Back to Journals » International Journal of Nanomedicine » Volume 16
Nanomaterials Enhance the Immunomodulatory Effect of Molecular Targeted Therapy
Authors Li Z ![]() , Liu Y, Fang X, Shu Z
, Liu Y, Fang X, Shu Z
Received 4 November 2020
Accepted for publication 23 January 2021
Published 1 March 2021 Volume 2021:16 Pages 1631—1661
DOI https://doi.org/10.2147/IJN.S290346
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Zhongmin Li, Yilun Liu, Xuedong Fang, Zhenbo Shu
Department of Gastrointestinal Colorectal and Anal Surgery, China-Japan Union Hospital of Jilin University, Changchun, 130033, People’s Republic of China
Correspondence: Zhenbo Shu Email [email protected]
Abstract: Molecular targeted therapy, a tumor therapy strategy that inhibits specific oncogenic targets, has been shown to modulate the immune response. In addition to directly inhibiting the proliferation and metastasis of tumor cells, molecular targeted drugs can activate the immune system through a variety of mechanisms, including by promoting tumor antigen processing and presentation, increasing intratumoral T cell infiltration, enhancing T cell activation and function, and attenuating the immunosuppressive effect of the tumor microenvironment. However, poor water solubility, insufficient accumulation at the tumor site, and nonspecific targeting of immune cells limit their application. To this end, a variety of nanomaterials have been developed to overcome these obstacles and amplify the immunomodulatory effects of molecular targeted drugs. In this review, we summarize the impact of molecular targeted drugs on the antitumor immune response according to their mechanisms, highlight the advantages of nanomaterials in enhancing the immunomodulatory effect of molecular targeted therapy, and discuss the current challenges and future prospects.
Keywords: nanomaterial, molecular targeted therapy, immunomodulation, immunotherapy, cancer therapy
Introduction
Molecular targeted therapy is a tumor treatment strategy based on the cellular and molecular levels that can be used to identify specific oncogenic targets of tumor cells and kill them by blocking the signaling pathways associated with cell growth, proliferation, and metastasis.1 Compared with traditional cytotoxic chemotherapy, molecular targeted therapy specifically kills tumor cells without affecting normal cells, thus effectively reducing side effects. Molecular targeted therapy has become the standard treatment for many malignant tumors, and a variety of drugs targeting different pathways are developed every year. However, the transient duration of the response and the development of drug resistance greatly limit the therapeutic efficacy and clinical application of molecular targeted drugs.2 For example, for non-small cell lung cancer (NSCLC) patients with endothelial growth factor receptor (EGFR) mutations, acquired resistance develops an average of 9.2–14.7 months after receiving EGFR tyrosine kinase inhibitor (TKI) treatment.3 The mechanism of drug resistance may be related to the activation of compensatory pathways, the mutation of target genes, tumor heterogeneity, changes in the tumor microenvironment (TME), and the epithelial-mesenchymal transformation of tumor cells.2 A major strategy to overcome therapeutic resistance is to develop a new generation of targeted drugs or multitarget drugs. It has been disappointing that even some widely used multitarget drugs, such as sorafenib, face problems of limited therapeutic efficacy. Another strategy to improve antitumor efficacy is to combine targeted therapy with other treatment strategies such as chemotherapy. For example, the results from a Phase III clinical trial confirmed that the progression-free survival of NSCLC patients with EGFR mutations treated with gefitinib + chemotherapy was significantly longer than that of patients receiving gefitinib alone.4 Therefore, combination therapy is a promising strategy to improve the effectiveness of the targeted therapies.
In recent years, immunotherapy has become a new strategy for cancer treatment following targeted therapy. Since the first programmed cell death protein-1 (PD-1) monoclonal antibody, Opdivo (nivolumab), was approved by the United States Food and Drug Administration (FDA) in 2014, immune checkpoint blockade (ICB) targeting PD-1/programmed cell death-ligand 1 (PD-L1) and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) have become the standard treatments for a variety of malignant tumors, including melanoma, kidney cancer, and lung cancer.5 Immunotherapy has opened up a new era of tumor treatment, and it has also provided more options for combination therapy. For example, radiotherapy, chemotherapy, and photodynamic therapy (PDT) can induce immunogenic death (ICD) of tumor cells, and their combination with immunotherapy can produce stronger antitumor immune effects. Compared with targeted therapy, immunotherapy causes a longer-lasting response but a lower response rate. Most tumor patients fail to respond to ICBs, possibly due to the insufficient recognition of tumor antigens, inadequate immune cell infiltration into tumors, various immunosuppressive factors in the TME, and the expression of alternative immune checkpoints.6 Studies have shown that molecular targeted drugs not only kill tumor cells directly but also exert immunomodulatory effects, which can affect multiple stages of the immune response.7 The impact of molecular targeted drugs on the immune system is due to their targeting of certain proteins related to immune activation or suppression, such as vascular endothelial growth factor (VEGF)/VEGF receptor (VEGF-R), Ras/Raf/mitogen-activated protein kinase (MAPK), nuclear transcription factor-κB (NF-κB), and phosphatidylinositol 3-kinase (PI3K)–Akt–mammalian target of rapamycin (mTOR) pathway.8 The combination of these two treatment modalities may further improve antitumor efficacy. A series of clinical trials are underway to assess the safety and efficacy of this combination therapy.9
Molecular targeted drugs can exert immunomodulatory effects through many different mechanisms. First, some molecular targeted drugs can increase the expression of tumor antigens and promote antigen presentation by antigen presenting cells, thus inducing a stronger antitumor immune response. For example, a BRAF inhibitor alone (vemurafenib) or in combination with MEK inhibitors (dabrafenib + trametinib) increased the expression of tumor antigens in patients with metastatic melanoma, which enhanced the antitumor ability of cytotoxic T cells.10 Second, studies have shown that some molecular targeted drugs can also induce the ICD of tumor cells, thus stimulating tumor-specific immune responses. Liu et al demonstrated that crizotinib, a TKI targeting ALK, c-MET, and ROS1 for the treatment of NSCLC, is a potential ICD inducer and can be used to enhance the efficacy of chemotherapy and ICB-based immunotherapy.11 Third, for some “cold” tumors, the failure of ICB is attributed to the lack of sufficient tumor-infiltrating T cells, which can be increased by molecular targeted drugs. For example, tumor blood vessels are characterized by abnormal morphology and impaired perfusion function, which hinders the infiltration of T cells into tumors. A recent study showed that low-dose antiangiogenic therapy with the anti-VEGFR2 antibody DC101 promoted the normalization of tumor blood vessels and increased the infiltration of CD8+ T cells, thereby enhancing the effect of PD-1 blockade in breast cancer mouse models.12 In addition, MEK inhibitors can prevent the death of CD8+ T cells that is induced by chronic T cell receptor (TCR) stimulation, and their cytotoxic activity is retained.13 Fourth, as major components of innate immunity, natural killer (NK) cells play a crucial role in antitumor immunotherapy. Influencing the activity of NK cells may be another important mechanism by which molecular targeted drugs can regulate the immune response. Hage et al found that sorafenib triggered NK cell-induced cytotoxicity of hepatocellular carcinoma (HCC) cells by inducing the pyrolysis of macrophages.14 Finally, molecular targeted drugs can alleviate the immunosuppressive TME by reducing the number of myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor associated macrophages (TAMs) in tumors, thereby increasing the efficacy of immunotherapy. Studies have found that the activation of the Wnt/β-catenin pathway is closely related to the number of Tregs in the tumor, and blocking this pathway reduces the infiltration of Tregs and overcomes tumor resistance to ICB.15 Moreover, molecular targeted therapy can downregulate the expression of immune checkpoint molecules, thereby attenuating the immunosuppression induced by the TME.16
For most molecular targeted drugs, such as TKIs, poor water solubility, rapid elimination in vivo, and serious side effects caused by off-target action seriously restrict their efficacy and clinical application. Fortunately, nanomaterials can effectively solve these problems and enhance the immunomodulatory effect of molecular targeted drugs.17 Nanomaterials can improve the water solubility of molecular targeted drugs and increase their accumulation in tumor sites through the enhanced permeability and retention (EPR) effect. At the same time, through targeted ligand modification, nanomaterials can specifically deliver molecular targeted drugs to immune cells, thereby improving the effectiveness of triggering immune effects. In addition, some stimulus-responsive nanomaterials can release drugs under the stimulation of acidic pH and high levels of glutathione (GSH), reactive oxygen species (ROS) or enzymes in the TME, thus enabling the specific release of drugs in the tumor. Finally, nanomaterials provide an effective platform for the codelivery of molecular targeted drugs and other immunotherapeutic drugs, helping to enhance synergistic antitumor effects and reduce side effects.
In this article, we review the immunomodulatory effects of molecular targeted drugs based on their mechanisms, and explain the effectiveness of applying nanomaterials to further enhance the immune effect (Table 1). In addition, the challenges and prospects in this field are highlighted.
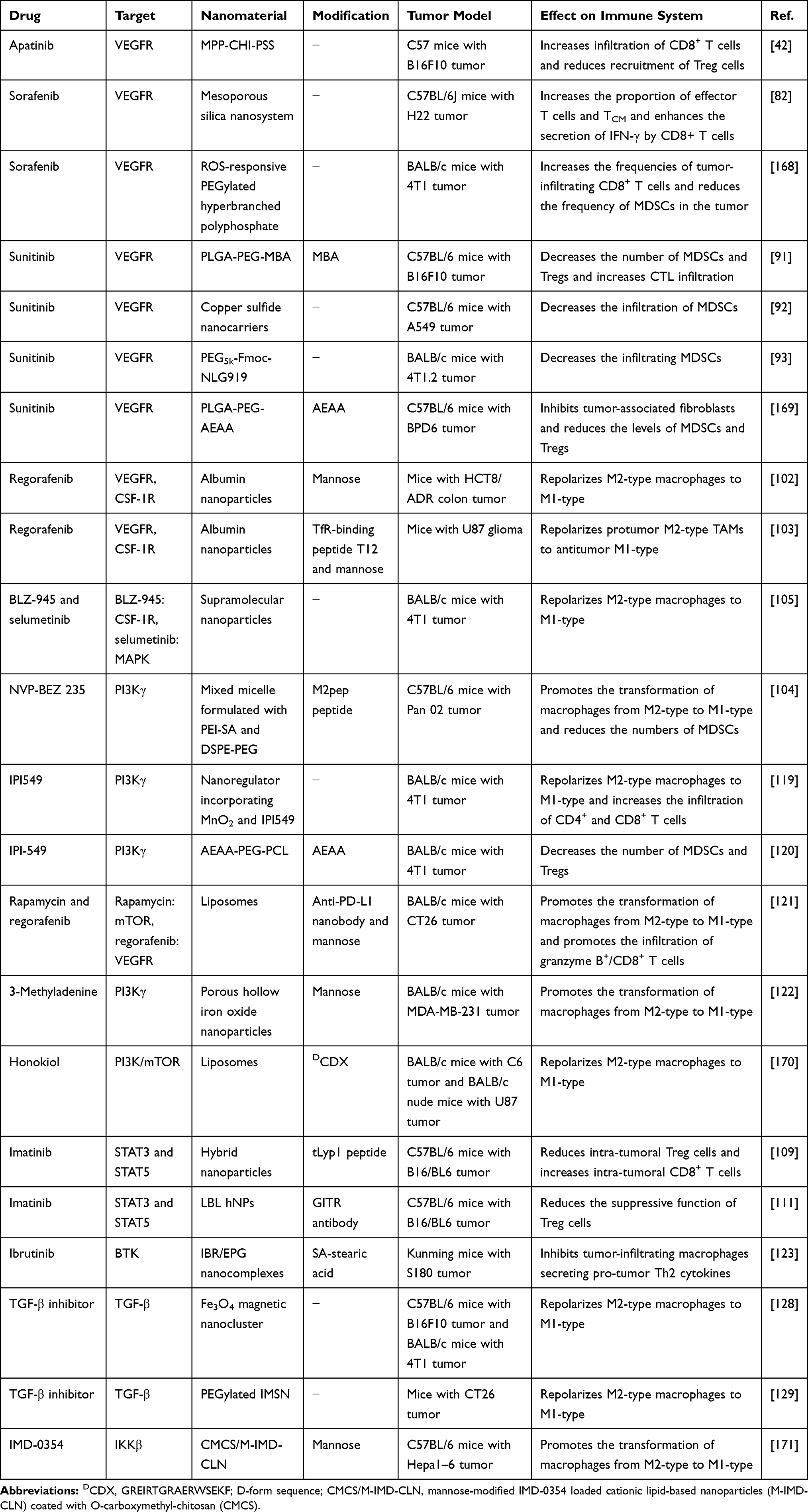 |
Table 1 Summary of Representative Molecular Targeted Drugs with Immunomodulatory Effects Delivered Through Nanomaterials |
Nanomaterial-Enhanced Immunomodulatory Effects of Molecular Targeted Therapy
To trigger an effective antitumor immune response, a series of cellular processes must be initiated, repeated, and expanded in the cancer-immunity cycle.18 In the first step, tumor antigens produced during oncogenesis are released, and subsequently captured and presented by dendritic cells (DCs). Then, stimulated by some proinflammatory signals, DCs are activated and present antigens to T cells, thus promoting the differentiation of naive T cells into effector T cells. Next, effector T cells migrate from the draining lymph node to the tumor site and infiltrate into the tumor bed. Finally, effector T cells specifically recognize tumor antigens and kill the tumor cells, thereby releasing tumor antigens and initiating another cancer-immunity cycle. Failure of any step in this cycle prevents the immune system from effectively eliminating tumor cells, leading to immune escape. Molecular targeted therapy affects various steps of immune activation, and nanomaterials can further enhance the immune response (Scheme 1). We introduce these immunomodulatory effects in detail according to their mechanisms.
 |
Scheme 1 Schematic illustration of the nanomaterial-enhanced immunomodulatory effect of molecular targeted drugs. |
Promoting Tumor Antigen Processing and Presentation
The recognition and presentation of tumor antigens by DCs are critical for initiating an antitumor immune response. Studies have shown that molecular targeted therapy can promote tumor antigen release and presentation. Correale et al reported that the anti-EGFR monoclonal antibody cetuximab enhanced the phagocytosis of colon cancer cells by DCs.19 It has been confirmed that cetuximab can induce cross talk between NK cells and DCs to promote antigen presentation and DC maturation, thereby generating antigen-specific cytotoxic T lymphocytes (CTLs).20 Specifically, cetuximab binds to EGFR on tumor cells via the Fa region and FcγR on the NK cell surface via the Fc region, which promotes NK cell activation and kills tumor cells to produce antigens. Tumor antigens are then presented by DCs, and the CTLs are activated through cross-presentation. Mature DCs further activate NK cells by releasing cytokines such as interleukin-12 (IL-12), and activated NK cells further promote the maturation of DCs by secreting interferon-γ (IFN-γ).21 A similar effect was also observed with anti-human epidermal growth factor receptor-2 (anti-hHER2) monoclonal antibodies. In HER-2/neu transgenic tumor-bearing mice, anti-HER-2 monoclonal antibody increased the uptake of vaccine cells by DCs and promoted the expression of costimulatory molecules CD40, CD80, and CD86, thus triggering a stronger tumor-specific T cell response.22 Gall et al demonstrated that trastuzumab enhanced the uptake and cross-presentation of HER2-derived peptides by DCs, thereby generating a peptide-specific immune response.23 Moreover, trastuzumab deruxtecan (DS-8201a), an antibody–drug conjugate composed of an anti-hHER2 antibody and a derivative of the topoisomerase I inhibitor exatecan, increased the infiltration of DCs in tumors and promoted their maturation in CT26. WT-hHER2 tumor-bearing mice.24 DS-8201a also increased the expression of PD-L1 and major histocompatibility complex class I (MHC-I) in tumor cells. Compared with monotherapy, DS-8201a combined with anti-PD-1 antibody significantly prolonged the survival time of mice.
In addition to monoclonal antibodies, some small-molecule inhibitors can promote antigen presentation by DCs. Nefedova et al demonstrated that the activation of the Janus-activated kinase 2/signal transducers and activators of transcription 3 (JAK2/STAT3) pathway was closely related to the abnormal differentiation of DCs.25 They then found that JSI-124, a selective inhibitor of JAK2/STAT3, significantly promoted the differentiation of DCs and enhanced the therapeutic effect of tumor vaccines.26 In melanoma, oncogenic BRAFV600E mutations promote the rapid internalization and degradation of MHC-I on the tumor cell surface, which leads to immune evasion.27 Vemurafenib, a selective inhibitor of BRAFV600E, can increase the expression of MHC-I and MHC-II as induced by IFN-γ on the surface of homozygous BRAFV600E melanoma cells, thereby enhancing the recognition and killing of tumor cells by CTLs.28 In addition, EGFR TKIs have also been shown to increase IFN-γ-induced MHC expression, indicating their immunomodulatory potential.29
Although some of aforementioned molecular targeted drugs can enhance antigen presentation by promoting the differentiation and maturation of DCs or by increasing the expression of MHC molecules, insufficient aggregation at the tumor site and nonspecific targeting usually limit their effects. The application of nanomaterial-based drug delivery systems is expected to enhance their immunomodulatory effects.
Studies have shown that the expression of Wnt family member 5A (Wnt5a) in melanoma is closely related to DC tolerance and an immunosuppressive TME, possibly because Wnt5a increases the expression of indoleamine 2.3-dioxygenase (IDO).30 Since there is currently no specific molecular targeted drug for Wnt5a, Huang and coworkers designed a trimeric trap protein for Wnt5a and delivered it to the tumor with aminoethyl anisamide (AEAA)-modified cationic lipid-protamine-DNA nanoparticles (LPD NPs) (Figure 1).31 Since AEAA targets overexpressed sigma receptor 1 on the surface of melanoma cells, Wnt5a trap-loaded LPD NPs accumulated in melanoma tumor tissues and significantly reduced the level of Wnt5a. In the BPD6 tumor-bearing mouse model, Wnt5a trap-loaded LPD NPs combined with low-dose doxorubicin (DOX) effectively inhibited tumor growth and prolonged the median survival time of the mice to 65 days, which was significantly longer than the survival time of the mice treated with phosphate-buffered saline (PBS) (39 days). When CD4+ or CD8+ T cells were eliminated with antibodies before treatment, the therapeutic effect was partially impaired, indicating that the antitumor mechanism is related to immunity. Further studies showed that the combination therapy significantly increased the number of CD103+ DCs and promoted the maturation of DCs, thus facilitating tumor antigen presentation. In addition, the combination therapy did not cause significant weight loss or organ dysfunction in the mice, demonstrating its excellent biosafety. Further research is expected to promote antigen presentation and enhance the immune response by modulating other specific molecular pathways.
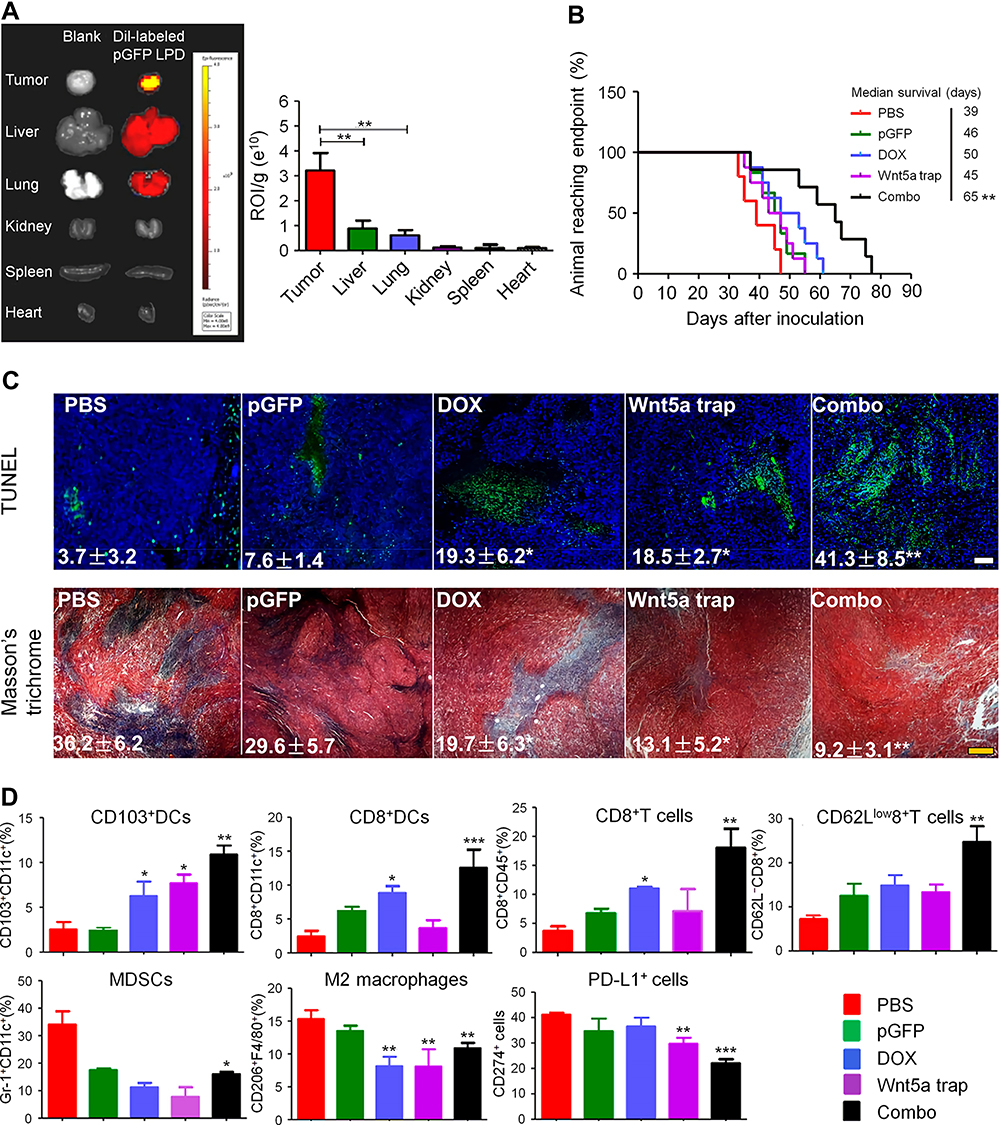 |
Figure 1 Wnt5A trap delivered to tumors by LPD NPs enhanced immunotherapy. (A) In vivo distribution of DiI-loaded LPD NPs, pGFP: control plasmid trap. (B) Survival of BPD6 tumor-bearing mice receiving different treatments. (C) TUNEL and Masson’s trichrome staining of tumor sections from BPD6 tumor-bearing mice in different treatment groups, scale bar: 300 µm. (D) Flow cytometry analysis of immune cells in tumor tissues. *P < 0.05, **P < 0.01, ***P < 0.001. Reproduced from Liu Q, Zhu HD, Tiruthani K, et al. Nanoparticle-mediated trapping of Wnt family member 5A in tumor microenvironments enhances immunotherapy for B-Raf proto-oncogene mutant melanoma. ACS Nano. 2018;12(2):1250-1261.31 Copyright 2018, American Chemical Society. |
Increasing Intratumoral T Cell Infiltration
T cells play key roles in tumor immunotherapy. DCs need to present tumor antigens to naive T cells to produce effector T cells. Therefore, sufficient T cells in tumor tissues are a guarantee for inducing effective antitumor immune effects. Solid tumors are classified as “hot” or “cold” tumors according to the extent to which lymphocytes can infiltrate them. “Hot” tumors are characterized by a large number of lymphocytes infiltrating the tumors and a high response rate to immunotherapy, while “cold” tumors have the opposite characteristics.32 Therefore, increasing T cell infiltration in tumors is an effective method to improve the response rate of immunotherapy. Some signaling pathways, such as VEGF/VEGFR, MAPK, PI3K–Akt–mTOR, and Wnt/β-catenin, have been shown to be associated with tumor T cell deficiency, and inhibition of these pathways contributes to increased T cell infiltration. We introduce the mechanisms and applications of targeted therapies that increase T lymphocyte infiltration via these pathways.
The VEGF/VEGFR Pathway
Peripheral immune cells infiltrate tumors mainly through the blood vessel network. The blood vessels inside a tumor are often abnormal and disordered, which limits the infiltration of immune cells. The hypoxic TME within solid tumors induces the secretion of a variety of proangiogenic factors, such as VEGF, platelet-derived growth factor (PDGF), angiogenin, and fibroblast growth factor, which promote rapid and abnormal tumor angiogenesis. Tumor blood vessels are often structurally and functionally abnormal and characterized by tortuosity, expansion, and uneven distribution, and adjacent endothelial cells are loosely connected to each other. In addition, the leakage of tumor blood vessels and the dysfunction of tumor lymphatic drainage led to an increase in interstitial fluid pressure in the TME. Abnormal blood vessels and increased interstitial fluid pressure prevent immune cells and antitumor drugs from entering the tumor cells from the circulatory system, thereby limiting their antitumor effects. In addition, proangiogenic factors such as VEGF in the TME can prevent the migration of immune cells by regulating the expression of adhesion molecules on the surface of immune cells and endothelial cells.33 Therefore, promoting the normalization of blood vessels is a promising strategy to increase immune cell infiltration.
Antiangiogenesis targeted therapy mainly prevents tumor growth and metastasis by inhibiting tumor angiogenesis and destroying tumor blood vessels. It has become the main treatment strategy for many advanced malignant tumors. Antiangiogenesis therapy helps to promote the normalization of tumor blood vessels, thus promoting the infiltration of immune cells and enhancing the therapeutic effect of immunotherapy.34 Recent studies have shown that the combination of anti-angiogenesis therapy and immunotherapy can improve the prognosis of cancer patients. Huang et al demonstrated that low doses of DC101, an anti-VEGFR2 antibody, dramatically increased the infiltration of CD4+ and CD8+ T cells in the tumors in MCaP0008 tumor-bearing mice.35 Furthermore, DC101 significantly enhanced the antitumor immune response induced by cancer vaccines in a model of MMTV-PyVT breast cancer. Similarly, bevacizumab, an anti-VEGF antibody, has also been shown to promote vascular normalization and promote the entry of T cells into tumors, thus enhancing the effectiveness of adoptive cell transfer–based immunotherapy.36 In another study, bevacizumab combined with atezolizumab increased the number of intratumoral CD8+ T cells and promoted the migration of antigen-specific T cells.37 Shigeta et al demonstrated that dual anti-PD-1/anti-VEGFR-2 therapy promoted the infiltration and activation of CD8+ T cells in HCC.38 Currently, a variety of clinical trials are ongoing or recruiting patients to evaluate the therapeutic effect of antiangiogenesis therapy combined with immunotherapy. A phase Ⅰ clinical trial showed that the combination of bevacizumab and ipilimumab increased the intratumoral trafficking of CD8+ T cells in patients with metastatic melanoma.39 A phase Ⅰb/Ⅱ clinical trial involving 48 patients with metastatic renal cell carcinoma (mRCC) showed that bevacizumab combined with pembrolizumab achieved an overall response rate of 60.9%, indicating that the combination therapy is active and effective.40
In addition to monoclonal antibodies, certain small-molecule inhibitors targeting the VEGF/VEGFR axis can also increase T cell infiltration. Zhao et al confirmed that low-dose apatinib (APA), a small-molecule TKI of VEGFR2, significantly increased the number of CD8+ T cells in the tumors of model mice with Lewis lung carcinoma.41 APA combined with anti-PD-L1 treatment effectively inhibited tumor growth, reduced lung metastasis, and prolonged the survival time of the tumor-bearing mice. However, some other factors in the TME may have attenuated the immune activation effect of antiangiogenic drugs. For example, lactic acid secreted by tumor cells promotes the proliferation of tumor endothelial cells (TECs) and secretes VEGF to restore abnormal growth of blood vessels. Lactic acid also inhibits the activity of T cells to aggravate immune suppression. To maximize the antitumor effect of antiangiogenesis therapy and immunotherapy, Yao and coworkers synthesized positively charged nanoparticles composed of methoxy polyethylene glycol (mPEG)-PLA (MPP) and cationic chitosan (CHI) to load APA and lonidamine (LND), which can reduce the production of lactic acid by inhibiting monocarboxylic acid transporter receptor 4.42 The nanoparticles were then modified with hyaluronic acid (HA) and polystyrene sulfonate (PSS) to form APA/MPP-CHI-PSS (APA/MCP) and LND/MPP-CHI-HA (LND/MCA) nanoparticles, respectively. After simultaneous intravenous injection, APA/MCP nanoparticles preferentially entered TECs and released APA to promote vascular normalization and LND/MCA nanoparticles were specifically internalized by tumor cells and released LND to prevent lactic acid production. The results showed that the pericyte coverage ratio of the tumor vasculature in the mice treated with jet-lagged nanoparticles (APA/MCP + LND/MCA) was as high as 69%, which was significantly higher than that of the other groups. The effect of vascular normalization was intense and durable, which helped to promote lymphocyte infiltration of the tumor site. In the mouse model with B16F10 tumor, jet-lagged nanoparticles + anti-PD-1 monoclonal antibody significantly inhibited tumor growth and increased the ratio of CD8+ T cell/Treg. The safety assessment showed that the nanomaterial-based combination treatment did not result in severe pathological damage to major organs or significant weight loss in the mice. This study provides a basis for the combination of antiangiogenesis therapy and immunotherapy based on nanomaterials.
The PI3K–Akt–mTOR Pathway
In many tumors, the PI3K/Akt/mTOR signaling pathway is overactivated, which is closely related to the proliferation, differentiation, migration, metabolism, and apoptosis of tumor cells.43 Drugs that target PI3K/Akt/mTOR not only kill tumor cells directly, but also have indirect immunomodulatory effects. Studies have shown that PI3K and mTOR determine the expression of adhesion and chemokine receptors, including CD62L and CCR7, on the T cell surface, thus regulating T cell trafficking.44 Borcoman et al demonstrated that mutations in the PIK3CA oncogene activate the PI3K pathway, which is closely related to the lack of immune cell infiltration in muscle-invasive bladder cancer.45 In PIK3CA-mutated model mice, the number of tumor-infiltrating immune cells in the mice treated with BKM120, a PI3K inhibitor, was significantly higher than that in the untreated mice. Moreover, BKM120 increased the sensitivity of the tumors to anti-PD-1 treatment. One of the main reasons for the activation of the PI3K/Akt/mTOR pathway is the loss of the expression of the tumor suppressor PTEN, a lipid phosphatase that can inhibit the function of PI3K signaling. Peng et al reported that the loss of PTEN in tumor cells reduced the migration of T cells into tumors and impaired the function of T cells.46 GSK2636771, a small-molecule inhibitor of PI3Kβ, combined with anti-PD-1 antibody remarkably increased the number of tumor-infiltrating CD8+ and CD4+ T lymphocytes in tumor-bearing mice. In another study, the combination of AZD8055, an ATP-competitive inhibitor of mTOR, and a CD40 agonist increased the infiltration of CD8+ T cells and enhanced their activation and proliferation in a mRCC mouse model.47 These results suggest that the inhibition of the PI3K pathway contributes to overcoming tumor resistance to immunotherapy.
The PI3K/Akt/mTOR and MAPK pathways are two important targets affecting cancer immunotherapy, and inhibiting these two pathways helps to further improve the therapeutic effect of immunotherapy. However, this combination therapy will inevitably increase side effects and toxicity to naive systemic T cells. Nanomaterials have exhibited some unique advantages in mediating combination therapy.48 First, nanomaterials can simultaneously load multiple drugs for combination therapy and increase their accumulation in tumors. Second, nanomaterials can specifically deliver different drugs to tumor cells or other target cells to reduce off-target effects. Third, stimulus-responsive nanomaterials can realize the selective release of drugs both spatially and temporally. In addition, multifunctional nanomaterials enable multimodal imaging for monitoring their therapeutic effects. Finally, some protective nanomaterials can reduce the side effects of combination therapy.
To achieve more efficient and safe combination therapy, Ramesh et al prepared supramolecular nanotherapeutics (DiLNs) to codeliver inhibitors of PI3K and MAPK.49 They synthesized kinase inhibiting amphiphiles by coupling free inhibitors of PI3K (PI103) or MAPK (selumetinib) to cholesterol hemisuccinate. Then, kinase-inhibiting amphiphiles, L-a-phosphatidylcholine, and DSPE-PEG-Amine self-assembled into DiLNs. Due to sustained drug release, the DiLNs inhibited the proliferation of D4M cells (BRAFV600E melanoma cells) and TOV21G cells (clear cell ovarian carcinoma cells) more effectively than free drugs, and they showed little cytotoxicity to T cells even at high drug concentrations. The tumor volume of the mice receiving DiLNs + anti-PD-L1 treatment was significantly lower than that of the groups receiving other treatments in D4M tumor-bearing mice, and the combined treatment induced increased cytotoxic T cell infiltration. This study provides a new strategy for the treatment of melanoma.
The Wnt/β-Catenin Pathway
The Wnt/β-catenin pathway plays an important role in cell proliferation, differentiation, apoptosis, and adhesion, and its abnormal activation is related to the occurrence and development of tumors. Recent studies have shown that this pathway is also an important cause of tumor immune evasion.50,51 In patients with melanoma, the Wnt/β-catenin pathway is closely related to prognosis.52,53 Spranger et al demonstrated that the activation of Wnt/β-catenin prevented T cell infiltration in melanoma.54 Mechanistically, by downregulating the expression of the CCL4 gene, the activated Wnt/β-catenin pathway inhibited the recruitment of CD103+ DCs, which secrete chemokine (C-X-C motif) ligand 9 (CXCL9) and CXCL10 to promote the infiltration of CD8+ T cells.55 In addition to melanoma, Wnt/β-catenin is associated with immune exclusion in other tumors. By analyzing The Cancer Genome Atlas, Luke et al found that the activation of β-catenin signaling was observed in the non-T-cell-inflamed subset of 28 tumors.56 The level of β-catenin protein was inversely correlated with the expression of T-cell-inflamed genes. Therefore, the activation of the Wnt/β-catenin signaling pathway may be an important mechanism leading to failure of the tumor response to ICB therapy.
Targeting the Wnt/β-catenin signaling pathway has shown great value in the treatment of tumors. However, no effective inhibitors are currently in clinical use, which may be due to the nonspecific targeting of most drugs.57 Therefore, it is of great significance to develop new compounds that can specifically inhibit the tumor Wnt pathway. Nanomaterials can deliver drugs to tumor tissues through active or passive targeting, providing opportunities for improving immunotherapy by regulating Wnt pathways. Carnosic acid (CA) is a promising inhibitor of the Wnt/β-catenin pathway and it can block the interaction between β-catenin and BCL9.58 However, its high hydrophobicity and toxicity to normal tissues limit its clinical application. To overcome these problems, Liu et al designed a size-tuned nanocluster (CAcluster) assembled by CA and H1 peptide, the Achilles’ Heel of β-catenin (Figure 2).59 Specifically, gadolinium ion (Gd3+), H1, and 2-mercaptoimidazole (MM)-modified mercapto PEG formed Gd−H1/PEG through thiol-induced conjugation. Then CA induced Gd−H1/PEG to self-assemble into a clustered supramolecular nanostructure. MM endows CAcluster with tumor specificity, and the Gd−thiol conjugation makes it responsive to GSH. After intraperitoneal injection, CAcluster effectively accumulated at the tumor site, and the accumulation of CAcluster in the tumor was 5-fold that of CAcluster without MM modification. In vivo and in vitro experiments confirmed that CAcluster significantly inhibited the Wnt/β-catenin pathway. Further studies showed that the number of tumor-infiltrating CD4+ and CD8+ T cells in B16F10 tumor-bearing mice treated with CAcluster was more than 10-fold that of the control group. Moreover, CAcluster remarkably improved the tumor response to PD-1/PD-L1 ICB in B16F10 and MC38 tumor-bearing mice without causing obvious weight loss.
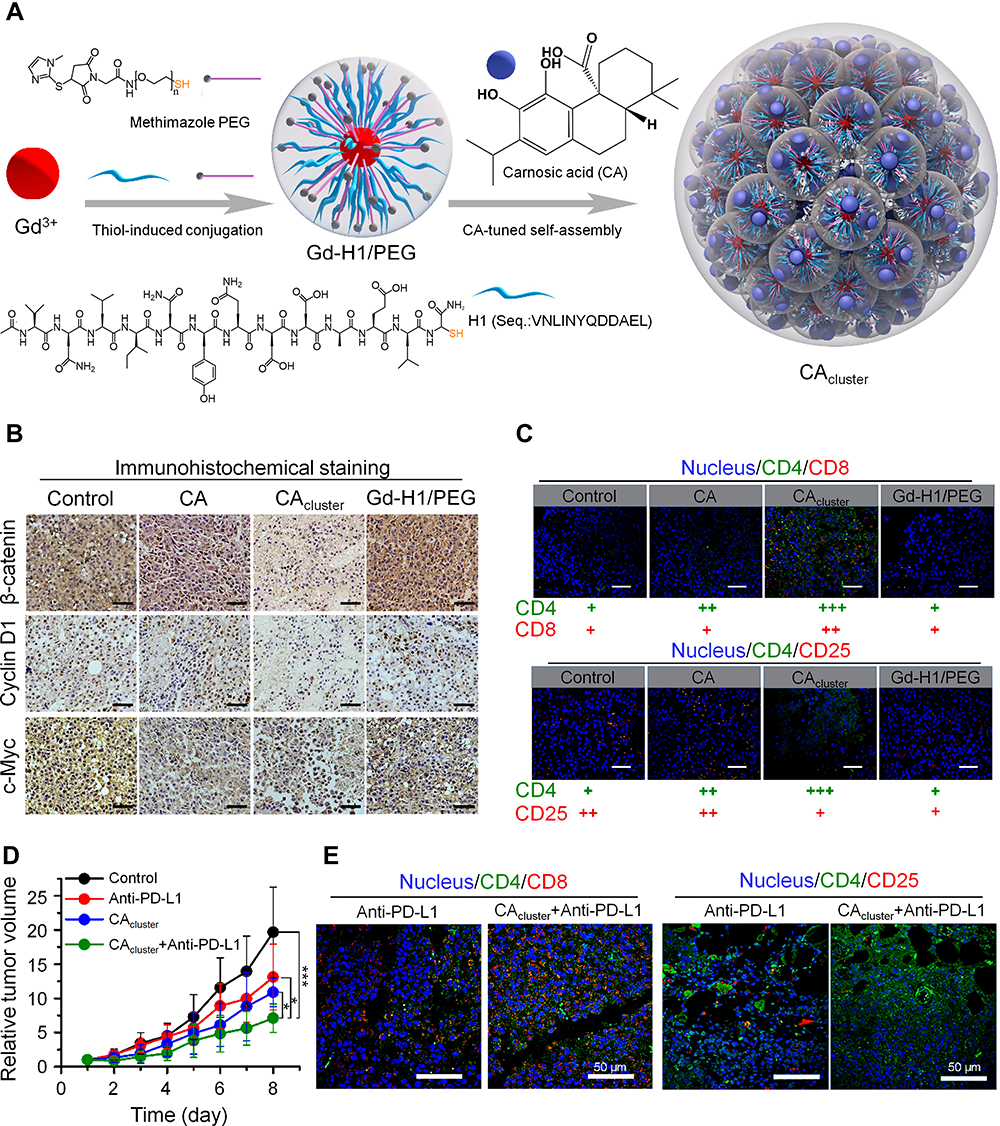 |
Figure 2 Self-assembled nanoclusters targeting β‑catenin enhanced tumor immunotherapy. (A) Schematic illustration of the preparation of CAcluster. (B) Immunohistochemical staining of β-catenin, Cyclin D1, and c-Myc of tumor sections from B16F10 tumor-bearing mice in different treatment groups, scale bar: 100 µm. (C) Immunofluorescence staining of CD4/CD8 and CD4/CD25 of tumor sections from mice receiving different treatments, scale bar: 50 µm. (D) Relative tumor volumes of B16F10 tumor-bearing mice in different treatment groups. (E) Immunofluorescence staining of CD4/CD8 and CD4/CD25 of tumor sections from mice receiving different treatments, scale bar: 50 µm. *P < 0.05, ***P < 0.001. Reproduced from Liu JX, Yan J, Yang SQ, et al. Biomimetic and self-assembled nanoclusters targeting beta-catenin for potent anticancer therapy and enhanced immunotherapy. Nano Lett. 2019;19(12):8708–8715.59 Copyright 2019, American Chemical Society. |
In another study, He et al fabricated Au–peptide nanohybrids (pParticles) via the copolymerization of HAuCl4 and a β-catenin/Bcl9 inhibitor (BBI).60 Then, the pParticle self-assembled into a size-switchable and pH-responsive nanocluster (pCluster) triggered by poly-L-lysine. In the acidic TME, pCluster accumulated at the tumor site due to the EPR effect and disintegrated into small nanoparticles, which contributed to deep penetration and cellular internalization. The results showed that pClusters effectively inhibited tumor growth and metastasis by blocking the Wnt/β-catenin pathway in many different tumor models. Moreover, pClusters combined with a PD-1/PD-L1 inhibitor achieved synergistic antitumor effects in MC38 tumor-bearing model mice, mainly due to the significant increase in the number of tumor-infiltrating CD8+ T cells induced by the pClusters. Biosafety assessments showed that pClusters did not cause serious side effects, such as severe hemocytosis or organ damage, in the mice. These results indicate that blocking the Wnt/β-catenin pathway with nanomedicine has broad prospects for clinical transformation, but more research is needed to further evaluate its efficacy and safety.
Other Pathways
Several other signaling pathways have also been shown to be involved in T cell infiltration. Wilmott et al found that the number of tumor-infiltrating CD4+ and CD8+ T cells increased significantly in metastatic melanoma patients treated with selective BRAF inhibitors, but the mechanism needs to be further clarified.61 In triple-negative breast cancer (TNBC), activation of the RAS/MAPK pathway is closely related to a decrease in tumor-infiltrating lymphocytes.62 Ashizawa et al reported that STX-0119, a STAT3 inhibitor, promoted the accumulation of lymphocytes at the tumor site in the mouse model with U87 glioma.63 Similar effects were observed in humanized mouse models with pancreatic cancer.64 The number of tumor-infiltrating CD8+ T cells in the mice treated with STX-0119 or anti-PD-1 antibody was significantly higher than that of the control group. However, STX-0119 combined with anti-PD-1 antibody dramatically reduced the infiltration of lymphocytes. Moreover, the antitumor effect of the combination therapy was weaker than that of monotherapy. The reason for the failure of this study is not clear. It might be that the combination therapy promoted the apoptosis of effector T cells. This study suggests that the combination of molecular targeted drugs and ICB needs careful evaluation.
The tumor stroma plays an important role in tumor occurrence, development, and treatment resistance. The dense extracellular matrix (ECM) of solid tumors constitutes a physical barrier around the tumor, which severely limits the penetration of antitumor drugs and the infiltration of immune cells in tumor tissue.65 For example, activated T cells accumulate in loose ECM regions in the tumor tissues of lung cancer, whereas few T cells infiltrate the dense fibronectin and collagen areas.66 Therefore, targeting the ECM of the tumor may be a novel strategy to increase the infiltration of immune cells. Studies have shown that the CXCL12/CXCR4 axis may be associated with the migration of fibrocytes to the lung and cause lung fibrosis,67 and the high expression of CXCL12/CXCR4 is an important factor for poor prognosis for patients with lung cancer. To improve the effect of immunotherapy, Li et al synthesized a CXCR4-inhibiting nanocomplex to treat lung cancer (Figure 3).68 First, they prepared a fluorinated bioreducible polymer (FX) that can antagonize the activity of CXCR4. Second, paclitaxel was encapsulated in human serum albumin to form HP nanoparticles. Then FX was attached to the surface of HP to obtain the FX@HP nanocomplex. Finally, siPD-L1 was packed into the FX@HP nanocomplex layer by layer through electrostatic interactions to obtain the FX/siPD-L1@HP nanocomplex. In vitro experiments confirmed that this nanocomplex can effectively antagonize CXCR4 and silence the expression of PD-L1. The median survival time of mice receiving FX/siPD-L1@HP nanocomplex treatment was 55 days and 53 days in the mouse model with orthotopic and metastatic lung cancer, respectively, which were significantly longer than that of the other treatment groups. Mechanistically, the CXCR4 antagonistic polymer FX markedly reduced the expression of collagen and α-smooth muscle actin by blocking CXCR4, which facilitated the infiltration of T cells into tumors. Therefore, inhibiting tumor fibrosis is an effective way to enhance immunotherapy.
 |
Figure 3 CXCR4-inhibiting nanocomplex relieved the immunosuppressive TME of pulmonary tumors and enhanced anti–PD-L1 immunotherapy. (A) Schematic illustration of the synthesis and mechanism of FX/siPD-L1@HP. (B) Flow cytometry analysis of CD4+ and CD8+ T cells and percentage of tumor-infiltrating CD8+ T cells of tumors in LLC tumor-bearing mice receiving different treatments. (C) α-SMA immunofluorescence staining and picrosirius red staining of tumor sections from mice receiving different treatments, scale bar: 100 µm. **P < 0.01. Reproduced from Li ZT, Wang YX, Shen YX, Qian CG, Oupicky D, Sun MJ. Targeting pulmonary tumor microenvironment with CXCR4-inhibiting nanocomplex to enhance anti-PD-L1 immunotherapy. Sci Adv. 2020;6(20):eaaz9240.68 © The Authors, some rights reserved; exclusive licensee AAAS. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC) http://creativecommons.org/licenses/by-nc/4.0/. |
Enhancing T Cell Activation and Function
The function and differentiation of T cells are also important for antitumor immunotherapy. T cells need to differentiate into effector T cells and memory T cells to exert a strong and lasting antitumor effect. Studies have found that many molecular targeted drugs can regulate the differentiation and function of T cells.
The PI3K–Akt–mTOR Pathway
The PI3K–Akt–mTOR pathway not only can inhibit the infiltration of T cells, but can also affect the differentiation of memory T cells. mTOR mainly includes two protein complexes, mTORC1 and mTORC2, and they have different functions and downstream signals. Araki et al demonstrated that rapamycin, a mTOR inhibitor, enhanced the immune response of T cells in vaccinated mice and nonhuman primates.69 Rapamycin can increase the number of memory precursors in the expansion phase of T-cell immunity and accelerate the differentiation of memory T cells in the contraction phase. In another study, Pollizzi et al found that mTORC1 affected the function of effector T cells, and mTORC2 regulated the generation of memory T cells.70 The inhibition of mTORC2 activity led to metabolic reprogramming of T cells, thereby enhancing the formation of CD8+ memory T cells. mTOR may determine the differentiation of naive T cells into effector T cells or memory T cells by regulating the expression of T-bet and Eomesodermin.71 Although mTOR inhibitors, including rapamycin and temsirolimus, promote effector T cell activation and memory T cell differentiation, they also have some immunosuppressive effects. They can inhibit the proliferation of activated T cells, promote the differentiation of Tregs, and inhibit the function of DCs.72,73 Therefore, the immune activation effect of inhibiting PI3K–Akt–mTOR alone is limited, and mTOR pathway inhibitors need to be combined with other immunotherapy strategies to achieve a synergistic antitumor effect.
The combination of mTOR inhibitors and tumor vaccines has achieved satisfactory antitumor effects.74,75 The immunomodulatory effect of mTOR inhibitors may be related to the dosage and frequency of their administration. A low dose of rapamycin seems to be more conducive to the differentiation of T cells into central memory T cells (TCMs), which can produce a large number of antigen-specific effector T cells after being stimulated by antigens. However, TCMs induced by rapamycin usually have a short half-life in the body, and therefore continuous administration of rapamycin is required.76 Moreover, rapamycin is hydrophobic and often needs to be delivered to lymph nodes with other vaccines. To overcome these hurdles, Jewell and coworkers encapsulated rapamycin in poly (lactide-co-glycolide) (PLGA) microparticles to promote the generation of TCMs.77 Rapamycin microparticles (Rapa MPs), with a drug loading rate of 17.3 ± 0.68 μg rapamycin/mg particle, achieved slow and sustained drug release and were effectively internalized by DCs. When cocultured with lipopolysaccharide-stimulated DC/transgenic CD8+ T cells, a low dose of Rapa MPs (0.1 μg/mL) increased the proportion of TCMs from 4.30% (in the untreated group) to 19.57%. The proportion was significantly reduced in the high dose drug treatment group (1 μg/mL). In vivo experiments showed that low dose of Rapa MPs increased the proportion of antigen-specific TCMs when combined with ovalbumin, a model antigen. This study indicates that the inhibition of the mTOR pathway via biomaterials can help improve the efficacy of cancer vaccines.
The Ras-Raf-MAPK Pathway
The Ras-Raf-MAPK signaling pathway is an important signal transduction system that mediates extracellular signals to trigger intracellular responses. It is critical for regulating various physiological processes such as cell growth, differentiation, apoptosis, and malignant transformation. In addition, this pathway is a key factor in mediating immune suppression. It engages downstream of TCR-mediated signaling, such as the MAPK cascade, calcium regulation, and NF-κB activation, thereby providing various targets for modulating the activity and function of T cells. Some early studies have shown that blocking the MAPK signaling pathway can inhibit the initiation, proliferation, and survival of T cells, but the effect seems to be short lived.78,79 Recent studies suggest that the selective inhibition of the Ras/Raf/MAPK pathway can enhance the activation and function of T cells. On the one hand, MEK inhibitors can prevent the exhaustive apoptosis of tumor-resident CD8+ T cells.13 On the other hand, MEK inhibitors or BRAFV600E inhibitors can increase the expression of melanocyte differentiation antigens, thereby enhancing their recognition by T cells.80 A series of clinical trials have been conducted to combine MEK inhibitors or BRAF inhibitors with immunotherapy. A phase Ⅰ/Ⅱ study showed that the combination therapy of dabrafenib, trametinib, and pembrolizumab increased the rate of long-lasting antitumor responses in patients with BRAF-mutant melanoma.81 The results from other clinical trials are also promising.
Sorafenib, a multikinase inhibitor targeting Raf, VEGFR, and PDGF receptor (PDGFR), can effectively inhibit tumor proliferation and tumor angiogenesis. Sorafenib has become the standard treatment for many malignant tumors, including HCC, but its poor water solubility and high side effects limit its clinical application. To further improve the therapeutic effect, Yang et al developed an indocyanine green (ICG) and sorafenib-loaded mesoporous silica nanosystem ((ICG+S)@mSiO2) to realize synergetic photothermal therapy (PTT) and immunoenhanced therapy for HCC.82 With the ICG: sorafenib ratio at 1:3, their drug loading efficiencies (DLEs) were 10.3% and 9.75%, respectively. The prepared (ICG+S)@mSiO2 nanoparticles showed excellent fluorescence imaging and photothermal properties. Upon irradiation with a 2.5 W cm–1808 nm laser, the (ICG+S)@mSiO2 nanoparticles rapidly increased in temperature from 20.0 °C to 52.3 °C in 600 s. In addition, the (ICG+S)@mSiO2 nanoparticles were effectively internalized by H22 cells. The average tumor weight of mice treated with (ICG+S)@mSiO2 + 808 nm irradiation was significantly lower than that of mice treated with sorafenib or ICG + 808 nm irradiation in H22 tumor-bearing mice. Moreover, no significant organ damage was observed in the mice treated with the (ICG+S)@mSiO2 nanoparticles. Further experiments revealed that sorafenib increased the proportion of effector T cells and TCMs and enhanced the secretion of IFN-γ by CD8+ T cells, thus amplifying the antitumor effect of the combination therapy.
In recent years, a variety of inorganic and organic nanomaterials have been developed for PTT, including quantum dots, nanoflakes, and nanocrystals, which have different photothermal conversion efficiency (PCE), stability, biodegradability, and biocompatibility. Two-dimensional (2D) nanomaterials have attracted increasing attention for use in PTT.83 Due to their high surface-to-volume ratio, easy surface modification, high drug-loading capacity, and strong accumulation at tumor sites, these 2D nanomaterials have broad application prospects for mediating the combination of PTT with molecular targeted therapy, immunotherapy, and chemotherapy. In addition to PTT, nanomaterials have also been widely used in PDT.84 First, nanomaterials can improve the water solubility and stability of photosensitizers (PSs). Second, nanomaterials can deliver PSs specifically to tumor sites and increase their accumulation. Third, some nanomaterials, such as upconversion nanomaterials, are able to improve the penetration of PDT. Moreover, some nanomaterials, including semiconducting nanomaterials, can intrinsically generate ROS to enhance PDT. Finally, in addition to delivering PSs, multifunctional nanomaterials can be loaded with imaging agents and other therapeutic drugs to achieve multimodal treatment. These nanomaterials are expected to further amplify the immunomodulatory effect of molecular targeted therapy by combining multiple therapeutic strategies.
Modulating the Immunosuppressive TME
The immunosuppressive TME is an important factor restricting the effect of immunotherapy. In the TME, immunosuppressive cells such as MDSCs, Tregs, and TAMs; immunosuppressive cytokines, such as IL-10 and transforming growth factor-β (TGF-β); and negative costimulatory molecules, such as PD-1 and CTLA-4, can inhibit the activity of effector T cells, thereby promoting the immune evasion of tumor cells. Some molecular targeted therapy drugs can effectively reverse the immunosuppressive TME, making it beneficial to immunotherapy. Nanomaterials help enhance the immunomodulatory effect and provide an effective platform for combined molecular targeted therapy and immunotherapy.
The VEGF/VEGFR Pathway
VEGF and its receptors play important roles in the formation of the immunosuppressive TME. Excessive VEGF secretion can lead to abnormal tumor blood vessels and cause tumor hypoxia, which increase the accumulation of MDSCs and Tregs in tumors, promote the polarization of macrophages toward M2- phenotype, and upregulate the expression of PD-L1 on the surface of tumor cells.85,86 In addition, VEGF can promote the infiltration of immunosuppressive cells into tumors. VEGF binding to VEGFR activates the JAK2/STAT3 signaling pathway, promoting the expansion and recruitment of MDSCs.87 Tumor-infiltrating MDSCs can also induce the infiltration and expansion of Tregs by secreting TGF-β, further promoting the formation of an immunosuppressive TME. Therefore, some small-molecule inhibitors targeting VEGFR, such as sunitinib and sorafenib, have been shown to be effective in reducing the number of MDSCs and Tregs. Ko et al reported that sunitinib significantly reduced the accumulation of MDSCs in the tumors of patients with RCC.88 Some studies have demonstrated that sunitinib enhances the antitumor immunity of cancer vaccines by relieving immunosuppression.89,90 Combining these small-molecule inhibitors with immunotherapy is expected to provide a new strategy for refractory malignant tumors.
To deliver sunitinib specifically to tumor tissues, Huo et al encapsulated sunitinib into anisamide-modified poly-lactic glycolic-acid-poly (ethylene glycol) (PLGA-PEG-MBA) polymeric micelles (SUNb-PMs).91 The drug loading content (DLC) and DLE of sunitinib were as high as 14.2% and 94.3%, respectively. As sigma receptors are highly expressed on the surface of melanoma cells, MBA, a targeting ligand of sigma receptors, was used to increase the accumulation of the nanomedicine in tumors. The results showed that the accumulation of SUNb-PMs at the tumor site was from 2- to 4-fold that of sunitinib oral suspension, SUNOS. In B16F10 tumor-bearing mice, SUNb-PMs significantly enhanced the antitumor effect of cancer vaccines without causing significant weight loss or severe organ dysfunction. The tumor volume of mice receiving SUNb-PM + vaccine treatment was significantly lower than that of mice receiving SUNOS + vaccine treatment, confirming the advantage of the targeted nano-delivery system. Further studies showed that although cancer vaccine monotherapy increased the number of tumor-infiltrating CD8+ T cells, the proportion of MDSCs and Tregs in tumors was also significantly increased, thus inhibiting the function of CTLs. Fortunately, SUNb-PMs effectively eliminated MDSCs and Tregs and restored the CTL activity. Moreover, the SUNb-PM + vaccine treatment remarkably reduced the secretion of IL-6 and IL-10. Other studies have also confirmed that nanomaterials effectively enhance the immunomodulatory effect of sunitinib, and the combination of sunitinib and other drugs achieved better antitumor effects.92,93
The CSF-1/CSF-1R Pathway
Colony stimulating factor-1 (CSF-1) is a cytokine that plays an important role in the survival, proliferation, differentiation, and function of macrophages.94 Studies have shown that CSF-1 is highly expressed in some tumors and is associated with poor prognosis and distant metastasis.95,96 CSF-1 receptor (CSF-1R) is specifically expressed on the surface of TAMs and monocytes, and inhibiting CSF-1R helps to deplete TAMs and promote their polarization from M2-type to M1-type. CSF-1/CSF-1R has become a target for the development of antitumor drugs. Pyonteck et al reported that CSF-1R inhibitors reprogrammed TAMs from M2-type to M1-type and inhibited the tumor growth of glioblastoma.97 In another study, CSF-1/CSF-1R blockade not only reduced the number of TAMs, but also promoted their polarization into types that favor antigen presentation and T cell activation, thus improving the therapeutic efficacy of ICB in pancreatic cancer models.98 However, blocking CSF-1R alone has limited antitumor effects, and off-target effects severely limit the application of CSF-1R inhibitors.99
Thanks to nanomaterials, some studies have successfully delivered CSF-1R inhibitors specifically to macrophages or combined them with other drugs, resulting in promising antitumor effects. Qian et al developed anti-CSF-1R small interfering RNA (siRNA)-loaded M2-type TAM dual-targeting nanoparticles (M2NPs) to treat melanoma (Figure 4).100 Considering that SR-B1 is highly expressed on the surface of M2-type TAMs and apolipoprotein A1 (ApoA 1) can bind to SR-B1 specifically, they linked two targeting units of ApoA 1 (α-peptide and M2pep) with a GSG linker to form α-M2pep. Then α-M2pep, phospholipids, and DiR-BOA (a near-infrared fluorescent dye) formed a core-shell structured lipid nanoparticle (M2NP) based on the amphiphilicity of the α-peptide. Finally, cholesterol-modified anti-CSF-1R siRNA (chol-siCD115) was inserted on the surface of the M2NPs. Twenty-four hours after intravenous injection, M2-type TAMs captured significantly more M2NPs than M1-type TAMs in B16 tumor-bearing mice. Further studies confirmed that M2NPs effectively delivered siCD115 to M2-type TAMs and decreased the expression of CSF-1R. Mice treated with M2NP-siCD115 showed the most obvious inhibition of tumor growth. Flow cytometry showed that M2NP-siCD115 reduced the number of M2-type TAMs by 52% compared with PBS. In addition, M2NP-siCD115 restored the activity of tumor-infiltrating CD8+ T cells. Finally, M2NP-siCD115 exhibited superior biosafety and biocompatibility.
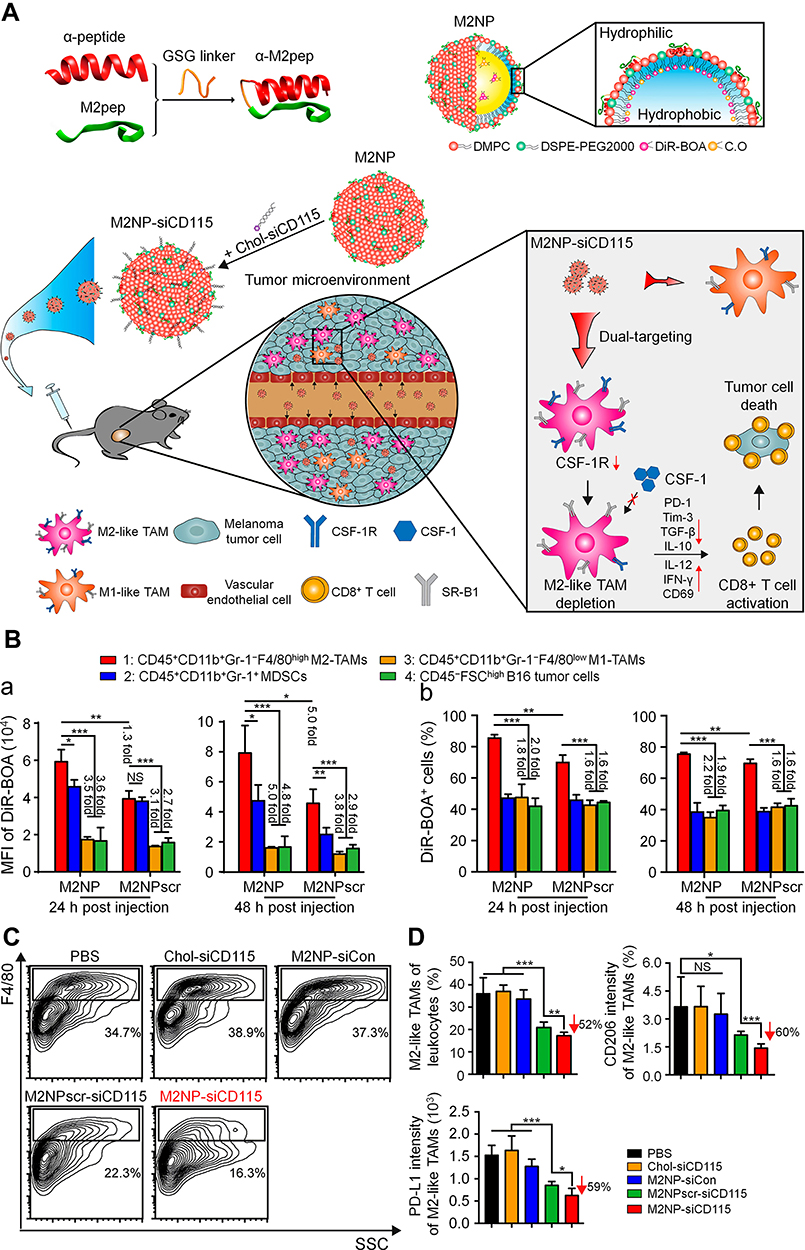 |
Figure 4 Targeted elimination of M2-type TAMs by M2NPs for the treatment of melanoma. (A) Schematic illustration of the synthesis and mechanism of M2NPs. (B) a. MFI of DiR-BOA in different cells after intravenous injection. b. Proportions of different cells that phagocytized M2NPs or M2NPscrs (M2NPscrambles) after intravenous injection. (C) Representative flow cytometry analysis of M2-type TAMs in the tumor of B16 tumor-bearing mice, siCon: control siRNA. (D) Proportion of M2-type TAMs in tumor-infiltrating leukocytes and expression of CD206 and PD-L1 in M2-type TAMs in mice receiving different treatments. *P < 0.05, **P < 0.01, ***P < 0.001. Reproduced from Qian Y, Qiao S, Dai YF, et al. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano. 2017;11(9):9536–9549.100 Copyright 2017, American Chemical Society. |
Regorafenib is a multikinase inhibitor targeting angiogenic kinases, CSF-1R, and oncogenic kinases including KIT, RET, and BRAF. Therefore, regorafenib not only can inhibit tumor growth by inhibiting angiogenesis and cell proliferation but can also potentially regulate macrophages. The disulfiram/copper complex (DSF/Cu) can promote the apoptosis of tumor cells by inducing the production of ROS.101 Huang and coworkers developed DSF/Cu and regorafenib-loaded mannosylated albumin nanoparticles (Man-BSA NPs) to treat drug-resistant tumors.102 They demonstrated that secreted protein acidic and rich in cysteine (SPARC), an albumin-binding protein, and mannose receptors were highly expressed on the surface of drug-resistant colon cancer cells and M2-type macrophages. Therefore, the prepared Man-BSA NPs could inhibit both the cancer cells and M2-type macrophages. The DLC and DLE were 1.43% and 48.58% for regorafenib and 2.69% and 85.76% for DSF/Cu, respectively. The Man-BSA NPs achieved high tumor accumulation and deep intratumoral penetration, indicating the effectiveness of the dual-targeting strategy. The tumor inhibition rate of the mice treated with Man-BSA NPs was 85.2%, which was significantly higher than that of the mice treated with free drugs. Man-BSA NPs reduced the proportion of M2-type macrophages to 22%, which constituted an important mechanism of their antitumor effect. This group also designed another similar dual-targeting nanoplatform modified with a transferrin receptor (TfR)-binding peptide T12 and mannose to deliver DSF/Cu and regorafenib.103 The prepared T12/Man-BSA NPs effectively inhibited the growth of glioma due to the synergistic antitumor effect of chemotherapy and macrophage-directed immunotherapy. Compared with free drugs, Man-BSA NPs did not cause obvious hepatotoxicity or pathological damage to other organs.
These studies indicate that the strategy of targeting and regulating macrophages through nanomaterials is feasible and effective. In addition, some studies have used nanomaterials to simultaneously deliver CSF-1R inhibitors and other inhibitors of related pathways, such as the PI3K and MAPK pathways.104,105 The dual-target nanomedicine inhibited M2-type macrophages more efficiently, providing a new option for macrophage-targeted immunotherapy.
The JAK-STAT Pathway
The JAK/STAT signaling pathway is a cytokine-stimulated signaling pathway that is involved in many important biological processes such as cell proliferation, differentiation, apoptosis, and immune regulation. Imatinib is a multikinase inhibitor targeting BCR-ABL, KIT, and PDGFR. It has been widely used to treat chronic myeloid leukemia and gastrointestinal stromal tumors. It has been shown that imatinib can inhibit the activation of the transcription factors STAT3 and STAT5 in Tregs and reduce the expression of Foxp3, thereby preventing the immunosuppressive function of Tregs.106 In addition, STAT1 and STAT3 are also involved in regulating the expression of IDO in tumors.107 Balachandran et al demonstrated that imatinib induced the apoptosis of Tregs and restored the activity of CD8+ T cells in tumors by inhibiting the expression of IDO.108 Therefore, imatinib can make the TME conducive to immunotherapy by inhibiting the activity of Tregs and it is a potential immune activator.
Poor water solubility and cytotoxicity to normal cells limit the application of imatinib. To enhance the targeting of imatinib to Tregs, Qu et al encapsulated imatinib into tLyp1 peptide-conjugated hybrid nanoparticles (tLyp1-hNPs), which were composed of a PLGA core and lipid layer.109 When the initial dose input was 10.0%, the DLC and DLE of imatinib were 8.9% and 85.0%, respectively. The tLyp1 peptide shows high affinity for the neuropilin-1 (Nrp1) receptor, which is highly expressed on Tregs and rarely expressed on effector T cells in tumors.110 Moreover, they found that Nrp1 was expressed at low levels in melanoma cells. Therefore, tLyp1 modification endows nanoparticles with the ability to target Tregs and prevents off-target effects. The results showed that the cellular uptake of tLyp1-hNPs by Tregs was significantly higher than that by B16 cells and CD8+ T cells, indicating the excellent targeting ability of the tLyp1 peptide. Moreover, the differentiation of Tregs treated with tLyp1-hNPs was 18.0%, which was significantly lower than that of Tregs treated with free imatinib. They also demonstrated that imatinib inhibited the phosphorylation of STAT3 and STAT5 and downregulated the expression of Foxp3 in Tregs, and tLyp1-hNPs further enhanced these effects. In the B16/BL6 tumor-bearing mouse model, the group treated with tLyp1-hNPs + anti-CTLA-4 showed the smallest tumor volume, indicating a synergistic antitumor effect. Compared with the control group, the combination therapy did not cause significant weight loss in mice. Flow cytometry confirmed that tLyp1-hNPs + anti-CTLA-4 dramatically reduced the number of Tregs in the tumors and increased the ratio of CD8+ T cell/Treg, compared with monotherapy.
In another study, they prepared glucocorticoid-induced TNF receptor family-related protein (GITR)-modified layer-by-layer hybrid nanoparticles to load imatinib and IR-780 (LBL hNPs) (Figure 5).111 The maximum DLCs of imatinib and IR-780 were 13% and 10%, respectively. Upon irradiation with a 1.0 W cm–1808 nm laser, the temperature of the LBL hNPs rapidly increased to 67 °C in 80 s. In contrast, free IR-780 was heated only to 53 °C under the same conditions. GITR also increased the targeting of nanoparticles to Tregs, and the LBL hNPs achieved satisfactory tumor inhibition through a combination of PTT, PDT, and Treg modulation. These studies suggested that nanomaterials can increase the biocompatibility and targeting of imatinib, thus effectively blocking the immunosuppressive function of Tregs.
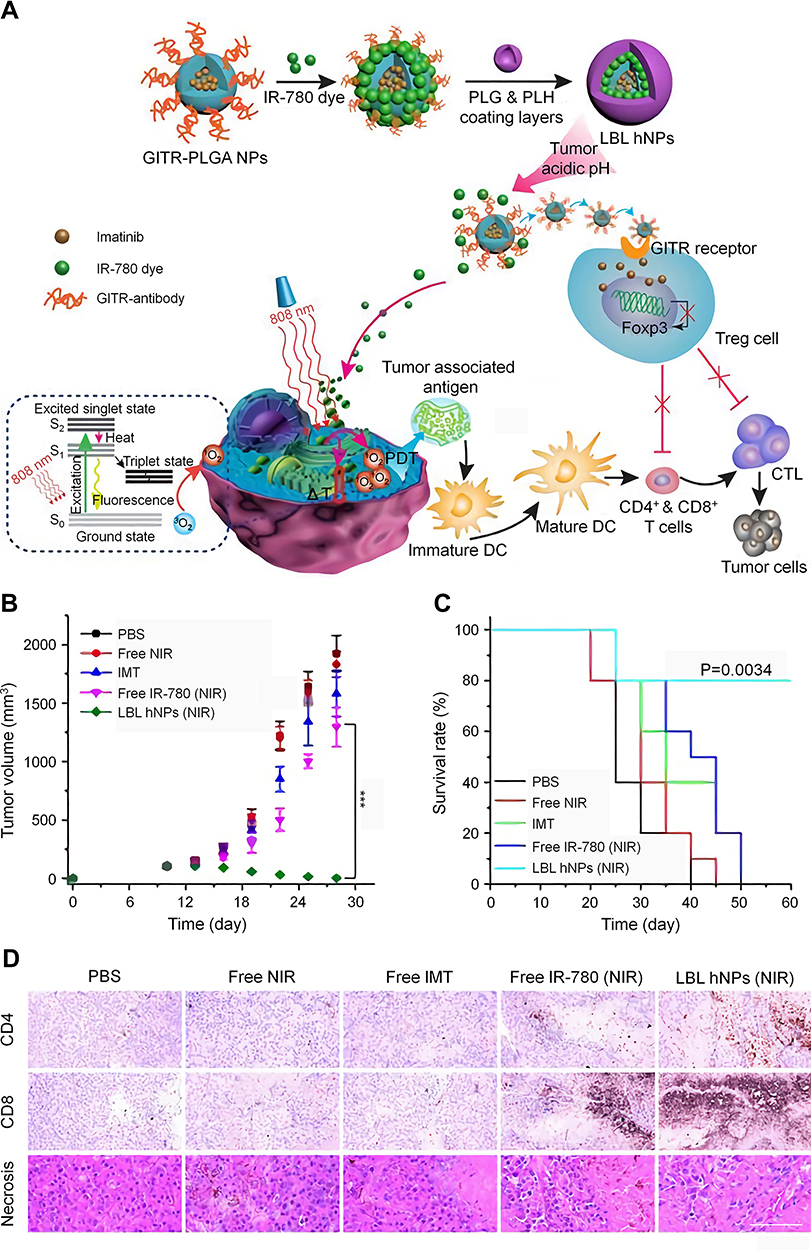 |
Figure 5 Combination of NIR therapy and regulatory T cell modulation mediated by LBL hNPs. (A) Schematic illustration of the synthesis and mechanism of LBL hNPs.Tumor volumes (B) and survival rates (C) of B16BL/6 tumor-bearing mice in different treatment groups. (D) Immunohistochemical staining of CD4+ T cells and CD8+ T cells and histopathological evaluation of tumor necrosis of tumor sections from mice in different treatment groups, scale bar: 120 µm. ***P < 0.001. Reproduced from Ou W, Jiang L, Thapa RK, et al. Combination of NIR therapy and regulatory T cell modulation using layer-by-layer hybrid nanoparticles for effective cancer photoimmunotherapy. Theranostics. 2018;8(17):4574-4590.111 Copyright 2018, Ivyspring International Publisher. |
The PI3K–Akt–mTOR Pathway
In addition to affecting the infiltration and differentiation of T cells, the PI3K–Akt–mTOR pathway is an important factor closely related to the formation of an immunosuppressive TME. Activation of the PI3K–Akt–mTOR pathway can promote the polarization of macrophages from M1-type to M2-type.112 For example, PI3Kγ is a key switch that regulates the polarization of macrophages, and it may induce immunosuppression by activating the mTOR-S6Kα-C/EBPβ pathway and inhibiting the NF-κB pathway.113 Inhibition of PI3K has been shown to promote the transformation of macrophages into the pro-inflammatory M1-type, thus attenuating the immunosuppressive TME.114 Qin et al reported that a PI3Kγ inhibitor reduced the number of M2-type macrophages, attenuating the immunosuppression caused by vascular disrupting agents, which significantly enhanced the antitumor effect of the combination therapy.115 mTOR can promote the differentiation of macrophages into M2-type by upregulating p-STAT3 and IL-10.116,117 Moreover, PI3K–Akt–mTOR pathway is also involved in the recruitment and activation of MDSCs. Foubert et al demonstrated that PI3Kγ promoted the migration of MDSCs to tumor tissues by activating integrin α4, thereby promoting tumor progression.118
A series of studies have shown that the use of nanomaterials to deliver drugs targeting PI3K–Akt–mTOR can enhance antitumor effects by reshaping the TME.12,119,120,122 The combination of PI3K–Akt–mTOR inhibitors with hypoxia-attenuating therapy,119 anti-fibrosis therapy,120 or anti-angiogenesis therapy121 further enhances immunomodulatory effects. Yu et al prepared a multifunctional tumor immune microenvironment nanoregulator (BSA-MnO2-IPI-549) by encapsulating IPI-549, a small-molecule PI3Kγ inhibitor, and MnO2 nanoparticles in BSA (Figure 6).119 In the acidic TME, MnO2 catalyzed H2O2 to produce oxygen to alleviate hypoxia, while the degradation of MnO2 led to the collapse of the nanoregulator and thus triggered the release of IPI-549. In addition, the produced Mn2+ provided tumor-specific magnetic resonance imaging (MRI). In the 4T1 tumor-bearing mouse model, BSA-MnO2-IPI549 promoted the polarization of macrophages to M1-type, reduced the number of Tregs, increased the infiltration of CD8+ T cells, and downregulated the expression of PD-L1, thereby significantly inhibiting tumor growth and prolonging the survival time of mice. In addition, BSA-MnO2-IPI549 did not cause significant weight loss, organ damage, or changes in serum biochemical parameters, indicating its superior biosafety.
 |
Figure 6 Multifunctional nanoregulator composed of MnO2 particles and IPI549 reshaped tumor immune microenvironment and enhanced immunotherapy. (A) Schematic illustration of the mechanism by which BSA-MnO2-IPI549 nanoregulator reshapes the tumor immune microenvironment and provides MRI-guided immunotherapy. (B) Immunofluorescence and immunohistochemical staining of the expressions of macrophage markers in tumor sections from 4T1 tumor-bearing mice receiving different treatments at day 18, BM: BSA-MnO2, BI: nanoparticles loaded with IPI549, BMI: BSA-MnO2-IPI549, scale bar: 100 µm. (C) Proportion of M1-type (CD11b+/F4/80+/CD16/32+) macrophages in tumor tissues of mice in different treatment groups detected by flow cytometry. **P < 0.01, ***P < 0.001. Reproduced from Yu M, Duan XH, Cai YJ, et al. Multifunctional nanoregulator reshapes immune microenvironment and enhances immune memory for tumor immunotherapy. Adv Sci. 2019;6(16):1900037.119 © 2019 The Authors. Published by WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim. |
Facilitated by targeting ligands, nanomaterials can specifically deliver molecular targeted drugs to tumor cells or other target cells. Jiang et al synthesized AEAA-PEG-polycaprolactone (AEAA-PEG-PCL) to load IPI-549 and silibinin (SLB), separately.120 SLB is an anti-fibrotic drug that targets tumor-associated fibroblasts (TAFs), and its antitumor effect is considered to be immune-mediated. AEAA is used to increase the targeting of nanoparticles because it is a ligand for the sigma receptor, which is highly expressed in TAFs and cancer cells. The DLC and DLE were 15.8% and 91.4% for the IPI-549 NPs and 12.9% and 76.9% for the SLB NPs, respectively. The results showed that SLB NP combined with IPI-549 NP treatment induced a more significant reduction in Tregs and MDSCs than either monotherapy in a 4T1 tumor-bearing mouse model, indicating the synergistic effect of attenuated immunosuppression. Moreover, no significant weight loss or organ damage was observed in the combination treatment group. In another study, Huang and coworkers developed a dual-targeting liposomal system (t-LRR) to codeliver rapamycin and regorafenib.121 As PD-L1 and mannose receptors were overexpressed in TAMs and cancer cells, the liposomal system was modified with PD-L1 nanobodies and mannose ligands for targeting. The DLC and DLE were 2.3% and 95.2% for rapamycin and 0.6% and 93.0% for regorafenib, respectively. The prepared t-LRR showed increased cellular uptake by M2-type macrophages and CT26 cells. In a CT26 tumor-bearing mouse model, t-LRR dramatically increased the proportion of M1-type macrophages and promoted the infiltration of CD8+ T cells, thus realizing the effective regulation of the TME. Therefore, it is of great significance to use nanomaterials to specifically and systematically regulate the tumor immune microenvironment.
Other Pathways
Ibrutinib (IBR), a small-molecule inhibitor of Bruton’s tyrosine kinase (BTK), has shown great potential in the treatment of B-cell lymphoma. As BTK is overexpressed in TAMs, IBR can also be used to eliminate TAMs. However, the rapid clearance of BTK in vivo and low levels of accumulation in tumors limit its clinical application. To solve the above problems, Qiu el al. synthesized IBR-loaded nanocomplexes modified with sialic acid (SA)–stearic acid conjugates to deliver IBR specifically to TAMs.123 As Siglec-1, the binding receptor of SA, is overexpressed on the surface of TAMs, SA endows nanocomplexes with the ability to target TAMs. The prepared nanocomplexes (SA/IBR/EPG) showed prolonged blood circulation, effective internalization by TAMs, and increased tumor accumulation. The tumor inhibition index of mice in the SA/IBR/EPG treatment group was significantly higher than that of mice in the other groups in S180 tumor-bearing model mice. Further studies demonstrated that SA/IBR/EPG remarkably reduced the cytokines secreted by TAMs, including IL-10 and TGF-β. This study verified the effectiveness and feasibility of using molecularly targeted drugs to regulate macrophages.
TGF-β is one of the most important immunosuppressive cytokines in the TME, and it can promote tumor progression and immune escape in a variety of ways.124 TGF-β not only inhibits the proliferation and activation of CD8+ T cells,125 but also activates Foxp3 through the TGF-β-Smad3 pathway, thereby promoting the differentiation of naive CD4+ T cells into Tregs.126 In addition, TGF-β promotes the differentiation of M2-type macrophages, which can secrete TGF-β to further aggravate immunosuppression.127 Several studies using nanomaterials to combine TGF-β inhibitors with other treatments have achieved satisfactory results.128,129 Xu et al designed an immunomodulation-enhanced nanozyme-based tumor catalytic therapy strategy by loading a TGF-β inhibitor in PEGylated iron manganese silicate nanoparticles (IMSN-PEG-TI NPs) (Figure 7).129 The DLC of this TGF-β inhibitor was 3.40%. In the acidic TME, Fe and Mn ions were released from the IMSN-PEG-TI NPs and catalyzed H2O2 decomposition to produce hydroxyl radicals (•OH) and oxygen. Moreover, IMSN-PEG-TI NPs promoted the transformation of macrophages from M2-type to M1-type and induced ferroptosis. In a CT26 tumor-bearing mouse model, the tumor inhibition rate of the mice treated with IMSN-PEG-TI NPs was 87.5%, which was significantly higher than that of the mice treated with IMSN nanozyme or TGF-β inhibitor. Increased M1-type macrophage proportions and CD8+ T cell/Treg ratios were observed in the IMSN-PEG-TI treatment group, indicating effective regulation of the tumor immune microenvironment. A safety assessment showed that IMSN-PEG-TI NPs did not cause significant toxicity to major organs. Therefore, TGF-β is an effective target for attenuating immunosuppression and is worthy of further study.
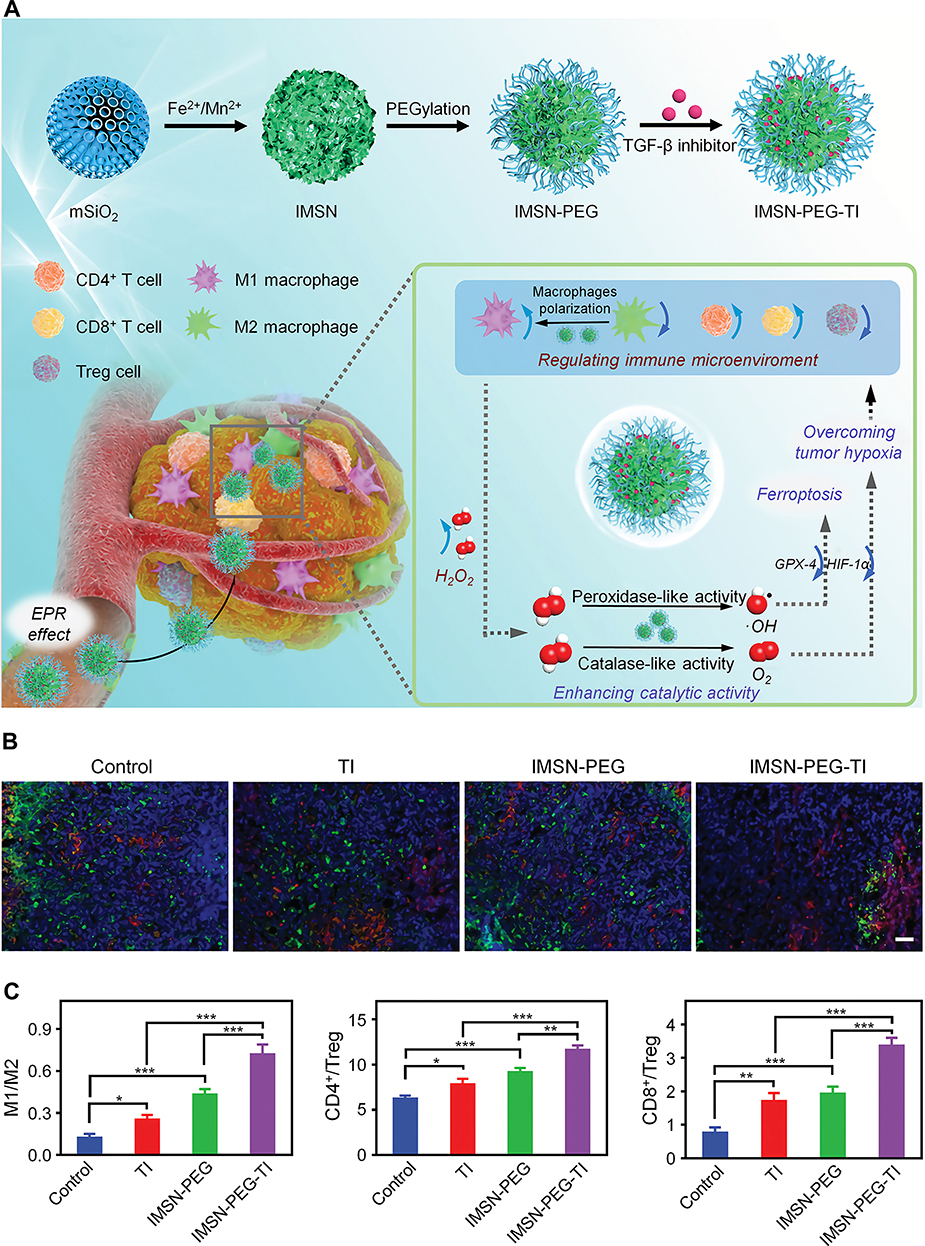 |
Figure 7 Immunomodulation-enhanced nanozyme-based catalytic therapy strategy for cancer therapy. (A) Schematic illustration of the synthesis and antitumor mechanism of IMSN-PEG-TI. (B) Immunofluorescence images of M1-type (red) and M2-type (green) macrophages in tumor sections from CT26 tumor-bearing mice receiving different treatments, scale bar: 100 µm. (C) The ratios of M1/M2 macrophages, CD4+/Treg cells, and CD8+/Treg cells in tumors of CT26 tumor-bearing mice in different treatment groups detected by flow cytometry. *P < 0.05, **P < 0.01, ***P < 0.001. Reproduced from Xu BL, Cui Y, Wang WW, et al. Immunomodulation-enhanced nanozyme-based tumor catalytic therapy. Adv Mater. 2020:2003563.129 Copyright 2020, Wiley-Blackwell. |
Conclusions and Perspectives
Traditional cancer treatment modalities such as chemotherapy and radiotherapy mainly inhibit tumor progression by directly killing tumor cells. With breakthroughs in immunotherapy in recent years, researchers have begun to focus on killing tumor cells by activating the body’s immune cells. In addition to the effectiveness of ICBs, studies have shown that other cancer treatments can also affect the immune system. In other words, the antitumor effects of these treatments may depend to some extent on the activation of the immune system. Molecular targeted therapy specifically inhibits tumor progression and metastasis by blocking some oncogenic targets. An increasing number of studies have shown that molecular targeted drugs not only can inhibit tumor cells, but can also regulate immune cells. In this article, we review the regulatory effects of molecular targeted drugs on the immune system according to their mechanisms. Molecular targeted drugs activate immunity mainly through the following mechanism: promoting antigen processing and presentation, increasing tumor-infiltrating T cells, enhancing the activation and function of T cells, and attenuating the immunosuppression of the TME. Molecular targeted drugs show great potential in regulating the immune system, providing a basis for the combination of molecular targeted therapy and immunotherapy. Some clinical trials have confirmed the effectiveness of this combined treatment model, which will provide new strategies for refractory cancers.
The molecular targeted drugs with immunomodulatory effects mentioned in this article, such as apatinib, rapamycin, and regorafenib, have been widely used clinically. As standard antitumor drugs, their effectiveness and safety have been confirmed. Several clinical trials have suggested that their combination with immunotherapy can exert synergistic antitumor effects and improve the prognosis of cancer patients.130 As the mechanism of immune regulation is further clarified, we believe that molecular targeted therapy will play an increasingly important role in the treatment of cancer.
Poor water solubility, rapid elimination in vivo, lack of effective accumulation in tumors, and serious side effects limit the clinical application of molecular targeted drugs. Moreover, they cannot fully activate the antitumor immune response when used alone. Nanomaterials effectively solve these problems and amplify the immune activation effect of molecular targeted drugs. First, nanomaterials improve the water solubility and biocompatibility of drugs, prolong their blood circulation time, and prevent their premature release. Second, nanomaterials increase the accumulation of drugs in tumor tissues through the EPR effect or active targeting. In particular, nanomaterials can deliver drugs specifically to target immune cells by targeting ligand modifications, thereby avoiding affecting other cells. As mentioned above, SR-B1, PD-L1, mannose receptor, and Siglec-1 are highly expressed on the surface of TAMs, so their ligands effectively increase the targeting of nanomedicine to macrophages. In addition, under the stimulation of the TME, stimulus-responsive nanomaterials can specifically release drugs at the tumor site. Finally, nanomaterials provide a platform for molecular targeted therapy to be combined with other immunotherapy strategies, such as ICBs, tumor vaccines, and IDO enzyme inhibitors. Although the combination therapy based on molecular targeted therapy has achieved some exciting effects in clinical practice, the increased side effects are often unbearable for patients. Nanomaterials can help to improve the synergistic antitumor effect of combination therapy and reduce side effects.
Although advanced nanomaterials have made encouraging progress in enhancing the immunomodulatory effects of molecular targeted therapy, there are still some issues that need to be addressed in this field. First, the mechanism of molecular targeted drugs affecting the immune response needs to be further explored and clarified. For example, some studies have shown that BRAF inhibitors and MEK inhibitors promote T cell activation by regulating the AKT and TCR signaling pathways,131 while Erkes et al demonstrated that they enhance the antitumor T cell response by inducing pyroptosis.132 For some molecular targeted drugs, their effects on the immune system are controversial. A recent study showed that vemurafenib played an immune-activating role in the early stage of BRAF-V600E mutant melanoma by downregulating the expression of PD-L1 and promoted the immune escape of tumor cells in the late stage by inducing the expression of galectin-1.133 Moreover, some molecular targeted drugs target multiple kinases and can affect a variety of immune cells. Therefore, clarifying the specific mechanism of molecular targeted drugs in regulating immunity can provide a sufficient basis for their combination with immunotherapy and contribute to the design of more reasonable and effective combination therapy strategies.
Second, the immunomodulatory effects of molecular targeted drugs are closely related to the dose administered. Low doses of antiangiogenic drugs promote normalization of tumor blood vessels, thereby increasing immune cell infiltration and reducing immunosuppressive cells. However, high-dose and long-term administration of antiangiogenic drugs lead to excessive pruning of blood vessels and aggravate tumor hypoxia, thereby causing immunosuppression.35,134 For example, most studies have shown that a low dose of sorafenib (≤30 mg/kg) in preclinical models increases the infiltration of immune cells or enhances the function of T cells, while a high dose of sorafenib (>30 mg/kg) negatively regulates the immune microenvironment, for example, by increasing the expression of PD-L1 and recruiting MDSCs and Tregs.135 Therefore, when antiangiogenic drugs and immunotherapy are combined to treat tumors, low doses are currently recommended. Similarly, the immunomodulatory effect of rapamycin is also dose-dependent. A low dose of rapamycin enhances the function of DCs and promotes the production of TCMs, while a high dose leads to the opposite effect.77 Therefore, dose is an important factor that needs to be considered carefully and comprehensively when using nanomaterials to deliver molecular targeted drugs for immune regulation.
Third, several new molecular signaling pathways have been found to be related to immunity and have the potential to be combined with immunotherapy. For example, some studies have shown that molecules related to epigenetic modification affect the immunogenicity of tumors and the function of antitumor immune cells.136,137 In melanoma, enhancer of zeste homologue 2 (EZH2), a histone methyltransferase, promoted tumor cell dedifferentiation and loss of immunogenicity, and inhibition of EZH2 and anti-CTLA-4 therapy synergistically inhibited tumor growth.138 The combination of epigenetic regulation and immunotherapy is expected to further enhance the antitumor effect of therapies, and some related clinical trials have been carried out.139 Poly (ADP-ribose) polymerase (PARP) inhibitors, which promote the apoptosis of tumor cells by inhibiting DNA damage repair, have been approved by the FDA for the treatment of ovarian cancer.140 In small cell lung cancer and TNBC, the PARP inhibitor olaparib promotes the recruitment and activation of CD8+ T cells by activating the stimulator of interferon genes (STING) pathway, thus enhancing the antitumor immune response.141,142 A recent phase Ⅰ/Ⅱ study found that olaparib combined with durvalumab showed promising antitumor effects in patients with germline BRCA-mutated metastatic breast cancer.143 Understanding the immunomodulatory activity of these new pathways will help to develop novel combinations of molecular targeted therapy and immunotherapy and optimize synergistic antitumor effects through the use of nanomaterials.
Fourth, molecular targeted therapy still faces some other challenges. Although the immunomodulatory effect of certain molecular targeted drugs has been demonstrated, the effect is not sufficient to induce a strong antitumor immune response. Therefore, it is necessary to combine molecular targeted therapy with other immunotherapy strategies, such as ICBs and tumor vaccines. More preclinical trials are needed to evaluate the optimal combination modality. At the same time, some inherent problems of molecular targeted therapy need to be considered. For example, drug resistance is a major problem that limits the application of molecularly targeted drugs. Combination therapy and the exploration of new therapeutic targets may be options for resolving drug resistance. In addition, screening suitable patients at the gene level and molecular level to achieve precise targeted therapy of tumors is also a research focus in the future.
In addition, the specific mechanism of the cellular uptake of nanoparticles needs to be further clarified. Cellular uptake is an important factor closely related to the drug delivery efficiency of nanoparticles. Understanding the mechanism of this process helps to achieve selective and efficient uptake of nanoparticles, thereby enhancing the therapeutic effect of the delivered drugs and avoiding off-target effects. According to previous studies, the mechanisms by which nanoparticles are internalized by cells include endocytic pathways and non-endocytic pathways.144 The endocytic pathway is considered to be the main mechanism for the uptake of nanoparticles, and it can be further classified into phagocytosis, clathrin-mediated endocytosis, caveolae-mediated endocytosis, and micropinocytosis. During these processes, however, nanoparticles are inevitably trapped in endosomes and subsequent lysosomes. The acidic environment and multiple degrading enzymes in lysosomes can lead to the degradation or inactivation of certain drugs, thus reducing their bioavailability. To this end, various endosome disruption strategies have been developed to achieve lysosomal escape. For example, Guo and coworkers prepared a photosensitizing monofunctional Pt complex, Pt-BDPA, to enhance the antitumor effect of the Pt complex.145 Under photoirradiation, Pt-BDPA induced the generation of ROS to destroy lysosomes, thereby achieving lysosome escape. ROS also reduced intracellular GSH levels, which favored the stability of Pt-BDPA and contributed to its accessibility to nuclear DNA. In addition, the physicochemical properties of nanoparticles, such as their composition, size, shape, surface charge, and surface modification, significantly affect their cellular uptake.146 A recent study showed that the corona composition also affected the internalization of nanoparticles.147 Therefore, it is necessary to design the next generation of nanocarriers by comprehensively considering the above factors to achieve specific and efficient drug delivery.
Furthermore, the appropriate strategy for preparing nanomaterials is another issue that needs to be considered. The synthesis strategies of nanomaterials are generally classified into two main categories: top–down and bottom–up approaches. Specifically, the top-down methods include mechanical exfoliation, chemical exfoliation, sonication exfoliation, and intercalation exfoliation, while the bottom–up methods include chemical vapor deposition, liquid-phase method, and solid-phase method.148 The top-down strategy is characterized by simple preparation process, controllable geometry, and preservation of the structure of the original material. However, this strategy is limited to precursors with layered structures, and non-selective cutting often results in poor control of the size and morphology of the nanomaterial.149 In contrast, the bottom–up strategy offers high controllability of the morphology and high-quality products. Unfortunately, it requires a complex preparation process and the resultant products tend to aggregate. Therefore, these factors need to be carefully considered when preparing multifunctional nanomaterials, especially 2D nanomaterials, to enhance the immunomodulatory effect of the molecular targeted drugs.
Finally, determining the best way to realize successful clinical application is the key problem that all biomedical nanomaterials need to solve. With the development of nanotechnology, nanomaterials have shown broad application prospects in biomedical fields such as medical diagnosis, drug delivery, gene therapy, and tissue engineering. Fortunately, some nanomaterials have already been approved for clinical applications. In 1995, Doxil, a PEG-modified liposome loaded with DOX, became the first nanomedicine approved by the FDA for clinical use.150 Since then, a variety of nanoformulations have entered clinical trials. However, only 15 nanomedicines have been approved for clinical cancer treatment worldwide so far, indicating that the clinical transformation of nanomedicine has a long way to go. Although the efficacy and safety of nanomaterials have been demonstrated in most preclinical models, the physiological structure of the human body is more complex than models. There are certain potential risks in the therapeutic application of nanomaterials that need to be paid attention to. After intravenous injection, nanomaterials will interact with the components in the blood and can produce a series of immunotoxic effects.151 At the same time, nanomaterials may accumulate non-selectively in some major organs, causing hepatotoxicity, nephrotoxicity, or cardiotoxicity. Moreover, certain nanomaterials, such as inorganic nanomaterials, cannot be degraded or metabolized in the body, resulting in long-term accumulation in the body. Notably, certain nanomaterials, such as carbon nanotubes, are even considered to be carcinogenic.152 Therefore, the biodistribution and metabolism, interaction with biological components, biomarkers to monitor therapeutic efficacy, and long-term safety of nanomedicine in the human body require further careful assessment. In addition, ways to achieve large-scale production and quality control are other key issues that needs to be considered for nanomedicines.
Nanomaterials can serve not only as drug delivery carriers, but also as modulators of specific molecular pathways, or they can have immunomodulatory functions. As nanomaterials are easily phagocytized by macrophages, some nanomaterials such as gold nanoparticles,153 silver nanoparticles,154 iron oxide nanoparticles,155 and carbon-based nanomaterials can affect the polarization of macrophages and thus the secretion of cytokines without drug loading. Chen et al demonstrated that gadolinium endohedral metallofullerenols (Gd@C82(OH)22) promoted the production of proinflammatory cytokines (such as tumor necrosis factor-α and IL-1β) by macrophages by activating the NF-κB pathway and NLRP3 inflammasomes.156,157 Moreover, some bioactive nanomaterials, such as polyethyleneimine (PEI)158 and mesoporous silica,159 can promote the recruitment and activation of immune cells, and their combination with immunotherapy drugs can activate immunity more effectively. Luo et al prepared ultra-pH-sensitive nanoparticles (PC7A NPs) and demonstrated that PC7A NPs enhanced tumor antigen presentation and triggered antitumor immune effects by activating the STING pathway.160 These bioactive nanomaterials are promising for further amplifying the effects of combined molecular targeted therapy and immunotherapy.
Since PTT or PDT can induce the ICD of tumors, molecular targeted therapy combined with these therapies can also exert a synergistic antitumor effect. Traditional photothermal conversion agents face problems with low photothermal conversion efficiency, poor biocompatibility, and poor biodegradability, etc., which greatly limit their applications. Nanomaterial-based photothermal conversion agents effectively solve these problems and provide opportunities for combining photothermal treatment with other treatments.161 For example, emerging 2D nanomaterials containing MXenes,162 antimonene,163,164 or black phosphorus165,166 have been widely used in PTT due to their superior biocompatibility and higher photothermal effects. In addition, these nanomaterials usually have high drug loading capacity and can achieve controlled drug release upon irradiation.167 Therefore, these advanced nanomaterials are expected to mediate the combination of PTT and molecular targeted therapy more effectively and activate the antitumor immune response.
Overall, the correlation between molecular signaling pathways and the immune system is gradually being discovered and attracting increased attention. Nanomaterials have shown great potential and advantages for amplifying the immunomodulatory effects of molecular targeted drugs. With the development of nanotechnology, molecular targeted drugs are expected to play greater roles in clinical tumor therapy.
Acknowledgments
We thank Professor Tianmeng Sun for his guidance on this work.
Disclosure
The authors declare that they have no conflict of interest.
References
1. Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction - a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006;3(8):448–457. doi:10.1038/ncponc0558
2. Schram AM, Chang MT, Jonsson P, Drilon A. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. 2017;14(12):735–748. doi:10.1038/nrclinonc.2017.127
3. Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018;29:I10–I19. doi:10.1093/annonc/mdx703
4. Hosomi Y, Morita S, Sugawara S, et al. Gefitinib alone versus gefitinib plus chemotherapy for non-small-cell lung cancer with mutated epidermal growth factor receptor: nej009 study. J Clin Oncol. 2020;38(2):115–123. doi:10.1200/JCO.19.01488
5. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi:10.1126/science.aar4060
6. Pitt JM, Vetizou M, Daillere R, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44(6):1255–1269. doi:10.1016/j.immuni.2016.06.001
7. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–251. doi:10.1038/nrc3237
8. Hughes PE, Caenepeel S, Wu LC. Targeted therapy and checkpoint immunotherapy combinations for the treatment of cancer. Trends Immunol. 2016;37(7):462–476. doi:10.1016/j.it.2016.04.010
9. Shin MH, Kim J, Lim SA, Kim J, Lee KM. Current insights into combination therapies with MAPK inhibitors and immune checkpoint blockade. Int J Mol Sci. 2020;21(7):2531. doi:10.3390/ijms21072531
10. Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–1231. doi:10.1158/1078-0432.CCR-12-1630
11. Liu P, Zhao LW, Pol J, et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat Commun. 2019;10:1486. doi:10.1038/s41467-019-09415-3
12. Li Q, Wang YF, Jia WJ, et al. Low-dose anti-angiogenic therapy sensitizes breast cancer to PD-1 blockade. Clin Cancer Res. 2020;26(7):1712–1724. doi:10.1158/1078-0432.CCR-19-2179
13. Ebert PJR, Cheung J, Yang YG, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity. 2016;44(3):609–621. doi:10.1016/j.immuni.2016.01.024
14. Hage C, Hoves S, Strauss L, et al. Sorafenib induces pyroptosis in macrophages and triggers natural killer cell-mediated cytotoxicity against hepatocellular carcinoma. Hepatology. 2019;70(4):1280–1297. doi:10.1002/hep.30666
15. Feng M, Jin JQ, Xia L, et al. Pharmacological inhibition of beta-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating T-reg cells. Sci Adv. 2019;5(5):eaau5240. doi:10.1126/sciadv.aau5240
16. Kondo K, Shaim H, Thompson PA, et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia. 2018;32(4):960–970. doi:10.1038/leu.2017.304
17. Wang QL, Zhang P, Li ZM, et al. Evaluation of polymer nanoformulations in hepatoma therapy by established rodent models. Theranostics. 2019;9(5):1426–1452. doi:10.7150/thno.31683
18. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi:10.1016/j.immuni.2013.07.012
19. Correale P, Botta C, Cusi MG, et al. Cetuximab ± chemotherapy enhances dendritic cell-mediated phagocytosis of colon cancer cells and ignites a highly efficient colon cancer antigen-specific cytotoxic T-cell response in vitro. Int J Cancer. 2012;130(7):1577–1589. doi:10.1002/ijc.26181
20. Lee SC, Srivastava RM, Lopez-Albaitero A, Ferrone S, Ferris RL. Natural killer (NK): dendritic cell (DC) cross talk induced by therapeutic monoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol Res. 2011;50(2–3):248–254. doi:10.1007/s12026-011-8231-0
21. Srivastava RM, Lee SC, Andrade PA, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19(7):1858–1872. doi:10.1158/1078-0432.CCR-12-2426
22. Kim PS, Armstrong TD, Song H, et al. Antibody association with HER-2/neu-targeted vaccine enhances CD8(+) T cell responses in mice through Fc-mediated activation of DCs. J Clin Invest. 2008;118(5):1700–1711. doi:10.1172/JCI34333
23. Gall VA, Philips AV, Qiao N, et al. Trastuzumab increases HER2 uptake and cross-presentation by dendritic cells. Cancer Res. 2017;77(19):5374–5383. doi:10.1158/0008-5472.CAN-16-2774
24. Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-targeting antibody-drug conjugate, trastuzumab deruxtecan (DS-8201a), enhances antitumor immunity in a mouse model. Mol Cancer Ther. 2018;17(7):1494–1503. doi:10.1158/1535-7163.MCT-17-0749
25. Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the Janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005;65(20):9525–9535. doi:10.1158/0008-5472.CAN-05-0529
26. Nefedova Y, Cheng PY, Gilkes D, et al. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol. 2005;175(7):4338–4346. doi:10.4049/jimmunol.175.7.4338
27. Bradley SD, Chen ZM, Melendez B, et al. BRAF (V600e) co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8(+) T-cell recognition of melanoma. Cancer Immunol Res. 2015;3(6):602–609. doi:10.1158/2326-6066.CIR-15-0030
28. Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAF (V600e) homozygous melanoma cells. Oncoimmunology. 2013;2(1):e22890. doi:10.4161/onci.22890
29. Pollack BP, Sapkota B, Cartee TV. Epidermal growth factor receptor inhibition augments the expression of MHC class I and II genes. Clin Cancer Res. 2011;17(13):4400–4413. doi:10.1158/1078-0432.CCR-10-3283
30. Holtzhausen A, Zhao F, Evans KS, et al. Melanoma-derived Wnt5a promotes local dendritic-cell expression of IDO and immunotolerance: opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol Res. 2015;3(9):1082–1095. doi:10.1158/2326-6066.CIR-14-0167
31. Liu Q, Zhu HD, Tiruthani K, et al. Nanoparticle-mediated trapping of Wnt family member 5A in tumor microenvironments enhances immunotherapy for B-Raf proto-oncogene mutant melanoma. ACS Nano. 2018;12(2):1250–1261. doi:10.1021/acsnano.7b07384
32. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218. doi:10.1038/s41573-018-0007-y
33. Hendry SA, Farnsworth RH, Solomon B, Achen MG, Stacker SA, Fox SB. The role of the tumor vasculature in the host immune response: implications for therapeutic strategies targeting the tumor microenvironment. Front Immunol. 2016;7:621. doi:10.3389/fimmu.2016.00621
34. Fukurnura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15(5):325–340. doi:10.1038/nrclinonc.2018.29
35. Huang YH, Yuan JP, Righi E, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. P Natl Acad Sci USA. 2012;109(43):17561–17566. doi:10.1073/pnas.1215397109
36. Shrimali RK, Yu ZY, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70(15):6171–6180. doi:10.1158/0008-5472.CAN-10-0153
37. Wallin JJ, Bendell JC, Funke R, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624. doi:10.1038/ncomms12624
38. Shigeta K, Datta M, Hato T, et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology. 2020;71(4):1247–1261. doi:10.1002/hep.30889
39. Hodi FS, Lawrence D, Lezcano C, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2(7):632–642. doi:10.1158/2326-6066.CIR-14-0053
40. Dudek AZ, Liu LC, Gupta S, et al. Phase Ib/II clinical trial of pembrolizumab with bevacizumab for metastatic renal cell carcinoma: BTCRC-GU14-003. J Clin Oncol. 2020;38(11):1138–1145. doi:10.1200/JCO.19.02394
41. Zhao S, Ren SX, Jiang T, et al. Low-dose apatinib optimizes tumor microenvironment and potentiates antitumor effect of PD-1/PD-L1 blockade in lung cancer. Cancer Immunol Res. 2019;7(4):630–643. doi:10.1158/2326-6066.CIR-17-0640
42. Jiang ZJ, Xiong H, Yang S, et al. Jet-lagged nanoparticles enhanced immunotherapy efficiency through synergistic reconstruction of tumor microenvironment and normalized tumor vasculature. Adv Healthc Mater. 2020;9(12):2000075. doi:10.1002/adhm.202000075
43. Vara JAF, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30(2):193–204. doi:10.1016/j.ctrv.2003.07.007
44. Sinclair LV, Finlay D, Feijoo C, et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol. 2008;9(5):513–521. doi:10.1038/ni.1603
45. Borcoman E, De La Rochere P, Richer W, et al. Inhibition of PI3K pathway increases immune infiltrate in muscle-invasive bladder cancer. Oncoimmunology. 2019;8(5):e1581556. doi:10.1080/2162402X.2019.1581556
46. Peng WY, Chen JQ, Liu CW, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6(2):202–216. doi:10.1158/2159-8290.CD-15-0283
47. Jiang Q, Weiss JM, Back T, et al. mTOR kinase inhibitor AZD8055 enhances the immunotherapeutic activity of an agonist CD40 antibody in cancer treatment. Cancer Res. 2011;71(12):4074–4084. doi:10.1158/0008-5472.CAN-10-3968
48. Xie ZJ, Fan TJ, An J, et al. Emerging combination strategies with phototherapy in cancer nanomedicine. Chem Soc Rev. 2020;49(22):8065–8087. doi:10.1039/D0CS00215A
49. Ramesh A, Natarajan SK, Nandi D, Kulkarni A. Dual inhibitors-loaded nanotherapeutics that target kinase signaling pathways synergize with immune checkpoint inhibitor. Cell Mol Bioeng. 2019;12(5):357–373. doi:10.1007/s12195-019-00576-1
50. Spranger S, Gajewski TF. A new paradigm for tumor immune escape: beta-catenin-driven immune exclusion. J Immunother Cancer. 2015;3:43. doi:10.1186/s40425-015-0089-6
51. Galluzzi L, Spranger S, Fuchs E, Lopez-Soto A. Wnt signaling in cancer immunosurveillance. Trends Cell Biol. 2019;29(1):44–65. doi:10.1016/j.tcb.2018.08.005
52. Nsengimana J, Laye J, Filia A, et al. Beta-catenin-mediated immune evasion pathway frequently operates in primary cutaneous melanomas. J Clin Invest. 2018;128(5):2048–2063. doi:10.1172/JCI95351
53. Massi D, Romano E, Rulli E, et al. Baseline beta-catenin, programmed death-ligand 1 expression and tumour-infiltrating lymphocytes predict response and poor prognosis in BRAF inhibitor-treated melanoma patients. Eur J Cancer. 2017;78:70–81. doi:10.1016/j.ejca.2017.03.012
54. Spranger S, Bao RY, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–U261. doi:10.1038/nature14404
55. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(5):711–723. doi:10.1016/j.ccell.2017.04.003
56. Luke JJ, Bao RY, Sweis RF, Spranger S, Gajewski TF. Wnt/beta-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res. 2019;25(10):3074–3083. doi:10.1158/1078-0432.CCR-18-1942
57. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13(7):513–532. doi:10.1038/nrd4233
58. de la Roche M, Rutherford TJ, Gupta D, et al. An intrinsically labile alpha-helix abutting the BCL9-binding site of beta-catenin is required for its inhibition by carnosic acid. Nat Commun. 2012;3:680. doi:10.1038/ncomms1680
59. Liu JX, Yan J, Yang SQ, et al. Biomimetic and self-assembled nanoclusters targeting beta-catenin for potent anticancer therapy and enhanced immunotherapy. Nano Lett. 2019;19(12):8708–8715. doi:10.1021/acs.nanolett.9b03414
60. He WX, Wang SM, Yan J, et al. Self-assembly of therapeutic peptide into stimuli-responsive clustered nanohybrids for cancer-targeted therapy. Adv Funct Mater. 2019;29(10):1807736. doi:10.1002/adfm.201807736
61. Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18(5):1386–1394. doi:10.1158/1078-0432.CCR-11-2479
62. Loi S, Dushyanthen S, Beavis PA, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22(6):1499–1509. doi:10.1158/1078-0432.CCR-15-1125
63. Akiyama Y, Nonomura C, Ashizawa T, et al. The anti-tumor activity of the STAT3 inhibitor STX-0119 occurs via promotion of tumor-infiltrating lymphocyte accumulation in temozolomide-resistant glioblastoma cell line. Immunol Lett. 2017;190:20–25. doi:10.1016/j.imlet.2017.07.005
64. Ashizawa T, Iizuka A, Maeda C, et al. Impact of combination therapy with anti-PD-1 blockade and a STAT3 inhibitor on the tumor-infiltrating lymphocyte status. Immunol Lett. 2019;216:43–50. doi:10.1016/j.imlet.2019.10.003
65. Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018;15(6):366–381. doi:10.1038/s41571-018-0007-1
66. Salmon H, Franciszkiewicz K, Damotte D, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122(3):899–910. doi:10.1172/JCI45817
67. Mehrad B, Burdick MD, Strieter RM. Fibrocyte CXCR4 regulation as a therapeutic target in pulmonary fibrosis. Int J Biochem Cell Biol. 2009;41(8–9):1708–1718. doi:10.1016/j.biocel.2009.02.020
68. Li ZT, Wang YX, Shen YX, Qian CG, Oupicky D, Sun MJ. Targeting pulmonary tumor microenvironment with CXCR4-inhibiting nanocomplex to enhance anti-PD-L1 immunotherapy. Sci Adv. 2020;6(20):eaaz9240. doi:10.1126/sciadv.aaz9240
69. Araki K, Turner AP, Shaffer VO, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460(7251):108–U124. doi:10.1038/nature08155
70. Pollizzi KN, Patel CH, Sun IH, et al. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest. 2015;125(5):2090–2108. doi:10.1172/JCI77746
71. Rao RR, Li QS, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8(+) T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32(1):67–78. doi:10.1016/j.immuni.2009.10.010
72. Wang Y, Wang XY, Subjeck JR, Shrikant PA, Kim HL. Temsirolimus, an mTOR inhibitor, enhances anti-tumour effects of heat shock protein cancer vaccines. Brit J Cancer. 2011;104(4):643–652. doi:10.1038/bjc.2011.15
73. Berezhnoy A, Castro I, Levay A, Malek TR, Gilboa E. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest. 2014;124(1):188–197. doi:10.1172/JCI69856
74. Li QS, Rao R, Vazzana J, et al. Regulating mammalian target of rapamycin to tune vaccination-induced CD8(+) T cell responses for tumor immunity. J Immunol. 2012;188(7):3080–3087. doi:10.4049/jimmunol.1103365
75. Diken M, Kreiter S, Vascotto F, et al. mTOR inhibition improves antitumor effects of vaccination with antigen-encoding RNA. Cancer Immunol Res. 2013;1(6):386–392. doi:10.1158/2326-6066.CIR-13-0046
76. Mineharu Y, Kamran N, Lowenstein PR, Castro MG. Blockade of mTOR signaling via rapamycin combined with immunotherapy augments antiglioma cytotoxic and memory T-cell functions. Mol Cancer Ther. 2014;13(12):3024–3036. doi:10.1158/1535-7163.MCT-14-0400
77. Gammon JM, Gosselin EA, Tostanoski LH, et al. Low-dose controlled release of mTOR inhibitors maintains T cell plasticity and promotes central memory T cells. J Control Release. 2017;263:151–161. doi:10.1016/j.jconrel.2017.02.034
78. DeSilva DR, Jones EA, Favata MF, et al. Inhibition of mitogen-activated protein kinase kinase blocks T cell proliferation but does not induce or prevent anergy. J Immunol. 1998;160(9):4175–4181.
79. D’Souza WN, Chang CF, Fischer AM, Li MQ, Hedrick SM. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J Immunol. 2008;181(11):7617–7629. doi:10.4049/jimmunol.181.11.7617
80. Boni A, Cogdill AP, Dang P, et al. Selective BRAF (V600e) inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70(13):5213–5219. doi:10.1158/0008-5472.CAN-10-0118
81. Ribas A, Lawrence D, Atkinson V, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med. 2019;25(6):936–940. doi:10.1038/s41591-019-0476-5
82. Yang H, Liu HS, Hou W, et al. An NIR-responsive mesoporous silica nanosystem for synergetic photothermal-immunoenhancement therapy of hepatocellular carcinoma. J Mater Chem B. 2020;8(2):251–259. doi:10.1039/C9TB01891C
83. Luo MM, Fan TJ, Zhou Y, Zhang H, Mei L. 2D black phosphorus-based biomedical applications. Adv Funct Mater. 2019;29(13):1808306. doi:10.1002/adfm.201808306
84. Chen JM, Fan TJ, Xie ZJ, et al. Advances in nanomaterials for photodynamic therapy applications: status and challenges. Biomaterials. 2020;237:119827. doi:10.1016/j.biomaterials.2020.119827
85. Schito L. Bridging angiogenesis and immune evasion in the hypoxic tumor microenvironment. Am J Physiol-Reg I. 2018;315(6):R1072–R1084. doi:10.1152/ajpregu.00209.2018
86. Noman MZ, Hasmim M, Lequeux A, et al. Improving cancer immunotherapy by targeting the hypoxic tumor microenvironment: new opportunities and challenges. Cells-Basel. 2019;8(9):1083. doi:10.3390/cells8091083
87. Vetsika E-K, Koukos A, Kotsakis A. Myeloid-derived suppressor cells: major figures that shape the immunosuppressive and angiogenic network in cancer. Cells-Basel. 2019;8(12):1647. doi:10.3390/cells8121647
88. Ko JS, Zea AH, Rin BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. doi:10.1158/1078-0432.CCR-08-1332
89. Kim H, Khanna V, Kucaba TA, et al. Combination of sunitinib and PD-L1 blockade enhances anticancer efficacy of TLR7/8 agonist-based nanovaccine. Mol Pharmaceut. 2019;16(3):1200–1210. doi:10.1021/acs.molpharmaceut.8b01165
90. Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T, Daemen T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology. 2015;4(3):e989764. doi:10.4161/2162402X.2014.989764
91. Huo MR, Zhao Y, Satterlee AB, Wang YH, Xu Y, Huang L. Tumor-targeted delivery of sunitinib base enhances vaccine therapy for advanced melanoma by remodeling the tumor microenvironment. J Control Release. 2017;245:81–94. doi:10.1016/j.jconrel.2016.11.013
92. Domvri K, Petanidis S, Anestakis D, et al. Dual photothermal MDSCs-targeted immunotherapy inhibits lung immunosuppressive metastasis by enhancing T-cell recruitment. Nanoscale. 2020;12(13):7051–7062. doi:10.1039/D0NR00080A
93. Chen YC, Sun JJ, Huang YX, Lu BF, Li S. Improved cancer immunochemotherapy via optimal co-delivery of chemotherapeutic and immunomodulatory agents. Mol Pharmaceut. 2018;15(11):5162–5173. doi:10.1021/acs.molpharmaceut.8b00717
94. Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front Immunol. 2014;5:489. doi:10.3389/fimmu.2014.00489
95. Zhu XD, Zhang JB, Zhuang PY, et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol. 2008;26(16):2707–2716. doi:10.1200/JCO.2007.15.6521
96. Richardsen E, Uglehus RD, Johnsen SH, Busund LT. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res. 2015;35(2):865–874.
97. Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. doi:10.1038/nm.3337
98. Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–5069. doi:10.1158/0008-5472.CAN-13-3723
99. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5:53. doi:10.1186/s40425-017-0257-y
100. Qian Y, Qiao S, Dai YF, et al. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano. 2017;11(9):9536–9549. doi:10.1021/acsnano.7b05465
101. Morrison BW, Doudican NA, Patel KR, Orlow SJ. Disulfiram induces copper-dependent stimulation of reactive oxygen species and activation of the extrinsic apoptotic pathway in melanoma. Melanoma Res. 2010;20(1):11–20. doi:10.1097/CMR.0b013e328334131d
102. Zhao PF, Yin WM, Wu AH, et al. Dual-targeting to cancer cells and M2 macrophages via biomimetic delivery of mannosylated albumin nanoparticles for drug-resistant cancer therapy. Adv Funct Mater. 2017;27(44):1700403. doi:10.1002/adfm.201700403
103. Zhao PF, Wang YH, Kang XJ, et al. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem Sci. 2018;9(10):2674–2689. doi:10.1039/C7SC04853J
104. Li M, Li MM, Yang YL, et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-gamma and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J Control Release. 2020;321:23–35. doi:10.1016/j.jconrel.2020.02.011
105. Ramesh A, Brouillard A, Kumar S, Nandi D, Kulkarni A. Dual inhibition of CSF1R and MAPK pathways using supramolecular nanoparticles enhances macrophage immunotherapy. Biomaterials. 2020;227:119559. doi:10.1016/j.biomaterials.2019.119559
106. Larmonier N, Janikashvili N, LaCasse CJ, et al. Imatinib mesylate inhibits CD4(+) CD25(+) regulatory T cell activity and enhances active immunotherapy against BCR-ABL (-) tumors. J Immunol. 2008;181(10):6955–6963. doi:10.4049/jimmunol.181.10.6955
107. Jung JY, Parson MG, Kraft JD, et al. Elevated interleukin-27 levels in human neonatal macrophages regulate indoleamine dioxygenase in a STAT-1 and STAT-3-dependent manner. Immunology. 2016;149(1):35–47. doi:10.1111/imm.12625
108. Balachandran VP, Cavnar MJ, Zeng S, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of IDO. Nat Med. 2011;17(9):1094–U1099. doi:10.1038/nm.2438
109. Ou W, Thapa RK, Jiang LY, et al. Regulatory T cell-targeted hybrid nanoparticles combined with immuno-checkpoint blockage for cancer immunotherapy. J Control Release. 2018;281:84–96. doi:10.1016/j.jconrel.2018.05.018
110. Bruder D, Probst-Kepper M, Westendorf AM, et al. Neuropilin-1: A surface marker of regulatory T cells. Eur J Immunol. 2004;34(3):623–630. doi:10.1002/eji.200324799
111. Ou W, Jiang L, Thapa RK, et al. Combination of NIR therapy and regulatory T cell modulation using layer-by-layer hybrid nanoparticles for effective cancer photoimmunotherapy. Theranostics. 2018;8(17):4574–4590. doi:10.7150/thno.26758
112. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017;198(3):1006–1014. doi:10.4049/jimmunol.1601515
113. Kaneda MM, Messer KS, Ralainirina N, et al. PI3K gamma is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437–442. doi:10.1038/nature19834
114. Locatelli SL, Careddu G, Serio S, et al. Targeting cancer cells and tumor microenvironment in preclinical and clinical models of hodgkin lymphoma using the dual PI3K delta/gamma inhibitor RP6530. Clin Cancer Res. 2019;25(3):1098–1112. doi:10.1158/1078-0432.CCR-18-1133
115. Qin HJ, Yu HY, Sheng JY, et al. PI3Kgamma inhibitor attenuates immunosuppressive effect of poly (l-glutamic acid)-combretastatin A4 conjugate in metastatic breast cancer. Adv Sci. 2019;6(12):1900327. doi:10.1002/advs.201900327
116. Chen W, Ma T, Shen XN, et al. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012;72(6):1363–1372. doi:10.1158/0008-5472.CAN-11-2684
117. Shan MH, Qin JF, Jin FJ, et al. Autophagy suppresses isoprenaline-induced M2 macrophage polarization via the ROS/ERK and mtor signaling pathway. Free Radical Bio Med. 2017;110:432–443. doi:10.1016/j.freeradbiomed.2017.05.021
118. Foubert P, Kaneda MM, Varner JA. PI3K gamma activates integrin alpha (4) and promotes immune suppressive myeloid cell polarization during tumor progression. Cancer Immunol Res. 2017;5(11):957–968. doi:10.1158/2326-6066.CIR-17-0143
119. Yu M, Duan XH, Cai YJ, et al. Multifunctional nanoregulator reshapes immune microenvironment and enhances immune memory for tumor immunotherapy. Adv Sci. 2019;6(16):1900037. doi:10.1002/advs.201900037
120. Jiang M, He KY, Qiu T, et al. Tumor-targeted delivery of silibinin and IPI-549 synergistically inhibit breast cancer by remodeling the microenvironment. Int J Pharmaceut. 2020;581:119239. doi:10.1016/j.ijpharm.2020.119239
121. Chen B, Gao A, Tu B, et al. Metabolic modulation via mTOR pathway and anti-angiogenesis remodels tumor microenvironment using PD-L1-targeting codelivery. Biomaterials. 2020;255:120187. doi:10.1016/j.biomaterials.2020.120187
122. Li K, Lu L, Xue CC, et al. Polarization of tumor-associated macrophage phenotype via porous hollow iron nanoparticles for tumor immunotherapy in vivo. Nanoscale. 2020;12(1):130–144. doi:10.1039/C9NR06505A
123. Qiu QJ, Li C, Song YZ, et al. Targeted delivery of ibrutinib to tumor-associated macrophages by sialic acid-stearic acid conjugate modified nanocomplexes for cancer immunotherapy. Acta Biomater. 2019;92:184–195. doi:10.1016/j.actbio.2019.05.030
124. Derynck R, Turley SJ, Akhurst RJ. TGF beta biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. 2021;18(1):9–34. doi:10.1038/s41571-020-0403-1
125. Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7(10):1118–1122. doi:10.1038/nm1001-1118
126. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9(2):194–202. doi:10.1038/ni1549
127. Gratchev A. TGF-beta signalling in tumour associated macrophages. Immunobiology. 2017;222(1):75–81. doi:10.1016/j.imbio.2015.11.016
128. Zhang F, Li F, Lu GH, et al. Engineering magnetosomes for ferroptosis/immunomodulation synergism in cancer. ACS Nano. 2019;13(5):5662–5673. doi:10.1021/acsnano.9b00892
129. Xu BL, Cui Y, Wang WW, et al. Immunomodulation-enhanced nanozyme-based tumor catalytic therapy. Adv Mater. 2020;32:2003563. doi:10.1002/adma.202003563
130. Liu JQ, Liu Q, Li Y, et al. Efficacy and safety of camrelizumab combined with apatinib in advanced triple-negative breast cancer: an open-label Phase II trial. J Immunother Cancer. 2020;8(1):e000696. doi:10.1136/jitc-2020-000696
131. Yue P, Harper T, Bacot SM, et al. BRAF and MEK inhibitors differentially affect nivolumab-induced T cell activation by modulating the TCR and AKT signaling pathways. Oncoimmunology. 2019;8(1):e1512456. doi:10.1080/2162402X.2018.1512456
132. Erkes DA, Cai W, Sanchez IM, et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 2020;10(2):254–269. doi:10.1158/2159-8290.CD-19-0672
133. Gorniak P, Wasylecka-Juszczynska M, Lugowska I, et al. BRAF inhibition curtails IFN-gamma-inducible PD-L1 expression and upregulates the immunoregulatory protein galectin-1 in melanoma cells. Mol Oncol. 2020;14(8):1817–1832. doi:10.1002/1878-0261.12695
134. Jung K, Heishi T, Incio J, et al. Targeting CXCR4-dependent immunosuppressive Ly6C (low) monocytes improves antiangiogenic therapy in colorectal cancer. P Natl Acad Sci USA. 2017;114(39):10455–10460. doi:10.1073/pnas.1710754114
135. Lin YY, Tan CT, Chen CW, Ou DL, Cheng AL, Hsu C. Immunomodulatory effects of current targeted therapies on hepatocellular carcinoma: implication for the future of immunotherapy. Semin Liver Dis. 2018;38(4):379–388. doi:10.1055/s-0038-1673621
136. Peng DJ, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of T (H)1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527(7577):249–253. doi:10.1038/nature15520
137. Nagarsheth N, Peng DJ, Kryczek I, et al. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. 2016;76(2):275–282. doi:10.1158/0008-5472.CAN-15-1938
138. Zingg D, Arenas-Ramirez N, Sahin D, et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 2017;20(4):854–867. doi:10.1016/j.celrep.2017.07.007
139. Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov. 2020;19(11):776–800. doi:10.1038/s41573-020-0077-5
140. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. New Engl J Med. 2016;375(22):2154–2164. doi:10.1056/NEJMoa1611310
141. Sen T, Rodriguez BL, Chen LM, et al. Targeting DNA damage response promotes antitumor immunity through sting-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9(5):646–661. doi:10.1158/2159-8290.CD-18-1020
142. Pantelidou C, Sonzogni O, Taveira MD, et al. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 2019;9(6):722–737. doi:10.1158/2159-8290.CD-18-1218
143. Domchek SM, Postel-Vinay S, Im SA, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (mediola): an open-label, multicentre, Phase 1/2, basket study. Lancet Oncol. 2020;21(9):1155–1164. doi:10.1016/S1470-2045(20)30324-7
144. Behzadi S, Serpooshan V, Tao W, et al. Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev. 2017;46(14):4218–4244. doi:10.1039/C6CS00636A
145. Xue XL, Qian CG, Fang HB, et al. Photoactivated lysosomal escape of a monofunctional Pt-II complex Pt-BDPA for nucleus access. Angew Chem Int Edit. 2019;58(36):12661–12666. doi:10.1002/anie.201906203
146. Murugan K, Choonara YE, Kumar P, Bijukumar D, Du Toit LC, Pillay V. Parameters and characteristics governing cellular internalization and trans-barrier trafficking of nanostructures. Int J Nanomed. 2015;10:2191–2206. doi:10.2147/IJN.S75615
147. Francia V, Yang KN, Deville S, Reker-Smit C, Nelissen I, Salvati A. Corona composition can affect the mechanisms cells use to internalize nanoparticles. ACS Nano. 2019;13(10):11107–11121. doi:10.1021/acsnano.9b03824
148. Zhou J, Yang TQ, Chen JJ, Wang C, Zhang H, Shao YH. Two-dimensional nanomaterial-based plasmonic sensing applications: advances and challenges. Coordin Chem Rev. 2020;410:213218. doi:10.1016/j.ccr.2020.213218
149. Ji XY, Kong N, Wang JQ, et al. A novel top-down synthesis of ultrathin 2D boron nanosheets for multimodal imaging-guided cancer therapy. Adv Mater. 2018;30(36):1803031. doi:10.1002/adma.201803031
150. He HL, Liu LS, Morin EE, Liu M, Schwendeman A. Survey of clinical translation of cancer nanomedicines-lessons learned from successes and failures. Acc Chem Res. 2019;52(9):2445–2461. doi:10.1021/acs.accounts.9b00228
151. Hannon G, Lysaght J, Liptrott NJ, Prina-Mello A. Immunotoxicity considerations for next generation cancer nanomedicines. Adv Sci. 2019;6(19):1900133. doi:10.1002/advs.201900133
152. Hansen SF, Lennquist A. Carbon nanotubes added to the sin list as a nanomaterial of very high concern. Nat Nanotechnol. 2020;15(1):3–4. doi:10.1038/s41565-019-0613-9
153. Mocan T, Matea C, Tabaran F, Iancu C, Orasan R, Mocan L. In vitro administration of gold nanoparticles functionalized with MUC-1 protein fragment generates anticancer vaccine response via macrophage activation and polarization mechanism. J Cancer. 2015;6(6):583–592. doi:10.7150/jca.11567
154. Raja MRC, Kumar VV, Srinivasan V, et al. ApAGP-fabricated silver nanoparticles induce amendment of murine macrophage polarization. J Mater Chem B. 2017;5(19):3511–3520. doi:10.1039/C6TB02095J
155. Zanganeh S, Hutter G, Spitler R, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11(11):986–994. doi:10.1038/nnano.2016.168
156. Chen ZY, Liu Y, Sun BY, et al. Polyhydroxylated metallofullerenols stimulate IL-1 beta secretion of macrophage through TLRs/MyD88/NF-kappa B pathway and NLRP3 inflammasome activation. Small. 2014;10(12):2362–2372. doi:10.1002/smll.201302825
157. Tang J, Chen Z, Sun B, et al. Polyhydroxylated fullerenols regulate macrophage for cancer adoptive immunotherapy and greatly inhibit the tumor metastasis. Nanomedicine. 2016;12(4):945–954. doi:10.1016/j.nano.2015.11.021
158. Li AW, Sobral MC, Badrinath S, et al. A facile approach to enhance antigen response for personalized cancer vaccination. Nat Mater. 2018;17(6):528–534. doi:10.1038/s41563-018-0028-2
159. Kim J, Li WA, Choi Y, et al. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat Biotechnol. 2015;33(1):64–U241. doi:10.1038/nbt.3071
160. Luo M, Wang H, Wang ZH, et al. A sting-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol. 2017;12(7):648–654. doi:10.1038/nnano.2017.52
161. Chen SY, Xing CY, Huang DZ, et al. Eradication of tumor growth by delivering novel photothermal selenium-coated tellurium nanoheterojunctions. Sci Adv. 2020;6(15):eaay6825. doi:10.1126/sciadv.aay6825
162. Xing CY, Chen SY, Liang X, et al. Two-dimensional mxene (Ti3C2)-integrated cellulose hydrogels: toward smart three-dimensional network nanoplatforms exhibiting light-induced swelling and bimodal photothermal/chemotherapy anticancer activity. Acs Appl Mater Inter. 2018;10(33):27631–27643. doi:10.1021/acsami.8b08314
163. Tao W, Ji XY, Zhu XB, et al. Two-dimensional antimonene-based photonic nanomedicine for cancer theranostics. Adv Mater. 2018;30(38):1802061. doi:10.1002/adma.201802061
164. Tao W, Ji XY, Xu XD, et al. Antimonene quantum dots: synthesis and application as near-infrared photothermal agents for effective cancer therapy. Angew Chem Int Edit. 2017;56(39):11896–11900. doi:10.1002/anie.201703657
165. Xing CY, Chen SY, Qiu M, et al. Conceptually novel black phosphorus/cellulose hydrogels as promising photothermal agents for effective cancer therapy. Adv Healthc Mater. 2018;7(7):1701510. doi:10.1002/adhm.201701510
166. Xie ZJ, Peng MH, Lu RT, et al. Black phosphorus-based photothermal therapy with aCD47-mediated immune checkpoint blockade for enhanced cancer immunotherapy. Light-Sci Appl. 2020;9(1):161. doi:10.1038/s41377-020-00388-3
167. Qiu M, Singh A, Wang D, et al. Biocompatible and biodegradable inorganic nanostructures for nanomedicine: silicon and black phosphorus. Nano Today. 2019;25:135–155. doi:10.1016/j.nantod.2019.02.012
168. Sun X, Cao ZY, Mao KR, et al. Photodynamic therapy produces enhanced efficacy of antitumor immunotherapy by simultaneously inducing intratumoral release of sorafenib. Biomaterials. 2020;240:119845. doi:10.1016/j.biomaterials.2020.119845
169. Zhu HD, Liu Q, Miao L, Musetti S, Huo MR, Huang L. Remodeling the fibrotic tumor microenvironment of desmoplastic melanoma to facilitate vaccine immunotherapy. Nanoscale. 2020;12(5):3400–3410. doi:10.1039/c9nr09610h
170. Zheng ZN, Zhang JX, Jiang JZ, et al. Remodeling tumor immune microenvironment (TIME) for glioma therapy using multi-targeting liposomal codelivery. J Immunother Cancer. 2020;8(2):e000207. doi:10.1136/jitc-2019-000207
171. Wang TQ, Zhang J, Hou T, Yin XL, Zhang N. Selective targeting of tumor cells and tumor associated macrophages separately by twin-like core-shell nanoparticles for enhanced tumor-localized chemoimmunotherapy. Nanoscale. 2019;11(29):13934–13946. doi:10.1039/C9NR03374B
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.