Back to Journals » OncoTargets and Therapy » Volume 7
Myelodysplastic disorders carrying both isolated del(5q) and JAK2V617F mutation: concise review, with focus on lenalidomide therapy
Authors Musto P, Simeon V, Guariglia R, Bianchino G, Grieco V, Nozza M, La Rocca F, Marziano G, Lalinga AV, Fabiani E, Voso MT, Scaravaglio P, Mecucci C, D'Arena G ![]()
Received 23 December 2013
Accepted for publication 28 January 2014
Published 13 June 2014 Volume 2014:7 Pages 1043—1050
DOI https://doi.org/10.2147/OTT.S59628
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Pellegrino Musto,1 Vittorio Simeon,2 Roberto Guariglia,3 Gabriella Bianchino,4 Vitina Grieco,4 Filomena Nozza,4 Francesco La Rocca,2 Gioacchino Marziano,1 Anna Vittoria Lalinga,5 Emiliano Fabiani,6 Maria Teresa Voso,6 Patrizia Scaravaglio,7 Cristina Mecucci,8 Giovanni D'Arena3
1Scientific Direction, 2Laboratory of Preclinical and Translational Research, 3Unit of Hematology and Stem Cell Transplantation, 4Laboratory of Clinical Research and Advanced Diagnostics, 5Pathology Unit, IRCCS, Centro di Riferimento Oncologico della Basilicata, Rionero in Vulture, Italy; 6Department of Hematology, Universita Cattolica del Sacro Cuore, Rome, Italy; 7Laboratory of Internal Medicine and Hematology, S Luigi Gonzaga Hospital, Orbassano, Italy; 8Hematology and Bone Marrow Transplantation Unit, University of Perugia, Perugia, Italy
Abstract: The concomitant presence of del(5q) and JAK2V617F mutation is an infrequent event which occurs in rare patients with peculiar cytogenetic, molecular, morphological and clinical features, resembling those of both myelodysplastic syndromes and myeloproliferative neoplasms. Lenalidomide may induce rapid, profound, and long-lasting responses in a subset of these patients. However, the mechanism(s) by which the drug acts in these conditions remain not completely elucidated. A new case report and a review of all cases published so far in this setting are provided. Furthermore, the possibility of categorizing – from a clinical, pathological, and biological point of view – for at least some of these patients as a potential distinct entity is discussed.
Keywords: myelodysplastic syndromes, myeloproliferative neoplasms, lenalidomide, del(5q), JAK2, World Health Organization
Introduction
The interstitial deletion of the long arm of chromosome 5 – del(5q) – is the most frequent cytogenetic abnormality occurring in myelodysplastic syndromes (MDS).1 It may be present as the sole karyotype aberrancy (sometimes within a clinical picture defined as “5q– syndrome” and characterized by a favorable prognosis) or variously combined with other single or multiple chromosomal alterations.2 Complex karyotypes including del(5q), especially if associated with the presence of marrow blasts, confer to MDS patients a poor prognosis.2,3 From a therapeutic point of view, lenalidomide induces high rates of hematological and cytogenetic responses in anemic, low/intermediate- 1-risk MDS patients with isolated del(5q).4
JAK2 is involved in cellular growth factor signaling, and deregulation of JAK2 by chromosomal aberrations may contribute to leukemogenesis. JAK2V617F mutation, an activating point mutation in the JH2 domain, is mostly seen in BCR/ABL1-negative myeloproliferative neoplasms (MPN), with a percentage of positive cases that is about 95% in polycythemia vera (PV) and 40%–60% in essential thrombocythemia (ET) and primary myelofibrosis (PMF).5 Among other myeloid neoplasms, it occurs with high frequency in refractory anemia with ring sideroblasts (RARS) associated with marked thrombocytosis – a provisional, mixed MDS/MPN disorder which frequently harbors the splicing machinery-associated SF3B1 mutation.6,7 JAK2V617F mutation is instead an infrequent event in other “atypical” MPN or in MDS.8 Lenalidomide has also shown activity in del(5q) negative myelofibrosis (MF) with JAK2V617F mutation, determining significant clinical responses and reduction of JAK2V617F allele burden in these patients.9,10
Along with RARS associated with marked thrombocytosis, the 2008 World Health Organization’s proposed revision of MDS classification also recognized other cases showing overlaps in morphological, clinical, laboratory, and genetic aspects; in particular, rare MDS with concomitant JAK2V617F mutation and isolated del(5q) were identified.6
Presented here is a brief review on this last topic, starting with the description of a paradigmatic patient who experienced a complete and long-lasting hematological, cytogenetic, and molecular response to lenalidomide.
Case presentation
In February 2010, an 84-year-old man with a previous history of acute myocardial infarctions and peptic ulcer was diagnosed as having both MDS with del(5q) and JAK2V617F-positive ET in another hospital. Because of relevant thrombocytosis (1,285×109/L), anemia (hemoglobin 80 g/L), and erythromelalgia of the lower limbs, he was initially given hydroxycarbamide (1 g daily orally) plus aspirin and packed red cell transfusions. Due to a progressive increase of platelet count and worsening of anemia, hydroxycarbamide was substituted by pipobroman (50 mg daily orally).
In September 2011, the patient was admitted to the authors’ institute. At entry, hemoglobin level was 66 g/L, mean corpuscular volume 100 fL, platelet count 1,021×10/L, and white blood cell count 4.6×109/L with normal differential. Uric acid was 9.4 mg/dL and a “cystic complex” was found in the right kidney by ultrasound and computed tomography scan. Physical examination did not reveal enlargement of liver or spleen. The bone marrow aspirate was found moderately hypercellular and containing numerous atypical megakaryocytes, mainly characterized by hypolobated nuclei (Figure 1A and B). Blast cells were <1% of global cellularity. No ring sideroblasts were evidenced by Perls’ staining. Histological bone marrow examination confirmed a marked hyperplasia of morphologically abnormal megakaryocytes, which were similar to those observed in the “5q– syndrome”, with slightly reduced erythropoiesis and granulopoiesis, blast cells <5%, and a weak reticulin network (Figure 1D–F).
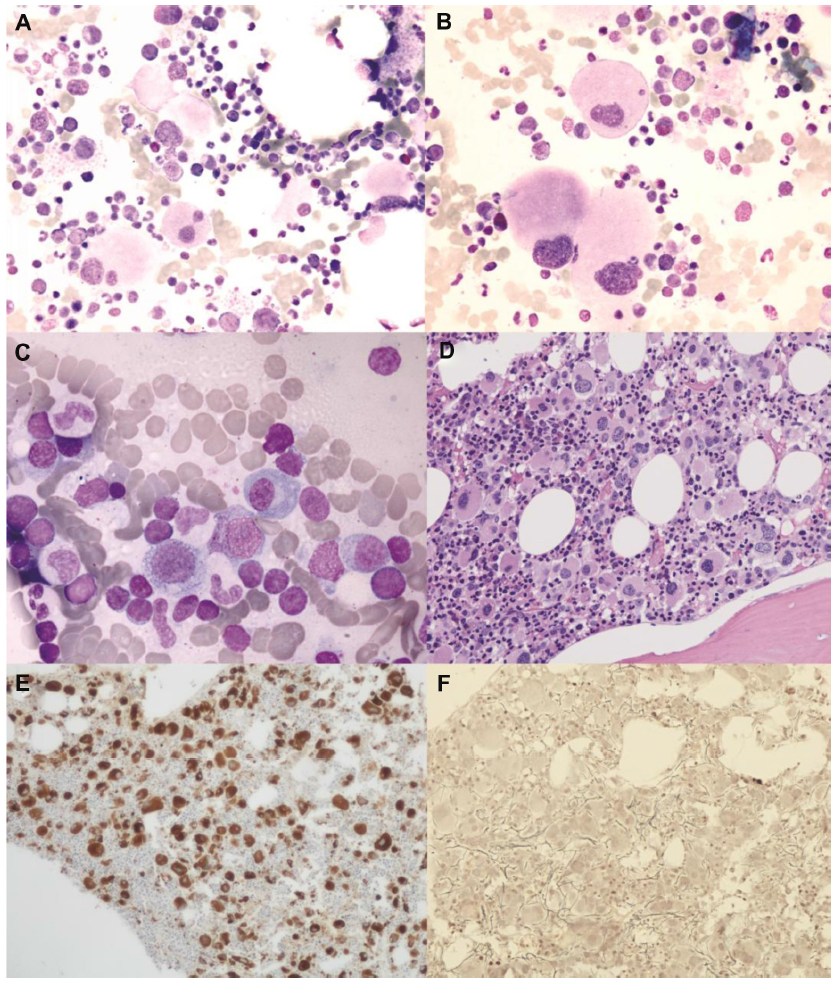 | Figure 1 (A and B) Light microscopy examination of marrow aspirate at baseline (May Grünwald–Giemsa) showing atypical, hypolobated megakaryocytes (A: original magnification ×50; B: original magnification ×100). (C) After 6 months of lenalidomide therapy, abnormal megakaryocytes disappeared and a scattered lymphocyte infiltration was seen (original magnification ×100). (D–F) Bone marrow trephine image at baseline demonstrating increased cellularity with numerous atypical hypolobated megakaryocytes (D: original magnification ×20, hematoxylin–eosin; E: original magnification ×10, immunoperoxidase staining for von Willebrand factor). Only a few reticulin fibers were present (F: original magnification ×20, reticulin staining). |
Both conventional cytogenetic and fluorescence in situ hybridization confirmed the presence of del(5q)(q13q31) as the unique chromosomal abnormality in 25% of metaphases and 50% of analyzed cells, respectively. At molecular level, BCR/ABL1 rearrangement was negative, while the JAK2V617F mutation was identified by quantitative reverse transcriptase polymerase chain reaction (26.28%). SF3B1 (exons 13, 14, 15, and 16), as well as SRSF2 (P95-), U2AF1 (S34), IDH1 (R132−), IDH2 (R140− and R172−), and DNAMT3A (R882−) genes were analyzed by Sanger sequencing (kinetic 3.1), as previously reported:11 all these genes were found to be not mutated.
According to the World Health Organization, a diagnosis of MDS with isolated del(5q) and JAK2V617F mutation was made. The patient initially received packed red cell transfusions and a treatment with anagrelide (500 mg/day); this drug, however, was poorly tolerated.
In October 2011, the patient started lenalidomide 10 mg daily orally for 21 days every 28 days on the basis of a national, named-patient program (data not shown). The drug was well tolerated and platelet count declined rapidly, while hemoglobin levels were raised just after the first month of therapy (hemoglobin 106 g/L and platelet count 231×109/L); a substantially normal whole blood cell count, including disappearance of macrocytosis, was reached 3 months later (Figure 2). Erythromelalgia of the lower limbs also disappeared during the first weeks of therapy.
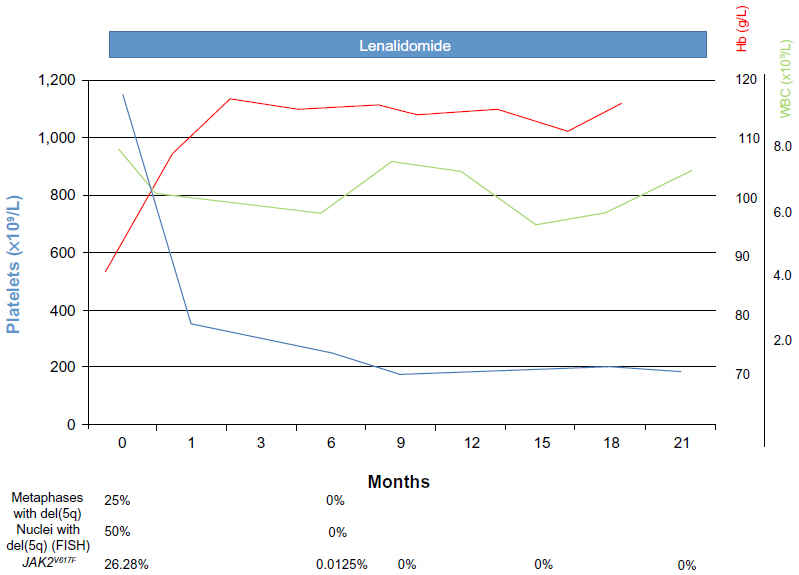 | Figure 2 Modifications of Hb, WBC, and platelet levels under lenalidomide therapy. Changes in del(5q) metaphases/nuclei and JAK2V617F allele burden, when available, are also indicated. |
After 6 months of lenalidomide, a complete morphological, cytogenetic, and molecular re-evaluation showed the absence of del(5q) in both cytogenetic and fluorescence in situ hybridization analysis, as well as a remarkable reduction of JAK2V617F mutation by quantitative polymerase chain reaction (0.0215%) (Figure 2). Sequential bone marrow aspirates documented progressive disappearance of atypical megakaryocytes, decrease of cellularity, and the scattered presence of focal polyclonal lymphocyte infiltration (Figure 1C). During the following months, hematological values remained substantially normal, while monitoring of JAK2V617F mutation evidenced a complete molecular remission (allele burden 0%) in two subsequent controls. At the last follow-up (August 2013), after about 2 years of lenalidomide therapy, hematological values were stable and JAK2V617F mutation undetectable.
Review
Concomitant presence of del(5q) and JAK2V617F mutation: incidence and origin
Looking to the scarce available literature, JAK2V617F mutation was first found in six MDS patients with isolated del(5q) – five with refractory cytopenia with multilineage dysplasia and one with RARS among a total of 97 MDS patients (6.2%), sixteen of whom had additional chromosomal abnormalities.12 JAK2V617F mutant cases showed a trend toward a higher platelet (3/6 >700×109/L) and white blood cell count, as well as a moderate to marked increase in bone marrow cellularity. No response to recombinant human erythropoietin was observed in two of these patients with red cell transfusion-dependent disease, while hydroxycarbamide or anagrelide were employed in two additional patients with elevated thrombocytosis.
In another series from the Mayo Clinic, JAK2V617F mutation was detected in five out of 78 (6.4%) MDS patients carrying isolated del(5q); allele burden ranged from 1%–10% and no significant relevance on clinical phenotype, blood counts, or prognosis was found.13 Interestingly, one of these patients also showed the presence of MPLW55L mutation. In a subsequent study, the same group found that the incidence of JAK2V617F by mutation screening was 14.5% in 138 patients with isolated del(5q), including higher risk MDS variants and some cases of acute myeloid leukemia or MPN.14 In one patient with PV, an additional mutation of JAK2 exon 12 was found. In this study, leukocytosis, higher blast count, and IDH mutation – but not JAK2V617F mutation – negatively affected the prognosis of patients with myeloid malignancies carrying isolated del(5q).
More recently, using a highly sensitive molecular method, Geyer et al found evidence of heterozygous JAK2V617F mutation in four out of 17 (23.5%) newly diagnosed MDS patients with del(5q).15 One of these patients had del(5q) as single anomaly (“5q– syndrome”). In another one with refractory cytopenia with multilineage dysplasia it was associated with trisomy 8. The remaining two patients (one with refractory anemia with blast excess-type 2 and one with therapy-related MDS) showed del(5q) or monosomy 5, respectively, within the context of a complex karyotype. Of note, marrow proliferative features were seen only in the first two patients.
Among MPN, del(5q) can be only rarely found in MF. A retrospective review of 939 cases identified only eight patients (0.8%) who carried del(5q), three of whom also showed JAK2V617F mutation.16 All three of these patients (one post-ET MF, one post-PV MF, and one PMF) had a complex karyotype and two of them had an excellent response to anti-JAK2 inhibitors. A literature review indicated that isolated del(5q) is also uncommon in JAK2V617F-positive ET.17 In these cases, the disease often shows mixed features of both ET and “5q– syndrome”. Interestingly, in a retrospective analysis performed within a single center in Hong Kong, the occurrence of JAK2V617F was found particularly high (three out of eight patients, 37.5%) in the Chinese female MDS population with isolated del(5q), thus suggesting a possible ethnic effect.18 Two of these patients had “5q– syndrome” and one had refractory cytopenia with multilineage dysplasia.
From a biological point of view, Ingram et al investigated a MDS patient with JAK2V617F mutation and isolated del(5q) by processing progenitor cell cultures (granulocyte/monocyte colony-forming units and erythropoietin-independent erythroid burst-forming units) as well as fractionated CD34+ progenitor cells, using cytogenetic and molecular approaches.12 Although the results of this report raise the potential that some CD34+ cells may harbor both genetic defects, genetic derivation of the clones was not investigated. Indeed, although these authors evidenced that JAK2V617F mutation may be an early event in these patients arising in a pluripotent stem cell and that del(5q) may be present in the same CD34+ progenitor cells, they also noticed that not all del(5q) cells are necessarily JAK2V617F mutant.
More recently, Sokol et al investigated the clonal origin of JAK2V617F mutation in a patient with del(5q) MDS presenting with thrombocytosis and normal hemoglobin.19 These authors detected the presence of both genetic abnormalities del(5q) and JAK2V617F early in the course of disease, before the development of anemia. Furthermore, although cytogenetic analysis revealed the del(5q) in 15%–30% of bone marrow metaphases, fluorescence in situ hybridization analysis of progenitor colonies identified no clonogenic cells harboring the cytogenetic abnormality despite demonstration of the JAK2V617F mutation. Interestingly, in this study JAK2V617F mutation was detected in at least two distinct multipotent stem cell progeny that lacked del(5q), suggesting that del(5q) and JAK2V617F existed in progenitors derived from genetically independent clones. The evidence of JAK2V617F clones lacking del(5q) implies independent acquisition of the genetic abnormalities in discordant clones; however, this does not completely exclude the possibility that both del(5q) and JAK2V617F clones may emerge as progeny of a genetically unstable hierarchal stem cell.
Effects of lenalidomide in patients with concomitant presence of del(5q) and JAK2V617F mutation
Lenalidomide is a very effective treatment for lower risk MDS with isolated del(5q). However, the efficacy of lenalidomide as a single agent in MDS with isolated del(5q) and JAK2V617F mutation has been described in only two patients so far (Table 1).20,21 Interestingly, in addition to good response to lenalidomide, the posted two cases and current case also shared morphological and clinical features, such as macrocytic anemia, remarkable thrombocytosis, no splenomegaly, hypercellular marrow with increased number of hypolobated megakaryocytes, no evidence of relevant fibrosis or ring sideroblasts, and a favorable outcome.
 | Table 1 Summary of published cases of hematological disorders with concomitant presence of del(5q) and JAK2V617F mutation treated with lenalidomide |
Regarding patients with other types of diagnosis (Table 1), a transient, near complete hematological response and a complete cytogenetic remission after lenalidomide monotherapy was observed in an elderly female patient with minimally differentiated acute myeloid leukemia arising from a JAK2V617F-positive, del(5)(q13q33) MDS.22 The treatment was administered for 5 months and was then followed by progression with clonal evolution. Lenalidomide therapy also induced cytogenetic and molecular remissions in three patients with JAK2V617F-positive MF (two with isolated del[5q] and one with chromosome 5 monosomy plus other cytogenetic abnormalities).23 Two of these patients had post-MPN MF and one had PMF. Marked improvement of anemia was seen in these subjects, one of whom had been already reported,9 while splenomegaly significantly reduced when present. One patient experienced a complete cytogenetic remission and two experienced a partial cytogenetic remission. Bone marrow JAK2V617F allele burden decreased in two patients. Remission was maintained after >1 year of treatment in two cases, while the third subject developed a picture of acute erythroleukemia which did not respond to retreatment with lenalidomide. Finally, one patient with post-PV MF, JAK2V617F mutation, and del(5q) in the context of a complex karyotype, did not respond at all to lenalidomide but had a dramatic improvement of cytopenia and splenomegaly after administration of CEP-701 anti-JAK2 inhibitor.16 Interestingly, complete hematological response, disappearance of constitutional symptoms and improvement in marrow fibrosis was observed after lenalidomide therapy in a 54-year-old female patient with post-ET MF carrying a double cytogenetic abnormality (del[5q] and t[1;12]) associated with MPL mutation but not JAK2V617F mutation. The response was longer than 3 years, although the patient did not achieve cytogenetic response and del(5q) abnormality was persistently detected in serial bone marrow analyses.16
Conclusion
The simultaneous presence of del(5q) and JAK2V617F mutation is a rare event which occurs prevalently in patients with a possibly “mixed” and still not well-defined MDS/MPN disorder. However, it would seem possible to identify some cases characterized by peculiar cytogenetic, molecular, morphological, and clinical findings (Table 2) and a relatively favorable prognosis (the first three patients of Table 1). The mechanism(s) by which lenalidomide seems to be particularly able to induce rapid and long-lasting responses in these conditions are still not completely understood. As in other neoplastic disorders, lenalidomide could suppress the malignant clone(s) by a direct cytotoxic effect or by an indirect immunomodulatory activity. According to the hypothesis of two genetic aberrations overlapping in the same clone,12 the efficacy of lenalidomide in suppressing the del(5q) clone would induce a parallel reduction of JAK2V617F allele burden. Alternatively, in the case of an independent acquisition of the two genetic abnormalities,19 a different activity of lenalidomide on the JAK2V617F mutated clone should be hypothesized as probably it occurs in patients with JAK2V617F-positive MF9,10 or RARS associated with marked thrombocytosis24,25 who may show variable clinical and molecular responses to lenalidomide but are not harboring del(5q). Both these mechanisms, however, could be not mutually exclusive.
 | Table 2 Main characteristics of myelodysplastic disorders with concomitant del(5q) and JAK2V617F mutation |
It is probably too early to categorize MDS with concomitant evidence of del(5q) and JAK2V617F mutation as a distinct entity and more work is needed to draw definitive conclusions. However, combined cytogenetic and molecular screening of these markers in patients who clinically correspond to those already described should be recommended in order to improve clinical management and biological knowledge in this field.
Acknowledgments
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Author contributions
PM and GD wrote the paper, followed the patient and analyzed the literature. VS, GM, and FLR collected and reviewed literature data. RG clinically followed the patient. GB, VG, EF, and MTV performed molecular studies. FN, PS, and CM performed cytogenetic studies. AVL performed histological studies. All authors gave substantial contributions to conception and design, took part in drafting the article or revising it critically for important intellectual content, gave final approval of the version to be published; and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved.
Disclosure
PM and MTV received honoraria from Celgene, the manufacturer of Revlimid (lenalidomide). CM received research funds from Celgene. The other authors report no conflicts of interest in this work.
References
Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30(8):820–829. | |
Mallo M, Cervera J, Schanz J, et al. Impact of adjunct cytogenetic abnormalities for prognostic stratification in patients with myelodysplastic syndrome and deletion 5q. Leukemia. 2011;25(1):110–120. | |
Germing U, Lauseker M, Hildebrandt B, et al. Survival, prognostic factors and rates of leukemic transformation in 381 untreated patients with MDS and del(5q): a multicenter study. Leukemia. 2012;26(6):1286–1292. | |
Fenaux P, Giagounidis A, Selleslag D, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with low-/intermediate-1-risk myelodysplastic syndromes with del5q. Blood. 2011;118(14):3765–3776. | |
Kiladjian JJ. The spectrum of JAK2-positive myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2012;2012:561–566. | |
Vardiman JW, Bennet JM, Bain BJ, Baumann I, Thiele J, Orazi A. Myelodysplastic/myeloproliferative neoplasms, unclassifiable. In: Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: International Agency of Research on Cancer; 2008:85–86. | |
Broséus J, Alpermann T, Wulfert M, et al. Age, JAK2(V617F) and SF3B1 mutations are the main predicting factors for survival in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia. 2013;27(9):1826–1831. | |
Steensma DP, Dewald GW, Lasho TL, et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood. 2005;106(4):1207–1209. | |
Tefferi A, Cortes J, Verstovsek S, et al. Lenalidomide therapy in myelofibrosis with myeloid metaplasia. Blood. 2006;108(4):1158–1164. | |
Quintás-Cardama A, Kantarjian HM, Manshouri T, et al. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol. 2009;27(28):4760–4766. | |
Voso MT, Fabiani E, Fianchi L, et al. Mutations of epigenetic regulators and of the spliceosome machinery in therapy-related myeloid neoplasms and in acute leukemias evolved from chronic myeloproliferative diseases. Leukemia. 2013;27(4):982–985. | |
Ingram W, Lea NC, Cervera J, et al. The JAK2 V617F mutation identifies a subgroup of MDS patients with isolated deletion 5q and a proliferative bone marrow. Leukemia. 2006;20(7):1319–1321. | |
Patnaik MM, Lasho TL, Finke CM, et al. WHO-defined “myelodysplastic syndrome with isolated del(5q)” in 88 consecutive patients: survival data, leukemic transformation rates and prevalence of JAK2, MPL and IDH mutations. Leukemia. 2010;24(7):1283–1289. | |
Patnaik MM, Lasho TL, Finke CM, et al. Isolated del(5q) in myeloid malignancies: clinicopathologic and molecular features in 143 consecutive patients. Am J Hematol. 2011;86(5):393–398. | |
Geyer JT, Verma S, Mathew S, et al. Bone marrow morphology predicts additional chromosomal abnormalities in patients with myelodysplastic syndrome with del(5q). Hum Pathol. 2013;44(3):346–356. | |
Takahashi K, Cortes J, Pierce S, Abruzzo L, Kantarjian H, Verstovsek S. Chromosome 5q deletion is extremely rare in patients with myelofibrosis. Leuk Res. 2013;37(5):552–555. | |
Wong KF, Yu PH, Wong WS. Essential thrombocythemia with deleted 5q – a genetic and morphologic hybrid? Cancer Genet Cytogenet. 2010;201(1):39–41. | |
Wong KF, Wong WS, Siu LL, Lau TC, Chan NP. JAK2 V617F mutation is associated with 5q- syndrome in Chinese. Leuk Lymphoma. 2009;50(8):1333–1335. | |
Sokol L, Caceres G, Rocha K, Stockero KJ, Dewald DW, List AF. JAK2(V617F) mutation in myelodysplastic syndrome (MDS) with del(5q) arises in genetically discordant clones. Leuk Res. 2010;34(6):821–823. | |
Azaceta G, Calasanz MJ, Dourdil V, Bonafonte E, Izquierdo I, Palomera L. Response to lenalidomide in a patient with myelodysplastic syndrome with isolated del(5q) and JAK2 V617F mutation. Leuk Lymphoma. 2010;51(10):1941–1943. | |
Nomdedeu M, Maffioli M, Calvo X, et al. Efficacy of lenalidomide in a patient with myelodysplastic syndrome with isolated del(5q) and JAK2V617F mutation. Leuk Res. 2011;35(9):1276–1278. | |
Mesa RA, Tefferi A, Li CY, Steensma DP. Hematologic and cytogenetic response to lenalidomide monotherapy in acute myeloid leukemia arising from JAK2(V617F) positive, del(5)(q13q33) myelodysplastic syndrome. Leukemia. 2006;20(11):2063–2064. | |
Tefferi A, Lasho TL, Mesa RA, Pardanani A, Ketterling RP, Hanson CA. Lenalidomide therapy in del(5)(q31)-associated myelofibrosis: cytogenetic and JAK2V617F molecular remissions. Leukemia. 2007;21(8):1827–1828. | |
Zhang M, You Y, Li X, et al. Response to lenalidomide of a patient with t(2;3)(p23;q29) and JAK2 non-mutated refractory anemia with ring sideroblasts and thrombocytosis. Leuk Lymphoma. 2013;54(7):1544–1546. | |
Huls G, Mulder AB, Rosati S, van de Loosdrecht AA, Vellenga E, de Wolf JT. Efficacy of single-agent lenalidomide in patients with JAK2 (V617F) mutated refractory anemia with ring sideroblasts and thrombocytosis. Blood. 2010;116(2):180–182. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.