")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Nanoemulsions as novel oral carriers of stiripentol: insights into the protective effect and absorption enhancement
Authors Lu R, Liu S, Wang Q, Li X
Received 28 April 2015
Accepted for publication 15 June 2015
Published 31 July 2015 Volume 2015:10(1) Pages 4937—4946
DOI https://doi.org/10.2147/IJN.S87471
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Lei Yang
Rong Lu,1 Shan Liu,1 Qilin Wang,1 Xia Li1,2
1School of Ocean, Shandong University, Weihai, 2School of Pharmaceutical Sciences, Shandong University, Jinan, People’s Republic of China
Abstract: Oral administration remains a significant challenge in regards to drugs with serious solubility and stability issues. This article aimed to investigate the suitability of nanoemulsions as oral carriers of stiripentol (STP), an acid-labile drug, for enhancement of stability and bioavailability. STP-loaded nanoemulsions (STP-NEs) were prepared by using a solvent-diffusion/ultrasonication technique. STP-NEs were characterized in a variety of ways such as by particle size, entrapment efficiency, in vitro drug release, and transmission electron microscopy. A bioavailability study was performed in rats after oral administration of either STP-NEs, or commercial formulation (Diacomit®). The resultant nanoemulsions were 146.6 nm in particle size with an entrapment efficiency of 99.47%. It was demonstrated that nanoemulsions significantly improved the biochemical stability and bioavailability of STP. The bioavailability of STP-NEs was up to 206.2% relative to Diacomit®. Nanoemulsions fabricated from poly(ethylene glycol) monooleate/medium-chain triglycerides exhibited excellent performance in drug stabilization and absorption enhancement. The results suggest that STP-NEs are a promising means to solve the problems associated with stability and solubility of STP.
Keywords: stiripentol, nanoemulsions, stability, intestinal permeability, bioavailability
Introduction
Stiripentol (STP) is an orphan drug that treats Severe Myoclonic Epilepsy of Infancy (SMEI) also known as Dravet syndrome.1 It has also been shown that STP could be used to treat super-refractory status epilepticus.2 Sada et al reported that STP and its analog could target lactate dehydrogenase (LDH) to potently suppress seizures of SMEI by inhibiting the metabolic pathway of lactate mediated by LDH.3 STP is highly insoluble in water, with a solubility of 49.2 μg/mL.4 It chemically possesses the property of ethylene alcohol, and is extremely unstable in acidic milieu. Poor solubility along with gastric instability largely limits its oral bioavailability and clinical efficacy. To achieve effective therapeutic concentration a high dose is required, with a maximum dose of up to 4 g adopted in clinical practice. Therefore, it is necessary to ameliorate the current plight by developing a suitable drug delivery system which is favorable in avoiding drug waste and reducing adverse reactions as a result of high dose.
The dosage forms of STP on the current market are mainly formulated in capsules and dry suspensions. Apart from convenient storage, conventional solid formulations are generally lackluster in oral delivery. To improve the dissolution and/or bioavailability, a variety of novel solid formulations have been attempted, including solid dispersions,5,6 cyclodextrin inclusion complexes,7,8 and various solidified nanocarriers.9–11 However, these formulations possess an inferior capability of resisting drug degradation in harsh conditions, such as low gastric pH, digestive stress, and first-pass metabolism. This is because of rapid dissolution, or release from these systems, which easily result in considerable exposure of the drug to the gastrointestinal (GI) tract. For unstable drugs in the GI lumen, it is required that the delivery system protects the integrity of the drug from degradation, and in addition benefit from absorption enhancement. In this respect, some versatile nanocarriers have shown the potential of protecting labile drugs, such as PEGylated lipid nanoparticles,12,13 polymeric micelles or nanoparticles,14–16 and liposomes.17,18 Although micelles assembled from mPEG-PCL have been explored to orally deliver STP,4 the material used is rather expensive and the micellar system easily dissociates upon dilution of physiological fluids. To date, the protective effect of nanoemulsions on acid-labile drugs has not been fully investigated.
Nanoemulsions are oil-in-water emulsion composed of surfactant, oil, and water, with a droplet diameter at the nanoscale. Generally, a large quantity of surfactants (~weight is 20% of the oil phase), is needed to obtain nanoscale emulsions, which potentially raises the safety risk of the drug delivery system, especially for long-term use. Another disadvantage held by conventional nanoemulsions is the remarkable lipolysis in the GI tract. Lipids, like oils and triglycerides, are readily digested by pancreatic lipases, resulting in drug precipitation and variable bioavailability.19 To enhance the oral delivery, an effective approach is to disguise the carrier with PEGylation.20 PEGylated materials possess several advantages, such as being less toxic, strong solvent power for solubilization, and protect the drug or carriers from degradation.21 Those PEGylated materials with surfactant property may be ponderable to prepare nanoemulsions instead of the common surfactants.
In this study, nanoemulsions made up of poly(ethylene glycol) monooleate (PM) and medium-chain triglycerides, were designed to enhance the gastric stability of STP by masking proton attack, and enhancing the bioavailability by improving the intestinal absorption (Figure 1). The emulsion system contains no co-surfactant (eg, glycerin) which is regularly required to reduce the interfacial tension. Furthermore, PM has the underlying ability to change the digestion of nanoemulsions. STP-loaded nanoemulsions (STP-NEs) were prepared using a solvent-diffusion/ultrasonication technique and characterized by transmission electron microscopy (TEM) (Philips Tecnai-10, Philips, Amsterdam, the Netherlands), in vitro release, and stability test. The performance of STP-NEs in bioavailability enhancement was tested in rats following oral administration with a commercial formulation (Diacomit®).
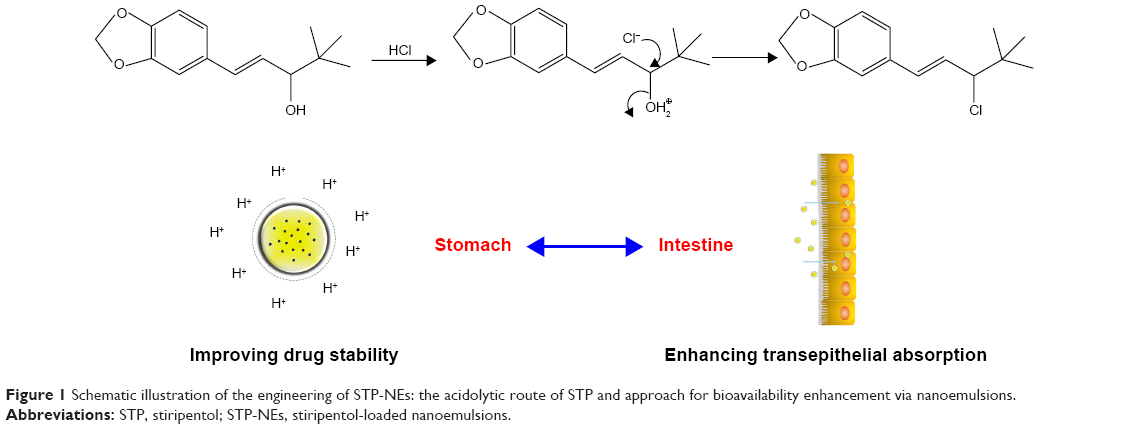 | Figure 1 Schematic illustration of the engineering of STP-NEs: the acidolytic route of STP and approach for bioavailability enhancement via nanoemulsions. |
Materials and methods
Materials
STP was obtained from Haiku Pharmaceutical Science & Technology Co., Ltd. (Guangzhou, People’s Republic of China). Diclofenac sodium (internal standard) was purchased from Aladdin Industrial Inc. (Shanghai, People’s Republic of China). Medium-chain triglycerides (MCT) were kindly gifted by Gattefosse (Saint-Priest Cedex, France). PM (Mn ~1,200) was purchased from Aoke Chemicals (Shanghai, People’s Republic of China). Deionized water was prepared by a Milli-Q water purifier (EMD Millipore, Billerica, MA, USA). High-performance liquid chromatograph (HPLC)-grade methanol was obtained from Merck KGaA (Darmstadt, Germany). All other chemicals were of analytical grade and used as received.
Preparation of STP-NEs
STP-NEs were prepared by the technique of solvent-diffusion/ultrasonic homogenization. STP, PM and MCT were dissolved in absolute ethanol and then rapidly injected into water using a syringe. The components were spontaneously assembled into emulsions upon the solvent diffusion into the aqueous phase. After this, the resultant coarse emulsions were homogenized with an ultrasonic probe for 2 minutes (400 watt) to produce the final nanoemulsions. The residual ethanol (5% v/v) was reserved in the system as a preservative. Factors affecting the properties of nanoemulsions were investigated, including the ratios of PM/MCT and drug/excipients. The produced nanoemulsions were stored in a refrigerated cabinet at 4°C ready for use.
Characterization of STP-NEs
The particle size of STP-NEs was measured by a Zetasizer Nano ZS (Malvern, UK) at 25°C. To measure the particle size, nanoemulsions were diluted and transferred into a disposable cuvette. The sample was then subjected to laser diffraction after equilibration for 120 seconds. The mean particle size was acquired using the build-in software in the Zetasizer Nano ZS based on dynamic light scattering.
TEM was used to probe the morphology of STP-NEs. STP-NEs with 50-fold dilution were dropped on a carbon-coated copper grid and fixed by vacuum drying. The anchored droplets were then subjected to TEM observation and micrographs were collected at an acceleration voltage of 100 kV.
The entrapment efficiency (EE) and drug load (DL) of STP-NEs were determined by separating the free STP from the emulsion system. Briefly, the freshly prepared nanoemulsions were centrifuged at 5,000× g to remove the free bulk drug. The refined STP-NEs then proceeded to centrifugal filtration to further remove free STP by a centrifugal filter device (Amicon® Ultra-0.5, molecular weight cut-off 10 K, EMD Millipore). The concentration of STP entrapped in mixed micelles was quantified by the HPLC established below. The EE and DL of STP-NEs were calculated according to the following equations:
| (1) |
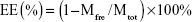
| (2) |
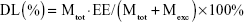
where Mfre and Mtot are the amounts of free and total STP in the nanoemulsions quantified on HPLC, respectively; Mexc is the amount of all excipients used in the formulation quantified on weight.
Quantification of STP
The concentration of STP in nanoemulsions and other samples was determined by Agilent 1100 series HPLC system (Santa Clara, CA, USA) equipped with a quaternary pump, a degasser, an autosampler, a column heater, and an ultraviolet detector. STP was separated by a C18 column (Agilent-Bonus-RP, 5 μm, 4.6 mm ×150 mm) guarded with a precolumn at 40°C. The injection volume was 20 mL, and the eluates were monitored at 254 nm. The mobile phase consisted of 78% methanol and 22% water pumped at a flow rate of 1.0 mL/min.
Acidolytic destabilization experiment
Simulated gastric fluid (ie, 0.1 M HCl with no pepsin) was used to check the acidolysis of free STP and STP-NEs. An aliquot of STP methanol solution or STP-NEs, equal to 0.5 mg STP, was introduced into 10 mL simulated gastric fluid and maintained at 37°C. At the time of 0.25, 0.5, 1, 1.5, 2, 3, and 4 hours, 1 mL of sample solution was withdrawn and immediately replenished by the same volume of fresh medium. The concentration of retained STP was analyzed by HPLC.
In vitro release study
The release of STP from nanoemulsions was investigated using the dialysis method.22 Aliquots of STP-NEs containing 250 mg STP from a dialysis bag (molecular weight cut-off 14,000) were added into 900 mL of pH 6.8 phosphate buffer, or 0.1 M HCl solution, in which 0.25% (w/v) sodium dodecyl sulfate was involved as solubilizers. At predetermined time points, 5 mL of the sample was withdrawn and immediately displaced with the same volume of fresh medium. STP concentration was determined by HPLC as described in the Quantification of STP section, and the percentage of drug release was calculated as mean ± SD (n=3).
Stability test of STP-NEs
For the evaluation of stability, STP-NEs were stored in ambient temperature for 1 month. The changes in particle size and drug content as a function of time were observed. STP-NEs encapsulated in a penicillin bottle were placed on the experiment desk under ambient temperature. The particle size of STP-NEs were monitored every day for 8 days, and then every other day thereafter. Three batches of STP-NEs, treated as particle size sample, were used to quantify the drug content at day 30 with the original value (day 0) as reference. The stability of STP-NEs was outlined by particle size distribution and the percentage of the remaining drug.
Bioavailability study
All animal experiments were conducted according to the protocols for animal experiments launched by the Experimental Animal Ethical Committee of Shandong University, which reviewed and approved the study. Sprague Dawley rats (220 g ±20 g) were fasted overnight (12 hours) before drug administration, but were allowed free access to water. Rats were randomly grouped into STP-NEs (n=5), and the commercial formulation group (Diacomit®), (n=5). The rats were dosed with the two preparations, STP-NEs and the conventional suspensions prepared from the commercial formulation, by gavage at the dose of 27 mg/kg. Blood was gathered into the heparinized tubes from the jugular vein at 0.5, 1, 2, 4, 6, 8, and 12 hours after administration. The blood samples were then centrifuged at 5,000× g for 5 minutes to collect the plasma.
To quantify the plasma STP, a deproteinization procedure was used to recover STP from the plasma. Briefly, 100 μL of plasma was mingled with 400 μL of methanol, and supplemented with 10 μL of diclofenac sodium (100 μg/mL), as an internal standard. After alternate vortex (1 minute) and sonication (15 minutes), the samples were centrifuged at 10,000× g for 10 minutes to precipitate the insoluble components. The extraction step was performed twice. The supernatant fraction was transferred into new tubes followed by evaporation at 30°C for 5 hours under vacuum to remove the solvent using a Concentrator Plus (Eppendorf, NY, USA). The residue were reconstituted in 100 μL of 78% methanol for HPLC analysis.
In situ single-pass intestinal perfusion
To investigate the effect of nanoemulsions on membrane permeability of STP, in situ single-pass intestinal perfusion was performed as described in previous studies.23,24 Sprague Dawley rats were fasted for 12 hours (free access to water) prior to the experiment. The rats were sacrificed by an intraperitoneal injection of 20% urethane (1.0 g/kg). The intestines were exposed by a midline abdominal incision. The segments of duodenum, jejunum, and ileum were identified and cannulated with silicone tubes (φ2.5×4 mm). Krebs–Ringer buffer (pH 7.4) was gently injected through the inlet tube to wash the intestinal contents. Perfusion solutions, equivalent to 45 μg/mL of STP, were prepared by diluting STP-NEs or STP ethanol solution into Krebs–Ringer buffer. After pre-perfusion for 30 minutes, the perfusate (approximately 3 mL) were collected every 15 minutes, up to 120 minutes. Finally, the diameter and length of the intestinal segments were measured. Net water flux was calibrated by weight with the sham-operated group in the perfusion experiment. The effective permeability coefficient (Peff) was calculated based on the inlet and outlet concentrations according to the equation below:
| (3) |
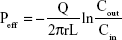
where Q denotes the flow rate (0.2 mL/min), r is the radius of the intestine (cm), L is the length of the perfused intestinal segment (cm), and Cin and Cout are the inlet and outlet concentrations of STP, respectively.
Cytotoxic evaluation
The cytotoxicity of nanoemulsions was checked by assessing the viability of Caco-2 cells in the presence of different concentration of nanoemulsions using MTT assay. Cell culture followed the same method as described in a previous study.18 Caco-2 cells were cultured for 48 hours and were triply washed with PBS (pH 7.4). STP-NEs with various PM levels were then introduced to the cells and incubated for 24 hours at 37°C. After that time, 20 μL of MTT solution (5 mg/mL) was dripped thoroughly into each well and continued to incubate for 4 hours. To dissolve the purple formazan crystals, 200 μL of dimethyl sulfoxide was used. The ultraviolet absorbance of each well was measured at 490 nm. The cell viability was calculated according to the equation:
Cell viability (%) = (Atri/Acon) ×100% | (4) |
where Atri was the absorbance of the cells treated with STP-NEs, and Acon was the absorbance treated with the culture medium.
Results and discussion
Preparation and characterization of STP-NEs
It was convenient and economical to prepare STP-NEs by solvent diffusion/homogenization. The procedure of preparation was rather straightforward. All ingredients (ie, drug, PM, and MCT) were dissolved in ethanol and then rapidly injected into water, followed by an ultrasonic processing. The particle size of carriers can reflect the formulation performance. It was demonstrated that smaller nanoparticles are more easily taken up by the absorptive epithelia.25,26 The factors affecting the particle size of STP-NEs included the ratios (w/w) of PM/MCT and drug/excipients. The influence of these variables on the particle size are shown in Figure 2. The ratio of PM/MCT had a certain effect on the droplet diameter of emulsions. In the case of blank formulation, high ratio of PM resulted in smaller nanoemulsions (Figure 2A), which conformed to the rule that more surfactant is favorable to the reduction of particle size. Otherwise, the incorporation of drug exerted negative effect on particle size. The particle size of STP-NEs decreased with the ratio of drug/excipients (Figure 2B). This could be explained by the increased viscosity or consistency of the system, due to more drug incorporation into the oil phase, thereby enhancing the interfacial tension upon emulsifying. Of note, most of formulations possessed an acceptable polydispersity index (PDI) below 0.3, indicating good suitability of the preparation process.
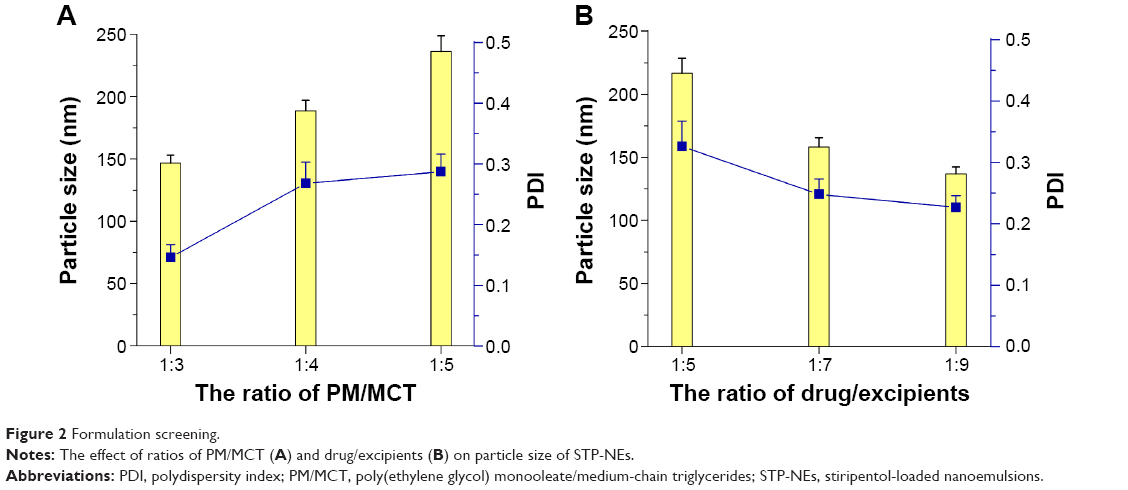 | Figure 2 Formulation screening. |
Considering the particle size and drug load, the formulation was finally fixed to PM/MCT at 1:3 and drug/excipients at 1:7, respectively, which consisted of 100 mg STP, 250 mg PM, and 450 mg MCT. The above components were dissolved in 1 mL absolute ethanol and then formulated into 20 mL water. The resulting STP-NEs had a particle size of 142.5 nm with a PDI value of 0.190 (Figure 3A). The ζ potential was determined to be −33.7 mv, a hallmark of stable colloidal systems. STP-NEs showed an excellent entrapment efficiency of 99.47%, and the drug load was up to 12.43%. STP-NEs appeared opalescent and exhibited blue light scatting upon dilution (Figure 3B). In addition, the prepared STP-NEs were near-spherical as revealed by TEM (Figure 3C). The particle size gauged from the scale bar (0.5 μm) was consistent with the hydrodynamic size measured by the Nano ZS analyzer.
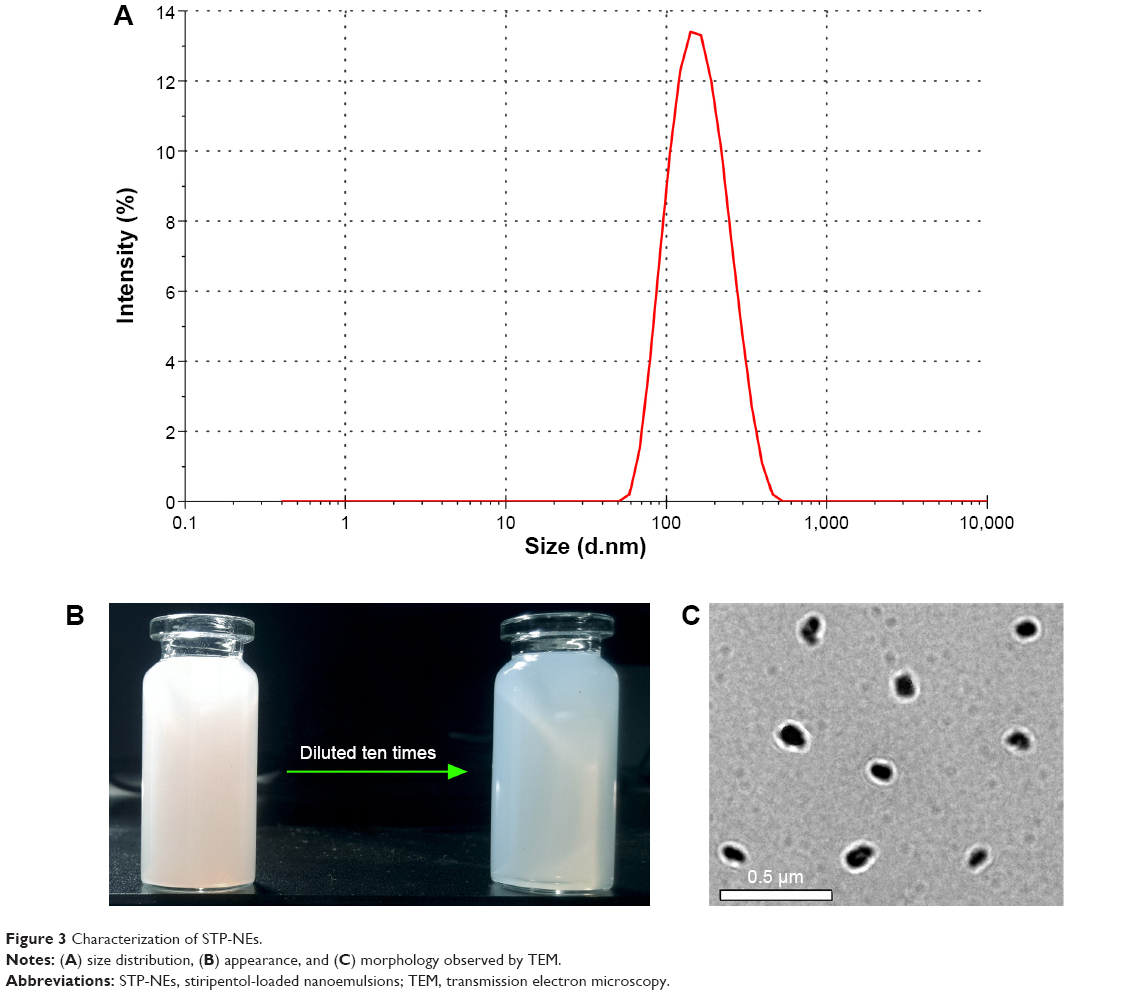 | Figure 3 Characterization of STP-NEs. |
Anti-acid stability of STP-NEs
Figure 4 shows the survival profiles of free STP and entrapped STP in nanoemulsions in simulated gastric fluid. It could be seen that significant acidic hydrolysis took place for free STP due to direct exposure to H+. Although the transformation was decelerated after 0.5 hour as a result of H+ depletion, the cumulative loss of the parent drug was more than 45% after 4 hours. However, STP-NEs almost did not undergo acidolysis, and the retained percentage of STP was kept well above 98%. The minor loss came from the destabilization of free STP that intrinsically dissolved in the external aqueous phase of emulsions. It was clear that the protective effect of nanoemulsions on STP was evident. The encapsulation of STP into the inner oil phase of nanoemulsions effectively shielded the attack of the proton, suggesting that nanoemulsions are suitable carriers for oral delivery of STP.
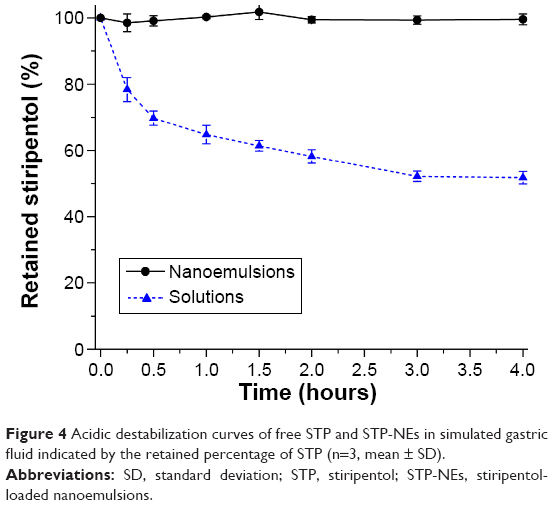 | Figure 4 Acidic destabilization curves of free STP and STP-NEs in simulated gastric fluid indicated by the retained percentage of STP (n=3, mean ± SD). |
Drug release
The release of STP from nanoemulsions is shown in Figure 5. The release increased as a function of time both in pH 6.8 buffer, and in 0.1 M HCl. STP could be continuously released into the media in a mode of slowness, which observed the principle of passive diffusion. The release pattern of STP-NEs was also consistent with those from previous studies.27,28 However, the release of STP-NEs was somewhat different in pH 6.8 buffer and in 0.1 M HCl. The accumulative release of STP was 11.56% in pH 6.8 buffer and 5.23% in 0.1 M HCl within 24 hours. Decreased release in 0.1 M HCl than in pH 6.8 buffer might be ascribed to the chemical conversion of a portion of STP. High lipophilicity (logP 2.94) should be responsible for the insignificant release of STP. From the release kinetics, it can be observed that the majority of STP was maintained in nanoemulsions in the first 4 hours. This nature of release allowed the STP-NEs to survive in the harsh gastric environment.
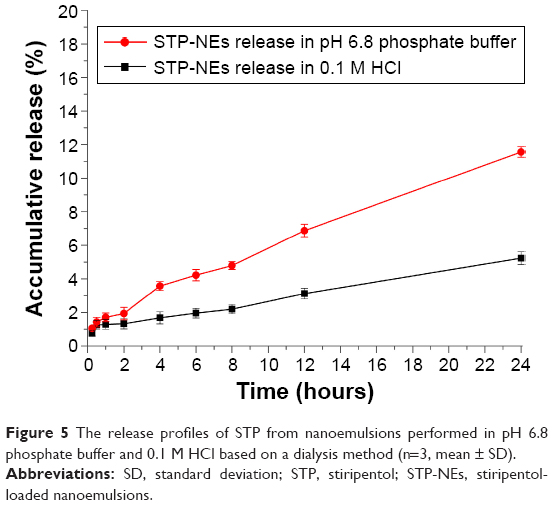 | Figure 5 The release profiles of STP from nanoemulsions performed in pH 6.8 phosphate buffer and 0.1 M HCl based on a dialysis method (n=3, mean ± SD). |
Stability of STP-NEs
Figure 6 shows the changes of particle size plus PDI of STP-NEs as a function of storage time. There were no significant differences in particle size between various appointed time and the origin (paired t-test, P>0.05). Although the PDI fluctuated somewhat day to day, the value was below 0.3, demonstrating an acceptable physical stability. The drug content in nanoemulsions at day 30 was determined to be 99.22%±0.65% of the initial level, which was indicative of good chemical stability. Taken together, STP-NEs showed satisfactory physicochemical stability.
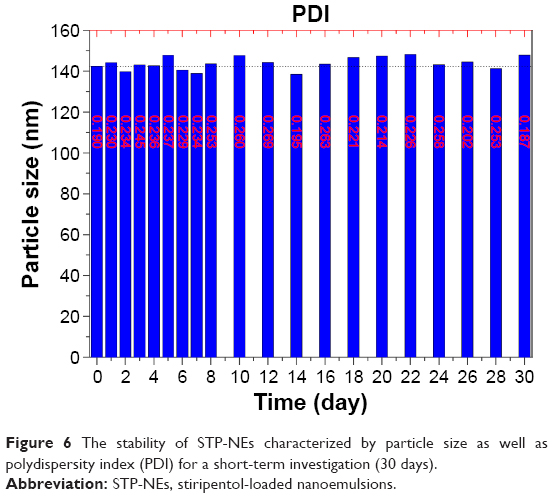 | Figure 6 The stability of STP-NEs characterized by particle size as well as polydispersity index (PDI) for a short-term investigation (30 days). |
Enhanced bioavailability
The plasma STP concentration versus time plots are presented in Figure 7 with pharmacokinetic parameters, processed by the WinNonlin software (Certara USA, Inc., Princeton, NJ, USA), are summarized in Table 1. For the commercial formulation (dry suspensions), the oral absorption was significantly lower than the home-made nanoemulsions. The maximum concentration (Cmax) and area under the plasma STP concentration–time curve (AUC)0–t were 2.02 μg/mL and 10.21 μg·h/mL, respectively. In comparison with the commercial formulation, STP-NEs produced higher STP levels at various time points with the Cmax of 6.16 μg/mL and AUC0–t of 21.06 μg·h/mL. Both the absorption rate and extent of STP were enhanced after oral administration of STP-NEs. Analysis of variance (ANOVA) showed that there were significant differences in Cmax and AUC0–t between STP-NEs and Diacomit® (n=5, P<0.05). The relative bioavailability of STP-NEs was up to 206.2% compared to the conventional suspensions. In terms of peak time (Tmax) and plasma half-time (T1/2), STP-NEs and Diacomit® possessed similar values of approximately 1 and 2.5 hours, respectively, indicating that STP could be rapidly absorbed and eliminated following administration.
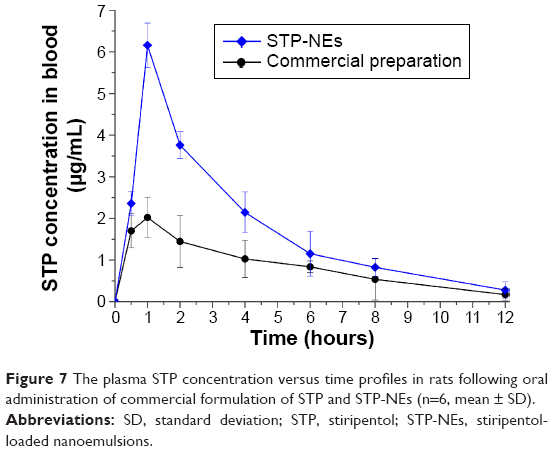 | Figure 7 The plasma STP concentration versus time profiles in rats following oral administration of commercial formulation of STP and STP-NEs (n=6, mean ± SD). |
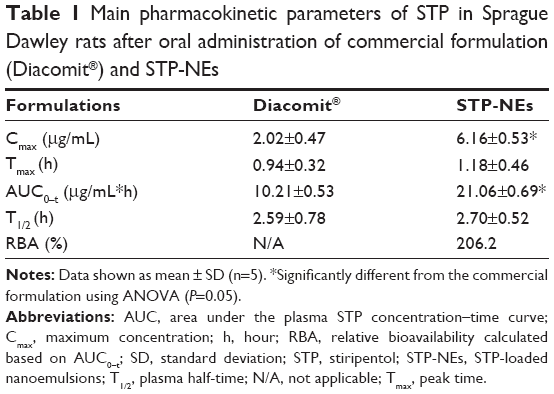 | Table 1 Main pharmacokinetic parameters of STP in Sprague Dawley rats after oral administration of commercial formulation (Diacomit®) and STP-NEs |
By contrast with the micellar system,4 our prepared nanoemulsions yielded higher blood STP concentrations and bioavailability. As known, lipid-based formulations have many advantages in enhancing the oral bioavailability of poorly soluble drugs by several mechanisms, such as solubilizing drug in the intestinal milieu, increasing intestinal lymphatic transport of drug (thereby reducing first-pass effect), and altering the drug disposition by enterocytes.29 It was reported that PEG materials having a molecular weight over 2,000 are difficult to absorb by the epithelial membrane through paracellular transport or macropinocytosis.30 Thus, micelles entering into the absorptive epithelia, as intact nanoparticles, are less feasible. To date, the absorption mechanism of micelles has been elusive. The digestion–absorption route is considered as the essential mechanism of nanoemulsions absorption, by which the lipid/oil constituents of nanoemulsions are digested by pancreatic lipases on the apical side of enterocytes and taken up by enterocytes subsequently.31,32 In addition, the uptake of nanoemulsions into the enterocytes can be fulfilled via clathrin- or caveolae-mediated endocytosis and macropinocytosis.33 Besides, MCT used in the nanoemulsions formulation has been proved to be able to enhance the permeability of the intestinal membrane.34 These advantages render nanoemulsions more suitable to orally deliver the highly lipophilic STP.
Intestinal permeability
The Peff of free STP and STP-NEs are given in Table 2. STP itself has good permeability in various intestinal segments with Peff >1×10−5 cm/s, a value implying good intestinal absorption.35 There were no significant differences in Peff of free STP among three intestinal segments. However, the entrapment of STP into nanoemulsions not only failed to improve the Peff of STP, but also decreased it to some extent. This can be accounted for by the complete drug molecule exposure to the intestine in the case of solution formulation and encapsulation effect of the drug in the case of nanoemulsions. The concentration of STP for perfusion was lower, close to the saturated concentration (49.2 μg/mL), which allowed STP molecules to be fully accessible to the intestinal mucosa. For STP-NEs, the drug release can be negligible relative to STP solution, causing the STP to be unable to be absorbed quickly in the form of molecules. Moreover, the ligation of intestinal segments blocked the entry of digestive enzymes. These factors rendered STP-NEs transport across the intestine slower than free STP. Free STP appeared to be more permeable in the intestinal epithelia than STP-NEs. This seemed to be in conflict with the significantly enhanced bioavailability of STP-NEs. It was suggestive that other factors should be involved in the overall absorption of STP-NEs, such as pre-enterocyte digestion, post-enterocyte processing of nanoemulsions, and first-pass metabolism of the drug. Interestingly, the Peff of STP-NEs decreased from the proximal intestine to the distal intestine. The underlying reason for such a decrease was possibly related to the differential permeability of intestinal segments to MCT. Although the permeability of STP was not improved by STP-NEs, the oral bioavailability has been substantially enhanced. The perfusion experiment reversely demonstrated the positive role of nanoemulsions in enhancing the oral absorption of STP.
 | Table 2 The effective intestinal permeability (Peff) of STP and STP-NEs evaluated by the in situ single-pass intestinal perfusion (n=3) |
Cytotoxicity
For pediatric medication, a major concern is the safety of preparations. The toxicity of nanoemulsions generally comes from the use of surfactants in large quantity. In this study, PM is the exclusive substance of surfactant, and thus a cytotoxic test was performed on the variable of PM concentration. Figure 8 shows the cell viability of Caco-2 cells after treatment with STP-NEs. There was no obvious cytotoxicity observed for STP-NEs at various concentrations of PM. The cell viability remained above 96% after 24 hours incubation, demonstrating the low cytotoxicity of STP-NEs. Emulsions are a favorite dosage form that children like to take. The low toxicity and popularity make STP-NEs rather applicable as a pediatric formulation.
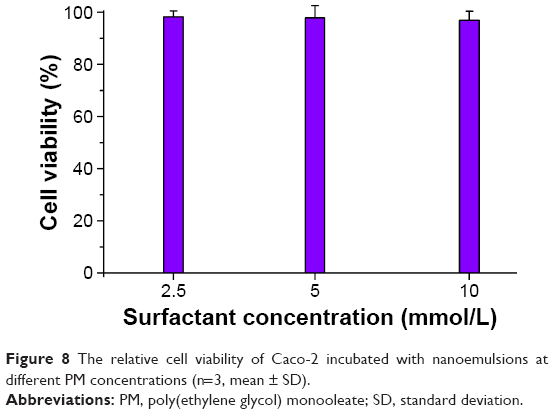 | Figure 8 The relative cell viability of Caco-2 incubated with nanoemulsions at different PM concentrations (n=3, mean ± SD). |
Conclusion
In this study, a novel nanoemulsions formulation based on the excipients of MCT and PM was developed for oral delivery of STP. The nanoemulsions prepared by solvent-diffusion/ultrasonic homogenization possessed a small particle size (<200 nm) and slow drug release, both in pH 6.8 buffer and 0.1 M HCl. STP-NEs exhibited good protection of STP from acidic destabilization and had acceptable physiochemical stability. The bioavailability of STP was significantly enhanced through the nanoemulsion system. Furthermore, the nanoemulsions were fairly safe owing to the use of PM, a low-toxic surfactant excipient. This article demonstrated the suitability of nanoemulsions as oral delivery carriers of STP.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81273532). The authors are thankful to the financial assistance from the Shandong Provincial Natural Science Foundation (ZR2011HQ028).
Disclosure
The authors report no conflicts of interest in this work.
References
Chiron C. Stiripentol. Expert Opin Investig Drugs. 2005;14(7): 905–911. | ||
Strzelczyk A, Kortland LM, Knake S, Rosenow F. Stiripentol for the treatment of super-refractory status epilepticus. Acta Neurol Scand. Epub 2015. | ||
Sada N, Lee S, Katsu T, Otsuki T, Inoue T. Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science. 2015; 347(6228):1362–1367. | ||
Zhang X, Wang H, Zhang T, Zhou X, Wu B. Exploring the potential of self-assembled mixed micelles in enhancing the stability and oral bioavailability of an acid-labile drug. Eur J Pharm Sci. 2014;62: 301–308. | ||
Zhang X, Sun N, Wu B, Lu Y, Guan T, Wu W. Physical characterization of lansoprazole/PVP solid dispersion prepared by fluid-bed coating technique. Powder Technol. 2008;182(3):480–485. | ||
Yun F, Kang A, Shan J, et al. Preparation of osthole-polymer solid dispersions by hot-melt extrusion for dissolution and bioavailability enhancement. Int J Pharm. 2014;465(1–2):436–443. | ||
Zhang X, Wu D, Lai J, Lu Y, Yin Z, Wu W. Piroxicam/2-hydroxypropyl-beta-cyclodextrin inclusion complex prepared by a new fluid-bed coating technique. J Pharm Sci. 2009;98(2):665–675. | ||
Liu H, Yang G, Tang Y, et al. Physicochemical characterization and pharmacokinetics evaluation of beta-caryophyllene/beta-cyclodextrin inclusion complex. Int J Pharm. 2013;450(1–2):304–310. | ||
Tian Z, Yi Y, Yuan H, et al. Solidification of nanostructured lipid carriers (NLCs) onto pellets by fluid-bed coating: Preparation, in vitro characterization and bioavailability in dogs. Powder Technol. 2013;247: 120–127. | ||
Lei Y, Qi J, Nie S, et al. Solid self-nanoemulsifying cyclosporine A pellets prepared by fluid-bed coating: stability and bioavailability study. J Biomed Nanotechnol. 2012;8(3):515–521. | ||
Bachar M, Mandelbaum A, Portnaya I, et al. Development and characterization of a novel drug nanocarrier for oral delivery, based on self-assembled beta-casein micelles. J Control Release. 2012; 160(2):164–171. | ||
Yuan H, Chen CY, Chai GH, Du YZ, Hu FQ. Improved transport and absorption through gastrointestinal tract by PEGylated solid lipid nanoparticles. Mol Pharm. 2013;10(5):1865–1873. | ||
Zhang X, Chen G, Zhang T, Ma Z, Wu B. Effects of PEGylated lipid nanoparticles on the oral absorption of one BCS II drug: a mechanistic investigation. Int J Nanomedicine. 2014;9:5503–5514. | ||
Xu W, Ling P, Zhang T. Polymeric micelles, a promising drug delivery system to enhance bioavailability of poorly water-soluble drugs. J Drug Deliv. 2013;2013:1–15. | ||
Malathi S, Nandhakumar P, Pandiyan V, Webster TJ, Balasubramanian S. Novel PLGA-based nanoparticles for the oral delivery of insulin. Int J Nanomedicine. 2015;10:2207–2218. | ||
Tariq M, Alam MA, Singh AT, Iqbal Z, Panda AK, Talegaonkar S. Biodegradable polymeric nanoparticles for oral delivery of epirubicin: in vitro, ex vivo, and in vivo investigations. Colloids Surf B Biointerfaces. 2015;128:448–456. | ||
Niu M, Lu Y, Hovgaard L, Wu W. Liposomes containing glycocholate as potential oral insulin delivery systems: preparation, in vitro characterization, and improved protection against enzymatic degradation. Int J Nanomedicine. 2011;6:1155–1166. | ||
Zhang X, Qi J, Lu Y, He W, Li X, Wu W. Biotinylated liposomes as potential carriers for the oral delivery of insulin. Nanomedicine. 2014; 10(1):167–176. | ||
Porter CJ, Kaukonen AM, Boyd BJ, Edwards GA, Charman WN. Susceptibility to lipase-mediated digestion reduces the oral bioavailability of danazol after administration as a medium-chain lipid-based microemulsion formulation. Pharm Res. 2004;21(8):1405–1412. | ||
Feeney OM, Williams HD, Pouton CW, Porter CJ. ‘Stealth’ lipid-based formulations: Poly(ethylene glycol)-mediated digestion inhibition improves oral bioavailability of a model poorly water soluble drug. J Control Release. 2014;192:219–227. | ||
Zhang X, Wang H, Ma Z, Wu B. Effects of pharmaceutical PEGylation on drug metabolism and its clinical concerns. Expert Opin Drug Metab Toxicol. 2014;10(12):1691–1702. | ||
Zhou X, Zhang X, Ye Y, et al. Nanostructured lipid carriers used for oral delivery of oridonin: an effect of ligand modification on absorption. Int J Pharm. 2015;479(2):391–398. | ||
Nozawa T, Imai T. Prediction of human intestinal absorption of the prodrug temocapril by in situ single-pass perfusion using rat intestine with modified hydrolase activity. Drug Metab Dispos. 2011;39(7): 1263–1269. | ||
Zhang X, Zhang T, Zhou X, et al. Enhancement of oral bioavailability of tripterine through lipid nanospheres: preparation, characterization, and absorption evaluation. J Pharm Sci. 2014;103(6):1711–1719. | ||
He C, Yin L, Tang C, Yin C. Size-dependent absorption mechanism of polymeric nanoparticles for oral delivery of protein drugs. Biomaterials. 2012;33(33):8569–8578. | ||
Gaumet M, Gurny R, Delie F. Localization and quantification of biodegradable particles in an intestinal cell model: The influence of particle size. Eur J Pharm Sci. 2009;36(4–5):465–473. | ||
Sun D, Wei X, Xue X, et al. Enhanced oral absorption and therapeutic effect of acetylpuerarin based on D-alpha-tocopheryl polyethylene glycol 1000 succinate nanoemulsions. Int J Nanomedicine. 2014;9: 3413–3423. | ||
Zhao L, Wei Y, Fu J, Huang Y, He B, Zhou Y. Nanoemulsion improves the oral bioavailability of baicalin in rats: in vitro and in vivo evaluation. Int J Nanomedicine. 2013;8:3769–3779. | ||
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. | ||
He YL, Murby S, Warhurst G, et al. Species differences in size discrimination in the paracellular pathway reflected by oral bioavailability of poly(ethylene glycol) and D-peptides. J Pharm Sci. 1998;87(5): 626–633. | ||
Qian C, Decker EA, Xiao H, McClements DJ. Nanoemulsion delivery systems: influence of carrier oil on beta-carotene bioaccessibility. Food Chem. 2012;135(3):1440–1447. | ||
Trevaskis NL, Charman WN, Porter CJ. Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Del Rev. 2008;60(6):702–716. | ||
You X, Xing Q, Tuo J, Song W, Zeng Y, Hu H. Optimizing surfactant content to improve oral bioavailability of ibuprofen in microemulsions: just enough or more than enough? Int J Pharm. 2014; 471(1–2):276–284. | ||
Lindmark T, Kimura Y, Artursson P. Absorption enhancement through intracellular regulation of tight junction permeability by medium chain fatty acids in Caco-2 cells. J Pharmacol Exp Ther. 1998; 284(1):362–369. | ||
Zakeri-Milani P, Valizadeh H, Tajerzadeh H, et al. Predicting human intestinal permeability using single-pass intestinal perfusion in rat. J Pharm Pharm Sci. 2007;10(3):368–379. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.