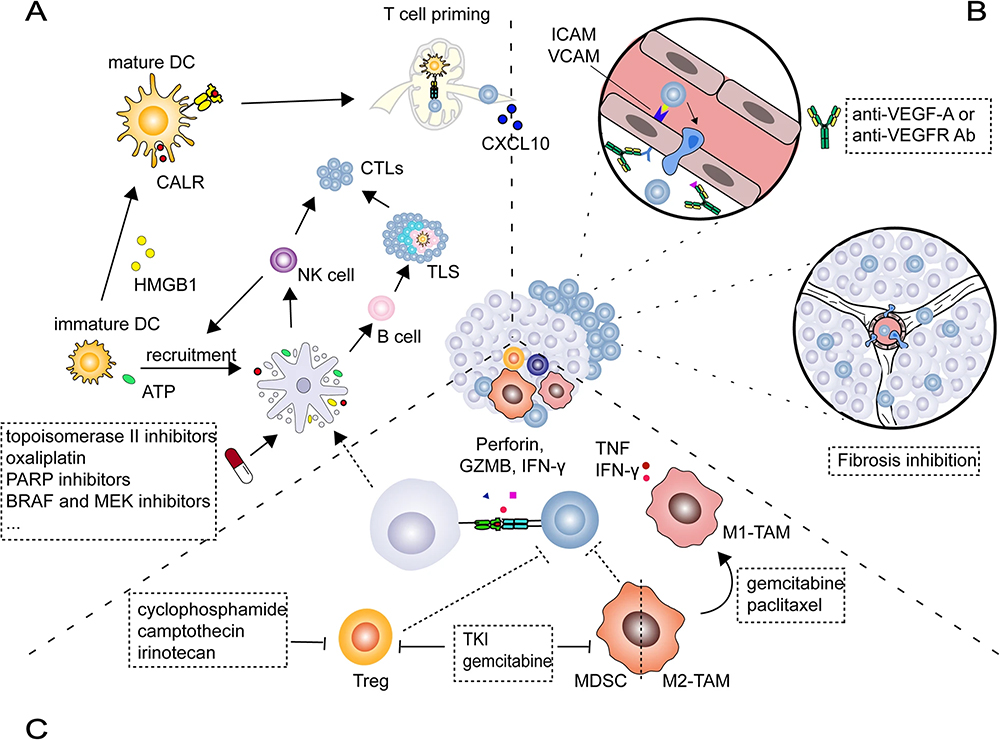 |
Figure 7 Chemotherapy drugs drive the cancer immune cycle. (A) chemotherapy drugs induce ICD; (B) promote CD8+ T cell infiltration; (C) inhibit immunosuppressive cells. Reprinted with permission from Li JY, Chen YP, Li YQ, Liu N, Ma J. Chemotherapeutic and targeted agents can modulate the tumor microenvironment and increase the efficacy of immune checkpoint blockades. Mol Cancer. 2021;20(1):27. doi:10.1186/s12943-021-01317-7. Copyright © 2021.134 |
ICD spontaneously releases abundant antigens and adjuvants, providing an exemplary opportunity to establish effective in situ vaccination.151 Induction of ICD by chemotherapeutics in combination with ICI, cytokines, and immune adjuvants has been shown to have significant antitumor efficacy.148 Wang et al administered a single intra-tumor injection of an ICD-inducing agent, mitochondria-targeting fenofibric acid (Mito-FFa), into 4T1 tumor models and found that it promoted CRT exposure and IFN-I secretion while triggering tumor-specific CD8 T cell responses to both primary and metastatic cancers.152 Peng et al prepared a redox-responsive polymer micellar-based nanovaccine encapsulating both DOX and the adjuvant TLR7/8 agonist R848, resulting in a 6-fold increase in CD8+ T cells in the TME.153
Promote the Infiltration of Tumor Killer Cells
In addition to stimulating the production of CTLs by inducing ICDs, some chemotherapy agents can also enhance the ability of CTLs to enter the tumor center (Figure 7B). The microenvironment of some solid tumors, such as pancreatic ductal adenocarcinoma (PDAC), has a highly fibrotic interstitial and extensive infiltrating immunosuppressive cell population. High stromal density not only affects the delivery of cytotoxic drugs, but also causes T cells to be blocked at the tumor edge. What’s more, many myeloid cell infiltrations may further lead to dysfunction of the CTLs. Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase. It is considered a key factor in the pathogenesis of fibrosis and immunosuppressive TME. Studies have shown that FAK inhibitors can promote CTLs to invade tumors by reducing fibrotic interstitium. Moreover, FAK inhibitors may reduce the proportion of immune-suppressed myeloid cells by inhibiting the PI3K/AKT/JAK/STAT3 and p38/JNK pathways, thereby enhancing CD8+ T cell-mediated killing of cancer cells.136 Vascular endothelial growth factor (VEGF) is a secretion factor that specifically acts on endothelial cells to stimulate angiogenesis, and has been shown to have immunosuppressive function.134 Recent studies have found that anti-angiogenic drugs can significantly increase CTL infiltration by normalizing immature blood vessels. Molecular and cellular analysis showed that bevacizumab (anti-VEGF) treatment increased Th1 chemokine-related T cell transport, tumor MHC-I protein expression, and tumor-specific T cell clonal infiltration.138 It has also been shown that the combination of ICBs and anti-angiogenic drugs further promotes the normalization of blood vessels, which allows not only CTLs but also DCs and B cells to infiltrate and accumulate.154 Carbonic anhydrase XII is a transmembrane zinc metalloenzyme that promotes tumor progression. It can induce macrophages to produce a large amount of chemokine (C-C motif) ligand 8 (CCL8) and enhance the epithelial-mesenchymal transition (EMT) of cancer cells. Some researchers have found that CAXII inhibitors (CAXIIis) can increase the apoptosis of macrophages and improve the proportion of CD8+ T cells in total tumor lymphocytes. In addition, overexpression of CAXII may mediate the accumulation of M2-TAMs in tumor tissues by regulating protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) and CCL8. This suggests that CAXIIis plays an important role in the survival and function of M2 subtype TAMs.140,141
Downregulation of the Immunosuppressive Microenvironment
Barnett Rosenberg discovered that cisplatin has antitumor activity in 1969, followed by his suggestion that cisplatin may alter the immunogenicity of tumor cells by eliminating immunosuppressive molecules, was confirmed by subsequent studies.155–157 TIME consists of several inhibitory immune cells, including tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs) (Figure 7C). Besides, several soluble inhibitors, such as indoleamine 2,3- dioxygenase (IDO) and TGF-β, which activate immunosuppressive pathways and thus suppress antitumor immunity. Tregs are a subpopulation of CD4+ T cells whose primary role is to inhibit the progression of autoimmune diseases by balancing peripheral tolerance to autoantigens. As early as the last century, it was found that cyclophosphamide (Cy) promotes overexpression of immunity against L5178Y lymphoma by eliminating suppressor T cells, and that combined immunotherapy leads to complete tumor regression and results in long-term survival.129 Paclitaxel potentiates the antitumor effects of the TLR9 agonist CpG, leading to delayed tumor growth in renal cell carcinoma, RENCA, and EG7 thymoma attributable to a reduction in Treg numbers in a TLR4 non-dependent manner and preferentially affecting circulating Tregs expressing high levels of FoxP3.127 MDSCs are consist of myeloid progenitor cells and heterogeneous immature myeloid cells that promote cancer progression by suppressing antitumor immune responses. Gemcitabine remarkably lowered the number of MDSCs in spleens and tumors and increased the proportions of CD4/CD8 T cells, NK cells, macrophages, and B cells in mice.130,158 In addition, 5-fluorouracil (5-FU), docetaxel, and sunitinib were all found to have the ability to deplete MDSCs.131,132,139 As an important component of the tumor microenvironment, TAMs affect tumor growth, angiogenesis, metastasis, and chemotherapy resistance. TAMs are mainly divided into M1 type and M2 type. M1 macrophages are generally considered tumor-killing macrophages, while M2 macrophages are immunosuppressive and can promote tissue repair and tumorigenesis. Regorafenib (REG) is an oral small-molecule inhibitor that effectively blocks a variety of protein kinases, including VEGF receptor (VEGFR) 1, 2, and 3, TIE2, KIT, RET, RAF1, BRAF, and others. REG has effective anti-angiogenesis and immunomodulatory properties. It can significantly inhibit the recruitment of TAMs and induce persistent M1 TAMs polarization. REG may also reduce the number of Tregs by inhibiting VEGFA/VEGFR signaling. Researchers examined the combined application effect of REG and aPD1 in a mouse CRC model. The sustained tumor suppression induced by REG + aPD1 may be due to their synergistic immunomodulatory effects, which lead to the activation of cytotoxic T cells.142 In addition, REG can effectively inhibit JAK1/2-STAT1 and MAPK signaling by targeting the RET-SRC axis, subsequently reducing IFNγ-induced PD-L1 and IDO1 expression to promote anti-tumor immunity.143
Clinical Trials of Chemoimmunotherapy
Combination therapy can reduce the dose of chemotherapy drugs and lower the likelihood of tumor resistance and metastasis. Due to the strength of CIT, many clinical trials have shown promising results in cancer treatment (Table 3), and some combination therapy modalities have been approved by the FDA (Figure 8). For example, two CT-based randomized controlled trials (RCTs) trials for endometrial carcinoma (EC) have recently progressed: RUBY and NRG-GY018.159,160 In RUBY, the MMRd subjects progression-free survival (PFS) at 24 months was 61.4% (95% CI, 46.3–73.4) in the dostarlimab group and 15.7% (95% CI, 7.2–27.0) in the placebo group (HR for progression or death, 0.28; 95% CI, 0.16–0.50; P <0.001). The MMRp subjects PFS in both groups were 28.4% (95% CI, 21.2–36.0) and 18.8% (95% CI, 12.8–25.7), respectively (HR for disease progression or death, 0.76; 95% CI, 0.99–0.98). In GY018, Kaplan-Meier estimated that the ratio of pembrolizumab-treated MMRd subjects with no disease progression or mortality at 12 months was 74% vs 38% (HR 0.30; 95% CI, 0.19–0.48; P < 0.001). Median PFS was 13.1 months in the pembrolizumab group and 8.7 months in the placebo group (HR 0.54; 95% CI, 0.41–0.71; P < 0.001). Based on the results of these studies, the NCCN guidelines include the combination therapy of pembrolizumab/carboplatin/paclitaxel and dostarlimab/carboplatin/paclitaxel as the first-class primary treatment option for stage III or IV endometrial cancer. July 31, 2023, the FDA approved dostarlimab in combination with carboplatin and paclitaxel for primary or recurrent mismatch repair deficiency (MMRd) EC.161
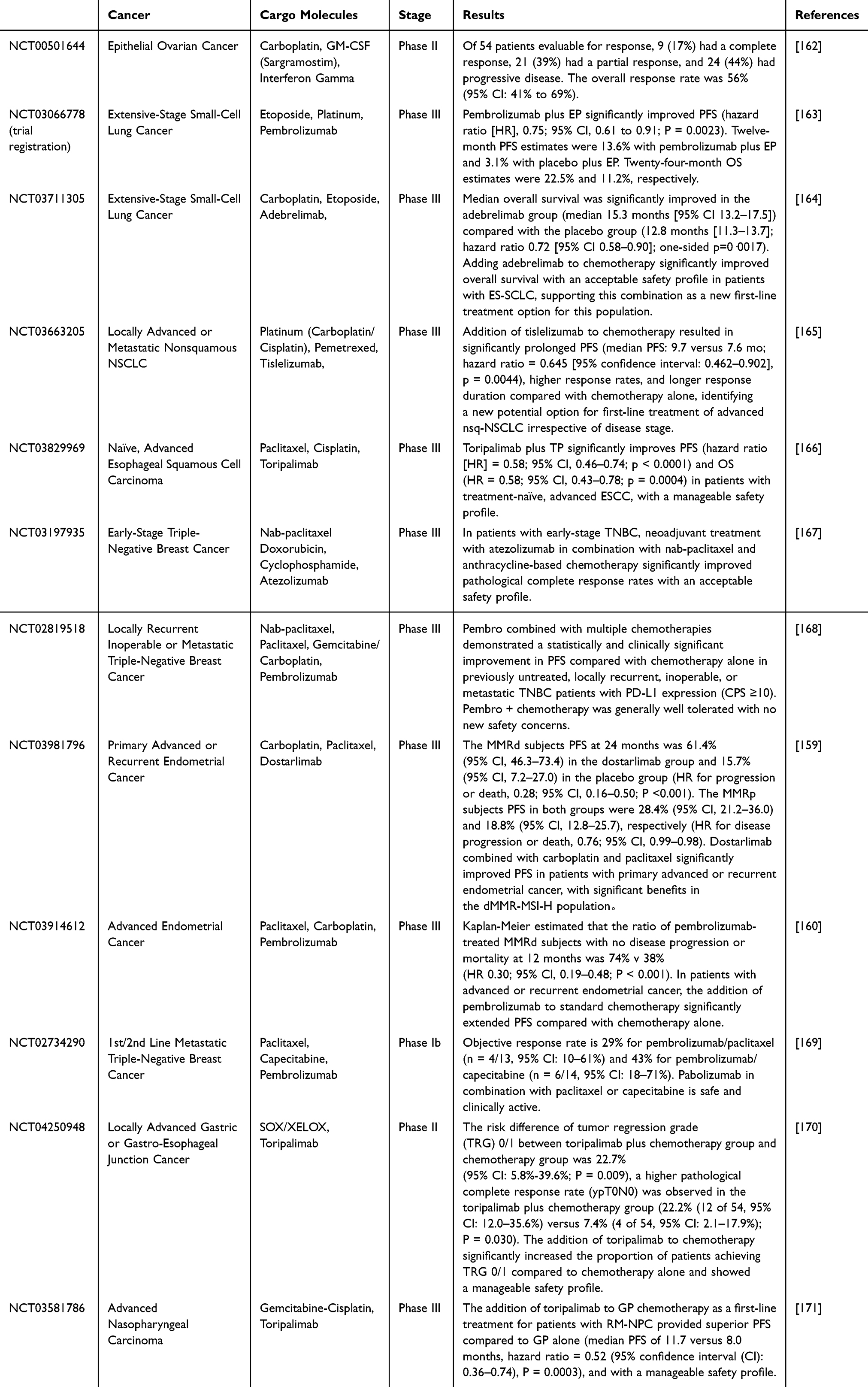 |
Table 3 Clinical Combination Trials |
 |
Figure 8 A timeline for FDA-approved products for chemoimmunotherapy from 2020 to 2024. Abbreviations: ES-SCLC, extensive-stage small-cell lung cancer; NSCLC, non-small cell lung cancer; TNBC, triple-negative breast cancer; ESCC, esophageal squamous cell carcinoma; BTC, biliary tract cancer; UC, urothelial cancer; GC, gastric cancer; AEG, adenocarcinoma of esophagogastric junction; ESCA, esophageal adenocarcinoma; GEJ, gastroesophageal junction; NPC, nasopharyngeal carcinoma. |
Nanoparticle-Mediated Chemoimmunotherapy
Although CIT is a promising way to achieve tumor cure and reduce recurrence rates, conventional dosing strategies may reduce efficacy. Under physiological conditions, low circulation time, insufficient accumulation, inadequate penetration in tumor tissues, and induction of toxicity can all affect clinical use. Nanotechnology can address some of these limitations by reducing the dose and frequency of administration, which provides a new path to safety for CIT. Over the past decade, multifunctional nanomaterial-based delivery vehicles NDDSs have become increasingly popular, and their versatility and adjustability make them promising in solving various cancer problems. Kuai et al prepared a DOX-loaded synthetic high-density lipoprotein (sHDL) simulated nanodiscs. Compared with anti-PD-1 and free DOX+anti-PD-1, the expansion rate of tumor-specific CD8+ T cell in mice treated with sHDL-DOX+anti-PD-1 was increased 8-fold and 4-fold, respectively, and the survival rate was 88% in situ colon cancer metastasis model. In contrast, the response rate in the free DOX+anti-PD-1 group was only 13%, which demonstrates the potential of nanotechnology in CIT.124
Advantages of NDDSs
Multi-Drug Co-Delivery
The differences in solubility, biostability, pharmacokinetics, and tissue distribution patterns of the drugs make the delivery of chemotherapy-immunotherapy a bottleneck. From hydrophobic small molecules (such as most inhibitors and chemotherapeutic agents) to hydrophilic macromolecules (such as antibodies and nucleic acids), the design of corresponding NDDSs can better achieve drug loading for drugs with different physical and chemical properties (such as molecular weight, log P and pKa). A variety of carrier materials are used for the co-delivery of hydrophobic drug combinations, including polymers, micelles, silica, etc.172 Polymer-drug couplings can also increase the solubility of hydrophobic drugs and protect them from degradation. If the physicochemical properties of the two drugs are significantly different, then liposomes with the hydrophobic lipid bilayer and hydrophilic core, polymer NPs prepared by the double emulsion method, or carrier-free self-assembled nanoparticles can be selected.173,174
Phagocytosis of the endothelial system, protein crown construction, blood clearance, and rapid enzyme degradation hinder the delivery of free chemotherapeutic and immunotherapeutic agents to the tumor site. Due to the loss of rapid blood elimination and non-targeted distribution, large doses are often required, which can accumulate stronger toxicity in the body.175 NDDSs can significantly improve the efficiency of loaded drugs and reduce toxicity. Despite all that, Unprotected nanoparticles in the bloodstream are often rapidly removed, which may affect the drug’s circulation time in the body.176 Many formulations employ the attachment of polyethylene glycol (PEGylation) to improve circulation time while protecting NP from enzymatic hydrolysis or antibody clearance. Another approach is to coat the surface of NPs with platelet membranes, erythrocyte membranes, and other membrane-like structures, making NPs “invisible” to escape the clearance mechanisms in vivo and providing more opportunities for NPs to reach target tissues.176,177
Compared to the direct mixing of free drugs, the most important advantage of NPs for multi-drug delivery is to deliver encapsulated drugs at a defined dose ratio.178 Some drug combinations show the best synergistic activity at specific dose ratios. It has been found that cytarabine and daunorubicin exhibit the highest synergistic frequency and the lowest antagonistic frequency at the molar ratio of 5:1 (designated CPX-351). When free cytarabine and daunorubicin are injected, the drugs are rapidly cleared, and the circulating drug ratio is inconsistent with that encountered by the tumor cells. After injection of CPX-351, the proportion of drugs in plasma changed little within 24 hours. This shows that NDDSs can ensure the optimal proportion of drug action to the greatest extent.179
Precise Targeting
Tumor targeting based on NDDSs is usually achieved through two main mechanisms, namely passive targeting and active targeting. Passive targeting is mainly achieved by the enhanced permeability and retention (EPR) effect, which are the accumulation of some molecules or particles in tumor tissues more easily than in healthy tissues due to the leaky tumor vascular system and damaged lymphatic vessels. This will facilitate the selective distribution of nanoparticles with diameters of 100–400 nm in tumor tissues, thus improving drug efficacy and reducing systemic side effects.180 Active targeting depends on modifying nanoparticles with ligands (eg, antibodies or peptides), and targeting ligands specifically recognizes receptors overexpressed at pathological sites. Currently, commonly used receptors include the EGFR receptor, transferrin receptor (TfR1), folate receptor (FR), and integrins receptor.181 It can be divided into three categories according to the target, including the peripheral immune system, the TIME, and cancer cells.23
Secondary lymphoid organs, including the lymph nodes and spleen, together make up the peripheral immune system, which is critical in tumor immune regulation because antigen presentation occurs in these cellular compartments. Intradermally or subcutaneously injected nanoparticles flow into lymph nodes, and antigens carried by NPs are more effectively absorbed by APCs than small molecules and soluble vaccines.23 Kuai et al prepared nanodiscs physically loaded with CpG and peptide antigen (Ag) and demonstrated that sHDL nanodiscs significantly improved co-delivery of Ag/adjuvant to lymphatic organs. Remarkably, in clinical trials, nanodiscs induced neoantigen-specific CTLS at a rate 47 times higher than that of soluble vaccines and even 31 times higher than the strongest adjuvants, such as CpG in Montanide.182
Nanomedicines are designed to target and modulate TIME. Based on the interaction between tumor cells and the immune system, targeting TIME provides a new perspective for tumor therapy. Macrophage mannose receptor (MMR, CD206) is significantly expressed on TAMs, so it is widely used to target TAMs. Secreted protein acidic and rich in cysteine (SPARC) is also highly expressed on TAMs and has emerged as a potential therapeutic target for albumin nanoparticle systems.183 Some researchers have found that leucine-rich repeat containing 15 (LRRC15) peptide is overexpressed on cancer-associated fibroblasts (CAFs) in many solid tumors, and the small peptide FH has a high binding affinity with tenascin C secreted by CAFs, which provides implications for corresponding targeting strategies.184 Chen et al developed anti-CD47 antibody-functionalized CaCO3 nanoparticles. In acidic and inflammatory TME, CaCO3 particles can gradually lysate and release coated aCD47, thus promoting the activation of M1-like TAM.185 Lu et al combined IDO inhibitor indoximod (IND) with oxaliplatin in lipid-coated mesoporous silica NPs and achieved cancer cure in mouse models of pancreatic ductal adenocarcinoma.186
Nanomedicines that target tumor cells are often designed to induce ICDs and enhance cancer immune circulation. Researchers encapsulated OXA into amphiphilic diblock copolymer PLGA nanoparticles and found that the NP-OXA group had considerably higher therapeutic effects than the free OXA group and could induce more production of tumor-infiltrating activated cytotoxic T lymphocytes. This suggests that chemotherapeutic agents can be more effectively targeted to cancer cells by nanoformulations while avoiding systemic toxicity induced by free chemotherapeutic agents.187 Moreover, effective NDDSs are not limited to carrying therapeutic drugs to target cells but also attempt to deliver drugs to different subcellular sites. Direct delivery of NDDSs to sub-organelles is likely to allow NDDSs to bypass the subcellular barrier and concentrate the drug around the target of the action, thereby reducing resistance. Such as alkylation of the piperidine segment can be targeted at lysosome, E3/19k of adenovirus, phosphotetrapeptide (4p) has been used to build NPs that target the endoplasmic reticulum (ER). Several anticancer drugs act within the nucleus, including cisplatin, camptothecin, and doxorubicin, which induce ICD by causing DNA damage.188 Nuclear localization signal sequences (NLSs) (such as SV40 T antigen, adenovirus, and TAT peptide) are the most classical ligands for nuclear transport of exogenous nanoparticles via the α/β pathway of importin protein. Another method is to open the nuclear membrane with the help of cell-penetrating peptides (CPPs). For example, the newly synthesized, more permeable CB5005 has been found to have a unique affinity for brain gliomas.189 Mitochondria are the main places that provide energy for life activities, and they are involved in intracellular signal transduction and apoptosis regulation. Mitochondrial targeting is usually achieved through two different targeting ligands: one is mitochondrial targeting small molecules, such as triphenylphosphine (TPP), dequalinium chloride (DQA), F16, and rhodamine. The other is mitochondria-targeting bioactive molecules, including mitochondrial penetrating peptides (MPPs), SS peptides, mitochondrial targeting sequences (MTSs), cysteine-rich peptides (CRPs), and ER signaling peptides.190
Promote Immune Response
Most biomedical applications of nanocarriers in CIT focus on drug delivery, with little attention paid to their potential positive stimulative function on the immune system. Some nanomaterials can be used as adjuvants to promote DC maturation by enhancing APC antigen presentation or changing the type of immune response. It has been shown that mesoporous silica acts as an adjuvant, stimulating immune response.191 Luo et al found that the free Fe3O4 NPs group showed significant immunotherapeutic ability to stimulate dendritic cells and activated macrophages. This suggests that Fe3O4 nanoformulations not only serve as delivery tools to protect the drug from degradation but also act as a powerful enhancer to promote immune responses.192 Superparamagnetic iron oxides can induce M2-TAMs to M1-TAMs, and lead to cell apoptosis through the Fenton reaction.193 In addition, some polymers also exhibit adjuvant properties. Chitosan promotes DC maturation and enhances Th-1 immune response by inducing IFN production.194 PLGA can induce cytoplasmic antigen delivery and enhance MHC-I antigen presentation.195 Polyethyleneimine (PEI) can increase the cytotoxicity of T lymphocytes and promote Th1/Th2 activation, thus inhibiting tumor growth.196 The polypeptide carrier poly-L-lysine (PLL) has also been used as a coating material to enhance the efficacy of DNA vaccines.197
Applying an external energy field to inorganic NPs can enhance the tumor-killing effect induced by chemotherapy drugs to amplify the ICD. Many inorganic NPs, including iron oxide, Au NPs, CuS NPs, GO, molybdenum disulfide (MoS2) nanosheets, or carbon nanotubes, can further kill tumor by magnetic fluid hyperthermia (MFH), PTT, PDT.198 Interestingly, due to the inherent physicochemical properties of some nanomaterials, similar effects can be achieved even without chemotherapy drugs. Liu et al developed a ferrimagnetic vortex domain iron oxide nanoring and graphene oxide (FVIOs-GO) hybrid nanoparticle as a magneto thermodynamic (MTD) agent. In hypoxic tumor microenvironments below 40 °C, FVIOs-GO nanoparticles can significantly amplify ROS levels under alternating magnetic fields (AMF). FVIOs-GO mediated MTD can induce ICD. There 83% of 4T1 cells released the CRT, TAMs were polarized from M2 phenotype to M1 phenotype, and T lymphocyte infiltration was increased in TME.199
The Unique Properties of NDDSs - Stimulus Responsiveness
In order to further enhance the accumulation at tumor sites, maintain the plasma concentration of drugs, and reduce their side effects, vectors that can be designed to control the spatial and temporal release of the drug are becoming increasingly popular. Based on differences between cancer and normal cells, stimuli-sensitive intelligent vectors that can be affected by various internal and external stimuli have been studied in detail. Endo-stimuli-responsive nanoparticles (en-srNPs) respond to pH changes, redox potential, and enzyme activation, while exo-stimuli-responsive nanoparticles (ex-srNPs) respond to heat, magnetic field, light, etc. The combination of dual or multiple stimuli also increases their feasibility as smart nanocarriers.
PH-Mediated en-srNPs
Normal physiological conditions have a pH of approximately 7.4, the microenvironment of solid tumors is weakly acidic (pH≈6.8), and the lysosome is more acidic (pH≈5.5).200 This situation is mainly due to the inability of the abnormal vascular network to provide adequate blood supply to all cells within the tumor, the rapid proliferation of cancer cells leading to an inadequate supply of oxygen and nutrients, and the energy expenditure of glycolysis (rather than oxidative phosphorylation) increases biosynthetic function, resulting in an increased rate of lactate production, which is also known as the Warburg effect. There are two main approaches to achieving controlled release of anticancer drugs through pH-responsive self-assembled NPs. One is to exploit the breakage of acid-unstable chemical bonds, such as hydrazones and acetals; the other is to use basic or acidic groups (amino, phosphate, or carboxyl) in the polymer, pH-mediated swelling behavior induced by protonation is considered to be the main factor for controlling drug delivery. Besides, acrylic acid, methacrylic acid, maleic anhydride, and N, N-dimethylamino ethyl methacrylate are all pH-sensitive monomers; and many functional groups, including benzoic imine bonds, carboxylic acid, and phenylboronic acid, also exhibit pH-responsive shifts when pH changes.180,201 Zhu et al utilized the multimolecular adsorption capacity of CaCO3 to prepare a poly (ethylene glycol) -β-poly (lactate-co glycolic acid) (PLGA-PEG) nanoparticle (DNCaNPs) co-loaded with DOX and aNLG919 (an IDO1 inhibitor). In the slightly acidic tumor environment, the NP shell is removed to release drug-carrying Ca NPs, which can cause effective ICD, while limiting immunosuppressive kynuridine production by inhibiting IDO1.202
Redox-Mediated en-srNPs
Reactive oxygen species (ROS) and reduced glutathione (GSH) are more expressed in cancer tissues than in normal tissues, the redox-responsive NP collapses with sensitivity to GSH or ROS, thus resulting in controlled release of the drug. ROS are highly reactive chemicals containing oxygen free radicals, including superoxide radical (O2-) hydroxyl radical (•OH), and hydrogen peroxide (H2O2). In order to adjust to high levels of intracellular ROS, tumor cells usually develop adequate amounts of adaptive antioxidants to keep redox homeostasis. These antioxidants are mainly divided into two groups, which are enzymes (eg, catalase, superoxide dismutase, and glutathione peroxidase) and reducing agents (eg, GSH). Therefore, increasing ROS production and/or decreasing ROS clearance to disrupt redox homeostasis should be effective in amplifying intracellular oxidative stress and thus treating cancer more effectively.203 ROS can be produced by a variety of methods, such as PDT, biochemical reactions, and molecular drugs.204,205 One such reaction is the Fenton reaction, which uses ferrous ions to convert H2O2 to •OH in situ. A variety of iron-containing preparations, including iron oxide nanoparticles, iron nano metallic glasses, and metal polyphenol networks (MPNs) have been used as catalysts for the Fenton reaction to induce cell death.205 Tan et al designed a GSH-reactive mixed micellar nanoparticle based on camptothecin (CPT) prodrug (siPD-L1@HM-CPT). siPD-L1@HM-CPT improved intratumoral CD8+ T cell infiltration by strong ICD and sustained ICB, and demonstrated the favorable anti-tumor immune response in B16-F10 and 4T1 tumor models.206
Enzyme-Mediated eEn-srNPs
Enzymes are an essential component of bio-nanotechnology, which have excellent bio-cognitive capabilities and superior catalytic properties. In general, the aberrant expression of enzymes observed in tumors provides some ideas for developing enzyme-active bond-modified nanocarriers. Enzymes that play a major role in tumors include proteases, lipases, oxidoreductases, etc.207 Once exposed to the enzymes, enzyme-responsive NPs will release drugs at the target site due to the breakage of the ester bond or peptide fragment. Matrix metalloproteinases (MMPs) are proteases that catabolize extracellular matrix components. They have been reported to be overexpressed in many tumors, where MMP-2 and MMP-9 are primarily used for enzymatic drug release.208 Zhou et al designed OXA prodrug and pegylated photosensitizer (PS) as prodrug vesicles. The PEG crown of the prodrug vesicle is stripped by matrix metalloproteinase-2 (MMP-2) at the tumor site, and its surface charge changes from negative to positive, allowing deep tumor penetration and enhancing cell uptake. Under laser irradiation, the prodrug vesicles induce ROS production and promote the instantaneous release of the drug. The nano-vesicles can enhance anti-tumor immunity by ICD, and inhibit tumor immune evasion by αCD47, which can effectively inhibit tumor growth/metastasis and prevent tumor recurrence.209
Thermal-Mediated ex-srNPs
When the ambient temperature is elevated by exposure to certain energy sources such as laser or ultrasound above the lower critical solution temperature (LCST), the thermally responsive NPs undergo a phase transition, and the encapsulated drug is released.180 In addition, some photothermal materials produce photothermal conversion effects under NIR, which may directly kill tumor cells and improve the TME. Zheng et al prepared Her2-DOX-superparamagnetic iron oxide nanoparticles (SPIOs)@PLGA@ Au nanoparticles (Her2-DOX-SPIOs@PLGA@Au), the results suggest that PTT can reshape the TME by reducing the amount of CAFs and, enhance the accumulation of nanomedicines.210
Magnetic Field-Mediated ex-srNPs
Magnetic nanoparticles are nanomaterials composed of magnetic elements (eg, Co, Fe, Ni, Ti), and their compounds, which are usually classified as pure metals, metal oxides, and magnetic nanocomposites. In among thereinto, iron oxide NPs (typically Fe2O3 or Fe3O4) are widely used because of their low toxicity.211 The function of magnetically responsive NPs can be divided into two treatments. On the one hand, when a permanent magnetic field is applied, NPs are magnetically targeted to accumulate in the desired tissue and are guided to achieve a controlled release of cargo. On the other hand, the NP generates induced heat and elevates the temperature at local positions when an alternating magnetic field is applied externally. When NP binds to a thermosensitive or photosensitive material, it eventually leads to cancer cell death. Qin et al prepared super magnetic iron oxide nanoparticles (SPIOs) and encapsulated the immune adjuvant R848 and phase change agent PFP in PLGA shells (RPPs). The primary tumor was injected with mixed nanoparticles SPIOs + RPPs. Under magnetothermal stimulation, the nanodroplets underwent the liquid-gas transition and produced microbubbles in situ. The combination of MHT and cavitation effect enhances ICD and facilitates the release of R848. Ultimately, bioactive DAMP combined with R848 creates the highly immunogenic TME that activates both DC and CTL to combat tumor proliferation or metastasis.212
Light-Mediated ex-srNPs
When exposed to a variety of light irradiation including ultraviolet (UV), visible light and NIR, the photoswitchable groups undergo a succession of shape changes and electronic conformations that enable the breakdown of light-mediated ex-srNPs and the controlled release of drugs. One way to use high-energy ultraviolet lasers to ablate the covalent bonds of the polymer that are non-specifically. Another way is to embed metal clusters or dyes (photosensitivities) in NPs to generate local thermal energy as they absorb light. Thermal shock causes the vector to be destroyed and the drug is released.213 Gong et al prepared nanoparticles that co-delivered DOX, photosensitizer Indocyanine Green (ICG), and angiotensin II receptor blocker valsartan (VAL) (PEG-VAL&ICG@RNPs). The results showed that in 4T1 tumor-bearing mice, the PEG-VAL&DOX&ICG@RNPs group could effectively inhibit tumor growth by down-regulating the expression of α smooth muscle actin in CAFs, inducing DCs maturation and promoting infiltration of cytotoxic T lymphocytes.214
Although independent stimulus responses to NPs have many advantages, they cannot overcome all obstacles. Therefore, different combinations of endogenous or exogenous stimuli should be investigated. In particular, for unique multi-stimuli-responsive nanoparticles (m-srNPs) that respond to stimulus signals simultaneously or sequentially, including pH/photon, pH/ROS, pH/GSH, etc., the effect of m-srNPs on TME is more significant than that of single stimulus-responsive nanoparticles.201 The intelligent m-srNPs with multiple responsiveness provide flexible strategies for the precise control of cancer therapeutics.
Common Carriers for Combination Therapy
NDDSs can simultaneously load multiple therapeutic agents into the defined carrier, enabling synergistic delivery of antigen+adjuvant or chemotherapy/phototherapy/immunotherapy “all-in-one” combination therapy. Numerous NDDSs have been designed, such as lipid nanocarriers, polymeric carriers, inorganic NPs, and biomimetic nanoparticles (Table 4).
 |
Table 4 Carrier-Mediated Combinations of Chemotherapeutic and Immunological Agents |
Lipid-Based NPs
Lipid-based nanoparticles are mainly separated into liposomes and lipid nanoparticles (LNPs). Compared to other nano-delivery systems, lipid-based NPs maintain high solubility in the aqueous phase while reducing systemic toxicity, and these strengths make lipid NPs one of the commonly approved nano-drug types.231
Liposomes are composed of phospholipids and cholesterol bilayer, with hydrophilic drugs encapsulated within the core and hydrophobic drugs encapsulated within the lipid bilayer. Since the FDA approval of the liposome formulation Doxil® in 1995, many new liposome formulations have been developed, entered clinical trials, and were approved by the FDA for clinical use.181 Liu et al reported that 5-carboxyl-8-hydroxyquinoline (IOX1) and DOX co-delivered nanoliposomes (IPLD) reduced the growth of various murine tumors, and offered long-term immunological memory effects on tumor recurrence. IOX1 inhibits the tumor histone demethylase Jumonji domain-containing 1A (JMJD1A), thereby down-regulating β-catenin and PD-L1. This study provides an antibody-independent mode that interrupts the PD-1/PD-L1 checkpoint.232 Kim et al prepared nanoliposomes containing DOX and IDO1 siRNA (Aptm [DOX/IDO1]), which significantly reduced tumor metastasis by by ICD and reversal of IDO1-mediated immunosuppressive TME.217
LNPs include solid lipid nanoparticles (SLN), nanostructured lipid vectors, cationic lipid-nucleic acid complexes, etc. SLNs can be simply mass-produced without organic solvents, and the reduced molecular mobility in the solid state enables SLNs to control the release of their drug.233 Kim et al designed a solid lipid nanoparticle (DSLN) loaded with docetaxel (DTX), which was lidded with glycocholic acid-chondroitin sulfate conjugate (CSG) (DSLN-CSG).216 Guo et al used nanoprecipitation technology to make aminoethyl anisamide (AEAA)-targeted PEGylated lipid nanoparticle (Nano-Folox). FOLFOX is a combination strategy of folinic acid (FnA), 5-Fu, and oxaliplatin (OXP) that has been used as a standard treatment for colorectal cancer (CRC). The nanoparticle showed good chemoimmunotherapeutic activity in mice with colorectal cancer in situ.215
Polymer NPs
Natural polymers are composed of macromolecules from nature and can be divided into two main types: protein polymers, such as gelatin and collagen; and polysaccharides, like Chitosan (CS), glycogen, starch, and cellulose. The synthesized polymers are biocompatible and biodegradable, including PEG, PLGA, PLA, PGA, poly (vinyl alcohol) PVA, polyphosphoesters (PPEs), PLL and poly (ε-caprolactone) (PCL).234
Chitosan is a naturally cationic polymer with a variety of attractive properties such as renewable, hydrophilic, biodegradable, biocompatible, non-toxic, etc. Its structure bears active functional groups that allow for a variety of chemical reactions at the biomolecular level, which can improve the physicochemical properties and functions.235 Gu et al prepared an in situ therapeutic composite stent (DOX-JQ1@Gel) to allow the local release of DOX and JQ1 (small molecule inhibitors of the extra terminal protein BRD4 and bromodomain). DOX-JQ1@Gel induces immunogenic tumor phenotype, and JQ1-mediated PD-L1 checkpoint-blocking accumulation contributes to triggering anti-tumor immune responses. This study suggests that local treatment produces systemic anti-cancer immune responses and can inhibit tumor growth at a distance.236
Polymeric micelles are made up of amphiphilic block copolymers that spontaneously self-assemble in aqueous solutions, and hydrophobic drugs can be chemically coupled or physically encapsulated in micelle nuclei. Da Silva et al used a solvent volatilization-extraction method to synthesize biocompatible poly (lactic-co-glycolic acid)-PEG NPs for the loading of NIR dyes, doxorubicin cytostatic agent, polyinosinic-polycytidylic acid (poly (I:C)), R848, and CCL20 chemokine. The NPs can increase the levels of lymphocytes and CD8+ T cells circulating in the tumor and blood, enhancing the immune response and thereby eradicating the tumor.223 Ge et al prepared triblock copolymeric micelles carrying CPT and TLR7/8 agonist 1-(4-(aminomethyl) benzyl)-2-butyl-1H-imidazo[4,5-c] quinoline-4-amine (IMDQ) (NanoPCPT+PIMDQ), a dual-responsive polymeric drug conjugate that responds to pH/GSH in the TME, releasing CPT to kill tumor cells, while PIMDQ activates DCs and increases CTL infiltration. Both CPT down-regulation of Foxp3+Treg and PIMDQ-induced repolarization of M2 macrophages alleviate TIME, which provides a new idea for synergistic tumor chemoimmunotherapy.224 Sunitinib (SUN) is a multi-target receptor tyrosine kinase inhibitor that the FDA has approved as an anti-angiogenic drug. It can not only inhibit the increase of tumor extracellular matrix components but also reshape the immunosuppressive TME and reduce the level of suppressive MDSCs and Tregs. Qin et al prepared polymeric micelles comprised of poly (styrene-co-maleic anhydride) (SMA) by combining PTX and SUN, the drug ratio of 1:5 (PTX: SUN), which enhanced tumor immunogenicity by promoting the ICD response and significantly modulates immunosuppressive factors in TME, inducing synergistic effects.237
Dr. Donald Tomalia first shared groundbreaking results on the dendrimer of poly (amidoamine) (PAMAM) in 1985, and researchers began applying dendrimers to drug delivery systems in the late 1990s. Two approaches were used for drug delivery of dendrimers, physical encapsulation and nanoconstruct. In the former approach, the drug is physically wrapped in the dendrimer through non-covalent interactions, while in the latter method, the drug is covalently coupled to the dendrimer.238 Lee et al reported an approach to targeting CIT based on aptamer-dendrimer couples, in which an RNA aptamer that specifically binds prostate-specific membrane antigen (PSMA) acts as a targeting ligand. It uses plasmid-DOX complexes, in which plasmids carrying unmethylated CpG act as both immunostimulants and carriers for DOX (DOX@Apt•dONT-DEN). The results showed that the cytokines TNF-α, IL-1β, IL-12, and IL-6 in the DOX@Apt•dONT-DEN treatment group were considerably higher than those in the control group.226 Tong et al bind N, N-dipentylethyl moieties and monomethoxylpoly (ethylene glycol) to PAMAM dendrimer and introduced the prodrug hydrophobic Gem in the cavity of the dendrimer macromolecule. The nanoparticle regulates the infiltration frequency of TAMs, cytotoxic T cells, MDSCs, and Tregs by enhancing the expression of PD-L1. Besides, the combination of PD-1 antibodies can further improve the therapeutic effect.225
Metallic and Inorganic NPs
In addition to stability, ease of synthesis, modifiability, inertness, magnetic and optical properties make inorganic NPs attractive for imaging and ablation of malignant tissues. Several inorganic materials have been used to synthesize nanostructured materials such as silica, iron, and gold. The inorganic NPs are precise and can be designed into a wide range of sizes, structures, and geometries, including nanospheres, nanorods, nanostars, nanoshells, and nanocages.239
At present, among various nanoparticles, mesoporous silica nanoparticles (MSNs) are considered to be one of effective drug delivery systems. Various micro silicas with regular mesopores have been widely used, such as SBA-15, MCM-41, MCM-48, hollow MSNs, and core-shell MSNs.240 Liu et al prepared irinotecan-loaded silicasome coated with lipid bilayer. The alkalinity of irinotecan neutralizes lysosomal acidity in PDAC cells and triggers associated downstream cascade events, including endoplasmic reticulum stress, autophagy inhibition, ICD, and PD-L1 expression. Furthermore, it was demonstrated that silicasome combined with anti-PD-1 greatly improved survival and was much better than anti-PD-1 combined with free irinotecan or Onivyde.241 All-trans retinoic acid (ATRA), DOX, and IL-2 were co-wrapped by Kong et al to construct lipid-coated hollow mesoporous silica nanoparticle (dHMLB), which exerts synergistic antitumor effects by remodeling the tumor immune microenvironment. The dHMLB promotes the increase of IL-12 and IFN-γ while down-regulating the expression of MDSCs, IL-10, and TGF--β, exerting a benign regulatory effect on the TME.227 Sun et al constructed hollow mesoporous silica-coated gold nanoparticles (Au@HMnMSns) doped with manganese in the ovate form of tadpoles as a biodegradable cascade catalytic nanoreactor, which loaded with DOX and Aspirin (ASA) to generate intra-tumoral highly toxic hydroxyl radicals to promote ICD induction and DC cell maturation. Meanwhile, the tadpole ellipsoidal structure of Au@HMnMSNs can prevent the deactivation of gold NPs because of protein adsorption in organisms and promote further GOx-like catalysis.228
Iron oxide nanoparticles (IONPs) can respond to magnetic fields, allowing therapeutic nanomedicines to act as excellent contrast agents for MRI. The surface of IONPs is typically coated with silica to promote biocompatibility, reduce toxicity, and provide stability under physiological conditions.239 Some investigators have prepared prodrug-modified pegylated phospholipid micellar coated IONPs loaded with cisplatin (Pt (IV)-IONPs and poly (I:C) -Pt (IV)-IONPs), which has shown a much greater ability to activate dendritic cells compared to free cisplatin and poly (I:C).230 Mu et al prepared an ultra-small (8 nm) iron oxide core with a layer of negatively charged PEGylation silica to contain endoglin-binding peptide (EBP), DOX, and poly I:C (IONP-DOX-Poly I:C-EBP). The unique structure formed by these NPs relies on the alternating accumulation of positively charged DOX and negatively charged poly I:C, without the help of additional polyelectrolytes. The nanoparticles not only markedly inhibited tumor growth and metastasis but also significantly extended the survival of mice with drug-resistant triple-negative breast cancer (TNBC).229
AuNPs exhibit a distinct photophysical property due to the free electron resonance generated when the cloud of free electrons is illuminated by resonant frequency photons, known as the localized surface plasmon resonance (LSPR). In addition, the tunable shape and size, the flexibility to fabricate different morphologies, and the easy functionalization of ligands via Au-S chemical bonding make AuNPs hopeful drug carriers for disease therapy.242 Zheng et al prepared Her2-DOX- SPIOs@PLGA@Au nanoparticles (Her2-DSG NPs). Gold shell-mediated photothermal effects can reshape the TME by reducing CAFs while promoting DOX release. Moreover, the NPs can enable photoacoustic (PA)/MRI dual-mode imaging for diagnosis and identification of Her2-positive breast cancer.210
Bionic NPs
Surface functionalization by chemical coupling or non-covalent bonding to the particle surface is typically single, utilizing only known biological interactions. In reality, however, effective biological interfaces are often multifactorial, and the mechanisms involved still need to be fully elucidated.243 Membrane camouflage nanoparticles (CM-NPs) are an exciting development in bionanotechnology that camouflage the synthetic core through the naturally derived cell membrane that includes platelets, red blood cells, neutrophils, macrophages, or cancer cells. Cell membrane coatings can preserve the complex composition of the complete cell surface on nanoparticles without the need to identify individual membrane components. Due to the inherent self-recognition properties, cell adhesion properties, and homing capabilities of cell membrane coatings, CM-NPs have the potential to inhibit reticuloendothelial system (RES) elimination and immune clearance, prolonged somatic circulation, and targeted drug delivery.220
Erythrocyte membrane-coated nanoparticles are a novel method to increase the circulating half-life of nanoparticles. As the most abundant blood cells in the bloodstream, erythrocytes provide a large number of coating materials for the functionalization of drug carriers. Moreover, mature erythrocytes lack nuclei and various organelles, which is highly beneficial for their extraction and purification. The long cycling effect of erythrocytes depends on the mutual recognition between the “self-tagging” protein CD47 on the surface of erythrocytes and the signal-regulating protein SIRPα expressed by phagocytes.244 Li et al prepared an acid-responsive “core-shell” nanoparticle (RBC-SLip) for tumor-targeted therapy. Due to the outer coating derived from the red blood membrane (RBCm), it is stable under physiological conditions. The levels of DC mature cells, NK cells, CLTs, and tumor-infiltration CD4+ T cells in the RBC-SLip/PTX group were higher than those in the normal saline group. These results suggest that RBC-SLip/PTX does improve immune inhibitory TME, and explain the beneficial effect of enhancing immune response.220 Wang et al developed a hemoglobin-poly (ε-caprolactone) (Hb-PCL) conjugated self-assembled bionanosome system V(Hb)@DOX for delivering DOX and reprogramming the TIME hypoxic environment. The Hb portion binds to endogenous plasma haptoglobin (Hp) and targets M2-type TAMs via the specific receptor CD163. In addition, O2 released from Hb attenuates the hypoxic environment of the tumor, thereby further enhancing the anti-tumor immune effect by reducing the recruitment of M2-type macrophages.221
Macrophages play a role in TME and therefore directly influence tumor progression. Macrophage-coated NPs promote RES escape, tumor homing, and intercellular adhesion. After completing their task, the extracellular microenvironment stimulates the macrophage cell membrane to undergo morphological changes and shed the coating.245 Yang et al co-wrapped short-hairpin RNA (shRNA) targeting protein tyrosine phosphatase nonreceptor type 2 (Ptpn2) and DOX with M1 macrophages membrane (M1HD@RPR), NPs escape from RES was improved, and tumor site concentration was increased. M1HD@RPR directly killed cancer cells and stimulated ICD, while down-regulating Ptpn2 expression, promoting M1 macrophage polarization and dendritic cell maturation, and realizes the inhibition of primary melanoma and lung metastasis.219
Multi-membrane functionalized biomimetic nanoparticles were prepared using fused cell membranes, which combined the natural properties of different cells. The hybrid film-coated NPs keep the protein markers of each source cell and combine both of these unique functions.246 Hao et al developed a chemoimmunotherapeutic vector based on C6 cell membranes and DC membranes to manufacture hybrid membrane-coated DTX nanosuspensions (DNS-[C6&DC]m). The homologous targeting of tumor cell membranes is a key driver for smart targeted drug delivery. Moreover, because of the specialized antigen presentation properties of DCm, the antigen can be presented to the body’s immune system to facilitate, and the synergistic effect of CIT increases the anti-tumor effect.222
Immune Cocktail Therapy
“Cocktail therapy” was first proposed by the Chinese-American scientist David Ho in response to the frequent drug resistance of AIDS patients. It refers to the combination of three or more antiretroviral drugs to treat AIDS. The combination of multiple drugs is more effective and less susceptible to resistance than a single drug. In recent years, researchers have referred to similar combination therapies for other diseases as “cocktail therapy”. In 2020, Tian et al proposed an innovative “immune cocktail therapy” that combines immune checkpoint therapy with other treatments to modulate multiple stages of the cancer immune cycle using an intelligent nano-delivery system. They found that the acid-responsive shPD-L1 + DOX NP group achieved only 60.6% tumor inhibition in the mouse B16F10 model, which is not insufficient to achieve effective antitumor therapy. After that, the researchers added sperm molecule 1 plasmid (pSpam1) expressing HAase to eliminate the dense tumor extracellular matrix (ECM). The results showed that DOX NPs plus (shPD-L1 + Spam1) NPs induced 97.3% tumor reduction and longer median survival compared with the PBS group. This indicates that immune cocktail therapy has excellent anti-tumor efficiency.247 Yu et al developed a local cocktailization scheme. The researchers combined DOX/OXA and imiquimod (R837) in the drug excipient alginate (ALG) (DOX/R837/ALG or OXA/R837/ALG). Anti-PD-L1 is added to the injection during administration. Due to the presence of Ca2+ in the tumor, ALG can rapidly form hydrogels. After in situ gelation, the therapeutic drug is released continuously. The results showed that in the CT26 tumor model, OXA/R837/ anti-PD-L1 /ALG local treatment could eliminate the primary tumor, inhibit distant tumors, and further prevent tumor recurrence. Using another cocktail formulation with DOX in place of OXA, this localized CIT showed exceptional efficacy in an in-situ mouse model of breast cancer with spontaneous metastasis, as well as in an in-situ brain tumor model with a single injection.248 Chen et al reported a homologous targeted cocktail formulation to achieve photothermally-chemotherapy-immune synergistic therapy. They prepared a cancer cell membrane-coated PLGA nanosphere (M@P-PDR) containing the photothermal transmitter Prussian blue (PB), DTX, and R837. The results showed that with the help of R837, the maturation rate of dendritic cells was 4.34 times higher than that of the control group. In addition, DTX polarizes the M2-TAMs to the M1 phenotype. The expansion rate of CTLs increased from 17.33% to 35.5%. Primary tumors and metastases were significantly suppressed. This suggests that nano-targeted cocktail therapy is a promising approach to promote tumor regression and combat metastasis/recurrence.249 Overall, any strategy that promotes the positive shift in key parts of the cancer immune cycle could be a tool for future tumor immune cocktail therapy.
Conclusion and Perspective
Understanding immunotherapy options, including immune checkpoint suppression, adoptive immunotherapy, cancer vaccines, oncolytic virotherapy and cytokine therapy, can advance the future of CIT. It has been shown that chemotherapeutic drugs regulate immune effect through four main mechanisms: inhibiting immune checkpoint expression, inducing ICD, promoting tumor killer cell infiltration, and down-regulating immunosuppressive immune cells. These unique and potent modes can promote the synergistic effect of tumor CIT. A number of functional NDDS have been designed for CIT and these NDDS show a number of benefits, including increased solubility and bioavailability of the drug, extended circulation time in vivo, and improved pharmacokinetic behavior. In addition, NDDSs can deliver multiple drugs to specific sites by stimulating responsiveness and modifying targeting ligands to achieve precise targeting. Some special preparation structures, such as core-shell structures, allow chemical agents and immunomodulators to be transported to tumor cells and immune microenvironment respectively. The drugs can be released slowly in the fixed proportion, which improves the efficiency of treatment.
Although NDDSs have demonstrated excellent value in improving anti-cancer efficacy, only a few have made it to clinical trials, with most discontinued in vivo and in vitro. Most NPs face similar challenges. First, most studies have focused on cells and animal models, but the large tumor differences between mice and humans have led to differences in preclinical/clinical trial results for NDDSs.250 Due to the complexity of human tumor immunity, it is difficult for preclinical model inoculated tumor cell lines to simulate the influence of tumor immune background. Moreover, most of the models are subcutaneous tumor bearing, and this approach does not well reflect the tumor immune response shaped by long-term human tumor co-existence. Therefore, the selection of clinically relevant animal models is more challenging. The definition of preclinical models is also being updated and expanded, and some researchers believe that the genetically engineered mouse model is currently one of the most representative models of human disease.251 Secondly, from the perspective of commercial transformation, NDDSs with simple prescription, mature preparation process and good biocompatibility have higher clinical transformation prospect. Most NPs for in vivo and in vitro studies are typically produced in small batches, and formulations based on multi-drug combinations, triggering responses, will exhibit greater design complexity. Mass production is not always feasible due to equipment, cost, process complexity, etc. Therefore, in the design stage, more consideration should be given to practical application and cost-effective preparation of NDDSs with robust production capacity to increase the opportunity of commercialization.174 Another challenge is the need to optimize the combination of chemoimmunotherapies. Combination regimens typically involve multiple drugs, so they are complex and need to be properly designed. These types of regimens will require optimization of drug dosage and timing to achieve maximum benefit. The overall goal is to avoid overlapping toxicities, maximize synergies, and minimize the possibility of drug resistance. It has been proved that drug combinations show the best synergistic activity at a specific dose ratio. Metronomic doses have also been shown to enhance immunotherapy activity, and some drugs may only need to be given a few times over the course of treatment.4,179 Therefore, further investigation of the timing, sequence, and dosage of multiple drugs may provide a design paradigm for the development of CIT-based NDDSs.
Most CIT combination therapy formulations approved by the FDA in the last five years are still focused on combining chemotherapy drugs with immune checkpoint inhibitors. For immune checkpoint blocking, “cold tumors” are a huge obstacle. Immune cocktail therapy can regulate the cancer immune cycle through various mechanisms, transform cold TME into hot TME, promote T cell/dendritic cell initiation and maturation, significantly increase peripheral CD8+ T cell infiltration, induce strong immune memory effect, and effectively prevent tumor recurrence/metastasis. Therefore, immune cocktail therapy that combines chemotherapy drugs with immune checkpoint inhibitors and suitable immune adjuvants holds great promise. Depending on the properties of the material, a variety of materials have certain immune adjuvant effects, such as mesoporous silica, some iron preparations, and polymer materials. Many inorganic materials can amplify ICD after exposure to magnetic fields and light. We believe that the use of such materials as drug delivery vehicles can perform functions other than drug delivery and may enhance the therapeutic efficacy of CIT. In addition, we also noticed some biological materials. Cell drug loading systems can be carried out in three ways: co-loading, adsorption, and coating. Cells and their derivatives can escape the surveillance of the body’s immune system, and have good biocompatibility, inherent tissue targeting ability, inherent biodegradability, high drug loading ability, and natural ability to cross biological barriers. Therefore, we believe that biomimetic nanomaterials have broad development potential.
While this is still an emerging field and most of the current treatments are experimental at best, success in the laboratory paves the way for the clinical translation of these treatments. In conclusion, CIT still has many obstacles in oncology treatment, and NDDS may play a key role in facilitating better planning of long-term cancer treatment from a CIT perspective.
Acknowledgments
We would like to express our sincere gratitude to Professor Yifei Guo for her invaluable guidance and assistance. We thank BioRender (www.biorender.com) for expert assistance in the pattern drawing. Graphical Absrtact Created with BioRender.com.
Funding
This research was funded by CAMS Innovation Fund for Medical Sciences (No. 2022-I2M-2-002, No. 2021-I2M-1-031, No. 2021-I2M-1-071), Hainan Province Science and Technology Special Fund (No. ZDYF2023SHFZ140).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12–49. doi:10.3322/caac.21820
2. Nussbaumer S, Bonnabry P, Veuthey JL, Fleury-Souverain S. Analysis of anticancer drugs: a review. Talanta. 2011;85(5):2265–2289. doi:10.1016/j.talanta.2011.08.034
3. Perez-Herrero E, Fernandez-Medarde A. Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015;93:52–79. doi:10.1016/j.ejpb.2015.03.018
4. Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens J. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol. 2019;30(2):219–235. doi:10.1093/annonc/mdy551
5. Wu Q, Yang Z, Nie Y, Shi Y, Fan D. Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 2014;347(2):159–166. doi:10.1016/j.canlet.2014.03.013
6. Nurgali K, Jagoe RT, Abalo R. Editorial: adverse effects of cancer chemotherapy: anything new to improve tolerance and reduce sequelae?. Front Pharmacol. 2018;9:245. doi:10.3389/fphar.2018.00245
7. Bukowski K, Kciuk M, Kontek R. Mechanisms of multidrug resistance in cancer chemotherapy. Int J Mol Sci. 2020;21(9):3233. doi:10.3390/ijms21093233
8. Davern M, Lysaght J. Cooperation between chemotherapy and immunotherapy in gastroesophageal cancers. Cancer Lett. 2020;495:89–99. doi:10.1016/j.canlet.2020.09.014
9. Koury J, Lucero M, Cato C, et al. Immunotherapies: exploiting the immune system for cancer treatment. J Immunol Res. 2018;2018:9585614. doi:10.1155/2018/9585614
10. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154–158.
11. Zhu H, Liu Y, Yi X, et al. Novel biomimetic mesoporous silica nanoparticle system possessing targetability and immune synergy facilitates effective solid tumor immuno-chemotherapy. Biomater Adv. 2023;144:213229. doi:10.1016/j.bioadv.2022.213229
12. Mu W, Chu Q, Liu Y, Zhang N. A review on nano-based drug delivery system for cancer chemoimmunotherapy. Nanomicro Lett. 2020;12(1):142. doi:10.1007/s40820-020-00482-6
13. Bagherifar R, Kiaie SH, Hatami Z, et al. Nanoparticle-mediated synergistic chemoimmunotherapy for tailoring cancer therapy: recent advances and perspectives. J Nanobiotechnology. 2021;19(1):110. doi:10.1186/s12951-021-00861-0
14. Apetoh L, Ladoire S, Coukos G, Ghiringhelli F. Combining immunotherapy and anticancer agents: the right path to achieve cancer cure?. Ann Oncol. 2015;26(9):1813–1823. doi:10.1093/annonc/mdv209
15. Gilham DE, Debets R, Pule M, Hawkins RE, Abken H. CAR-T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med. 2012;18(7):377–384. doi:10.1016/j.molmed.2012.04.009
16. Wing A, Fajardo CA, Posey AJ, et al. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res. 2018;6(5):605–616. doi:10.1158/2326-6066.CIR-17-0314
17. He C, Tang Z, Tian H, Chen X. Co-delivery of chemotherapeutics and proteins for synergistic therapy. Adv Drug Deliv Rev. 2016;98:64–76. doi:10.1016/j.addr.2015.10.021
18. Akao T, Kimura T, Hirofuji YS, et al. A poly (gamma-glutamic acid)-amphiphile complex as a novel nanovehicle for drug delivery system. J Drug Target. 2010;18(7):550–556. doi:10.3109/10611861003599453
19. Zolnik BS, Gonzalez-Fernandez A, Sadrieh N, Dobrovolskaia MA. Nanoparticles and the immune system. Endocrinology. 2010;151(2):458–465. doi:10.1210/en.2009-1082
20. Kennedy LB, Salama A. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70(2):86–104. doi:10.3322/caac.21596
21. Liu Y, Zheng P. Preserving the CTLA-4 checkpoint for safer and more effective cancer immunotherapy. Trends Pharmacol Sci. 2020;41(1):4–12. doi:10.1016/j.tips.2019.11.003
22. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33(17):1974–1982. doi:10.1200/JCO.2014.59.4358
23. Shi Y, Lammers T. Combining Nanomedicine and Immunotherapy. Acc Chem Res. 2019;52(6):1543–1554. doi:10.1021/acs.accounts.9b00148
24. Camacho LH. CTLA-4 blockade with ipilimumab: biology, safety, efficacy, and future considerations. Cancer Med. 2015;4(5):661–672. doi:10.1002/cam4.371
25. Hosseini A, Gharibi T, Marofi F, Babaloo Z, Baradaran B. CTLA-4: from mechanism to autoimmune therapy. Int Immunopharmacol. 2020;80:106221. doi:10.1016/j.intimp.2020.106221
26. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood. 2018;131(1):58–67. doi:10.1182/blood-2017-06-741033
27. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–569. doi:10.1084/jem.174.3.561
28. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. doi:10.1126/science.271.5256.1734
29. Lee HT, Lee SH, Heo YS. Molecular interactions of antibody drugs targeting PD-1, PD-L1, and CTLA-4 in immuno-oncology. Molecules. 2019;24(6). doi:10.3390/molecules24061190
30. Zito CR, Kluger HM. Immunotherapy for metastatic melanoma. J Cell Biochem. 2012;113(3):725–734. doi:10.1002/jcb.23402
31. Keam SJ. Tremelimumab: first Approval. Drugs. 2023;83(1):93–102. doi:10.1007/s40265-022-01827-8
32. Khoja L, Day D, Wei-Wu CT, Siu LL, Hansen AR. Tumour- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol. 2017;28(10):2377–2385. doi:10.1093/annonc/mdx286
33. Janjigian YY, Shitara K, Moehler M, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398(10294):27–40. doi:10.1016/S0140-6736(21)00797-2
34. Zhang H, Dai Z, Wu W, et al. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J Exp Clin Cancer Res. 2021;40(1):184. doi:10.1186/s13046-021-01987-7
35. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi:10.1126/science.aar4060
36. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol. 2016;34(1):539–573. doi:10.1146/annurev-immunol-032414-112049
37. Wu Q, Jiang L, Li SC, He QJ, Yang B, Cao J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol Sin. 2021;42(1):1–9. doi:10.1038/s41401-020-0366-x
38. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413.
39. Hirose T, Yamamoto S, Kato K. Emerging data on nivolumab for esophageal squamous cell carcinoma. Expert Rev Gastroenterol Hepatol. 2021;15(8):845–854. doi:10.1080/17474124.2021.1948836
40. Suzman DL, Agrawal S, Ning YM, et al. FDA approval summary: atezolizumab or pembrolizumab for the treatment of patients with advanced urothelial carcinoma ineligible for cisplatin-containing chemotherapy. Oncologist. 2019;24(4):563–569. doi:10.1634/theoncologist.2018-0084
41. Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–2017. doi:10.1056/NEJMoa1414428
42. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi:10.1056/NEJMoa1504627
43. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–319. doi:10.1056/NEJMoa1411087
44. Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi:10.1056/NEJMoa1510665
45. Ferris RL, Blumenschein GJ, Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–1867. doi:10.1056/NEJMoa1602252
46. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–1191. doi:10.1016/S1470-2045(17)30422-9
47. Finkelmeier F, Waidmann O, Trojan J. Nivolumab for the treatment of hepatocellular carcinoma. Expert Rev Anticancer Ther. 2018;18(12):1169–1175. doi:10.1080/14737140.2018.1535315
48. Argenziano G, Fargnoli MC, Fantini F, et al. Identifying candidates for immunotherapy with cemiplimab to treat advanced cutaneous squamous cell carcinoma: an expert opinion. Ther Adv Med Oncol. 2022;14:17488992. doi:10.1177/17588359211066272
49. Apolo AB, Ellerton J, Infante JR, et al. Avelumab treatment of metastatic urothelial carcinoma (mUC) in the phase 1b JAVELIN solid Tumor study: updated analysis with ≥6 months of follow-up in all patients. Ann Oncol. 2017;28:v300–v301. doi:10.1093/annonc/mdx371.010
50. Powles T, O’Donnell PH, Massard C, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a Phase 1/2 open-label Study. Jama Oncol. 2017;3(9):e172411. doi:10.1001/jamaoncol.2017.2411
51. Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 2017;389(10064):67–76. doi:10.1016/S0140-6736(16)32455-2
52. Mohsenzadegan M, Bavandpour P, Nowroozi MR, et al. The potential of T cell immunoglobulin and mucin-domain containing-3 (Tim-3) in designing novel immunotherapy for bladder cancer. Endocr Metab Immune Disord Drug Targets. 2021;21(12):2131–2146. doi:10.2174/1871530321666210310142141
53. Shiravand Y, Khodadadi F, Kashani S, et al. Immune checkpoint inhibitors in cancer therapy. Curr Oncol. 2022;29(5):3044–3060. doi:10.3390/curroncol29050247
54. Chen Y, Hu H, Yuan X, Fan X, Zhang C. Advances in immune checkpoint inhibitors for advanced hepatocellular carcinoma. Front Immunol. 2022;13:896752. doi:10.3389/fimmu.2022.896752
55. Guedan S, Ruella M, June CH. Emerging cellular therapies for cancer. Annu Rev Immunol. 2019;37(1):145–171. doi:10.1146/annurev-immunol-042718-041407
56. Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–1680. doi:10.1056/NEJM198812223192527
57. Zhao Y, Deng J, Rao S, et al. Tumor Infiltrating Lymphocyte (TIL) therapy for solid tumor treatment: progressions and challenges. Cancers. 2022;14(17):4160. doi:10.3390/cancers14174160
58. Zacharakis N, Chinnasamy H, Black M, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. 2018;24(6):724–730. doi:10.1038/s41591-018-0040-8
59. Stevanovic S, Draper LM, Langhan MM, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. 2015;33(14):1543–1550. doi:10.1200/JCO.2014.58.9093
60. Tran E, Robbins PF, Lu YC, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375(23):2255–2262. doi:10.1056/NEJMoa1609279
61. Sun Y, Li F, Sonnemann H, et al.. Evolution of CD8(+) T cell receptor (TCR) engineered therapies for the treatment of cancer. Cells-Basel. 2021;10(9):2379.
62. Manfredi F, Cianciotti BC, Potenza A, et al. TCR redirected T cells for cancer treatment: achievements, hurdles, and goals. Front Immunol. 2020;11:1689. doi:10.3389/fimmu.2020.01689
63. Liu Y, Yan X, Zhang F, et al. TCR-T immunotherapy: the challenges and solutions. Front Oncol. 2021;11:794183. doi:10.3389/fonc.2021.794183
64. Getts D, Hofmeister R, Quintas-Cardama A. Synthetic T cell receptor-based lymphocytes for cancer therapy. Adv Drug Deliv Rev. 2019;141:47–54. doi:10.1016/j.addr.2019.04.002
65. Zhang R, Zhang Z, Liu Z, et al. Adoptive cell transfer therapy for hepatocellular carcinoma. Front Med. 2019;13(1):3–11. doi:10.1007/s11684-019-0684-x
66. Fuchsl F, Krackhardt AM. Adoptive cellular therapy for multiple myeloma using CAR- and TCR-transgenic T cells: response and resistance. Cells-Basel. 2022;11(3):410.
67. Pan K, Farrukh H, Chittepu V, Xu H, Pan CX, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022;41(1):119. doi:10.1186/s13046-022-02327-z
68. Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med. 2017;68(1):139–152. doi:10.1146/annurev-med-062315-120245
69. Ma S, Li X, Wang X, et al. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. 2019;15(12):2548–2560. doi:10.7150/ijbs.34213
70. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–28. doi:10.1038/nm.4441
71. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi:10.1056/NEJMoa1707447
72. Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726–1737. doi:10.1056/NEJMoa1817226
73. Lin H, Cheng J, Mu W, Zhou J, Zhu L. Advances in Universal CAR-T Cell Therapy. Front Immunol. 2021;12:744823. doi:10.3389/fimmu.2021.744823
74. Moretti A, Ponzo M, Nicolette CA, Tcherepanova IY, Biondi A, Magnani CF. The past, present, and future of non-viral CAR T cells. Front Immunol. 2022;13:867013. doi:10.3389/fimmu.2022.867013
75. Chiang CL, Coukos G, Kandalaft LE. Whole tumor antigen vaccines: where are we?. Vaccines. 2015;3(2):344–372. doi:10.3390/vaccines3020344
76. Gonzalez FE, Gleisner A, Falcon-Beas F, Osorio F, Lopez MN, Salazar-Onfray F. Tumor cell lysates as immunogenic sources for cancer vaccine design. Hum Vaccin Immunother. 2014;10(11):3261–3269. doi:10.4161/21645515.2014.982996
77. Kroll AV, Fang RH, Jiang Y, et al.. Nanoparticulate delivery of cancer cell membrane elicits multiantigenic antitumor immunity. Adv Mater. 2017;29(47). doi:10.1002/adma.201703969
78. Zhang X, Cui H, Zhang W, Li Z, Gao J. Engineered tumor cell-derived vaccines against cancer: the art of combating poison with poison. Bioact Mater. 2023;22:491–517. doi:10.1016/j.bioactmat.2022.10.016
79. Igarashi Y, Sasada T. Cancer vaccines: toward the next breakthrough in cancer immunotherapy. J Immunol Res. 2020;2020:5825401. doi:10.1155/2020/5825401
80. Tornesello AL, Tagliamonte M, Buonaguro FM, Tornesello ML, Buonaguro L. Virus-like particles as preventive and therapeutic cancer vaccines. Vaccines. 2022;10(2):227. doi:10.3390/vaccines10020227
81. Mohsen MO, Zha L, Cabral-Miranda G, Bachmann MF. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin Immunol. 2017;34:123–132. doi:10.1016/j.smim.2017.08.014
82. Hills RA, Howarth M. Virus-like particles against infectious disease and cancer: guidance for the nano-architect. Curr Opin Biotechnol. 2022;73:346–354. doi:10.1016/j.copbio.2021.09.012
83. Kines RC, Thompson CD, Spring S, et al. Virus-like particle-drug conjugates induce protective, long-lasting adaptive antitumor immunity in the absence of specifically targeted tumor antigens. Cancer Immunol Res. 2021;9(6):693–706. doi:10.1158/2326-6066.CIR-19-0974
84. Kamolratanakul S, Pitisuttithum P. Human papillomavirus vaccine efficacy and effectiveness against cancer. Vaccines. 2021;9(12):1413. doi:10.3390/vaccines9121413
85. Stanley M. Tumour virus vaccines: hepatitis B virus and human papillomavirus. Philos Trans R Soc Lond B Biol Sci. 2017;372(1732):20160268. doi:10.1098/rstb.2016.0268
86. Nooraei S, Bahrulolum H, Hoseini ZS, et al. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J Nanobiotechnol. 2021;19(1):59. doi:10.1186/s12951-021-00806-7
87. Kwak M, Leick KM, Melssen MM, Slingluff CJ. Vaccine strategy in melanoma. Surg Oncol Clin N Am. 2019;28(3):337–351. doi:10.1016/j.soc.2019.02.003
88. Knutson KL, Schiffman K, Cheever MA, Disis ML. Immunization of cancer patients with a HER-2/neu, HLA-A2 peptide, p369-377, results in short-lived peptide-specific immunity. Clin Cancer Res. 2002;8(5):1014–1018.
89. Aurisicchio L, Ciliberto G. Genetic cancer vaccines: current status and perspectives. Expert Opin Biol Ther. 2012;12(8):1043–1058. doi:10.1517/14712598.2012.689279
90. Yang B, Jeang J, Yang A, Wu TC, Hung CF. DNA vaccine for cancer immunotherapy. Hum Vaccin Immunother. 2014;10(11):3153–3164. doi:10.4161/21645515.2014.980686
91. Rinaldi M, Signori E, Rosati P, et al. Feasibility of in utero DNA vaccination following naked gene transfer into pig fetal muscle: transgene expression, immunity and safety. Vaccine. 2006;24(21):4586–4591. doi:10.1016/j.vaccine.2005.08.030
92. Lopes A, Vandermeulen G, Preat V. Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J Exp Clin Cancer Res. 2019;38(1):146. doi:10.1186/s13046-019-1154-7
93. Absalon J, Koury K, Gruber WC. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. reply. N Engl J Med. 2021;384(16):1578. doi:10.1056/NEJMc2036242
94. Jackson LA, Anderson EJ, Rouphael NG, et al. An mRNA vaccine against SARS-CoV-2 - preliminary report. N Engl J Med. 2020;383(20):1920–1931. doi:10.1056/NEJMoa2022483
95. Duan LJ, Wang Q, Zhang C, Yang DX, Zhang XY. Potentialities and challenges of mRNA vaccine in cancer immunotherapy. Front Immunol. 2022;13:923647. doi:10.3389/fimmu.2022.923647
96. Chakraborty C, Sharma AR, Bhattacharya M, Lee SS. From COVID-19 to cancer mRNA vaccines: moving from bench to clinic in the vaccine landscape. Front Immunol. 2021;12:679344. doi:10.3389/fimmu.2021.679344
97. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. Npj Vaccines. 2019;4(1):7. doi:10.1038/s41541-019-0103-y
98. Paston SJ, Brentville VA, Symonds P, Durrant LG. Cancer vaccines, adjuvants, and delivery systems. Front Immunol. 2021;12:627932. doi:10.3389/fimmu.2021.627932
99. Hailemichael Y, Dai Z, Jaffarzad N, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med. 2013;19(4):465–472. doi:10.1038/nm.3105
100. Ma L, Diao L, Peng Z, et al. Immunotherapy and prevention of cancer by nanovaccines loaded with whole-cell components of tumor tissues or cells. Adv Mater. 2021;33(43):e2104849.
101. Chen L, Zuo M, Zhou Q, Wang Y. Oncolytic virotherapy in cancer treatment: challenges and optimization prospects. Front Immunol. 2023;14:1308890. doi:10.3389/fimmu.2023.1308890
102. Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. 2023;20(3):160–177. doi:10.1038/s41571-022-00719-w
103. Andtbacka R, Collichio F, Harrington KJ, et al. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J Immunother Cancer. 2019;7(1):145. doi:10.1186/s40425-019-0623-z
104. Larocca CA, LeBoeuf NR, Silk AW, Kaufman HL. An update on the role of Talimogene Laherparepvec (T-VEC) in the treatment of melanoma: best practices and future directions. Am J Clin Dermatol. 2020;21(6):821–832. doi:10.1007/s40257-020-00554-8
105. Volovat SR, Scripcariu DV, Vasilache IA, et al. Oncolytic virotherapy: a new paradigm in cancer immunotherapy. Int J Mol Sci. 2024;25(2):1180. doi:10.3390/ijms25021180
106. Ferguson MS, Lemoine NR, Wang Y. Systemic delivery of oncolytic viruses: hopes and hurdles. Adv Virol. 2012;2012:805629. doi:10.1155/2012/805629
107. Zheng N, Fang J, Xue G, et al. Induction of tumor cell autosis by myxoma virus-infected CAR-T and TCR-T cells to overcome primary and acquired resistance. Cancer Cell. 2022;40(9):973–985. doi:10.1016/j.ccell.2022.08.001
108. Taheri B, Zarei-Behjani Z, Babaei A, Moradkhan FM. Extracellular vesicles: a trojan horse delivery method for systemic administration of oncolytic viruses. Regenerat Eng Transl Med. 2023;9(4):447–457. doi:10.1007/s40883-023-00295-0
109. Chen Y, Chen X, Bao W, Liu G, Wei W, Ping Y. An oncolytic virus-T cell chimera for cancer immunotherapy. Nat Biotechnol. 2024. doi:10.1038/s41587-023-02118-7
110. Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19(4):237–253. doi:10.1038/s41571-021-00588-9
111. Yang Y, Lundqvist A. Immunomodulatory effects of IL-2 and IL-15; implications for cancer immunotherapy. Cancers. 2020;12(12):3586. doi:10.3390/cancers12123586
112. Waldmann TA. Cytokines in Cancer Immunotherapy. Cold Spring Harb Perspect Biol. 2018;10(12):a028472. doi:10.1101/cshperspect.a028472
113. Muhammad S, Fan T, Hai Y, Gao Y, He J. Reigniting hope in cancer treatment: the promise and pitfalls of IL-2 and IL-2R targeting strategies. Mol Cancer. 2023;22(1):121. doi:10.1186/s12943-023-01826-7
114. Baldo BA. Side effects of cytokines approved for therapy. Drug Saf. 2014;37(11):921–943. doi:10.1007/s40264-014-0226-z
115. Deckers J, Anbergen T, Hokke AM, et al. Engineering cytokine therapeutics. Nat Rev Bioeng. 2023;1(4):286–303. doi:10.1038/s44222-023-00030-y
116. Zhang Y, Zhang C, He M, Xing W, Hou R, Zhang H. Co-expression of IL-21-Enhanced NKG2D CAR-NK cell therapy for lung cancer. BMC Cancer. 2024;24(1):119. doi:10.1186/s12885-023-11806-1
117. Levin AM, Bates DL, Ring AM, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484(7395):529–533. doi:10.1038/nature10975
118. Naing A, Infante JR, Papadopoulos KP, et al. PEGylated IL-10 (Pegilodecakin) induces systemic immune activation, CD8(+) T cell invigoration and polyclonal T cell expansion in cancer patients. Cancer Cell. 2018;34(5):775–791. doi:10.1016/j.ccell.2018.10.007
119. Halin C, Rondini S, Nilsson F, et al. Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature. Nat Biotechnol. 2002;20(3):264–269. doi:10.1038/nbt0302-264
120. Dholakia J, Cohen AC, Leath CR, Evans ET, Alvarez RD, Thaker PH. Development of delivery systems for local administration of cytokines/cytokine gene-directed therapeutics: modern oncologic implications. Curr Oncol Rep. 2022;24(4):389–397. doi:10.1007/s11912-022-01221-3
121. Tan L, Han S, Ding S, et al. Chitosan nanoparticle-based delivery of fused NKG2D-IL-21 gene suppresses colon cancer growth in mice. Int J Nanomed. 2017;12:3095–3107. doi:10.2147/IJN.S128032
122. Saxton RA, Glassman CR, Garcia KC. Emerging principles of cytokine pharmacology and therapeutics. Nat Rev Drug Discov. 2023;22(1):21–37. doi:10.1038/s41573-022-00557-6
123. McDonnell AM, Nowak AK, Lake RA. Contribution of the immune system to the chemotherapeutic response. Semin Immunopathol. 2011;33(4):353–367. doi:10.1007/s00281-011-0246-z
124. Kuai R, Yuan W, Son S, et al. Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci Adv. 2018;4(4):o1736. doi:10.1126/sciadv.aao1736
125. Mihich E. Modification of tumor regression by immunologic means. Cancer Res. 1969;29(12):2345–2350.
126. Han R, Ling C, Wang Y, et al. Enhancing HCC Treatment: innovatively combining HDAC2 inhibitor with PD-1/PD-L1 inhibition. Cancer Cell Int. 2023;23(1):203. doi:10.1186/s12935-023-03051-0
127. Vicari AP, Luu R, Zhang N, et al. Paclitaxel reduces regulatory T cell numbers and inhibitory function and enhances the anti-tumor effects of the TLR9 agonist PF-3512676 in the mouse. Cancer Immunol Immunother. 2009;58(4):615–628. doi:10.1007/s00262-008-0586-2
128. Yi C, Chen L, Lin Z, et al. Lenvatinib Targets FGF Receptor 4 to Enhance Antitumor Immune Response of Anti-Programmed Cell Death-1 in HCC. Hepatology. 2021;74(5):2544–2560. doi:10.1002/hep.31921
129. Awwad M, North RJ. Cyclophosphamide (Cy)-facilitated adoptive immunotherapy of a Cy-resistant tumour. Evidence that Cy permits the expression of adoptive T-cell mediated immunity by removing suppressor T cells rather than by reducing tumour burden. Immunology. 1988;65(1):87–92.
130. Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9(7–8):900–909. doi:10.1016/j.intimp.2009.03.015
131. Vincent J, Mignot G, Chalmin F, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–3061. doi:10.1158/0008-5472.CAN-09-3690
132. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. 2010;16(18):4583–4594. doi:10.1158/1078-0432.CCR-10-0733
133. Lee YM, Chen YH, Ou DL, et al. SN-38, an active metabolite of irinotecan, enhances anti-PD-1 treatment efficacy in head and neck squamous cell carcinoma. J Pathol. 2023;259(4):428–440. doi:10.1002/path.6055
134. Li JY, Chen YP, Li YQ, Liu N, Ma J. Chemotherapeutic and targeted agents can modulate the tumor microenvironment and increase the efficacy of immune checkpoint blockades. Mol Cancer. 2021;20(1):27. doi:10.1186/s12943-021-01317-7
135. Wang X, Liu Y, Xue C, et al. A protein-based cGAS-STING nanoagonist enhances T cell-mediated anti-tumor immune responses. Nat Commun. 2022;13(1):5685. doi:10.1038/s41467-022-33301-0
136. Jiang H, Hegde S, Knolhoff BL, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–860. doi:10.1038/nm.4123
137. Chen F, Gong M, Weng D, et al. Phellinus linteus activates Treg cells via FAK to promote M2 macrophage polarization in hepatocellular carcinoma. Cancer Immunol Immunother. 2024;73(1):18. doi:10.1007/s00262-023-03592-3
138. Wallin JJ, Bendell JC, Funke R, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7(1):12624. doi:10.1038/ncomms12624
139. Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. doi:10.1158/1078-0432.CCR-08-1332
140. Ning WR, Jiang D, Liu XC, et al.. Carbonic anhydrase XII mediates the survival and prometastatic functions of macrophages in human hepatocellular carcinoma. J Clin Invest. 2022;132(7). doi:10.1172/JCI153110
141. Han R, Li J, Hony J, et al. CAXII inhibitors: potential sensitizers for immune checkpoint inhibitors in HCC treatment. Front Immunol. 2023;14:1052657. doi:10.3389/fimmu.2023.1052657
142. Doleschel D, Hoff S, Koletnik S, et al. Regorafenib enhances anti-PD1 immunotherapy efficacy in murine colorectal cancers and their combination prevents tumor regrowth. J Exp Clin Cancer Res. 2021;40(1):288. doi:10.1186/s13046-021-02043-0
143. Wu RY, Kong PF, Xia LP, et al. Regorafenib promotes antitumor immunity via inhibiting PD-L1 and IDO1 expression in melanoma. Clin Cancer Res. 2019;25(14):4530–4541. doi:10.1158/1078-0432.CCR-18-2840
144. Han R, Ling C, Wang Y, Lu L. Enhancing HCC treatment: innovatively combining HDAC2 inhibitor with PD-1/PD-L1 inhibition. Cancer Cell Int. 2023;23(1):203. doi:10.1186/s12935-023-03051-0
145. Yi C, Chen L, Lin Z, et al. Lenvatinib targets FGF receptor 4 to enhance antitumor immune response of anti-programmed cell death-1 in HCC. Hepatology. 2021;74(5):2544–2560. doi:10.1002/hep.31921
146. Finn RS, Ikeda M, Zhu AX, et al. Phase Ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J Clin Oncol. 2020;38(26):2960–2970. doi:10.1200/JCO.20.00808
147. Ahmed A, Tait S. Targeting immunogenic cell death in cancer. Mol Oncol. 2020;14(12):2994–3006. doi:10.1002/1878-0261.12851
148. Li Z, Lai X, Fu S, et al. Immunogenic cell death activates the tumor immune microenvironment to boost the immunotherapy efficiency. Adv Sci. 2022;9(22):e2201734.
149. Ma X, Yang S, Zhang T, et al. Bioresponsive immune-booster-based prodrug nanogel for cancer immunotherapy. Acta Pharm Sin B. 2022;12(1):451–466. doi:10.1016/j.apsb.2021.05.016
150. Ren H, Yong J, Yang Q, et al. Self-assembled FeS-based cascade bioreactor with enhanced tumor penetration and synergistic treatments to trigger robust cancer immunotherapy. Acta Pharm Sin B. 2021;11(10):3244–3261. doi:10.1016/j.apsb.2021.05.005
151. Wang Y, Chen J, Duan R, et al. High-Z-sensitized radiotherapy synergizes with the intervention of the pentose phosphate pathway for in situ tumor vaccination. Adv Mater. 2022;34(13):e2109726.
152. Wang Y, Wang W, Gu R, et al. In situ vaccination with mitochondria-targeting immunogenic death inducer elicits CD8(+) T cell-dependent antitumor immunity to boost tumor immunotherapy. Adv Sci. 2023;10(20):e2300286.
153. Yan P, Luo Y, Li X, et al. A redox-responsive nanovaccine combined with A2A receptor antagonist for cancer immunotherapy. Adv Healthc Mater. 2021;10(21):e2101222. doi:10.1002/adhm.202101222
154. Shigeta K, Datta M, Hato T, et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology. 2020;71(4):1247–1261. doi:10.1002/hep.30889
155. Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Platinum compounds: a new class of potent antitumour agents. Nature. 1969;222(5191):385–386. doi:10.1038/222385a0
156. Rosenberg B. Fundamental studies with cisplatin. Cancer-Am Cancer Soc. 1985;55(10):2303–2306.
157. Chang CL, Hsu YT, Wu CC, et al. Dose-dense chemotherapy improves mechanisms of antitumor immune response. Cancer Res. 2013;73(1):119–127. doi:10.1158/0008-5472.CAN-12-2225
158. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–6721. doi:10.1158/1078-0432.CCR-05-0883
159. Mirza MR, Chase DM, Slomovitz BM, et al. Dostarlimab for primary advanced or recurrent endometrial cancer. N Engl J Med. 2023;388(23):2145–2158. doi:10.1056/NEJMoa2216334
160. Eskander RN, Sill MW, Beffa L, et al. Pembrolizumab plus chemotherapy in advanced endometrial cancer. N Engl J Med. 2023;388(23):2159–2170. doi:10.1056/NEJMoa2302312
161. Tubridy EA, Taunk NK, Ko EM. Treatment of node-positive endometrial cancer: chemotherapy, radiation, immunotherapy, and targeted therapy. Curr Treat Options Oncol. 2024;25(3):330–345. doi:10.1007/s11864-023-01169-x
162. Schmeler KM, Vadhan-Raj S, Ramirez PT, et al. A phase II study of GM-CSF and rIFN-gamma1b plus carboplatin for the treatment of recurrent, platinum-sensitive ovarian, fallopian tube and primary peritoneal cancer. Gynecol Oncol. 2009;113(2):210–215. doi:10.1016/j.ygyno.2009.02.007
163. Rudin CM, Awad MM, Navarro A, et al. Pembrolizumab or placebo plus etoposide and platinum as first-line therapy for extensive-stage small-cell lung cancer: randomized, double-blind, Phase III KEYNOTE-604 study. J Clin Oncol. 2020;38(21):2369–2379. doi:10.1200/JCO.20.00793
164. Wang J, Zhou C, Yao W, et al. Adebrelimab or placebo plus carboplatin and etoposide as first-line treatment for extensive-stage small-cell lung cancer (CAPSTONE-1): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2022;23(6):739–747. doi:10.1016/S1470-2045(22)00224-8
165. Xu J, Bai Y, Xu N, et al. Tislelizumab plus chemotherapy as first-line treatment for advanced esophageal squamous cell carcinoma and gastric/gastroesophageal junction adenocarcinoma. Clin Cancer Res. 2020;26(17):4542–4550. doi:10.1158/1078-0432.CCR-19-3561
166. Wang ZX, Cui C, Yao J, et al. Toripalimab plus chemotherapy in treatment-naive, advanced esophageal squamous cell carcinoma (Jupiter-06): a multi-center phase 3 trial. Cancer Cell. 2022;40(3):277–288. doi:10.1016/j.ccell.2022.02.007
167. Mittendorf EA, Zhang H, Barrios CH, et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet. 2020;396(10257):1090–1100. doi:10.1016/S0140-6736(20)31953-X
168. Cortes J, Cescon DW, Rugo HS, et al. KEYNOTE-355: randomized, double-blind, phase III study of pembrolizumab + chemotherapy versus placebo + chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer. J Clin Oncol. 2020;38(15_suppl):1000. doi:10.1200/JCO.2020.38.15_suppl.1000
169. Page DB, Pucilowska J, Chun B, et al. A phase Ib trial of pembrolizumab plus paclitaxel or flat-dose capecitabine in 1st/2nd line metastatic triple-negative breast cancer. NPJ Breast Cancer. 2023;9(1):53. doi:10.1038/s41523-023-00541-2
170. Yuan SQ, Nie RC, Jin Y, et al. Perioperative toripalimab and chemotherapy in locally advanced gastric or gastro-esophageal junction cancer: a randomized phase 2 trial. Nat Med. 2024;30(2):552–559. doi:10.1038/s41591-023-02721-w
171. Mai HQ, Chen QY, Chen D, et al. Toripalimab or placebo plus chemotherapy as first-line treatment in advanced nasopharyngeal carcinoma: a multicenter randomized phase 3 trial. Nat Med. 2021;27(9):1536–1543. doi:10.1038/s41591-021-01444-0
172. Zhu H, Zheng K, Boccaccini AR. Multi-functional silica-based mesoporous materials for simultaneous delivery of biologically active ions and therapeutic biomolecules. Acta Biomater. 2021;129:1–17. doi:10.1016/j.actbio.2021.05.007
173. Thakor P, Bhavana V, Sharma R, Srivastava S, Singh SB, Mehra NK. Polymer-drug conjugates: recent advances and future perspectives. Drug Discov Today. 2020;25(9):1718–1726. doi:10.1016/j.drudis.2020.06.028
174. Swetha KL, Maravajjala KS, Li SD, Singh MS, Roy A. Breaking the niche: multidimensional nanotherapeutics for tumor microenvironment modulation. Drug Deliv Transl Res. 2023;13(1):105–134. doi:10.1007/s13346-022-01194-7
175. Bian Q, Huang L, Xu Y, et al. A facile low-dose photosensitizer-incorporated dissolving microneedles-based composite system for eliciting antitumor immunity and the abscopal effect. Acs Nano. 2021;15(12):19468–19479. doi:10.1021/acsnano.1c06225
176. Zhang SQ, Fu Q, Zhang YJ, et al. Surface loading of nanoparticles on engineered or natural erythrocytes for prolonged circulation time: strategies and applications. Acta Pharmacol Sin. 2021;42(7):1040–1054. doi:10.1038/s41401-020-00606-z
177. Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–124. doi:10.1038/s41573-020-0090-8
178. Ma L, Kohli M, Smith A. Nanoparticles for combination drug therapy. Acs Nano. 2013;7(11):9518–9525. doi:10.1021/nn405674m
179. Tardi P, Johnstone S, Harasym N, et al. In vivo maintenance of synergistic cytarabine: daunorubicin ratios greatly enhances therapeutic efficacy. Leuk Res. 2009;33(1):129–139. doi:10.1016/j.leukres.2008.06.028
180. Li D, Zhang R, Liu G, Kang Y, Wu J. Redox-responsive self-assembled nanoparticles for cancer therapy. Adv Healthc Mater. 2020;9(20):e2000605. doi:10.1002/adhm.202000605
181. Amreddy N, Babu A, Muralidharan R, et al. Recent advances in nanoparticle-based cancer drug and gene delivery. Adv Cancer Res. 2018;137:115–170.
182. Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16(4):489–496. doi:10.1038/nmat4822
183. Zheng B, Bai Y, Chen H, et al. Targeted delivery of tungsten oxide nanoparticles for multifunctional anti-tumor therapy via macrophages. Biomater Sci. 2018;6(6):1379–1389. doi:10.1039/C8BM00218E
184. Martin JD, Miyazaki T, Cabral H. Remodeling tumor microenvironment with nanomedicines. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2021;13(6):e1730. doi:10.1002/wnan.1730
185. Chen Q, Wang C, Zhang X, et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat Nanotechnol. 2019;14(1):89–97. doi:10.1038/s41565-018-0319-4
186. Lu J, Liu X, Liao YP, et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat Commun. 2017;8(1):1811. doi:10.1038/s41467-017-01651-9
187. Zhao X, Yang K, Zhao R, et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials. 2016;102:187–197. doi:10.1016/j.biomaterials.2016.06.032
188. Pouton CW, Wagstaff KM, Roth DM, Moseley GW, Jans DA. Targeted delivery to the nucleus. Adv Drug Deliv Rev. 2007;59(8):698–717. doi:10.1016/j.addr.2007.06.010
189. Zhang L, Zhang Y, Tai L, et al. Functionalized cell nucleus-penetrating peptide combined with doxorubicin for synergistic treatment of glioma. Acta Biomater. 2016;42:90–101. doi:10.1016/j.actbio.2016.06.031
190. Wang H, Fang B, Peng B, et al. Recent advances in chemical biology of mitochondria targeting. Front Chem. 2021;9:683220. doi:10.3389/fchem.2021.683220
191. Gavas S, Quazi S, Karpinski TM. Nanoparticles for cancer therapy: current progress and challenges. Nanoscale Res Lett. 2021;16(1):173. doi:10.1186/s11671-021-03628-6
192. Luo L, Iqbal MZ, Liu C, et al. Engineered nano-immunopotentiators efficiently promote cancer immunotherapy for inhibiting and preventing lung metastasis of melanoma. Biomaterials. 2019;223:119464. doi:10.1016/j.biomaterials.2019.119464
193. Zanganeh S, Hutter G, Spitler R, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11(11):986–994. doi:10.1038/nnano.2016.168
194. Carroll EC, Jin L, Mori A, et al. The vaccine adjuvant chitosan promotes cellular immunity via DNA sensor cGAS-STING-dependent induction of type I interferons. Immunity. 2016;44(3):597–608. doi:10.1016/j.immuni.2016.02.004
195. Shen H, Ackerman AL, Cody V, et al. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117(1):78–88. doi:10.1111/j.1365-2567.2005.02268.x
196. Danaeifar M, Negahdari B, Eslam HM, et al. Polymeric nanoparticles for DNA vaccine-based cancer immunotherapy: a review. Biotechnol Lett. 2023;45(9):1053–1072. doi:10.1007/s10529-023-03383-x
197. Minigo G, Scholzen A, Tang CK, et al. Poly-L-lysine-coated nanoparticles: a potent delivery system to enhance DNA vaccine efficacy. Vaccine. 2007;25(7):1316–1327. doi:10.1016/j.vaccine.2006.09.086
198. Duan X, Chan C, Lin W. Nanoparticle-mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angew Chem Int Ed Engl. 2019;58(3):670–680. doi:10.1002/anie.201804882
199. Liu X, Yan B, Li Y, et al. Graphene oxide-grafted magnetic nanorings mediated magnetothermodynamic therapy favoring reactive oxygen species-related immune response for enhanced antitumor efficacy. Acs Nano. 2020;14(2):1936–1950. doi:10.1021/acsnano.9b08320
200. Sun D, Ding J, Xiao C, Chen J, Zhuang X, Chen X. pH-responsive reversible PEGylation improves performance of antineoplastic agent. Adv Healthc Mater. 2015;4(6):844–855. doi:10.1002/adhm.201400736
201. Zhang J, Lin Y, Lin Z, et al. Stimuli-responsive nanoparticles for controlled drug delivery in synergistic cancer immunotherapy. Adv Sci. 2022;9(5):e2103444.
202. Zhu Y, Yang Z, Dong Z, et al. CaCO(3)-assisted preparation of pH-responsive immune-modulating nanoparticles for augmented chemo-immunotherapy. Nanomicro Lett. 2020;13(1):29. doi:10.1007/s40820-020-00549-4
203. Dong Z, Feng L, Chao Y, et al. Amplification of tumor oxidative stresses with liposomal fenton catalyst and glutathione inhibitor for enhanced cancer chemotherapy and radiotherapy. Nano Lett. 2019;19(2):805–815. doi:10.1021/acs.nanolett.8b03905
204. Feng B, Hou B, Xu Z, Saeed M, Yu H, Li Y. Self-amplified drug delivery with light-inducible nanocargoes to enhance cancer immunotherapy. Adv Mater. 2019;31(40):e1902960. doi:10.1002/adma.201902960
205. Dai Y, Yang Z, Cheng S, et al.. Toxic reactive oxygen species enhanced synergistic combination therapy by self-assembled metal-phenolic network nanoparticles. Adv Mater. 2018;30(8). doi:10.1002/adma.201704877
206. Tan X, Zhou H, Wang C, Liu X, Yang X, Liu W. GSH-responsive camptothecin prodrug-based hybrid micellar nanoparticles enable antitumor chemo-immunotherapy by PD-L1 knockdown. Nano Res. 2023;16(1):834–848. doi:10.1007/s12274-022-4739-y
207. Kapalatiya H, Madav Y, Tambe VS, Wairkar S. Enzyme-responsive smart nanocarriers for targeted chemotherapy: an overview. Drug Deliv Transl Res. 2022;12(6):1293–1305. doi:10.1007/s13346-021-01020-6
208. Eatemadi A, Aiyelabegan HT, Negahdari B, et al. Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed Pharmacother. 2017;86:221–231. doi:10.1016/j.biopha.2016.12.021
209. Zhou F, Feng B, Yu H, et al. Tumor microenvironment-activatable prodrug vesicles for nanoenabled cancer chemoimmunotherapy combining immunogenic cell death induction and CD47 blockade. Adv Mater. 2019;31(14):e1805888. doi:10.1002/adma.201805888
210. Zheng D, Wan C, Yang H, et al. Her2-targeted multifunctional nano-theranostic platform mediates tumor microenvironment remodeling and immune activation for breast cancer treatment. Int J Nanomed. 2020;15:10007–10028. doi:10.2147/IJN.S271213
211. Cardoso VF, Francesko A, Ribeiro C, Banobre-Lopez M, Martins P, Lanceros-Mendez S. Advances in magnetic nanoparticles for biomedical applications. Adv Healthc Mater. 2018;7(5). doi:10.1002/adhm.201700845
212. Qin Q, Zhou Y, Li P, et al. Phase-transition nanodroplets with immunomodulatory capabilities for potentiating mild magnetic hyperthermia to inhibit tumour proliferation and metastasis. J Nanobiotechnology. 2023;21(1):131. doi:10.1186/s12951-023-01885-4
213. Diaz-Moscoso A, Ballester P. Light-responsive molecular containers. Chem Commun. 2017;53(34):4635–4652. doi:10.1039/C7CC01568B
214. Gong Y, Zhang J, Lu Y, Wan D, Pan J, Ma G. Light-activated arginine-rich peptide-modified nanoparticles for deep-penetrating chemo-photo-immunotherapy of solid tumor. Nano Res. 2023;16(7):9804–9814. doi:10.1007/s12274-023-5665-3
215. Guo J, Yu Z, Das M, Huang L. Nano codelivery of oxaliplatin and folinic acid achieves synergistic chemo-immunotherapy with 5-fluorouracil for colorectal cancer and liver metastasis. Acs Nano. 2020;14(4):5075–5089. doi:10.1021/acsnano.0c01676
216. Kim KS, Youn YS, Bae YH. Immune-triggered cancer treatment by intestinal lymphatic delivery of docetaxel-loaded nanoparticle. J Control Release. 2019;311–312:85–95. doi:10.1016/j.jconrel.2019.08.027
217. Kim M, Lee JS, Kim W, et al. Aptamer-conjugated nano-liposome for immunogenic chemotherapy with reversal of immunosuppression. J Control Release. 2022;348:893–910. doi:10.1016/j.jconrel.2022.06.039
218. Zhao Q, He X, Qin X, et al. Enhanced therapeutic efficacy of combining losartan and chemo-immunotherapy for triple negative breast cancer. Front Immunol. 2022;13:938439. doi:10.3389/fimmu.2022.938439
219. Zhang Y, Cai K, Li C, et al. Macrophage-membrane-coated nanoparticles for tumor-targeted chemotherapy. Nano Lett. 2018;18(3):1908–1915. doi:10.1021/acs.nanolett.7b05263
220. Li J, Tang W, Yang Y, et al. A programmed cell-mimicking nanoparticle driven by potato alkaloid for targeted cancer chemoimmunotherapy. Adv Healthc Mater. 2021;10(13):e2100311. doi:10.1002/adhm.202100311
221. Wang Y, Yu J, Luo Z, et al. Engineering endogenous tumor-associated macrophage-targeted biomimetic nano-RBC to reprogram tumor immunosuppressive microenvironment for enhanced chemo-immunotherapy. Adv Mater. 2021;33(39):e2103497. doi:10.1002/adma.202170304
222. Hao W, Cui Y, Fan Y, et al. Hybrid membrane-coated nanosuspensions for multi-modal anti-glioma therapy via drug and antigen delivery. J Nanobiotechnology. 2021;19(1):378. doi:10.1186/s12951-021-01110-0
223. Da SC, Camps M, Li T, et al. Effective chemoimmunotherapy by co-delivery of doxorubicin and immune adjuvants in biodegradable nanoparticles. Theranostics. 2019;9(22):6485–6500. doi:10.7150/thno.34429
224. Ge X, Hao Y, Li H, et al. Sequential acid/reduction response of triblock copolymeric nanomicelles to release camptothecin and toll-like receptor 7/8 agonist for orchestrated chemoimmunotherapy. J Nanobiotechnology. 2022;20(1):369. doi:10.1186/s12951-022-01577-5
225. Tong QS, Miao WM, Huang H, et al. A tumor-penetrating nanomedicine improves the chemoimmunotherapy of pancreatic cancer. Small. 2021;17(29):e2101208. doi:10.1002/smll.202101208
226. Lee IH, An S, Yu MK, Kwon HK, Im SH, Jon S. Targeted chemoimmunotherapy using drug-loaded aptamer-dendrimer bioconjugates. J Control Release. 2011;155(3):435–441. doi:10.1016/j.jconrel.2011.05.025
227. Kong M, Tang J, Qiao Q, et al. Biodegradable hollow mesoporous silica nanoparticles for regulating tumor microenvironment and enhancing antitumor efficiency. Theranostics. 2017;7(13):3276–3292. doi:10.7150/thno.19987
228. Sun K, Hu J, Meng X, et al. Reinforcing the induction of immunogenic cell death via artificial engineered cascade bioreactor-enhanced chemo-immunotherapy for optimizing cancer immunotherapy. Small. 2021;17(37):e2101897. doi:10.1002/smll.202101897
229. Mu QG, Lin G, Jeon M, et al. Iron oxide nanoparticle targeted chemo-immunotherapy for triple negative breast cancer. Mater Today. 2021;50:149–169. doi:10.1016/j.mattod.2021.08.002
230. Hernandez-Gil J, Cobaleda-Siles M, Zabaleta A, Salassa L, Calvo J, Mareque-Rivas JC. An iron oxide nanocarrier loaded with a Pt(IV) Prodrug and immunostimulatory dsRNA for combining complementary cancer killing effects. Adv Healthc Mater. 2015;4(7):1034–1042. doi:10.1002/adhm.201500080
231. Zhang Z, Yao S, Hu Y, Zhao X, Lee RJ. Application of lipid-based nanoparticles in cancer immunotherapy. Front Immunol. 2022;13:967505. doi:10.3389/fimmu.2022.967505
232. Liu J, Zhao Z, Qiu N, et al. Co-delivery of IOX1 and doxorubicin for antibody-independent cancer chemo-immunotherapy. Nat Commun. 2021;12(1):2425. doi:10.1038/s41467-021-22407-6
233. Tenchov R, Bird R, Curtze AE, Zhou Q. Lipid nanoparticles horizontal line from liposomes to mRNA vaccine delivery, a landscape of research diversity and advancement. Acs Nano. 2021;15(11):16982–17015. doi:10.1021/acsnano.1c04996
234. Maghsoudi S, Taghavi SB, Rabiee N, et al. Burgeoning polymer nano blends for improved controlled drug release: a review. Int J Nanomed. 2020;15:4363–4392. doi:10.2147/IJN.S252237
235. Abourehab M, Pramanik S, Abdelgawad MA, et al. Recent advances of chitosan formulations in biomedical applications. Int J Mol Sci. 2022;23(18):10975. doi:10.3390/ijms231810975
236. Gu J, Zhao G, Yu J, et al. Injectable pH-responsive hydrogel for combinatorial chemoimmunotherapy tailored to the tumor microenvironment. J Nanobiotechnology. 2022;20(1):372. doi:10.1186/s12951-022-01561-z
237. Qin T, Xu X, Zhang Z, et al. Paclitaxel/sunitinib-loaded micelles promote an antitumor response in vitro through synergistic immunogenic cell death for triple-negative breast cancer. Nanotechnology. 2020;31(36):365101. doi:10.1088/1361-6528/ab94dc
238. Chauhan AS. Dendrimers for drug delivery. Molecules. 2018;23(4):938. doi:10.3390/molecules23040938
239. Bayda S, Hadla M, Palazzolo S, et al. Inorganic nanoparticles for cancer therapy: a transition from lab to clinic. Curr Med Chem. 2018;25(34):4269–4303. doi:10.2174/0929867325666171229141156
240. Porrang S, Davaran S, Rahemi N, Allahyari S, Mostafavi E. How advancing are mesoporous silica nanoparticles? A comprehensive review of the literature. Int J Nanomed. 2022;17:1803–1827. doi:10.2147/IJN.S353349
241. Liu X, Jiang J, Liao YP, et al. Combination chemo-immunotherapy for pancreatic cancer using the immunogenic effects of an irinotecan silicasome nanocarrier plus anti-PD-1. Adv Sci. 2021;8(6):2002147. doi:10.1002/advs.202002147
242. Li W, Cao Z, Liu R, et al. AuNPs as an important inorganic nanoparticle applied in drug carrier systems. Artif Cells Nanomed Biotechnol. 2019;47(1):4222–4233. doi:10.1080/21691401.2019.1687501
243. Kroll AV, Fang RH, Zhang L. Biointerfacing and Applications of Cell Membrane-Coated Nanoparticles. Bioconjugate Chem. 2017;28(1):23–32. doi:10.1021/acs.bioconjchem.6b00569
244. Xia Q, Zhang Y, Li Z, Hou X, Feng N. Red blood cell membrane-camouflaged nanoparticles: a novel drug delivery system for antitumor application. Acta Pharm Sin B. 2019;9(4):675–689. doi:10.1016/j.apsb.2019.01.011
245. Oroojalian F, Beygi M, Baradaran B, Mokhtarzadeh A, Shahbazi MA. Immune cell membrane-coated biomimetic nanoparticles for targeted cancer therapy. Small. 2021;17(12):e2006484. doi:10.1002/smll.202006484
246. Dehaini D, Wei X, Fang RH, et al.. Erythrocyte-platelet hybrid membrane coating for enhanced nanoparticle functionalization. Adv Mater. 2017;29(16). doi:10.1002/adma.201606209
247. Wu J, Chen J, Feng Y, et al.. An immune cocktail therapy to realize multiple boosting of the cancer-immunity cycle by combination of drug/gene delivery nanoparticles. Sci Adv. 2020;6(40). doi:10.1126/sciadv.abc7828
248. Chao Y, Liang C, Tao H, et al. Localized cocktail chemoimmunotherapy after in situ gelation to trigger robust systemic antitumor immune responses. Sci Adv. 2020;6(10):z4204. doi:10.1126/sciadv.aaz4204
249. Chen Q, Zhang L, Li L, et al. Cancer cell membrane-coated nanoparticles for bimodal imaging-guided photothermal therapy and docetaxel-enhanced immunotherapy against cancer. J Nanobiotechnology. 2021;19(1):449. doi:10.1186/s12951-021-01202-x
250. Zhu X, Li S. Nanomaterials in tumor immunotherapy: new strategies and challenges. Mol Cancer. 2023;22(1):94. doi:10.1186/s12943-023-01797-9
251. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52(1):17–35. doi:10.1016/j.immuni.2019.12.011
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.