Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 5
MYC as therapeutic target in leukemia and lymphoma
Authors Cortiguera MG, Batlle-López A, Albajar M, Delgado MD, León J
Received 30 March 2015
Accepted for publication 12 May 2015
Published 22 July 2015 Volume 2015:5 Pages 75—91
DOI https://doi.org/10.2147/BLCTT.S60495
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor David Dingli
Maria G Cortiguera,1 Ana Batlle-López,1,2 Marta Albajar,1,2 M Dolores Delgado,1,3 Javier León1,3
1Institute of Biomedicine and Biotechnology of Cantabria (IBBTEC), CSIC-University of Cantabria, 2Department of Hemathology, Hospital Universitario Marqués de Valdecilla, 3Department of Molecular Biology, University of Cantabria, Santander, Spain
Abstract: MYC is a transcription factor that is involved in the expression of many genes. Deregulated MYC is found in about half of human tumors, being more prevalent in hematological neoplasms. Deregulation mechanisms include chromosomal translocation (particularly in lymphoma), amplification, and hyperactivation of MYC transcription. Here we review MYC involvement in the major types of leukemia and lymphoma. MYC rearrangements appear in all Burkitt lymphomas and are common in other lymphoma types, whereas in acute lymphoblastic leukemia, acute myeloid leukemia, lymphoproliferative, and myeloproferative diseases, they are less frequent. However, MYC overexpression is present in all types of hematological malignancies and often correlates with a worse prognosis. Data in leukemia-derived cells and in animal models of lymphomagenesis and leukemogenesis suggest that MYC would be a good therapeutic target. Several MYC-directed therapies have been assayed in preclinical settings and even in clinical trials. First, peptides and small molecules that interrupt the MYC–MAX interaction impair MYC-mediated tumorogenesis in several mouse models of solid tumors, although not yet in lymphoma and leukemia models. Second, there are a number of small molecules inhibiting the interaction of MYC–MAX heterodimers with DNA, still in the preclinical research phase. Third, inhibitors of MYC expression via the inhibition of BRD4 (a reader of acetylated histones) have been shown to control the growth of MYC-transformed leukemia and lymphoma cells and are being used in clinic trials. Finally, we review a number of promising MYC-mediated synthetic lethal approaches that are under study and have been tested in hematopoietic neoplasms.
Keywords: MYC, targeted therapy, leukemia, lymphoma
Introduction
Hematological neoplasms are the result of the malignant transformation of a hematopoietic cell at a specific stage of differentiation. Morphologic characterization has traditionally been the gold standard technique for the identification of the different types, but improvement on the molecular methodologies revealed that those subtypes are composed of many different molecular subtypes. In fact, many malignancies are nowadays classified based on a specific genetic abnormality or a transcriptional signature.1 There are, however, other nonspecific genetic aberrations, associated with particular biological and clinical implications. Such is the case of MYC, which is deregulated in many different subtypes of lymphoma and leukemia, sometimes as a primary event (eg, Burkitt lymphoma [BL]) or as a secondary event that usually implies aggressiveness and poor prognosis.
MYC (also called c-Myc) is an oncogenic transcription factor of the helix-loop-helix-leucine zipper family. MYC is deregulated in half of the human tumors, including leukemia and lymphoma.2–5 MYC forms dimers with MAX through the leucine zipper (LZ) domain. The MYC–MAX heterodimer is the active form which binds to specific DNA sequences (E-boxes, canonical sequence CACGTG) in the regulatory regions of target genes. The number of MYC-binding sites revealed by genome-wide technologies ranks between 7,000 and 15,000 in different models. Indeed, MYC is bound at one or more sites of the regulatory regions of 10%–15% of human genes and, as expected, there is a large number of MYC-regulated genes, reaching the staggering number of 1,000 genes in most models.6–8
The mechanism for MYC-mediated transactivation depends on the recruitment of complexes containing histone acetyltransferases.7,9 Recent work has shown that MYC is present at the promoter of nearly all active genes acting as an “amplifier” of the transcription intensity of genes already engaged in transcription.10 The mechanism is not well known, but the activating interaction of MYC with P-TEFb (positive transcription elongation factor b) likely plays an important role.11,12 However, the extent of MYC binding to chromatin depends on the level of MYC in the cell, and MYC overexpression provokes an “invasion” of new E-boxes, either in proximal promoters or at distal enhancers, so that a new set of genes are overexpressed. It is still unclear to what extent MYC contributes to the overexpression of these new “invaded” genes,13,14 but it is established that upon MYC induction or activation, the expression of a series of “MYC target genes” become overexpressed with respect to most other genes of the cell, whereas others (eg, the genes of cell cycle inhibitors CDKN1A and CDKN2B) are downregulated.14,15
In agreement with the large number of regulated genes, overexpression of MYC impinges on a series of functions that confer ample competitive advantages to the cell, such as cell cycle stimulation, nucleotide biosynthesis, differentiation impairment, energy production, protein synthesis and ribosome genesis, genomic instability, immortalization, and telomere maintenance or block of differentiation.3,7,8,15,16 All these combined functions contribute to – or trigger – the development of hematological neoplasia (Figure 1). Indeed, MYC oncogene was originally discovered as the oncogene carried by retroviruses that induced a myeloid neoplasm in chicken, ie, myelocytomatosis, and MYC was named after this tumor.17 Moreover, BL was the first human tumor where MYC deregulation was identified, due to a chromosomal translocation as described in “Neoplasms of mature B cells” section. In this review, we will first summarize MYC involvement in the major lymphoid and myeloid neoplasms and then the different approaches using MYC as a target in these neoplasms.
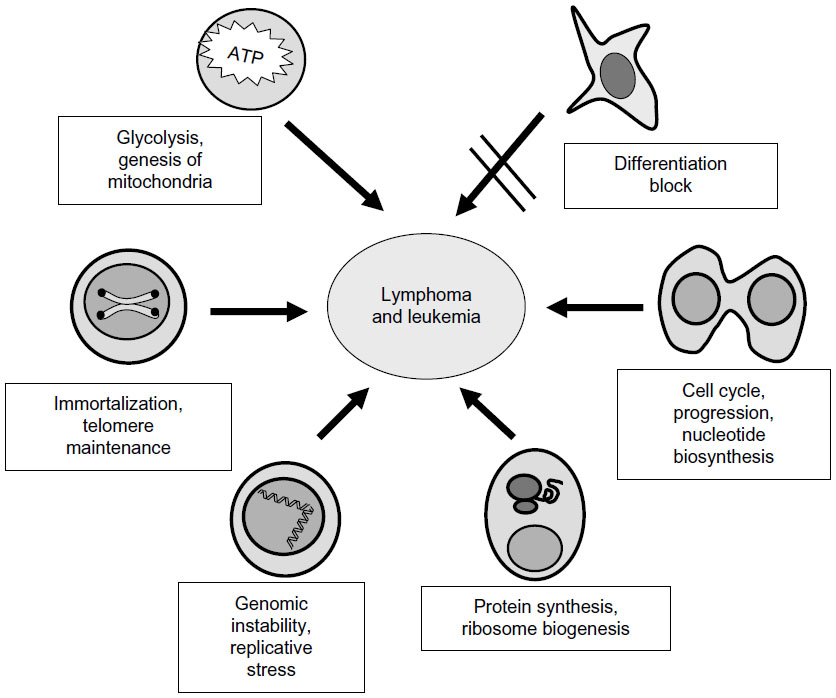 | Figure 1 Major biological activities elicited by deregulated MYC that contribute to the development and progression of leukemia and lymphoma. |
MYC and lymphoid neoplasms
The first animal model generated for MYC-driven cancer was the Eμ-Myc transgenic mouse, in which MYC expression is targeted to the lymphoid compartment by the immunoglobulin (Ig) heavy chain gene promoter and enhancer.18,19 The model demonstrated the ability of MYC to transform B cells in mice, although, as discussed in “Burkitt lymphoma” section, the tumors do not faithfully reproduce BL.
MYC plays key roles in different stages of the antigen- dependent B-cell differentiation process. On encounter with the antigen-dependent T-cell, the naïve B-cell moves to a follicle where it intensely proliferates to form a germinal center. MYC upregulation is essential to induce this migration, and indeed, MYC-deficient mice lack the ability to induce these germinal center reactions. Those lymphocytes that enter the dark zone of the germinal center start expressing BCL6, which in turn represses MYC expression. Once the somatic hypermutation (SHM) process is complete, those cells move to the light zone and are selected for the production of high affinity antibodies. Those B-cells that fail selection die by apoptosis, whereas those producing high affinity antibodies, will either return to the dark zone for a further round of SHM, which requires MYC expression, or exit the germinal center, either as an antibody producing cell (plasma cell) or as a memory B-cell (schematized in Figure 2). Germinal centers, therefore, contain highly proliferating B-cells that are undergoing mutations mediated by activation-induced deaminase (AID), and thus they might be predisposed to malignant transformation. In fact, a significant number of B-cell aggressive lymphomas emerge from these areas,20 and a considerable proportion of those will show MYC translocation. Interestingly, mice lacking AID do not develop IGH–MYC translocations.21 MYC involvement in lymphoid neoplasms is summarized in Table 1.
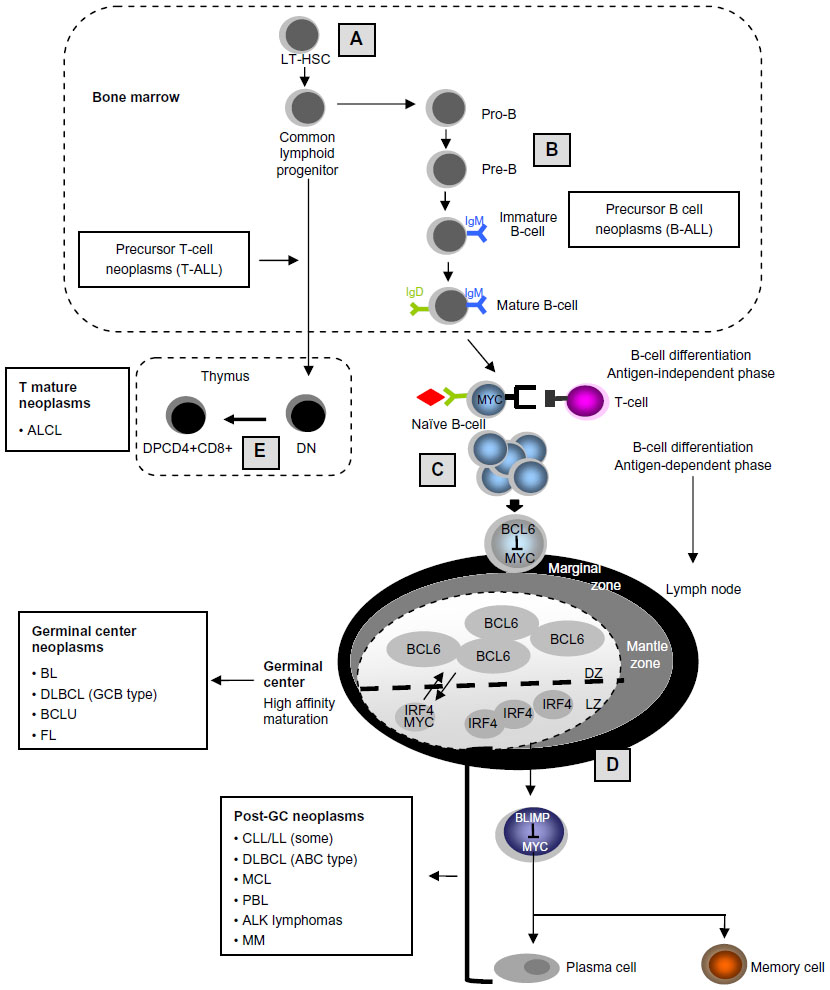 | Figure 2 MYC roles in lymphoid differentiation and in lymphoid neoplasms. |
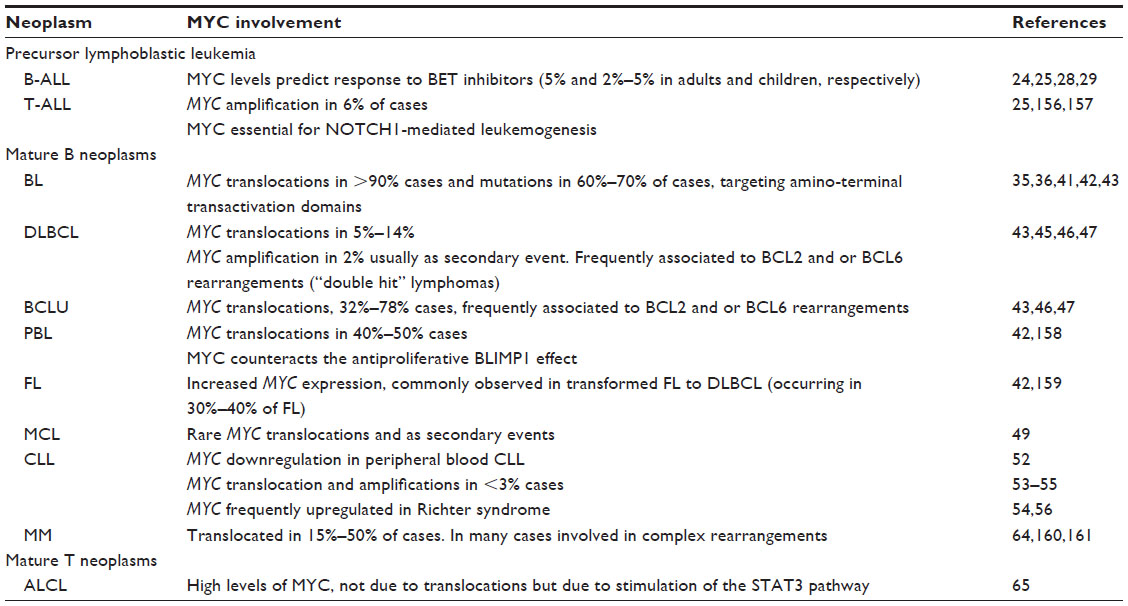 | Table 1 MYC in lymphoid neoplasms |
Neoplasms of lymphoid precursors
Neoplasms of T-cell precursors
Adult T-cell acute lymphoblastic leukemia (T-ALL) is associated with poor prognosis with standard chemotherapy-based regimens. MYC translocations are detected in 6% of T-ALLs, usually as secondary events, and associated with induction failure and relapse.22 Notch signaling pathway, which is deregulated in more than 50% of T-ALL, has been shown to directly upregulate MYC. Also, MYC binding to a NOTCH1-enhancer is required for NOTCH1-induced T-ALL.23 In vitro treatment of T-cell lines with valproic acid (a histone deacetylase inhibitor) led to downregulation of MYC in a dose-dependent manner and, specific inhibition of MYC function was shown to further increase cell death in those cell lines.24 Xenograft models have also demonstrated that MYC inhibition eliminates the leukemia-initiating cells (LICs) and inhibits growth of pediatric T-ALL cells.25 This effect might be more efficient when MYC inhibitors are used in combination with either chemotherapy regimens26 and/or with inhibitors of other pathways such as PI3K.27 Nevertheless, those combinations are yet to be assayed in specific subtypes.
Neoplasms of B-cell precursors
Pediatric B precursor acute lymphoblastic leukemia (ALL) is in most cases a curable disease with intensive chemotherapy. Still, 20%–30% of patients will undergo induction failure or relapse. Also, adult ALLs, usually associated with mixed lineage leukaemia (MLL) or BCR–ABL rearrangements, have a poor expectancy even with allogenic bone marrow transplantation. Approximately 2%–5% of ALLs show MYC rearrangements,28 and a number of B-cell acute lymphoblastic leukemia (B-ALL), while not having MYC gene abnormalities show high MYC expression.29 Cell cycle arrest is achieved using bromodomain and extra-terminal (BET) inhibitors in variety of MYC-expressing ALLs in vitro (see “Epigenetic-based MYC therapy: anti BRD4 drugs” section). Also, in vivo responses were observed in ALL xenograft models when BET inhibitors were used alone or in combination with dexamethasone.29
Neoplasms of mature B-cells
MYC deregulation may be observed in any type of mature lymphoid neoplasm, although it is more frequently observed in the aggressive lymphoma types. It may act as a driver abnormality such as in BL, but in many cases, appears as a secondary event, indicating clonal evolution and/progression. MYC is a frequent target of chromosomal translocations in lymphomas with different partners, the immunoglobulin heavy chain locus being the most common. As a result of these rearrangements, transcription of the unaltered MYC coding region is controlled by the regulatory sequences of the partner gene (promoter substitution), leading to deregulated MYC expression. Other mechanisms involved in MYC deregulated expression in neoplasms include amplification, insertional mutagenesis, and upregulation of certain signaling pathways that impinge in the hyperactivation of MYC promoter. Although MYC amplification has been described in many tumors and is often related to tumor progression, it must be noted that recent reports show that the coamplification of the adjacent gene PVT1, coding for a lncRNA, cooperates with MYC-driven tumorigenesis.30
Burkitt lymphoma
Animal models have shown us the relevance of MYC deregulation in B-cell malignancies. The lymphomas generated in the original Eμ-Myc transgenic mice (the first model for MYC-induced cancer) do not reproduce BL well,18,19 but additional transgenic mouse cell lines have been generated that better reproduce BL. These models for BL include models of mice carrying a single copy of the 240-kb IgH/c-Myc translocation region,31 mice carrying the murine Myc cDNA inserted in the IgH locus in a site that correspond to the human t(8;14) translocation break,32 mice with MYC linked to the 3′ IgH locus control region (3′ LCR),33 or mice with combined MYC overexpression and constitutive activation of the PI3K.34
Although BL is the most frequent subtype of lymphoma in children, in terms of total number of cases is more common in adults. IG–MYC rearrangements, while not being specific, constitute the hallmark of this lymphoma, being detected in more than 90% of cases, frequently as a single anomaly. More than 80% of cases will show a t(8;14)(q24;q32) (IGH-MYC), and the remaining cases will present either a t(8;22)(q24;q11) (IGL-MYC) or a t(2;8)(p12;q24) (IGK-MYC).35,36 Cases with no detectable translocation might represent failure to detect small rearrangements, but it is still a matter of debate whether true BL without MYC translocation exists. A non-rearranged-MYC-endemic BL type has been proposed to be the result of downregulation of a microRNA (hsa-mir-34b) that silences MYC expression.37 Recently, some atypical BL cases lacking MYC-rearrangement have been shown to present a peculiar pattern of chromosome 11q aberrations.38
Three subtypes of BL showing different clinical and biological features have been described: endemic, sporadic, and immunodeficiency-associated. The endemic is the most frequent type of lymphoma in children in Africa, and IGH–MYC in this subtype arise from either aberrant hypermutation process or might occur during the VDJ (variable, diverse, and joining gene segments) recombination process. In contrast, in sporadic and immunodeficiency-associated types, gene breakpoints generally affect the IGH switch regions.39
MYC deregulation is known to be necessary but not sufficient to induce the complete BL phenotype. While inducing proliferation, MYC also promotes apoptosis mainly through activation of both the p53 pathway and by inducing the expression of the proapoptotic gene BIM. BL cells have been shown to develop different mechanisms to counteract these MYC proapoptotic stimuli, such as the impairment of BIM function through direct and indirect mechanisms including p53 mutations, upregulation of MDM2, or p14ARF loss. Another mechanism to elude MYC-induced apoptosis is downregulation of the p27Kip mediated by LMP2A, an Epstein–Barr virus (EBV) protein.40 Sequencing analysis of BL patients have shown a low rate of mutations, but MYC is the most frequently mutated gene (up to 70% of cases).41 Approximately 60% of these mutations result in increased stability of the protein via reduced ubiquitin-mediated proteolysis.41–43
Additional transforming mechanisms have been described to contribute to the pathogenesis of the different subtypes of BL. EBV genome is detected in all endemic patients and has been shown to cooperate with MYC in the pathogenesis of this subtype.44 However, viral infection is only detected in 10% and 30% of sporadic and immunodeficiency-associated cases, respectively. In contrast, sequencing analysis have encountered mutations of the PI3K pathway (more frequently ID3/TCF3 mutations) in 70% of sporadic and immunodeficiency-associated BL, suggesting that this pathway plays key roles in those subtypes of BL. Interestingly, mice models, with constitutive activation of both MYC and PI3K, develop a lymphoma which morphologically and clinically resembles human BL.34
MYC in other aggressive mature B-cell lymphomas
Classification of aggressive lymphomas is becoming very complex, since many entities that in the past were thought to be single diseases are in reality composed of different types. We will now review MYC involvement in the major types of aggressive lymphomas.
Diffuse large B-cell lymphoma (DLBCL) is the most frequent subtype of mature B-cell lymphoma in western countries. MYC expression is detected in virtually all DLBCLs, but the number of positive cells vary from one case to another. Cells carrying rearrangements or amplifications of MYC frequently show a high fraction of MYC-expressing cells (~70%), whereas only 30% of the lymphomas with less positive cells (30%–40%) present MYC gene alterations. Overall, MYC gene rearrangements constitute the third most common aberrancy in this type of lymphoma (6%–15% of cases) and confer a bad prognosis.42,45 They are usually secondary events that appear in the context of complex karyotypes and are more frequently detected in DLBCL with a germinal center B-cell (GCB) phenotype.45,46
B-cell lymphoma unclassifiable (BCLU) with features intermediate between DLBCL and BL is a provisional entity described in the latest version of the WHO classification to designate those cases that share both clinical and biological characteristics between DLBCL and the BL, but that cannot be clearly assigned to any of those categories.1 Those lymphomas frequently resemble BL, have been proven to be very aggressive, and show poor response to conventional treatments. MYC rearrangements have been reported in 30%–70% of BCLU cases.47,48
Double hit/triple hit lymphomas refers to those aggressive lymphomas which have simultaneous MYC with BCL2 and/or BCL6 rearrangements. The phenotype of these lymphomas is heterogeneous, sometimes having a DLBCL appearance (2%–12%) while others have a BCLU phenotype (32%–78%),47 but they all show an aggressive course and appear to be resistant to conventional chemotherapy regimens. New therapeutic approaches using small molecule inhibitors that target MYC and BCL2 are currently under investigation.46 MYC rearrangement predicted an inferior outcome in aggressive lymphomas in most studies, but it is not yet entirely clear if this is due to the MYC rearrangement itself or because 50%–80% of MYC-translocated DLBCL cases harbor dual or even triple translocations also targeting BCL2 and/or BCL6.45–47 The prognostic implication of MYC in those patients is difficult to establish since the diagnostic, phenotypic, and cytogenetic criteria, together with therapeutic approaches are very heterogeneous in the different published series.47
An increasing interest on the “double-expressor” (DE) large B-cell lymphomas, defined by most groups to have approximately 40% MYC and 50%–70% BCL2 cells by immunohistochemistry has been recently described. These lymphomas have more frequently a non-GCB phenotype and whether the identification of these lymphomas helps to prognostically stratify aggressive lymphomas is not clear yet.47
Lymphomas with plasmablastic differentiation include a variety of lymphomas which show a plasmatic gene expression profiling (with upregulation of PRMD1/BLIMP1 and XBP1). All of these together constitute aggressive lymphomas with very poor response to conventional treatments. Among these cases, plasmablastic lymphoma shows MYC rearrangement in up to 50% of cases, frequently with the IGH and in context of complex karyotypes. MYC rearrangements have been shown to be involved in the pathogenesis of the disease maybe by repressing the antiproliferative effect of BLIMP1.42 Plasmablastic lymphomas have been proven to respond poorly to conventional CHOP (rituximab–cyclophosphamide, doxorubicin, vincristine, and prednisone). Whether these types of lymphomas might benefit by adding MYC inhibitors requires to be investigated.
MYC in low-grade mature B-cell neoplasms
Mantle cell lymphoma (MCL) is a lymphoproliferative disease characterized by a monoclonal proliferation of lymphocytes that usually bears an IGH–CCND1 translocation. Deregulation of CCND1 (cyclin D1) has been shown not to be sufficient to induce lymphomas, and cooperation with other oncogenes as MYC is linked to the pathogenesis of MCL. MCL blastoid variants frequently show p16INK4a deletion and overexpression of CDK4 and MYC. Consistently, an animal model expressing MYC and a mutant CDK4, which is resistant to p16 inhibition, develops a lymphoproliferative disease with overexpression of CCND1 that resemble MCL, including CCND1 overexpression.49
Transformation of follicular lymphoma (FL) to a higher grade DLBCL occurs in 10%–60% of the cases. One of the genetic abnormalities involved in this process is MYC deregulation.50
Regarding MYC expression, chronic lymphocytic leukemia (CLL) is an exception in human cancer. Although there are conflicting reports as to the mRNA expression,51,52 our laboratory showed that MYC protein expressions in peripheral blood of CLL are clearly below that of healthy lymphocytes from blood or tonsils, and the small fraction of cases with detectable MYC (less than 20%) do not show a difference in the course of the disease.52 Amplification and rearrangements of MYC are rare in CLL (less than 3%), but when they occur, they correlate with a poor prognosis and aggressive disease.53–55 Also, in CLL transformation to high-grade lymphoma, known as Richter syndrome, MYC upregulation is frequent, similar to other aggressive lymphomas.54,56 Thus, MYC gene abnormalities in low-grade lymphomas are usually secondary events that frequently appear in an event of progression or transformation, and thus are associated with an adverse prognosis.
Multiple myeloma
Rearrangements of the MYC oncogene are present in 15%–50% of primary human multiple myelomas (MMs), in many cases involved in complex rearrangements,57,58 and its activation seems to play a role in the progression of plasma cell neoplasms, particularly from monoclonal gammopathy of undetermined significance (MGUS) to plasma cell myeloma. Indeed, MYC rearrangements and overexpression are more frequent in MM than in MGUS57,59,60 and mark a more aggressive disease.57,61 Frequent upregulation of MYC is also observed in plasma cell leukemia, a monoclonal gammopathy which can evolve from MM.62 The involvement of MYC in MM is supported by the Vk*MYC transgenic mouse, that recapitulate the biological and clinical features of human MM. In these mice, MYC is under the control of the κ light chain gene.63 Moreover, MM is one of the neoplasms that respond to treatment with BRD4 inhibitors (see “MYC as a target in leukemia and lymphoma” section), leading to MYC downregulation.64
Neoplasms of mature T-cells
Anaplastic large cell lymphoma (ALCL) is a T-cell neoplasm that can be classified into two groups based on the presence or absence of ALK gene rearrangements. Both subtypes are known to express high levels of MYC, not due to translocations but due to stimulation of the STAT3 pathway. Moreover, pharmacologic inhibition of MYC induced ALCL cell apoptosis, and therefore, MYC inhibitors might be an effective treatment for ALCL.65
MYC and myeloid neoplasms
As compared to lymphoid neoplasms, MYC involvement in myeloid leukemia has been less studied. However, a myeloid tumor (myelocytomatosis) was the original tumor caused by MYC retroviruses in chicken, and the inhibition of myeloid cell differentiation was one of the first biological effects described for MYC.16 Moreover, MYC transgenic mice models reveal that MYC is an efficient oncogene inducing acute myeloid leukemia (AML),66,67 and mice with bone marrow repopulated with Myc overexpressing cells develop an AML-like disease.67 MYC deregulation has been found in most types of human myeloid neoplasms, and is reviewed in the following section and summarized in Table 2.
 | Table 2 MYC in myeloid neoplasms |
Acute myeloid leukemia
AML is a heterogeneous group of neoplasms affecting the myeloid lineage. MYC amplification and overexpression have been reported in AML. MYC rearrangements are rare in AML, and the mechanisms of MYC overexpression are not well known. Some leukemogenic transcription factors such as RUNX1-RUNX1T1 and PML-RARα induce MYC expression.68,69 MYC amplification in AML is infrequent, although double minute (dmin) chromosomes and homogeneous staining regions (hsr) including the region 8q24, where MYC maps, have been described in AML.70,71 MYC overexpression in AML induced resistance to chemotherapeutic drugs.72 Increased MYC levels were correlated with decreased microRNA-29 family expression in AML.73
Chronic myeloid leukemia
Chronic myeloid leukemia (CML) is a proliferative clonal disorder of hematopoietic stem cells that results in the expansion of mature myeloid cells that retain a capacity for differentiation. CML, in the absence of treatment, will progress from the initial chronic phase, to a blastic crisis phase, which is a secondary acute leukemia. BCR–ABL kinase has a central role in CML etiology.74,75 BCR–ABL upregulates MYC expression, which cooperates with BCR–ABL in transformation. Consistently, imatinib and other BCR–ABL inhibitors provoke downregulation of MYC.76–79 MYC mRNA levels are elevated in CML–blastic crisis80,81 and in chronic phase CML compared to healthy bone marrow samples.81–83 Our laboratory showed that MYC is upregulated during CML progression.81 High MYC expression correlates with poorer response to imatinib and progression to blastic crisis.81,84 MYC also induces genetic instability and blocks erythroid differentiation mediated by imatinib in CML-derived cells77,81 suggesting that MYC contributes to CML by acting at least at those two levels.
In the hematopoietic stem cells population, MYC controls the balance between hematopoietic stem cell self-renewal and differentiation.5 MYC also plays an important role in the establishment and maintenance of LICs. The interaction between the ubiquitin ligase Fbw7 and its substrate MYC controls the CML LIC homeostasis and has a role in CML initiation and progression.85,86
Myelodysplastic syndrome
The myelodysplastic syndromes (MDSs) are characterized by both an aberrant differentiation process with morphologic evidence of marrow dysplasia and an increased ineffective proliferation of the myeloid precursors in bone marrow, with enhanced risk of transformation to an AML. Gene expression profiles of CD34+ cells from MDS patients showed MYC as one of the most upregulated genes in these patients.87 In agreement, CD34+ cells from patients with trisomy 8 MDS showed upregulation of MYC mRNA.88 MYC amplification has also been found in MDS, but with low frequencies.
MYC as a target in leukemia and lymphoma
Given its pervasive involvement in leukemogenesis and lymphomagenesis, MYC would be an ideal oncoprotein target for therapy. The “oncogene addiction” is defined as the phenomenon by which some tumors exhibit a dependence on a single oncogenic protein or pathway for sustaining growth and proliferation.89 MYC addiction was demonstrated in animal models for lymphoma and myeloid leukemia, showing that inactivation of MYC results in sustained tumor regression.90 This fact and the overexpression found in many hematological neoplasms suggest that silencing or inactivation of MYC may be a sensible therapeutic strategy. Indeed, early studies established that genetically targeting MYC could control leukemogenesis. These studies showed that antisense-MYC oligonucleotides reduced the leukemia induced in vivo by cell lines derived from BL, CML, and AML.91,92 This was confirmed in different reports. In a recent report, MYC suppression by siRNA or pharmacologic approaches was shown to prevent leukemia initiation in mice by eliminating LICs of human T-cell ALL.25 Importantly, despite widespread expression of MYC in normal cells and its involvement in many biological processes, recent studies have demonstrated that long-term, whole-body inactivation of MYC in mouse models by expression of a dominant negative MYC form (Omomyc, see “MYC as a target in leukemia and lymphoma” section)93,94 or by treatment with a compound that repress MYC expression (JQ1)95 only provokes mild side effects.
Altogether, the data suggest that MYC inhibition could be a clinically feasible strategy for leukemia and lymphoma therapy. However, there also are some drawbacks when targeting MYC. First, no adverse effects of MYC inactivation have been detected in mouse models studied so far. Second, MYC being a transcriptional factor and not an enzyme, it lacks a pocket where small molecules can fit. Thus, like many other transcription factors, MYC has the reputation of being a “nondruggable” target. Despite that, several approaches have targeted MYC, either as a direct target or as an indirect target via synthetic lethal approaches. We will briefly review these approaches, which are also summarized in Table 3.
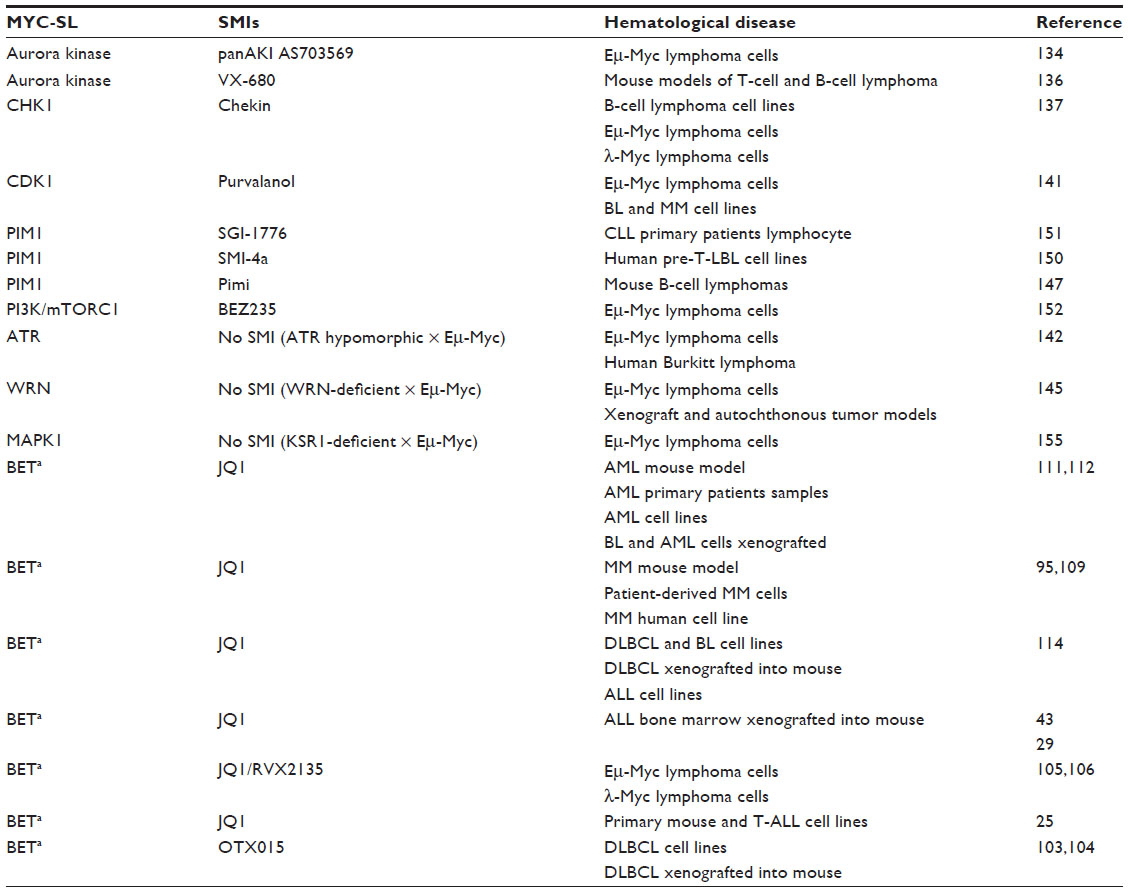 | Table 3 Myc synthetic lethal interactions |
Epigenetic-based MYC therapy: anti BRD4 drugs
Epigenetic mechanisms include histone postranslational modifications. Histone N-ter tails are rich in lysine residues which can be acetylated by histone lysine acetyltransferases. Acetylation neutralizes the positive charge of lysines and decreases the interaction between histones and DNA giving rise to a more open chromatin conformation, which is often associated to transcription factor accessibility and transcriptional activation. Acetyltransferases are forming part of large multiprotein complexes, and most of them have been implicated in cancer.96,97 Lysine acetylation is read by proteins containing specific interacting domains termed bromodomains (BRDs). BRD is a motif of 110 amino acids that binds the ε-aminoacetyl groups of nucleosomal histone lysines.98,99 The BRD and extraterminal (BET) proteins (BRD2, BRD3, BRD4, and BRDT) contain a double BRD in the N-terminal region and an extraterminal (ET) protein–protein interaction domain in the C-terminal region. BRD4 interacts and recruits P-TEFb to the core promoter of the active genes. P-TEFb is composed of cyclin T1 and CDK9, a kinase that phosphorylates the C-terminal domain (CTD) of the RNA polymerase II to allow transcription elongation11,100 (Figure 3). BRD4 aberrant expression or translocation has been found in different tumor types including AML.97 BRD2 and BRD4 have crucial roles in the control of cell cycle control in mammalian cells;101 thus, they have a promising potential as anticancer agents.
 | Figure 3 Scheme of the mechanism of action of BRD4 inhibitors as anti-MYC drugs. BRD4 is a reader of acetylated histones and promotes the activity of P-TEFb complex, composed by CDK9 and cyclin T1 (“Cyc.T” in the Figure). |
It has been demonstrated that MYC expression can be selectively regulated with BET inhibitors. The first one was JQ1, which shows strong affinity for the BRD4 family member thus inhibiting its activity.102 Other BET inhibitors such as OTX015 have shown their antiproliferative effects on lymphoma cells103 and have entered clinical trials.104 Lucas et al105 have described the inhibitor XD14, which shows a potent antiproliferative effect in leukemia cells. Another BET inhibitor RVX2135 inhibits proliferation of lymphoma cells from Eμ-Myc mice in vitro and in vivo.106 There are other BET inhibitors in preclinical studies, such as I-BET151, active against JAK2-dependent myeloproliferative neoplasms,107 but that those drugs truly target MYC has not been demonstrated.108
Recent studies in MM indicate that BET inhibitors are able to cause MYC downregulation in the context of translocated, amplified, or wild-type (WT) MYC alleles.109,110 Other reports show that the expression of MYC decreases in AML-derived cell lines with WT MYC, whereas cells with MYC amplification display relative resistance to the effect of BET inhibitors.111 Several studies have been performed on inhibiting BRD4, and hence MYC, in a range of hematological malignancies as AML,112 MLL-fusion leukemia;113 MM,95,109 ALL,43 B-cell lymphomas,106 BL,111 DLCBCL,114 and T-ALL.25 The most common biological effects of MYC downregulation upon BET inhibition is cell cycle arrest in G1 phase and apoptosis or senescence, but other effects such as terminal myeloid differentiation and elimination of leukemic stem cells have also been reported.95,109,114
One of the challenges is to understand how the inhibition of the activity of a general regulator such as BRD4 results in a selective effect on the expression of a small number of genes in specific cells.42,43,111 Several groups have demonstrated that in the case of MYC and other transcription factors, the specific effect is achieved because the BET inhibitor causes a depletion of BRD4 at the enhancers and superenhancers that drive the oncogene expression.95,109,114
Inhibition of MYC–MAX dimerization
In parallel to the repression of MYC expression with the BET inhibitors, other approaches specifically targeting MYC transactivation activities are under study (summarized in Figure 4). As noted earlier, MYC is only active when forming a dimer with MAX, suggesting that blocking the dimerization between MYC and MAX would be a good approach for inhibiting MYC function. Soucek et al115 constructed a MYC mutant, known as Omomyc, after identification of the molecular recognition site and induction of mutation of four amino acids at the LZ. Omomyc was able to sequester MYC and formed complexes with low binding efficiency to DNA, preventing the binding with MAX and inhibiting the function of MYC as a transcription factor (Figure 4B). Thus, Omomyc impairs MYC binding to E-boxes and changes MYC-dependent expression profile toward gene repression.116 Moreover, studies carried out in mouse models for some solid tumors (pancreas, skin, lung, and glioblastoma) reveal that MYC is required for full tumor development, even when tumor is triggered by other oncogenes.93,94,117,118 No data on Omomyc in lymphoma or leukemia model are available yet. As Omomyc is a peptide, its application in clinic might be difficult due to low biodisponibility and penetrance into the target cells. These problems will more likely be overcome with small molecules. However, the design of small molecules targeting the MYC–MAX interaction site is difficult due to the large interface between both proteins and because of the lack of structural “pockets” where small molecules could bind.
 | Figure 4 MYC inhibition strategies based on the interruption of the MYC–MAX dimerization. |
Despite these difficulties, attempts have been made to design small molecules which would inhibit MYC–MAX heterodimers (Figure 4C). In a screen of approximately 7,000 small organic molecules using FRET, two compounds were discovered to specifically inhibit MYC–MAX dimerization. These compounds Mycmycin-1 and Mycmycin-2 did not inhibit Jun dimerization and were a proof of concept to develop other molecules that specifically inhibit MYC-induced oncogenic transformation.119
A new series of compounds, 10058-F4 and 10074-G5, were discovered using a two-hybrid system.120 These compounds were able to inhibit both the growth of fibroblasts in vitro and growth of tumors in mice. The clinical applicability is still limited due to its low potency and its rapid degradation.121 The specificity of 10058-F4 and 10074-G5 was further corroborated by using a series of deletion and point mutations within the MYC bHLH-ZIP domain that resulted in the disruption of the heterodimer.122 Moreover, these compounds do not inhibit MAX homodimerization. Improvements of these drugs were achieved by adding chemical modifications and, as a result, they have enhanced growth inhibition of MYC-expressing cells in a manner that generally correlates with the compound ability to disrupt MYC–MAX association and DNA binding.123 Another study in nontransformed embryonal stem cells showed that these latter compounds results in loss of expression of MYC target genes but not of non-MYC target genes.124 The effect of 10054-F4 has been tested in AML cells, inhibiting leukemic proliferation, and inducing apoptosis through the mitochondrial pathway. Importantly, these effects were reproduced in primary AML cells.125 However, AML cells are partially resistant to 10054-F4 when they are in contact with bone marrow stroma.126 Another good model to test the effect of 10054-F4 is MM, which shows high deregulation of MYC as shown earlier. The MYC–MAX inhibitor 10054-F4 was effective on human MM cell lines and samples from patients and, although there was not a good correlation between sensitivity and MYC levels, cells expressing the highest levels of MYC tended to be more resistant to the treatment.64 All these results support the idea that targeting MYC dimerization is feasible. However, it may have the drawback that not all MYC functions depend on MAX.127–129
Inhibitors of the binding of MYC–MAX to DNA
Mo et al130 performed a cellular screening to identify substances that could be used to interfere with the MYC pathway (Figure 4D). Using cells with inducible MYC expression and WT expression, they found two compounds that selectively affected cell viability in MYC-overexpressing cells. They called the two compounds MYRA-A and MYRA-B (for MYC pathway response agents). MYRAs were more effective in human BL cells compared with other lymphoblastoid cell lines. Using three human cell lines with different levels of MYC (WT, null, or overexpressed), they found that MYRAs induced a high apoptotic state in cells overexpressing MYC, indicating that the effects of the compounds on cell viability is MYC-dependent.
MYRA-A interfered with the DNA binding of MYC–MAX (by electrophoretic mobility shift assays) but not with another E-box binding factor, USF, demonstrating that the inhibition was specific. Furthermore, they showed by coimmunoprecipitation that the MYC–MAX heterodimer remained intact after the treatment with MYRA-A. Recently, a new series of molecules interrupting the binding of MYC–MAX to DNA have been reported.131 These compounds inhibit MYC-dependent transactivation (by luciferase assays) in the μM range although MYC–MAX heterodimers remained intact.
MYC-mediated synthetic lethality
An alternative approach to target MYC is based on “synthetic lethality”. Synthetic lethal screens have been used to identify genes and pathways that are selectively activated by MYC in tumors, but not in nontumorigenic cells. Thus these molecules can be targeted with inhibitors to control MYC-driven malignancies. As expected from the multiplicity of the pathways in which MYC is involved, large series of putative synthetic lethal genes have been identified.132,133 We will review some of the MYC synthetic lethal interactions assayed in leukemia and lymphoma, which are also summarized in Table 3.
Aurora kinase inhibitors
MYC regulates aurora kinase A (AURKA) and B (AURKB) in the Eμ-Myc mouse model.134 Both kinases play a pivotal role in mitosis. Expression of MYC, but not other oncogenes, made the cells much more sensitive to Aurora kinase inhibitors (eg, AS703569), AURKB being the central target in this model. Another aurora kinase inhibitor, VX-680, was demonstrated to selectively kill the cells that overexpress MYC.135 Indeed, MYC expression levels may provide a biomarker to identify tumors that may be respond to aurora kinase B inhibitors. Moreover, the drug inhibited AURKB in vivo in mouse models that develop either B-cell or T-cell lymphomas in response to MYC overexpression.136 Furthermore, the lethal response is independent of p53-p21 pathway.136 This fact is relevant since TP53 is frequently mutated in different tumors and usually confers an adverse prognosis.
Chk1 inhibitors
One of the effects of MYC overexpression is to induce DNA replicative stress, which in turn activates CHK1 (checkpoint kinase 1). In cells from human and murine B-cell lymphomas, there is a correlation between MYC and CHK1 levels, although CHK1 seems to be an indirect target of MYC.137 Silencing of CHK1 with siRNA technology or inactivation with a small molecule (Chekin) results in selective death of MYC-overexpressing cells. These evidences turned CHK1 into an attractive therapeutic target. When tested in the λ-Myc mouse model, Chekin was able to induce a significantly slower disease progression followed by death in this lymphoma model.137
CDK1 inhibitors
The CDKs together with the cyclins form complexes that regulate cell cycle, both in neoplastic and normal cells. CDK1 is essential for mammalian cell division,138 and, as a matter of fact, is the only CDK required for cell cycling.139 Small molecule inhibitors have been developed against CDKs which induce cell cycle arrest in G2 phase.140 However, in MYC overexpressing cells, these drugs induce apoptosis,141 indicating that CDK1 inhibition is synthetically lethal on MYC expressing cells. Accordingly, a CDK1 inhibitor induces cell death in BL and MM cell lines depending on MYC levels, and CDK1 inhibition in Eμ-Myc mice results in extended survival.141
ATR inhibitors
Like CHK1, ATR kinase plays a pivotal role in replicative stress response. Myc-induced lymphomas in the Eμ-myc mice show a high level of replicative stress. The synthetic lethality between ATR and MYC has been demonstrated in a model of Eμ-myc mice crossed with mice with low ATR expression. In these mice, MYC-driven lymphomagenesis was suppressed.142 Preclinical data with highly specific ATR inhibitors have opened up the possibility of using them in synthetic lethality approaches.143
WRN inhibitors
WRN is a gene encoding a RecQ DNA helicase that is a direct transcriptional target of MYC. Even though WRN mutations have not been found in tumors, it has been reported that WRN is overexpressed in cancer cell lines from BL.144 Also, in BL cells, knock down of WRN impairs cell proliferation and increases apoptosis.144 In the same line of evidence, mutation of WRN in Eμ-Myc mouse models results in an increase in tumor-free survival and a delay in emergence of lethal lymphomas.145 These results demonstrate that using WRN as a target could result in an effective strategy not only to treat MYC-associated hematological diseases but also other MYC-associated cancers.
PIM kinases inhibitors
PIM kinases (1, 2, and 3) are involved in B-cell development and in hematologic malignancies.146,147 The PIM kinases, when coexpressed with MYC, provoke an acceleration of tumorigenesis.147 Given the fact that PIM1148 and MYC are overexpressed in lymphomas and that PIM1 is a coactivator of MYC,149 there has been an interest in developing PIM kinase inhibitors. A PIM kinase inhibitor (SMI-4a) kills several myeloid and lymphoid cell lines, with higher activity on T-cell lymphoblastic leukemia/lymphoma.150 Another inhibitor (SGI-1776) induces cytotoxicity in primary lymphocytes from CLL patients.151 Finally, a pan-Pim kinase inhibitor (Pimi) causes a reduction in mouse BL cell lines proliferation and a reduction in MYC-regulated transcripts.147
PI3K/TORC1 inhibitors
MYC-driven lymphomas demonstrate activation of mTORC1 and an endogenous DNA damage response. The small molecule BEZ235 inhibits both the PI3K-related DNA damage response kinases and mTORC1. This inhibitor shows a potent cytotoxic activity against Myc-driven B-cell lymphomas and BL-derived human cell lines bearing IG-cMYC translocations.152
MAPK inhibitors
The activation of the RAS-MEK-MAPK pathway results in MYC protein stabilization, which is mediated by the MAPK-dependent phosphorylation of a Ser residue in the MYC N-terminal region.153 Some reports suggest that MAPK inhibition may induce a synthetic lethal interaction with MYC. Indeed, the first example of oncogenic cooperation reported was that of MYC and RAS in the transformation of primary mouse fibroblasts.154 More recently, it has been shown that the impairment of RAS–MAPK pathway in mice deficient for KSR1 gene (encoding a scaffold protein of MAPK) results in a decrease in Myc-induced lymphomagenesis in a murine model.155
Conclusion
Deregulation of MYC oncogene is a pervasive finding in leukemia and lymphoma, in many cases inducing tumor progression and conferring poor prognosis. Cell culture studies and mouse transgenic models have shown that MYC plays a pivotal role in initiation and development of many types of hematological neoplasms. Thus, MYC would be a good therapeutic target in leukemia and lymphoma. As is the case for other transcription factors, the development of small molecules inhibiting MYC activity has been difficult. However, in recent years, different approaches targeting MYC have been described. These are based on the impairment of MYC expression (BET inhibitors), small molecules blocking MYC transactivation function, or synthetic lethal approaches. Altogether, the data suggest that MYC inhibition could be a clinically feasible strategy for leukemia and lymphoma therapy and that therapies targeting MYC are in sight.
Acknowledgments
The work in the authors’ laboratory is funded by grants SAF11-23796 from the Spanish Ministry of Industry and Innovation and ISCIII RETIC RD12/0036/0033 from the Spanish Ministry of Health. Funding was cosponsored by the European Union FEDER Program. We apologize to colleagues whose work has neither been cited in the form of their original papers (but in reviews) nor by unintentional omission.
Disclosure
MGC was supported by a Marcos Fernandez fellowship from Vistare and Leucemia-Linfoma Foundations. The authors report no other conflicts of interest in this work.
References
Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: World Health Organization; 2008. | |
Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. | |
Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med. 2014;4:1–24. | |
Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. | |
Delgado MD, Leon J. Myc roles in hematopoiesis and leukemia. Genes Cancer. 2010;1:605–616. | |
McMahon SB. Emerging concepts in the analysis of transcriptional targets of the MYC oncoprotein: are the targets targetable? Genes Cancer. 2010;1:560–567. | |
Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22:2755–2766. | |
Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–990. | |
Luscher B, Vervoorts J. Regulation of gene transcription by the oncoprotein MYC. Gene. 2012;494:145–160. | |
Wolf E, Lin CY, Eilers M, Levens DL. Taming of the beast: shaping Myc-dependent amplification. Trends Cell Biol. 2014;25:241–248. | |
Rahl PB, Young RA. MYC and transcription elongation. Cold Spring Harb Perspect Med. 2014;4:a020990. | |
Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–264. | |
Sabo A, Kress TR, Pelizzola M, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature. 2014;511:488–492. | |
Walz S, Lorenzin F, Morton J, et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature. 2014;511:483–487. | |
Bretones G, Delgado MD, Leon J. Myc and cell cycle control. Biochim Biophys Acta. 2015;1849:506–516. | |
Leon J, Ferrandiz N, Acosta JC, Delgado MD. Inhibition of cell differentiation: a critical mechanism for MYC-mediated carcinogenesis? Cell Cycle. 2009;8:1148–1157. | |
Sheiness D, Bishop JM. DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J Virol. 1979;31:514–521. | |
Adams JM, Harris AW, Pinkert CA, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. | |
Schmidt EV, Pattengale PK, Weir L, Leder P. Transgenic mice bearing the human c-myc gene activated by an immunoglobulin enhancer: a pre-B-cell lymphoma model. Proc Natl Acad Sci U S A. 1988;85:6047–6051. | |
Kuppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B-cell lymphomas. N Engl J Med. 1999;341:1520–1529. | |
Robbiani DF, Bothmer A, Callen E, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–1038. | |
La Starza R, Borga C, Barba G, et al. Genetic profile of T-cell acute lymphoblastic leukemias with MYC translocations. Blood. 2014;124:3577–3582. | |
Herranz D, Ambesi-Impiombato A, Palomero T, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20:1130–1137. | |
Mu Q, Ma Q, Lu S, et al. 10058-F4, a c-Myc inhibitor, markedly increases valproic acid-induced cell death in Jurkat and CCRF-CEM T-lymphoblastic leukemia cells. Oncol Lett. 2014;8:1355–1359. | |
Roderick JE, Tesell J, Shultz LD, et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood. 2014;123:1040–1050. | |
Loosveld M, Castellano R, Gon S, et al. Therapeutic targeting of c-Myc in T-cell acute lymphoblastic leukemia, T-ALL. Oncotarget. 2014;5:3168–3172. | |
Schubbert S, Cardenas A, Chen H, et al. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res. 2014;74:7048–7059. | |
Pui CH, Mullighan CG, Evans WE, Relling MV. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood. 2012;120:1165–1174. | |
Da Costa D, Agathanggelou A, Perry T, et al. BET inhibition as a single or combined therapeutic approach in primary paediatric B-precursor acute lymphoblastic leukaemia. Blood Cancer J. 2013;3:e126. | |
Tseng YY, Moriarity BS, Gong W, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–86. | |
Palomo C, Zou X, Nicholson IC, Butzler C, Bruggemann M. B-cell tumorigenesis in mice carrying a yeast artificial chromosome-based immunoglobulin heavy/c-myc translocus is independent of the heavy chain intron enhancer (Emu). Cancer Res. 1999;59:5625–5628. | |
Park SS, Kim JS, Tessarollo L, et al. Insertion of c-Myc into Igh induces B-cell and plasma-cell neoplasms in mice. Cancer Res. 2005;65:1306–1315. | |
Truffinet V, Pinaud E, Cogne N, et al. The 3′ IgH locus control region is sufficient to deregulate a c-myc transgene and promote mature B cell malignancies with a predominant Burkitt-like phenotype. J Immunol. 2007;179:6033–6042. | |
Sander S, Calado DP, Srinivasan L, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22:167–179. | |
Sanchez-Beato M, Sanchez-Aguilera A, Piris MA. Cell cycle deregulation in B-cell lymphomas. Blood. 2003;101:1220–1235. | |
Linch DC. Burkitt lymphoma in adults. Br J Haematol. 2012;156:693–703. | |
Leucci E, Cocco M, Onnis A, et al. MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol. 2008;216:440–450. | |
Salaverria I, Martin-Guerrero I, Wagener R, et al. A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood. 2014;123:1187–1198. | |
Neri A, Barriga F, Knowles DM, Magrath IT, Dalla-Favera R. Different regions of the immunoglobulin heavy-chain locus are involved in chromosomal translocations in distinct pathogenetic forms of Burkitt lymphoma. Proc Natl Acad Sci U S A. 1988;85:2748–2752. | |
Dittmer DP. Not like a wrecking ball: EBV fine-tunes MYC lymphomagenesis. Blood. 2041;123:460–461. | |
Vaque JP, Martinez N, Battle-Lopez A, et al. B-cell lymphoma mutations: improving diagnostics and enabling targeted therapies. Haematologica. 2014;99:222–231. | |
Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Blood. 2013;122:3884–3891. | |
Ott G. Impact of MYC on malignant behavior. Hematology Am Soc Hematol Educ Program. 2014;2014:100–106. | |
Shiramizu B, Barriga F, Neequaye J, et al. Patterns of chromosomal breakpoint locations in Burkitt’s lymphoma: relevance to geography and Epstein-Barr virus association. Blood. 1991;77:1516–1526. | |
Karube K, Campo E. MYC alterations in diffuse large B-cell lymphomas. Semin Hematol. 2015;52:97–106. | |
Dunleavy K. Double-hit lymphomas: current paradigms and novel treatment approaches. Hematology Am Soc Hematol Educ Program. 2014;2014:107–112. | |
Swerdlow SH. Diagnosis of ‘double hit’ diffuse large B-cell lymphoma and B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma: when and how, FISH versus IHC. Hematology Am Soc Hematol Educ Program. 2014;2014:90–99. | |
Kanagal-Shamanna R, Medeiros LJ, Lu G, et al. High-grade B cell lymphoma, unclassifiable, with blastoid features: an unusual morphological subgroup associated frequently with BCL2 and/or MYC gene rearrangements and a poor prognosis. Histopathology. 2012;61:945–954. | |
Vincent-Fabert C, Fiancette R, Rouand P, et al. A defect of the INK4-Cdk4 checkpoint and Myc collaborate in blastoid mantle cell lymphoma-like lymphoma formation in mice. Am J Pathol. 2012;180:1688–1701. | |
Lossos IS, Levy R. Higher grade transformation of follicular lymphoma: phenotypic tumor progression associated with diverse genetic lesions. Semin Cancer Biol. 2003;13:191–202. | |
Zhang W, Kater AP, Widhopf GF, et al. B-cell activating factor and v-Myc myelocytomatosis viral oncogene homolog (c-Myc) influence progression of chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2010;107:18956–18960. | |
Caraballo JM, Acosta JC, Cortes MA, et al. High p27 protein levels in chronic lymphocytic leukemia are associated to low Myc and Skp2 expression, confer resistance to apoptosis and antagonize Myc effects on cell cycle. Oncotarget. 2014;5:4694–4708. | |
Put N, Van Roosbroeck K, Konings P, et al. Chronic lymphocytic leukemia and prolymphocytic leukemia with MYC translocations: a subgroup with an aggressive disease course. Ann Hematol. 2012;91:863–873. | |
Huh YO, Lin KI, Vega F, et al. MYC translocation in chronic lymphocytic leukaemia is associated with increased prolymphocytes and a poor prognosis. Br J Haematol. 2008;142:36–44. | |
Rinaldi A, Mian M, Kwee I, et al. Genome-wide DNA profiling better defines the prognosis of chronic lymphocytic leukaemia. Br J Haematol. 2011;154:590–599. | |
Scandurra M, Rossi D, Deambrogi G, et al. Genomic profiling of Richter’s syndrome: recurrent lesions and differences with de novo diffuse large B-cell lymphomas. Hematol Oncol. 2010;28:62–67. | |
Sawyer JR. The prognostic significance of cytogenetics and molecular profiling in multiple myeloma. Cancer Genet. 2011;204:3–12. | |
Affer M, Chesi M, Chen WD, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia. 2014;28:1725–1735. | |
Xiao R, Cerny J, Devitt K, et al. MYC protein expression is detected in plasma cell myeloma but not in monoclonal gammopathy of undetermined significance (MGUS). Am J Surg Pathol. 2014;38:776–783. | |
Chng WJ, Huang GF, Chung TH, et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia. 2011;25:1026–1035. | |
Glitza IC, Lu G, Shah R, et al. Chromosome 8q24.1/c-MYC abnormality: a marker for high-risk myeloma. Leuk Lymphoma. 2015;56:602–607. | |
Chiecchio L, Dagrada GP, White HE, et al. Frequent upregulation of MYC in plasma cell leukemia. Genes Chromosomes Cancer. 2009;48:624–636. | |
Chesi M, Robbiani DF, Sebaq M, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13:167–180. | |
Holien T, Vatsveen TK, Hella H, Waage A, Sundan A. Addiction to c-MYC in multiple myeloma. Blood. 2012;120:2450–2453. | |
Weilemann A, Grau M, Erdmann T, et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood. 2015;125:124–132. | |
Xiang Z, Luo H, Payton JE, et al. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J Clin Invest. 2010;120:2109–2118. | |
Beverly LJ, Varmus HE. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene. 2009;28:1274–1279. | |
Rice KL, Hormaeche I, Doulatov S, et al. Comprehensive genomic screens identify a role for PLZF-RARalpha as a positive regulator of cell proliferation via direct regulation of c-MYC. Blood. 2009;114:5499–5511. | |
Muller-Tidow C, Steffen B, Cauvet T, et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol. 2004;24:2890–2904. | |
Sait SN, Qadir MU, Conroy JM, Matsui S, Nowak NJ, Baer MR. Double minute chromosomes in acute myeloid leukemia and myelodysplastic syndrome: identification of new amplification regions by fluorescence in situ hybridization and spectral karyotyping. Genes Chromosomes Cancer. 2002;34:42–47. | |
Bajaj R, Xu F, Xiang B, et al. Evidence-based genomic diagnosis characterized chromosomal and cryptic imbalances in 30 elderly patients with myelodysplastic syndrome and acute myeloid leukemia. Mol Cytogenet. 2011;4:3. | |
Pan XN, Chen JJ, Wang LX, et al. Inhibition of c-Myc overcomes cytotoxic drug resistance in acute myeloid leukemia cells by promoting differentiation. PLoS One. 2014;9:e105381. | |
Gong JN, Yu J, Lin HS, et al. The role, mechanism and potentially therapeutic application of microRNA-29 family in acute myeloid leukemia. Cell Death Differ. 2014;21:100–112. | |
Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer. 2008;8:341–350. | |
Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120:2254–2264. | |
Xie S, Lin H, Sun T, Arlinghaus RB. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene. 2002;21:7137–7146. | |
Gomez-Casares MT, Gracía-Alegria E, López-Jorge CE, et al. MYC antagonizes the differentiation induced by imatinib in chronic myeloid leukemia cells through downregulation of p27(KIP1). Oncogene. 2013;32:2239–2246. | |
Sawyers CL. The role of myc in transformation by BCR-ABL. Leuk Lymphoma. 1993;11:45–46. | |
Afar DE, Goga A, McLaughlin J, Witte ON, Sawyers CL. Differential complementation of Bcr-Abl point mutants with c-Myc. Science. 1994;264:424–426. | |
Beck Z, Bácsi A, Kovács E, et al. Changes in oncogene expression implicated in evolution of chronic granulocytic leukemia from its chronic phase to acceleration. Leuk Lymphoma. 1998;30:293–306. | |
Albajar M, Gómez-Casares MT, Llorca J, et al. MYC in chronic myeloid leukemia: induction of aberrant DNA synthesis and association with poor response to imatinib. Mol Cancer Res. 2011;9:564–576. | |
Diaz-Blanco E, Bruns I, Neumann F, et al. Molecular signature of CD34(+) hematopoietic stem and progenitor cells of patients with CML in chronic phase. Leukemia. 2007;21:494–504. | |
Nowicki MO, Pawlowski P, Fischer T, Hess G, Pawlowski T, Skorski T. Chronic myelogenous leukemia molecular signature. Oncogene. 2003;22:3952–3963. | |
Lucas CM, Harris RJ, Giannoudis A, Copland M, Slupsky JR, Clark RE. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood. 2011;117:6660–6668. | |
Reavie L, Buckley SM, Loizou E, et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell. 2013;23:362–375. | |
Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell. 2013;23:347–361. | |
Vasikova A, Belickova M, Budinska E, Cermak J. A distinct expression of various gene subsets in CD34+ cells from patients with early and advanced myelodysplastic syndrome. Leuk Res. 2010;34:1566–1572. | |
Sloand EM, Pfannes L, Chen G, et al. CD34 cells from patients with trisomy 8 myelodysplastic syndrome (MDS) express early apoptotic markers but avoid programmed cell death by up-regulation of antiapoptotic proteins. Blood. 2007;109:2399–2405. | |
Weinstein IB. Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science. 2002;297:63–64. | |
Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. | |
Skorski T, Nieborowska-Skorska M, Campbell K, et al. Leukemia treatment in severe combined immunodeficiency mice by antisense oligodeoxynucleotides targeting cooperating oncogenes. J Exp Med. 1995;182:1645–1653. | |
Skorski T, Nieborowska-Skorska M, Wlodarski P, Zon G, Iozzo RV, Calabretta B. Antisense oligodeoxynucleotide combination therapy of primary chronic myelogenous leukemia blast crisis in SCID mice. Blood. 1996;88:1005–1112. | |
Soucek L, Whitfield JR, Sodir NM, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504–513. | |
Annibali D, Whitfield JR, Favuzzi E, et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat Commun. 2014;5:4632. | |
Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. | |
Dawson MA, Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. | |
You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. | |
Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12:465–477. | |
Filippakopoulos P, Picaud S, Mangos M, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. | |
Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–488. | |
Prinjha RK, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol Sci. 2012;33:146–153. | |
Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. | |
Boi M, Gaudio E, Bonetti P, et al. The BET Bromodomain inhibitor OTX015 affects pathogenetic pathways in pre-clinical B-cell tumor models and synergizes with targeted drugs. Clin Cancer Res. 2015;21(7):1628–1638. | |
Herait P, Dombret H, Thieblemont C, et al. O7.3BET-bromodomain (BRD) inhibitor OTX015: final results of the dose-finding part of a phase I study in hematologic malignancies. Ann Oncol. 2015; (26 Suppl 2):ii10. | |
Lucas X, Wohlwend D, Hügle M, et al. 4-Acyl pyrroles: mimicking acetylated lysines in histone code reading. Angew Chem Int Ed Engl. 2013;52:14055–14059. | |
Bhadury J, Nilsson LM, Muralidharan SV, et al. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc Natl Acad Sci U S A. 2014;111:E2721–E2730. | |
Wyspianska BS, Bannister AJ, Barbieri I, et al. BET protein inhibition shows efficacy against JAK2V617F-driven neoplasms. Leukemia. 2014;28:88–97. | |
Lucas X, Gunther S. Targeting the BET family for the treatment of leukemia. Epigenomics. 2014;6:153–155. | |
Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. | |
Ott CJ, Kopp N, Bird L, et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120:2843–2852. | |
Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–16674. | |
Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. | |
Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. | |
Chapuy B, McKeown MR, Lin CY, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–790. | |
Soucek L, Helmer-Citterich M, Sacco A, Jucker R, Cesareni G, Nasi S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene. 1998;17:2463–2472. | |
Savino M, Annibali D, Carucci N, et al. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS One. 2011;6:e22284. | |
Sodir NM, Swiqart LB, Karnezis AN, et al. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011;25:907–916. | |
Soucek L, Nasi S, Evan GI. Omomyc expression in skin prevents Myc-induced papillomatosis. Cell Death Differ. 2004;11:1038–1045. | |
Shi J, Stover JS, Whitby LR, Vogt PK, Boger DL. Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg Med Chem Lett. 2009;19:6038–6041. | |
Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22:6151–6159. | |
Guo J, Parise RA, Joseph E, et al. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother Pharmacol. 2009;63:615–625. | |
Follis AV, Hammoudeh DI, Wang H, Prochownik EV, Metallo SJ. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem Biol. 2008;15:1149–1155. | |
Wang H, Hammoudeh DI, Follis AV, et al. Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther. 2007;6:2399–2408. | |
Rahl PB, Lin CY, Seila AC, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. | |
Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp Hematol. 2006;34:1480–1489. | |
Xia B, Tian C, Guo S, et al. c-Myc plays part in drug resistance mediated by bone marrow stromal cells in acute myeloid leukemia. Leuk Res. 2015;39:92–99. | |
Gallant P, Steiger D. Myc’s secret life without Max. Cell Cycle. 2009;8:3848–3853. | |
Wert M, Kennedy S, Palfrey HC, Hay N. Myc drives apoptosis in PC12 cells in the absence of Max. Oncogene. 2001;20:3746–3750. | |
Vaque JP, Fernández-Gracía B, Gracía-Sanz P, et al. c-Myc inhibits Ras-mediated differentiation of pheochromocytoma cells by blocking c-Jun up-regulation. Mol Cancer Res. 2008;6:325–339. | |
Mo H, Vita M, Crespin M, Henriksson M. Myc overexpression enhances apoptosis induced by small molecules. Cell Cycle. 2006;5:2191–2194. | |
Jung KY, Wang H, Teriete P, et al. Perturbation of the c-Myc-Max protein-protein interaction via synthetic α-helix mimetics. J Med Chem. 2015;58(7):3002–3024. | |
Toyoshima M, Howie HL, Imakura M, et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A. 2012;109:9545–9550. | |
Cermelli S, Jang IS, Bernard B, Grandori C. Synthetic lethal screens as a means to understand and treat MYC-driven cancers. Cold Spring Harb Perspect Med. 2014;4(3):a14209. | |
den Hollander J, Rimpi S, Doherty JR, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116:1498–1505. | |
Harrington EA, Bebbington D, Moore J, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–267. | |
Yang D, Liu H, Goga A, et al. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci U S A. 2010;107:13836–13841. | |
Hoglund A, Nilsson LM, Muralidharan SV, et al. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin Cancer Res. 2011;17:7067–7079. | |
Itzhaki JE, Gilbert CS, Porter AC. Construction by gene targeting in human cells of a “conditional’ CDC2 mutant that rereplicates its DNA. Nat Genet. 1997;15:258–265. | |
Santamaria D, Barrière C, Cerqueira A, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. | |
Gray N, Detivaud L, Doerig C, Meijer L. ATP-site directed inhibitors of cyclin-dependent kinases. Curr Med Chem. 1999;6:859–875. | |
Goga A, Yang D, Tward AD, Morgan DO, Bishop JM. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat Med. 2007;13:820–827. | |
Murga M, Campaner S, Lopez-Contreras AJ, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011;18:1331–1335. | |
Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. 2015;149:124–138. | |
Grandori C, Robinson KL, Galloway DA, Swisshelm K. Functional link between Myc and the Werner gene in tumorigenesis. Cell Cycle. 2004;3:22–25. | |
Moser R, Toyoshima M, Robinson K, et al. MYC-driven tumorigenesis is inhibited by WRN syndrome gene deficiency. Mol Cancer Res. 2012;10:535–545. | |
Cibull TL, Jones TD, Eble JN, et al. Overexpression of Pim-1 during progression of prostatic adenocarcinoma. J Clin Pathol. 2006;59:285–288. | |
Forshell LP, Li Y, Forshell TZ, et al. The direct Myc target Pim3 cooperates with other Pim kinases in supporting viability of Myc-induced B-cell lymphomas. Oncotarget. 2011;2:448–460. | |
Wang Z, Bhattacharya N, Weaver M, et al. Pim-1: a serine/threonine kinase with a role in cell survival, proliferation, differentiation and tumorigenesis. J Vet Sci. 2001;2:167–179. | |
Zippo A, De Robertis A, Serafini R, Oliviero S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat Cell Biol. 2007;9:932–944. | |
Lin YW, Beharry ZM, Hill EG, et al. A small molecule inhibitor of Pim protein kinases blocks the growth of precursor T-cell lymphoblastic leukemia/lymphoma. Blood. 2010;115:824–833. | |
Cheng F, Weidner-Glunde M, Varjosalo M, et al. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 2009;5:e1000324. | |
Shortt J, Martin BP, Newbold A, et al. Combined inhibition of PI3K-related DNA damage response kinases and mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood. 2013;121:2964–2974. | |
Thomas LR, Tansey WP. Proteolytic control of the oncoprotein transcription factor Myc. Adv Cancer Res. 2011;110:77–106. | |
Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. | |
Gramling MW, Eischen CM. Suppression of Ras/Mapk pathway signaling inhibits Myc-induced lymphomagenesis. Cell Death Differ. 2012;19:1220–1227. | |
Palomero T, Lim WK, Odom DT, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–18266. | |
Weng AP, Millholland JM, Yashiro-Ohtani Y, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–2109. | |
Montes-Moreno S, Montalban C, Piris MA. Large B-cell lymphomas with plasmablastic differentiation: a biological and therapeutic challenge. Leuk Lymphoma. 2012;53:185–194. | |
Lossos IS, Alizadeh AA, Diehn M, et al. Transformation of follicular lymphoma to diffuse large-cell lymphoma: alternative patterns with increased or decreased expression of c-myc and its regulated genes. Proc Natl Acad Sci U S A. 2002;99:8886–8891. | |
Avet-Loiseau H, Gerson F, Magrangeas F, et al. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood. 2001;98:3082–3086. | |
Shou Y, Martelli ML, Gabrea A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A. 2000;97:228–233. | |
Thomas L, Stamberg J, Gojo I, Ning Y, Rapoport AP. Double minute chromosomes in monoblastic (M5) and myeloblastic (M2) acute myeloid leukemia: two case reports and a review of literature. Am J Hematol. 2004;77:55–61. | |
Mathew S, Lorsbach RB, Shearer P, Sandlund JT, Raimondi SC. Double minute chromosomes and c-MYC amplification in a child with secondary myelodysplastic syndrome after treatment for acute lymphoblastic leukemia. Leukemia. 2000;14:1314–1315. | |
Storlazzi CT, Fioretos T, Surace C, et al. MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet. 2006;15:933–942. | |
Court EL, Smith MA, Avent ND, et al. DNA microarray screening of differential gene expression in bone marrow samples from AML, non-AML patients and AML cell lines. Leuk Res. 2004;28:743–753. | |
Larramendy ML, Niini T, Elonen E, et al. Overexpression of translocation-associated fusion genes of FGFRI, MYC, NPMI, and DEK, but absence of the translocations in acute myeloid leukemia. A microarray analysis. Haematologica. 2002;87:569–577. | |
Kawagoe H, Kandilci A, Kranenburg TA, Grosveld GC. Overexpression of N-Myc rapidly causes acute myeloid leukemia in mice. Cancer Res. 2007;67:10677–10685. | |
Ross ME, Mahfouz R, Onciu M, et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104:3679–3687. | |
Qian Z, Fernald AA, Godley LA, Larson RA, Le Beau MM. Expression profiling of CD34+ hematopoietic stem/progenitor cells reveals distinct subtypes of therapy-related acute myeloid leukemia. Proc Natl Acad Sci U S A. 2002;99:14925–14930. | |
Theophile K, Buesche G, Kreipe H, Bock O. The expression levels of telomerase catalytic subunit hTERT and oncogenic MYC in essential thrombocythemia are affected by the molecular subtype. Ann Hematol. 2008;87:263–268. | |
Thoennissen NH, Krug UO, Lee DH, et al. Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of Philadelphia chromosome-negative myeloproliferative neoplasms. Blood. 2010;115:2882–2890. | |
Poloni A, Serrani F, Berardinelli E, et al. Telomere length, c-myc and mad-1 expression could represent prognosis markers of myelodysplastic syndrome. Leuk Res. 2013;37:1538–1544. | |
Falantes JF, Trujillo P, Piruat JI, et al. Overexpression of GYS1, MIF, and MYC is associated with adverse outcome and poor response to azacitidine in myelodysplastic syndromes and acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2015;15(4):236–244. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.